

Abstract
Plant‐derived carbohydrates are an abundant renewable resource. Transformation of carbohydrates into new products, including amine‐functionalized building blocks for biomaterials applications, can lower reliance on fossil resources. Herein, biocatalytic production routes to amino carbohydrates, including oligosaccharides, are demonstrated. In each case, two‐step biocatalysis was performed to functionalize d‐galactose‐containing carbohydrates by employing the galactose oxidase from Fusarium graminearum or a pyranose dehydrogenase from Agaricus bisporus followed by the ω‐transaminase from Chromobacterium violaceum (Cvi‐ω‐TA). Formation of 6‐amino‐6‐deoxy‐d‐galactose, 2‐amino‐2‐deoxy‐d‐galactose, and 2‐amino‐2‐deoxy‐6‐aldo‐d‐galactose was confirmed by mass spectrometry. The activity of Cvi‐ω‐TA was highest towards 6‐aldo‐d‐galactose, for which the highest yield of 6‐amino‐6‐deoxy‐d‐galactose (67 %) was achieved in reactions permitting simultaneous oxidation of d‐galactose and transamination of the resulting 6‐aldo‐d‐galactose.
Keywords: amination, biocatalysis, carbohydrates, domino reactions, enzymes
Introduction
Given their wide availability and structural versatility, carbohydrates from plant cell walls are an important raw material for the production of new biobased products that reduce reliance on petroleum. To date, most applications of plant carbohydrates begin by deconstructing polysaccharides to monomers for fermentation to fuels and platform chemicals.1, 2, 3, 4, 5 Bifunctional molecules containing a terminal acid and an amine functionality are among the desired products, because they are key building blocks in the synthesis of different types of polymers.6, 7 For example, diacids, diamines, and AB monomers (e.g., molecules containing both carboxylic acid and amino groups) are required for polyamide synthesis,8 and diacids and diols for polyester synthesis.9, 10, 11, 12, 13
An emerging area of research aims to utilize the versatility and ensuing useful properties of native structures present in plant carbohydrates instead of degrading the structures to monomers.14, 15, 16, 17 Bifunctional carbohydrates (e.g., diacids, diamines, or AB monomers) from native carbohydrate structures are among the desired products. Besides enzymatic synthesis of amino sugars from activated sugar nucleotides and sugar phosphates,18, 19, 20 biocatalytic cascades to amino carbohydrates directly from biomass‐derived carbohydrates are still missing. If established, these pathways would create a new class of telechelic, amino‐functionalized building blocks that retain inherent attributes of native carbohydrate structures (e.g., biocompatibility, hydrophilicity), while being primed for assembly (e.g., through stable amide linkages) with other building blocks or polymers with complementary functionalities (e.g., carboxyl groups).15, 21, 22
Existing chemical pathways for carbohydrate amination include applications of 2,2,6,6‐tetramethylpiperidine‐1‐oxyl (TEMPO) to oxidize primary hydroxyl groups23 and sodium periodate to oxidize secondary hydroxyl groups,24 followed by reductive amination to assemble the corresponding amines.25 These routes, however, often require toxic transition metal catalysts, produce volatile organic compounds, and result in decreased polymer chain length.26 As a gentle alternative to chemical oxidation procedures, the galactose oxidase from Fusarium graminearum (FgrGaOx, UniProt: A0A2H3HJK8) has been used to introduce an aldehyde functionality at the C‐6 position of d‐galactose and in d‐galactose‐containing oligo‐ and polysaccharides.15, 27, 28 Specificity of FgrGaOx towards the C‐6 position of d‐galactose has been well documented,29 and the resulting oxidized positions have served as sites for further derivatization, including reductive amination, cross‐linking through acetal formation, and phosphorylation.15, 30, 31 Furthermore, we recently described the application of the oligosaccharide oxidase from Sarocladium strictum to permit amide bond formation with clickable monomers, leading to telechelic molecules from xylo‐oligosaccharides that are primed for reassembly through copper‐catalyzed azide–alkyne cycloaddition.22 Thus, chemo‐enzymatic routes to aminated carbohydrates have been demonstrated; however, fully biocatalytic cascades for amination of nonactivated carbohydrates are unprecedented yet highly desirable to simplify reaction pathways, increase sustainability, and to achieve greater control over reaction products.
Transaminases are pyridoxal 5′‐phosphate (PLP)‐dependent enzymes that catalyze the transfer of an amino group from primary amines acting as the amine donor to carbonyl compounds acting as amine acceptor.32 Briefly, transaminases operate through a ping‐pong, bi‐bi reaction in which the first half‐reaction involves the transfer of the amino group from the amine donor to the PLP cofactor. The deaminated amine donor (i.e., the respective ketone/aldehyde product) is then released, leaving the cofactor as pyridoxamine 5′‐phosphate (PMP). In the second half‐reaction, the amino group is transferred from enzyme‐bound PMP to the acceptor, regenerating the PLP cofactor and completing the transamination cycle. Transaminases have been functionally classified as α‐transaminases (α‐TAs) and ω‐transaminases (ω‐TAs), on the basis of amine donor and acceptor specificity. Whereas α‐TAs transfer amino groups from the α‐carbon atom of amino acids to α‐keto acids, ω‐TAs are more versatile as they do not require a carboxylate group in the amine acceptor and can donate the amino group to α‐keto acids as well as other ketones and aldehydes.32, 33, 34 In addition to substrate versatility, no requirement for cofactor regeneration or nucleotide sugars as substrates is a distinguishing advantage of ω‐TAs relative to other types of enzymes potentially capable of producing carbohydrate amines (e.g., reductive aminases).35, 36, 37 So‐called sugar transaminases have been shown to accept nucleotide sugars as amine donors and acceptors;19, 20 however, these are not suitable for direct amination of biomass‐derived carbohydrates.
Amines in general are greatly underrepresented in renewable biomass compared with the frequent need for amines in chemicals and polymer synthesis.38, 39 This makes biocatalytic production of amines from alcohols highly desirable, yet challenging, because no known single enzyme can catalyze such a transformation, and the chemical or chemo‐enzymatic methods involving oxidation and reductive amination often involve toxic chemicals and require complicated synthetic procedures.40, 41, 42 To date, ω‐TAs have been studied extensively for asymmetric synthesis of pharmaceuticals, which has been summarized in several reviews.41, 43, 44, 45 In this context, enzymatic pathways from primary and secondary alcohols to the corresponding amines, by utilizing oxidases or alcohol dehydrogenases coupled with an ω‐TA, have been demonstrated.46, 47, 48 By contrast, application of ω‐TAs for bioproduct development from renewable biomass has only been investigated in a few studies.49, 50 For example, Lerchner et al. showed the two‐step conversion of isosorbide to the corresponding diamine using an alcohol dehydrogenase and an ω‐TA.49, 51More recently, Dunbabin et al. demonstrated transaminase‐catalyzed production of furfurylamines from furfurals.52 On the other hand, the ability of FgrGaOx to oxidize C‐6 hydroxyl groups on galactose‐containing mono‐, oligo‐, and polysaccharide substrates has been shown to be an efficient way to produce aldehyde‐functionalized carbohydrates,27, 28 which might be accepted by ω‐TAs. Moreover, carbohydrate oxidoreductases with different regio‐ and substrate specificities beyond the oxidation of the primary C‐6 hydroxyl group (e.g., pyranose dehydrogenases) can help extend the range of available carbonyl‐containing carbohydrates to ketone‐functionalized carbohydrates, which are also potential substrates for ω‐TAs.53, 54, 55
The ω‐TA from Chromobacterium violaceum (Cvi‐ω‐TA, UniProt: Q7NWG4) is recognized as having a broad substrate range and has activity towards hydroxylated aldehydes such as d‐erythrose (1), glycolaldehyde, and glyceraldehyde.47, 48, 56 In the present study, Cvi‐ω‐TA was investigated for its potential to aminate aldol‐ and keto‐carbohydrates initially formed through oxidation by FgrGaOx or the pyranose dehydrogenase from Agaricus bisporus (AbiPDH1, UniProt: Q3L1D1),53 respectively. Cvi‐ω‐TA activity on oxidized carbohydrates was also compared against the M1 variant of the ω‐TA from Vibrio fluvialis (Vfl‐ω‐TA, UniProt: F2XBU9) engineered by the group of Bornscheuer for improved preference towards substrates other than pyruvate and generally improved activity in the neutral pH range.57 Our analysis demonstrates biocatalytic cascades to aminated cyclic carbohydrates, including oligosaccharides (Scheme 1). Corresponding pathways generate a new class of renewable telechelic molecules that were missing from the arsenal of building blocks to new biobased polymers.
Scheme 1.
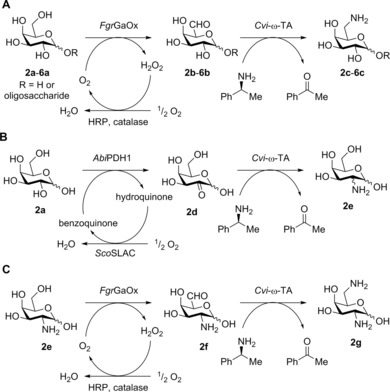
Biocatalytic cascades to aminated carbohydrates. A) Oxidation of a d‐galactosyl subunit on a carbohydrate molecule to 6‐aldo‐d‐galactosyl (2 b–6 b; see Table S1 for structures of 3 b–6 b) by FgrGaOx and subsequent amination of the aldehyde group to 6‐amino‐6‐deoxy‐d‐galactosyl (2 c–6 c) by Cvi‐ω‐TA. B) Oxidation of d‐galactose (2 a) to 2‐keto‐d‐galactose (2 d) by AbiPDH153 and subsequent amination of ketone 2 d to amine 2 e by Cvi‐ω‐TA. C) Oxidation of 2 e by FgrGaOx to bifunctional intermediate 2 f and putative amination of the aldehyde group to the diamine 2 g by Cvi‐ω‐TA. R=remaining oligosaccharide. Note: whereas the α‐configuration of galactose is drawn, both α and β isomers occur. The conformation of the C‐2 amino group in reaction product 2 e is unknown. Chiral (S)‐(−)‐PEA was used instead of a racemic mixture due to the strict stereoselectivity of Cvi‐ω‐TA.56 Abbreviations: AbiPDH1, pyranose dehydrogenase from Agaricus bisporus; Cvi‐ω‐TA, ω‐TA from Chromobacterium violaceum; FgrGaOx, galactose oxidase from Fusarium graminearum; HRP, horseradish peroxidase from horseradish; ScoSLAC, small laccase from Streptomyces coelicolor.
Results and Discussion
Activity of Cvi‐ω‐TA towards oxidized d‐galactose and d‐galactosamine
The yields of FgrGaOx and AbiPDH1 produced in Pichia pastoris were 108 and 3.9 mg L−1, respectively, and the yields of Cvi‐ω‐TA and Vfl‐ω‐TA M1 produced in E. coli were 115 and 98 mg L−1, respectively (Figure S1 in Supporting Information). These values are in the same range as previous reports describing the recombinant production of these enzymes.58, 59
Activity of Cvi‐ω‐TA towards the oxidized carbohydrates produced by FgrGaOx or AbiPDH1, and towards pyruvate and d‐erythrose (1), was measured by using the acetophenone assay.60 Here, Cvi‐ω‐TA exhibited significant activity toward aldehyde 2 b (160±1 U g−1), which, albeit lower than the Cvi‐ω‐TA activity measured toward pyruvate (2995±90 U g−1), was in the same order of magnitude as that toward 1, which is a preferred substrate of Cvi‐ω‐TA (Table 1; Scheme 1).56 Importantly, formation of hydrated and oxidized derivatives of 2 b in the reaction mixture effectively lowers the concentration of intermediate aldehyde 2 b in reactions involving d‐galactose (2 a) compared with pyruvate and 1.28 AbiPDH1 was previously shown to primarily target the C2 position of 2 a,53 and this suggests that 2‐ketogalactose (2 d) served as the main substrate for subsequent transamination by Cvi‐ω‐TA. Although the activity of Cvi‐ω‐TA toward 2 d was about 30 % of that measured with 2 b (Table 1), Cvi‐ω‐TA also showed activity toward this sugar substrate yielding amine 2 e in the multi‐enzyme cascade (Figure 1 B). Although a minor fraction of 2 a is expected to be in the open‐chain conformation, transamination of the C1 aldehyde was not observed by the acetophenone assay, nor were the corresponding products detected by high‐performance anion‐exchange chromatography with pulsed amperometric detection (HPAEC‐PAD) or mass spectrometry. Activity of Vfl‐ω‐TA M1 towards the FgrGaOx and AbiPDH1 products was tested, but was found to be less than 10 % of that of Cvi‐ω‐TA, which is why experiments with Vfl‐ω‐TA M1 were not continued.
Table 1.
Colorimetric activity assay of Cvi‐ω‐TA on selected substrates.
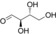
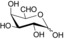
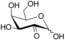
| Substrate | Structure | Activity±SD [U g−1][a] |
|---|---|---|
| d‐erythrose (1) |
|
700±20 |
| 6‐aldo‐d‐galactose (2 b) |
|
160±1 |
| 2‐keto‐d‐galactose (2 d) |
|
45±1 |
| 2‐amino‐2‐deoxy‐6‐aldo‐d‐galactose[b] (2 f) |
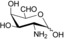
|
60±3 |
[a] Reaction conditions: V=200 μL, 10 mm amine acceptor substrate, 10 mm 1‐PEA, 20 μm PLP, 30 μg (2.9 μm) Cvi‐ω‐TA in 50 mm HEPES–NaOH buffer (pH 7.5) at 37 °C and 700 rpm. [b] Activity toward 2 f was measured at an amine acceptor concentration of 5 mm owing to the high background absorbance of the substrate at 245 nm. All measurements were conducted in triplicate at minimum.
Figure 1.
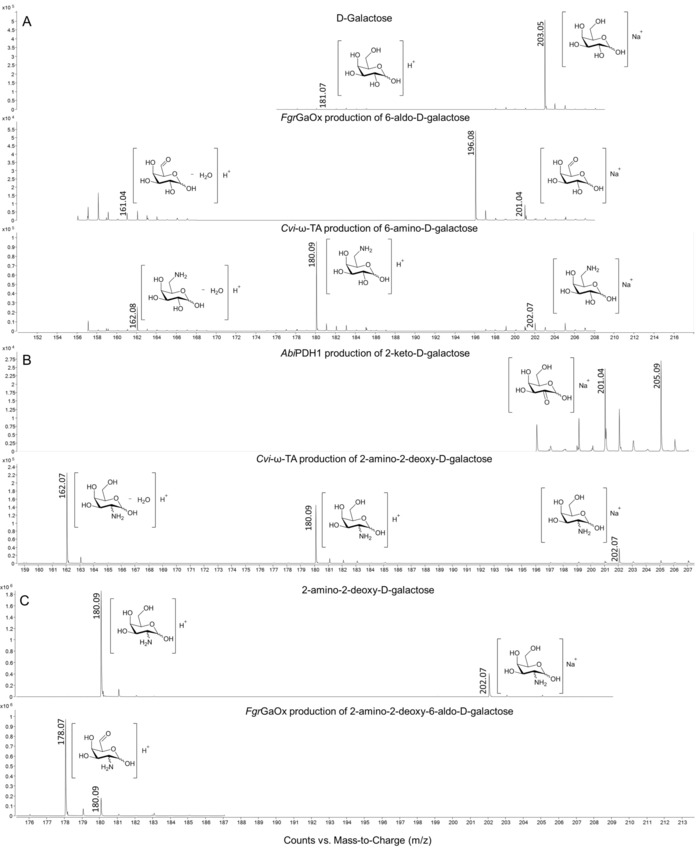
ESI‐Q‐TOF mass spectra of the conversion of A) d‐galactose (2 a) to 6‐amino‐6‐deoxy‐d‐galactose (2 c) expected from sequential action of FgrGaOx and Cvi‐ω‐TA; B) d‐galactose (2 a) to 2‐amino‐2‐deoxy‐d‐galactose (2 e) expected from sequential action of AbiPDH1 and Cvi‐ω‐TA; C) the expected 2‐amino‐2‐deoxy‐d‐galactose (2 e) from B) to 2‐amino‐2‐deoxy‐6‐aldo‐d‐galactose (2 f) through action of FgrGaOx. Similar spectra were collected from each of the three reaction replicates.
The purified FgrGaOx and Cvi‐ω‐TA, or AbiPDH1 and Cvi‐ω‐TA, were then tested in combination to establish a two‐step pathway to amino carbohydrates. Specifically, 2 a was oxidized to aldehyde 2 b by FgrGaOx and then treated with Cvi‐ω‐TA in an attempt to produce amine 2 c. Alternatively, d‐galactose was oxidized to ketone 2 d by AbiPDH1 and then treated with Cvi‐ω‐TA in an attempt to produce amine 2 e. In the case of each sequential reaction, a near‐quantitative yield for the oxidation of 2 a was confirmed by TLC before initiating the transaminase reaction (data not shown).
Mass spectrometric ESI‐Q‐TOF analysis verified the enzymatic production of both amines 2 c and 2 e. The masses of protonated and sodiated ion adducts of amines 2 c (Figure 1 A) and 2 e (Figure 1 B), as well as the expected isotopes, were all found for corresponding reaction mixture and confirmed their production through the oxidoreductase‐transaminase cascade reactions.
Having confirmed the production of 2 e, we ventured to produce diamine 2 g, which is expected to permit carbohydrate coupling through imine bond formation.61 The activity of FgrGaOx toward 50 mm 2 e was determined with the ABTS assay to be 50.1±4.9 U mg−1 enzyme, which is about 10 % of that of FgrGaOx toward 2 a. Formation of the corresponding bifunctional intermediate product 2 f was confirmed by ESI‐Q‐TOF MS (Figure 1 C), and nearly complete conversion in the subsequent transaminase reaction was verified by HPAEC‐PAD (Figure S3 in the Supporting Information). Despite this, diamine 2 g was not detected by mass spectrometry, possibly owing to the formation of unknown adducts or side reactions. Although depletion of intermediate 2 f through formation of imine derivatives can not be ruled out, Cvi‐ω‐TA accepted 2 f as a substrate, as shown by using the acetophenone assay (Table 1).
Quantification of 6‐amino‐6‐deoxy‐d‐galactose from d‐galactose
The comparison of different oxidized forms of 2 a showed that the highest Cvi‐ω‐TA activity was obtained with aldehyde 2 b (Table 1). Therefore, the sequential, two‐step enzymatic conversion of 2 a to amine 2 c was monitored by HPAEC‐PAD to quantify product formation (Figure 2).
Figure 2.
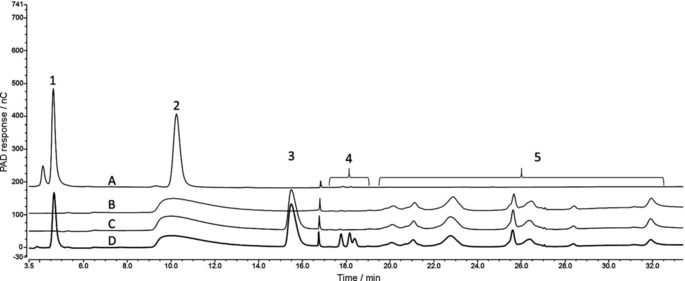
Staggered HPAEC‐PAD chromatograms tracking the conversion of 2 a to 2 c. A) 2 a and 2 c standards in ddH2O. B) FgrGaOx treatment of 20 mm 2 a in ddH2O (4 h at 30 °C, 700 rpm). C) Control experiment: incubation of the oxidation products (i.e., B) under the conditions of the transaminase reaction but without the addition of transaminase. D) Cvi‐ω‐TA treatment of 10 mm oxidation products containing aldehyde 2 b (i.e., B) [1.5 h at 37 °C, 700 rpm in 10 mm 1‐PEA, 20 μm PLP, 50 mm HEPES‐NaOH (pH 7.5)]. Prior to analysis, samples were diluted so that the total of the concentrations of 2 a along with oxidation and amination products was 90 μg mL−1. 1=amine 2 c (t R=4.7±0.1 min); 2=2 a in A (t R=10.3±0.1 min) and overlapping peaks of d‐galactose oxidation products in B, C and D; 3=HEPES; 4=side products formed in the transaminase reaction; 5=derivatives formed during the oxidation reaction (t R between 19.5–33.0 min).
As previously reported,28, 29, 62 oxidation of 2 a by FgrGaOx generated several different derivatives of the aldehyde group, including the hydrate (geminal diol) and the corresponding uronic acid due to further oxidation (data not shown). Accordingly, 2 a and chemically synthesized 2 c were used to generate standard curves to calculate substrate depletion in the oxidation reaction and product formation in the amination reaction (Figure S2 in Supporting Information). Nearly all (95±2 mol %) of 2 a was depleted in the sequential oxidation and amination reaction, in which the formation of 2 c from 2 a was 18±2 mol % prior to any optimization. Notably, calculating the formation of 2 c on the basis of the consumption of 2 a inevitably underestimates the efficiency of the amination step, as side reactions can occur after formation of intermediate 2 b,28 and so not all of this intermediate is available for the desired transamination step. In an attempt to reduce the formation of side products from 2 b by reducing the time 2 b remains in aqueous solution, the sequential, two‐step enzymatic reactions were instead performed simultaneously. Indeed, performing the oxidization and transamination steps simultaneously increased the formation of 2 c nearly 2.5‐fold (Table 2). A similar relative increase from sequential to simultaneous reaction was observed when l‐alanine was used as the amine donor, but the yields obtained with l‐alanine were roughly ten times lower than those with 1‐PEA as amine donor. This was consistent with the unfavorable equilibrium for this reaction.47, 48, 63 It is also notable that increasing the concentration of the PLP cofactor from 20 μm to 1 mm only moderately increased product yields in sequential reactions (Table 2) Because isopropylamine (IPA) is the preferred amine donor used by industry to push reaction equilibria towards the aminated product owing to its low cost and easy removal of the acetone byproduct,63 IPA was tested herein as a means to further increase the formation of 2 c from 2 b. However, replacing 1‐PEA by IPA resulted in undetectable product formation (data not shown), consistent with the comparatively high sensitivity of Cvi‐ω‐TA to IPA.64
Table 2.
Influence of reaction setup on the formation of 2‐amino‐2‐deoxy‐d‐galactose (2 c) from d‐galactose (2 a).[a]
| Amine donor | PLP concentration | Product (2 c) formation [mol %] | |
|---|---|---|---|
| sequential[b] | simultaneous[c] | ||
| 1‐pea | 20 μm | 18 | N/A[d] |
| 1‐pea | 1 mm | 27 | 67 |
| l‐Ala | 1 mm | 2.5 | 6.5 |
[a] Reaction conditions: 50 mm HEPES buffer containing 10 mm d‐galactose (2 a), 10 mm amine donor (1‐PEA or l‐Ala), and 20 μm or 1 mm PLP. Enzyme concentrations were 0.44 μm FgrGaOx, 0.53 μm catalase, 0.12 μm HRP, and 2.9 μm Cvi‐ω‐TA. [b] Sequential reactions proceeded for 4+1.5 h for the oxidation and transamination steps, respectively. [c] Simultaneous reactions proceeded for 5.5 h. Product (2 c) formation was quantified by HPAEC‐PAD. [d] Data not available.
Activity of Cvi‐ω‐TA toward selected oxidized 6‐aldo‐d‐galactosyl‐containing carbohydrates
Besides monosaccharide substrates, Cvi‐ω‐TA was tested on a series of d‐galactose‐containing oligosaccharides, each of which was first oxidized with FgrGaOx. As done for 2 a, near‐quantitative oxidation of each oligosaccharide was confirmed by TLC (data not shown), and subsequent Cvi‐ω‐TA activity toward each oxidized carbohydrate was measured by using the acetophenone assay with 1‐PEA as amine donor.
Cvi‐ω‐TA activity was detected for all tested oxidized oligosaccharides generated by using FgrGaO, including aldo‐melibiose (20±4 U mg−1), aldo‐lactose (50±6 U g−1), aldo‐raffinose (50±3 U g−1), and aldo‐xyloglucan oligosaccharides (32±4 U g−1). Whereas mass‐spectrometric options must be optimized to unequivocally confirm the identity of resulting products, substrate docking studies showed that all investigated saccharides dock similarly, with the catalytically relevant aldehyde group orientated towards the catalytically active exocyclic amino group of PMP (Figure S4 in the Supporting Information). Notably, comparison with the docked aldo‐xyloglucan oligosaccharide and the crystal structure of Vfl‐ω‐TA (PDB ID: 4E3Q) shows the beneficial architecture of the active site of Cvi‐ω‐TA towards oligosaccharides, since the active site of Cvi‐ω‐TA is more exposed (Figure S5).
Conclusions
We have demonstrated the application of two different fully biocatalytic cascades that employ carbohydrate oxidoreductases to transform specific hydroxyl groups into carbonyl groups, and Cvi‐ω‐TA to introduce amine functionality at the oxidized positions of the substrates. The pathways produced amino galactoses with two different regioselectivities: 1) the combination of FgrGaOx and Cvi‐ω‐TA yielded galactose derivatives aminated at the C‐6 position, and 2) the combination of AbiPDH1 with Cvi‐ω‐TA yielded galactose derivatives aminated at the C‐2 position. Production of 6‐amino‐6‐deoxy‐d‐galactosyl‐containing oligosaccharides through pathway 1 was also detected by acetophenone activity assay. Notably, a multistep synthetic route was required to synthesize the aminogalactose derivative used as an analytical standard in the current study, and this further highlights the benefits of the biocatalytic approach. Steps taken to maximize product formation included 1) establishing a simultaneous oxidation plus transaminase reaction to the aminated carbohydrate, 2) increasing PLP concentration, and 3) testing of different amine donors. The greatest gains in the formation of product 2 c were achieved by performing the oxidation and transamination steps simultaneously rather than sequentially, consistent with reduced formation of undesired side products from aldehyde intermediate 2 b.28 This work takes the first step in unlocking the potential of ω‐TAs for carbohydrate functionalization and thus expands the pool of building blocks available for new biobased materials.
Experimental Section
Materials
Yeast extract, yeast nitrogen base, and peptone were purchased from Lab M Ltd. (UK). d‐galactose, lactose, melibiose, and raffinose were of analytical grade and purchased from Sigma‐Aldrich. Xyloglucan oligosaccharides (hepta‐+octa‐+nonasaccharides) were purchased from Megazyme (O‐XGHON; Lot number 20509). 1,2,3,4‐Di‐O‐isopropylidene‐α‐d‐galactose, used for preparing the synthetic 6‐amino‐6‐deoxy‐d‐galactose used as a standard for product quantification, was purchased from Alfa Aesar. All other chemicals were of reagent grade, obtained from Sigma‐Aldrich (Germany), and used without further purification unless otherwise specified.
Production and purification of Fusarium graminearum galactose oxidase (FgrGaOx) and Agaricus bisporus pyranose dehydrogenase (AbiPDH1)
F. graminearum (FgrGaOx; d‐galactose:oxygen 6‐oxidoreductase, EC 1.1.13.9, CAZy family AA5_2) and A. bisporus pyranose dehydrogenase (AbiPDH1; pyranose:acceptor oxidoreductase, EC 1.1.99.29 CAZy family AA3_2) were heterologously expressed in Pichia pastoris KM71H. Genes (GenBank accession number: AH005781.2 coding FgrGaOx; KM851045 AbiPDH1) with C‐terminal 6×His tags were obtained from GenScript (New Jersey, USA) subcloned into pPICZαA or pPICZB vectors, respectively. Both enzymes were produced in shake‐flask cultivations, as previously described.69 Briefly, precultures were grown in up to 750 mL of buffered glycerol‐complex medium (BMGY; 100 mm potassium phosphate buffer, pH 6.0, 2 % (w/v) peptone, 1 % (w/v) yeast extract, 4×10−5 % (w/v) biotin, 1 % (v/v) glycerol) at 30 °C, 220 rpm. Methanol induction was performed over 4 d at 25 °C, 220 rpm, in buffered methanol‐complex medium (BMMY with 0.5 % (v/v) methanol), whereby 0.5 % (v/v) methanol was added every 24 h to replenish the inducer.
After induction and spinning down the cells, the supernatant was recovered, adjusted to pH 7.4, and filtered through a Sterivex‐GP 0.45 μm PES filter unit (Millipore, Germany). The filtrate was loaded directly onto 6 mL of Ni‐NTA resin (Qiagen, Germany) equilibrated in binding buffer (50 mm sodium phosphate buffer, pH 7.4, 500 mm NaCl, 20 mm imidazole) and packed in a XK‐16/10 column (GE Life Sciences, Germany). Bound protein was eluted with a linear gradient of 0–100 % Ni‐NTA elution buffer (50 mm sodium phosphate, pH 7.4 with 500 mm imidazole, 500 mm NaCl). Purified fractions were then exchanged to 50 mm phosphate buffer (pH 7.5) by using a 10 or 30 kDa Vivaspin 20 spin column (Sartorius AG, Germany).
FgrGaOx and AbiPDH1 were concentrated to 13.5 mg mL−1 and 1.8 mg mL−1, respectively, and stored at −80 °C until further use. Protein concentration was measured by the Bradford method (Bio‐Rad Laboratories, US), and protein purity was assessed by SDS‐PAGE (Figure S1).
Production and purification of the ω‐TAs (Cvi‐ω‐TA and Vfl‐ω‐TA M1)
A pET29a+plasmid containing the Cvi‐ω‐TA gene (GenBank: AAQ59697.1), obtained from GenScript, and the pET24b plasmid encoding Vfl‐ω‐TA M157 were transformed into chemically competent E. coli BL21. Selected E. coli transformants containing each plasmid were grown at 37 °C, 220 rpm in shake flasks containing 250 mL LB medium supplemented with 50 μg mL−1 kanamycin and 30 μg mL−1 chloramphenicol. When the OD600 reached 0.8–1, the E. coli transformant was induced to express the protein of interest by addition of 1 mm isopropyl‐β‐d‐thiogalactopyranoside (IPTG). After 15 h of induction at 30 °C, the cells were harvested by centrifugation (5000 g and 4 °C for 45 min). For each production, the cell pellet was suspended in 50 mL of 50 mm potassium phosphate buffer (pH 7.5) containing 0.1 mm PLP and lysed by using an Emulsiflex‐C3 French press (Avestin Inc., Canada) at 10 000 PSI for 20 min.
Immediately after cell lysis, the lysates were clarified by centrifugation at 15 000 g and 4 °C for 45 min. The supernatants each containing the soluble protein were filtered through Sterivex‐GP 0.22 μm PES filter units (Millipore). Cvi‐ω‐TA and Vfl‐ω‐TA M1, each containing a C‐terminal His tag, were purified to homogeneity by using Ni‐NTA resin as described above, except this time 0.1 mm PLP was added to the binding buffer and elution buffer. Purified fractions were then exchanged to 50 mm phosphate buffer (pH 7.5) with 0.1 mm PLP by using a 10 kDa Vivaspin 20 spin column (Sartorius AG, Germany). Cvi‐ω‐TA was concentrated to 9.5 mg mL−1 and Vfl‐ω‐TA M1 to 3.3 mg mL−1 and then stored at −80 °C until further use. Protein concentration was measured by the Bradford method (Bio‐Rad Laboratories, USA), and protein purity was assessed by SDS‐PAGE (Figure S1 in Supporting Information).
Galactose oxidase assay
The activity of FgrGaOx was measured by the chromogenic ABTS [2,2′‐azino‐bis(3‐ethylbenzothiazoline‐6‐sulfonic acid)] assay, originally described by Baron et al.,70 with modifications. The standard reaction mixture (final volume: 200 μL) contained 270 μg (31 μm, assuming purity) horseradish peroxidase (HRP, from horseradish, Sigma‐Aldrich, Germany), 2 mm ABTS, and 50 mm 2 a in 50 mm HEPES (4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid) buffer (pH 7.5); reactions were initiated by adding 5 μL of appropriately diluted enzyme sample to obtain initial rates of reaction. Absorbance was measured at 420 nm at 30 °C for 20 min in an Eon microplate reader (BioTek, US). Activity values were calculated on the basis of the extinction coefficient (36 000 L mol−1 cm−1 at 420 nm).71 Each reaction was performed in triplicate, at minimum.
Enzymatic production of oxidized carbohydrates
Enzymatic oxidation of d‐galactosyl‐containing substrates with FgrGaOx (reaction volume: 3 or 5 mL) was performed in 15 mL Cellstar tubes (Greiner BioOne) containing 20 mm d‐galactose equivalents of substrates (i.e., d‐galactose, melibiose, lactose, raffinose, or xyloglucan oligosaccharides), 150 μg (0.44 μm) FgrGaOx, 160 μg (0.53 μm, assuming purity and based on molecular weight of the catalase monomer) catalase (from bovine liver, Sigma‐Aldrich, Germany), and 27 μg (0.12 μm) HRP in 50 mm HEPES–NaOH (pH 7.5). Catalase was used as the primary means of removing hydrogen peroxide in the cascade, and HRP was included owing to its known ability to activate FgrGaOx.72 The effect is based on the ability of HRP to maintain the copper radical in the active site of FgrGaOx in the correct oxidation state (CuII).73 The enzyme concentrations were chosen on the basis of previous experience to achieve maximum oxidation of the galactosyl residues in each substrate.27, 28 Reaction mixtures were incubated for 4 h at 30 °C with shaking (700 rpm). Conversion of each oxidation reaction was evaluated by TLC (data not shown) on Macherey–Nagel precoated silica gel plates (TLC Silica gel 60 F254). AbiPDH1 oxidation of d‐galactose was performed as described by Sygmund and co‐workers,74 with minor modification. Specifically, reaction mixtures contained 58 μg (3.61 μm) of AbiPDH1, 50 mm d‐galactose, and 5 mm benzoquinone in double‐distilled water (ddH2O). The small laccase (ScoSLAC; 260 μg, 32.5 μm) from Streptomyces coelicolor was used to recycle the electron acceptor (benzoquinone), as shown in Scheme 1.75, 76
ω‐TA reactions and activity assays
After verifying that the oxidation reaction had reached maximum conversion (evaluated by TLC, data not shown), the activity of the purified Cvi‐ω‐TA towards oxidized carbohydrates was measured by chromogenic detection of acetophenone from 1‐phenylethylamine (1‐PEA).60 Activity of Vfl‐ω‐TA M1 towards aldehyde 2 b was also measured. Briefly, transaminase reactions (200 μL final volume) were carried out at 37 °C in 96‐well microtiter plates (96‐Well UV Microplate, Thermo Scientific, US) incubated in a plate reader (Biotek Eon, US). The reaction mixture contained 10 mm 1‐PEA (i.e., amine donor substrate), up to 10 mm 2 b, or 6‐aldo‐d‐galactosyl groups in oligosaccharide substrates, 20 μm PLP, and 30 μg (2.9 μm) of purified Cvi‐ω‐TA or Vfl‐ω‐TA M1. The reaction was buffered with 50 mm HEPES–NaOH (pH 7.5). In sequential reactions, products of the oxidation reactions were formed over 4 h as described above and then directly used as substrates in the transaminase reaction. Reactions (200 μL) in which the oxidized carbohydrate was substituted with 10 mm pyruvate and 30 ng ω‐TA was added served as positive controls, whereas reactions in which ω‐TA was substituted by ddH2O served as negative controls. Initial rates were determined by colorimetric detection of acetophenone over 30 min at 245 nm.60 Reaction mixtures were then transferred to a Thermomixer (Eppendorf, Germany) to continue incubation at 37 °C and 700 rpm for a further 1 h prior to product measurement by HPAEC‐PAD and ESI‐Q‐TOF mass spectrometry as described below. All reactions were performed in triplicate at minimum.
Reactions (500 μL) permitting simultaneous oxidation of galactose (2 a) to the aldehyde (2 b) and transamination of 2 b to the corresponding amine (2 c) were performed for 5.5 h at 30 °C and 700 rpm in 50 mm HEPES‐NaOH (pH 7.0), and the reaction mixtures contained 10 mm 2 a, 1 mm PLP, and 10 mm 1‐PEA or 10 mm l‐alanine. Enzyme concentrations were 0.44 μm FgrGaOx, 0.53 μm catalase, 0.12 μm HRP, and 2.9 μm Cvi‐ω‐TA. For each simultaneous oxidation–transamination reaction, a sequential oxidation–transamination reaction was performed under otherwise identical conditions, but by first running the oxidation reaction for 4 h, and then initiating the transaminase reaction by addition of PLP, 1‐PEA, and Cvi‐ω‐TA, and allowing the transamination reaction proceed for 1.5 h. Product formation was quantified by HPAEC‐PAD.
Confirmation of the oxidation and amination products by ESI‐Q‐TOF mass spectrometry
The following samples were analyzed by direct‐injection ESI‐Q‐TOF (Agilent 6530 Q‐TOF, Singapore): 1) 100 ppm 2 a, 2) product from FgrGaOx oxidation of 2 a containing up to 100 ppm 2 b, 3) product of the amination reaction containing up to 100 ppm of the prospective amine 2 c formed in the reaction (reaction conditions specified above). The product compounds were not isolated prior to analysis. Prior to dilution, samples 2) and 3) were desalted with AG 2‐X8 anion‐exchange resin (BioRad, US), and proteins removed by filtration with a Sartorius Vivaspin 500 spin column (10 000 kDa cutoff). Electrospray ionization was performed in positive mode, and nitrogen gas was used as both the nebulizing and drying gas. The following ionization parameters were used: the drying gas temperature was 250 °C, the drying gas flow was 3 L min−1, the capillary voltage was 3500 V, and nebulizer pressure was 103.4 kPa. Samples were injected directly to the ion source by the infusion pump at a flow rate of 250 μL min−1 by elution with 0.1 % formic acid in 50 % acetonitrile. Agilent Masshunter Qualitative Analysis (version B.07.00.Ink) was used for the data analysis. All samples were prepared and analyzed in triplicate.
Synthesis of 6‐amino‐6‐deoxy‐d‐galactopyranose trifluoroacetate salt (2 c⋅TFA)
Commercially available reagents were used without further purification. Column chromatography over silica gel was performed with Merck Millipore 60, 40–60 μm, 240–400 mesh silica gel. Reactions were monitored by TLC. Visualization of the TLC plates was achieved by UV light or staining with a basic potassium permanganate solution. 1H and 13C NMR spectra were recorded with a Bruker AV‐400 (Germany) instrument at 20 °C (see Supporting Information).
1,2,3,4‐Di‐O‐isopropylidene‐α‐d‐galactopyranose (1.5 g, 5.7 mmol) was dissolved in EtOAc (38 mL) and iodoxybenzoic acid (4.8 g, 17 mmol) was added. After complete oxidation, monitored by TLC, yielding 1,2,3,4‐di‐O‐isopropylidene‐α‐d‐galactohexodialdo‐1,5‐pyranose,77 the precipitate was removed by filtration and the crude reaction mixture was concentrated under reduced pressure. The crude product was purified by column chromatography (SiO2, cyclohexane/EtOAc 85:15 to 70:30), which afforded the product as a clear, viscous oil (1.00 g, 3.87 mmol, 67 %). R f=0.51 (n‐hexane/EtOAc 2:1).
1,2,3,4‐Di‐O‐isopropylidene‐α‐d‐galactohexodialdo‐1,5‐pyranose (250 mg, 0.97 mmol) was dissolved in a solution of ammonium acetate (800 mg, 10.4 mmol) in methanol (5 mL). After 15 min, NaCNBH3 (100 mg, 1.6 mmol) was added. The reaction mixture was stirred for 24 h at room temperature, after which all volatile substances were removed under reduced pressure. The residue was redissolved in water (15 mL) and extracted with EtOAc (10 mL). This organic layer was discarded. The aqueous phase was basified (pH>12) by addition of solid sodium hydroxide. The aqueous layer was extracted with EtOAc (3×10 mL), the combined organic layers were dried over anhydrous MgSO4, and the volatile substances were removed under reduced pressure. The product, 6‐amino‐6‐deoxy‐1,2,3,4‐di‐O‐isopropylidene‐α‐d‐galactopyranose, was obtained as clear, viscous oil (180 mg, 0.69 mmol, 71 %).
The protected amino sugar (35 mg, 0.14 mmol) was dissolved in deuterium oxide (0.7 mL), and trifluoroacetic acid (11 μL, 0.14 mmol) was added. After stirring for 24 h at room temperature, 1H NMR analysis confirmed completion of the deprotection and quantitative formation of the trifluoroacetate salt of 6‐amino‐6‐deoxy‐d‐galactopyranose. The thus‐obtained solution of the synthetic reference of 2 c (0.2 m, 0.14 mmol, 99 %) was used without further manipulation as stock solution (Supporting Information, section 1).
Quantification of reaction yields by HPAEC‐PAD
Amination reactions were performed as described above. Prior to analysis, the reaction samples for HPAEC‐PAD were diluted with ddH2O to 50–100 ppm of carbohydrate. Eluents used were 100 mm NaOH (A) and 100 mm NaOH with 1 m NaOAc (B). The chromatographic runs were performed with a Dionex CarboPac PA1 IC column and with a flow rate of 0.6 mL min−1, whereby 100 % eluent A was used for the first 5 min followed by 0–100 % eluent B over the next 50 min. Thermo Scientific Dionex Chromeleon 7 Chromatography Data System (version 7.2 SR4, Thermo Fisher Scientific) was used for data analysis. The conversion of d‐galactose in the oxidation reaction was calculated from decrease in area of the d‐galactose peak, and the yield of 6‐amino‐6‐deoxy‐d‐galactose was calculated on the basis of the peak area of the 6‐amino‐6‐deoxy‐d‐galactose standard 2 c⋅TFA (Figure S2).
Enzyme–substrate docking
For visualization of the substrates in the active site of the Cvi‐ω‐TA, a receptor was constructed by using the crystallized transaminase structure (PDB ID: 4A6T). The crystal structure was slightly altered by modifying the PLP–lysine complex structure to obtain PMP and the unbound K288 residue. Subsequently, the modified receptor was energy‐minimized by using the YASARA78, 79 built‐in energy‐minimization function. The R416 (flipping arginine) side chain was turned slightly upwards away from the PMP to gain space in the large binding pocket, since the sugar substrates did not contain charged residues.80, 81 The docking ligands 2 b (6‐aldo‐d‐galactose) and the corresponding 6‐aldo‐d‐galactosyl‐containing aldo‐lactose and aldo‐raffinose were constructed with the built‐in oligosaccharide building tool of YASARA, by using the β‐d‐conformation of each sugar. The docking ligand aldo‐melibiose was constructed by oxidizing the corresponding melibiose structure (PubChem Identifier: CID 11458). The XLLG molecule (Figure S6) was extracted as a ligand from the crystal structure PDB ID: 2VH9 and subsequently oxidized to obtain aldo‐XLLG. All ligands were energy‐minimized prior to the docking experiments. The docking was performed with YASARA by using the dock runensemble.mcr macro utilizing the VINA docking method with appropriate simulation cells covering the active site of the receptor. The flexible R416 residue was fixed in place. Plausible docking results were selected by evaluating orientation of the substrate to the PMP cofactor in the active site. In addition to the location and orientation of the substrate, binding energies and dissociation constants reported by YASARA were also considered (Table S2 in Supporting Information). Figures of the dockings were created with PyMOL (The PyMOL Molecular Graphics System, Version 2.2.0 Schrödinger, LLC).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This study was financially supported by the Academy of Finland (decision numbers 308996, 252183, and 298250) and the European Research Council (ERC) Consolidator Grant to E.R.M. (BHIVE‐648925). U.B. thanks the German Research Foundation (DFG, Grant No. Bo1862/16‐1) and the EU (Horizon2020, Grant No. 722610) for funding.
V. Aumala, F. Mollerup, E. Jurak, F. Blume, J. Karppi, A. E. Koistinen, E. Schuiten, M. Voß, U. Bornscheuer, J. Deska, E. R. Master, ChemSusChem 2019, 12, 848.
References
- 1.T. Werpy, G. Petersen, Top Value Added Chemicals from Biomass: Volume I—Results of Screening for Potential Candidates from Sugars and Synthesis Gas, National Renewable Energy Laboratory, Golden, CO, 2004, https://www.nrel.gov/docs/fy04osti/35523.pdf.
- 2. Delidovich I., Leonhard K., Palkovits R., Energy Environ. Sci. 2014, 7, 2803. [Google Scholar]
- 3. Gallezot P., Chem. Soc. Rev. 2012, 41, 1538–1558. [DOI] [PubMed] [Google Scholar]
- 4. Ruppert A. M., Weinberg K., Palkovits R., Angew. Chem. Int. Ed. 2012, 51, 2564–2601; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 2614–2654. [Google Scholar]
- 5. Mika L. T., Cséfalvay E., Németh Á., Chem. Rev. 2018, 118, 505–613. [DOI] [PubMed] [Google Scholar]
- 6. Harmsen P. F. H., Hackmann M. M., Bos H. L., Biofuels Bioprod. Biorefin. 2014, 8, 306–324. [Google Scholar]
- 7. Chung H., Yang J. E., Ha J. Y., Chae T. U., Shin J. H., Gustavsson M., Lee S. Y., Curr. Opin. Biotechnol. 2015, 36, 73–84. [DOI] [PubMed] [Google Scholar]
- 8. Froidevaux V., Negrell C., Caillol S., Pascault J. P., Boutevin B., Chem. Rev. 2016, 116, 14181–14224. [DOI] [PubMed] [Google Scholar]
- 9. Gunukula S., Anex R. P., Biofuels Bioprod. Biorefin. 2017, 11, 897–907. [Google Scholar]
- 10. Li J., Zheng X.-Y., Fang X.-J., Liu S.-W., Chen K.-Q., Jiang M., Wei P., Ouyang P.-K., Bioresour. Technol. 2011, 102, 6147–6152. [DOI] [PubMed] [Google Scholar]
- 11. Chen Y., Nielsen J., Curr. Opin. Biotechnol. 2013, 24, 965–972. [DOI] [PubMed] [Google Scholar]
- 12. Balaraman E., Fogler E., Milstein D., Chem. Commun. 2012, 48, 1111–1113. [DOI] [PubMed] [Google Scholar]
- 13. Arnaud S. P., Wu L., Chang M.-A. W., Comerford J. W., Farmer T. J., Schmid M., Chang F., Li Z., Mascal M., Faraday Discuss. 2017, 202, 61–77. [DOI] [PubMed] [Google Scholar]
- 14. Dusselier M., Mascal M., Sels B. F. in Selective Catalysis for Renewable Feedstocks and Chemicals (Ed.: K. M. Nicholas), Springer, Cham, 2014, pp. 1–40. [Google Scholar]
- 15. Xu C., Spadiut O., Araújo A. C., Nakhai A., Brumer H., ChemSusChem 2012, 5, 661–665. [DOI] [PubMed] [Google Scholar]
- 16. Edlund U., Ryberg Y. Z., Albertsson A.-C., Biomacromolecules 2010, 11, 2532–2538. [DOI] [PubMed] [Google Scholar]
- 17. Mizrahy S., Peer D., Chem. Soc. Rev. 2012, 41, 2623–2640. [DOI] [PubMed] [Google Scholar]
- 18. Skarbek K., Milewska M. J., Carbohydr. Res. 2016, 434, 44–71. [DOI] [PubMed] [Google Scholar]
- 19. van Straaten K. E., Ko J. B., Jagdhane R., Anjum S., Palmer D. R. J., Sanders D. A. R., J. Biol. Chem. 2013, 288, 34121–34130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hwang B.-Y., Lee H.-J., Yang Y.-H., Joo H.-S., Kim B.-G., Chem. Biol. 2004, 11, 915–925. [DOI] [PubMed] [Google Scholar]
- 21. Ban W., van Heiningen A., Cellul. Chem. Technol. 2011, 45, 57–65. [Google Scholar]
- 22. MacCormick B., Vuong T. V., Master E. R., Biomacromolecules 2018, 19, 521–530. [DOI] [PubMed] [Google Scholar]
- 23. Bragd P. L., van Bekkum H., Besemer A. C., Top. Catal. 2004, 27, 49–66. [Google Scholar]
- 24. Kristiansen K. A., Potthast A., Christensen B. E., Carbohydr. Res. 2010, 345, 1264–1271. [DOI] [PubMed] [Google Scholar]
- 25. Baxter E. W., Reitz A. B. in Org. React., Wiley, Hoboken, 2002, pp. 1–714. [Google Scholar]
- 26. Bragd P. L., Besemer A. C., van Bekkum H., Carbohydr. Res. 2000, 328, 355–363. [DOI] [PubMed] [Google Scholar]
- 27. Parikka K., Leppänen A. S., Pitkänen L., Reunanen M., Willför S., Tenkanen M., J. Agric. Food Chem. 2010, 58, 262–271. [DOI] [PubMed] [Google Scholar]
- 28. Parikka K., Master E., Tenkanen M., J. Mol. Catal. B 2015, 120, 47–59. [Google Scholar]
- 29. Parikka K., Tenkanen M., Carbohydr. Res. 2009, 344, 14–20. [DOI] [PubMed] [Google Scholar]
- 30. Parikka K., Ansari F., Hietala S., Tenkanen M., Food Hydrocolloids 2012, 26, 212–220. [Google Scholar]
- 31. Huang K., Parmeggiani F., Pallister E., Huang C. J., Liu F. F., Li Q., Birmingham W. R., Both P., Thomas B., Liu L., Voglmeir J., Flitsch S. L., ChemBioChem 2018, 19, 388–394. [DOI] [PubMed] [Google Scholar]
- 32. Steffen-Munsberg F., Vickers C., Kohls H., Land H., Mallin H., Nobili A., Skalden L., van den Bergh T., Joosten H. J., Berglund P., Höhne M., Bornscheuer U. T., Biotechnol. Adv. 2015, 33, 566–604. [DOI] [PubMed] [Google Scholar]
- 33. Malik M. S., Park E.-S., Shin J.-S., Appl. Microbiol. Biotechnol. 2012, 94, 1163–1171. [DOI] [PubMed] [Google Scholar]
- 34. Van Oosterwijk N., Willies S., Hekelaar J., Van Scheltinga A. C. T., Turner N. J., Dijkstra B. W., Biochemistry 2016, 55, 4422–4431. [DOI] [PubMed] [Google Scholar]
- 35. Knaus T., Böhmer W., Mutti F. G., Green Chem. 2017, 19, 453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aleku G. A., France S. P., Man H., Mangas-Sanchez J., Montgomery S. L., Sharma M., Leipold F., Hussain S., Grogan G., Turner N. J., Nat. Chem. 2017, 9, 961–969. [DOI] [PubMed] [Google Scholar]
- 37. Schrittwieser J. H., Velikogne S., Kroutil W., Adv. Synth. Catal. 2015, 357, 1655–1685. [Google Scholar]
- 38. Straathof A. J. J., Chem. Rev. 2014, 114, 1871–1908. [DOI] [PubMed] [Google Scholar]
- 39. Glueck S. M., Gümüs S., Fabian W. M. F., Faber K., Chem. Soc. Rev. 2010, 39, 313–328. [DOI] [PubMed] [Google Scholar]
- 40. Hollmann F., Arends I. W. C. E., Holtmann D., Green Chem. 2011, 13, 2285. [Google Scholar]
- 41. Guo F., Berglund P., Green Chem. 2017, 19, 333–360. [Google Scholar]
- 42. Guo Z., Liu B., Zhang Q., Deng W., Wang Y., Yang Y., Chem. Soc. Rev. 2014, 43, 3480. [DOI] [PubMed] [Google Scholar]
- 43. Fuchs M., Farnberger J. E., Kroutil W., Eur. J. Org. Chem. 2015, 6965–6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kelly S. A., Pohle S., Wharry S., Mix S., Allen C. C. R., Moody T. S., Gilmore B. F., Chem. Rev. 2018, 118, 349–367. [DOI] [PubMed] [Google Scholar]
- 45. Slabu I., Galman J. L., Lloyd R. C., Turner N. J., ACS Catal. 2017, 7, 8263–8284. [Google Scholar]
- 46. Tauber K., Fuchs M., Sattler J. H., Pitzer J., Pressnitz D., Koszelewski D., Faber K., Pfeffer J., Haas T., Kroutil W., Chem. Eur. J. 2013, 19, 4030–4035. [DOI] [PubMed] [Google Scholar]
- 47. Fuchs M., Tauber K., Sattler J., Lechner H., Pfeffer J., Kroutil W., Faber K., RSC Adv. 2012, 2, 6262. [Google Scholar]
- 48. Pickl M., Fuchs M., Glueck S. M., Faber K., ChemCatChem 2015, 7, 3121–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lerchner A., Achatz S., Rausch C., Haas T., Skerra A., ChemCatChem 2013, 5, 3374–3383. [Google Scholar]
- 50. Wu S., Zhou Y., Wang T., Too H.-P., Wang D. I. C., Li Z., Nat. Commun. 2016, 7, 11917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lerchner A., Daake M., Jarasch A., Skerra A., Protein Eng., Des. Sel. 2016, 29, 557–562. [DOI] [PubMed] [Google Scholar]
- 52. Dunbabin A., Subrizi F., Ward J. M., Sheppard T. D., Hailes H. C., Green Chem. 2017, 19, 397–404. [Google Scholar]
- 53. Volc J., Sedmera P., Halada P., Přikrylová V., Daniel G., Carbohydr. Res. 1998, 310, 151–156. [Google Scholar]
- 54. Peterbauer C. K., Volc J., Appl. Microbiol. Biotechnol. 2010, 85, 837–848. [DOI] [PubMed] [Google Scholar]
- 55. Martínez A. T., Ruiz-Dueñas F. J., Camarero S., Serrano A., Linde D., Lund H., Vind J., Tovborg M., Herold-Majumdar O. M., Hofrichter M., Liers C., Ullrich R., Scheibner K., Sannia G., Piscitelli A., Pezzella C., Sener M. E., Kilic S., van Berkel W. J. H., Guallar V., Lucas M. F., Zuhse R., Ludwig R., Hollmann F., Fernandez-Fueyo E., Record E., Faulds C. B., Tortajada M., Winckelmann I., Rasmussen J.-A., Gelo-Pujic M., Gutierre A., del Rio J. C., Rencoret J., Alcalde M., Biotechnol. Adv. 2017, 35, 815–831. [DOI] [PubMed] [Google Scholar]
- 56. Kaulmann U., Smithies K., Smith M. E. B. B., Hailes H. C., Ward J. M., Enzyme Microb. Technol. 2007, 41, 628–637. [Google Scholar]
- 57. Genz M., Vickers C., van den Bergh T., Joosten H. J., Dörr M., Höhne M., Bornscheuer U. T., Int. J. Mol. Sci. 2015, 16, 26953–26963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cassimjee K. E., Humble M. S., Miceli V., Colomina C. G., Berglund P., ACS Catal. 2011, 1, 1051–1055. [Google Scholar]
- 59. Mollerup F., Master E., Data Brief 2016, 6, 176–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schätzle S., Höhne M., Redestad E., Robins K., Bornscheuer U. T., Anal. Chem. 2009, 81, 8244–8248. [DOI] [PubMed] [Google Scholar]
- 61. van Wijk A., Siebum A., Schoevaart R., Kieboom T., Carbohydr. Res. 2006, 341, 2921–2926. [DOI] [PubMed] [Google Scholar]
- 62. Andberg M., Mollerup F., Parikka K., Koutaniemi S., Boer H., Juvonen M., Master E., Tenkanen M., Kruus K., Appl. Environ. Microbiol. 2017, 83, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Matcham G. W., Bowen A. R. S., Chim. Oggi 1996, 14, 20–24. [Google Scholar]
- 64. Dawood A. W. H., Weiß M. S., Schulz C., Pavlidis I. V., Iding H., de Souza R. O. M. A., Bornscheuer U. T., ChemCatChem 2018, 10, 951–955. [Google Scholar]
- 65. Savile C. K., Janey J. M., Mundorff E. C., Moore J. C., Tam S. S., Jarvis W. R., Colbeck J. C., Krebber A., Fleitz F. J., Brands J., Devine P. N., Huisman G. W., Hughes G. J., Science 2010, 329, 305–309. [DOI] [PubMed] [Google Scholar]
- 66. Gomm A., Lewis W., Green A. P., O'Reilly E., Chem. Eur. J. 2016, 22, 12692–12695. [DOI] [PubMed] [Google Scholar]
- 67. Martínez-Montero L., Gotor V., Gotor-Fernández V., Lavandera I., Adv. Synth. Catal. 2016, 358, 1618–1624. [Google Scholar]
- 68. Galman J. L., Slabu I., Weise N. J., Iglesias C., Parmeggiani F., Lloyd R. C., Turner N. J., Green Chem. 2017, 19, 361–366. [Google Scholar]
- 69. Mollerup F., Parikka K., Vuong T. V., Tenkanen M., Master E., Biochim. Biophys. Acta Gen. Subj. 2016, 1860, 354–362. [DOI] [PubMed] [Google Scholar]
- 70. Baron A. J., Stevens C., Wilmot C., Senereviratne K. D., Blakeley V., Dooley D. M., Phillips S. E., Knowles P. F., McPherson M. J., J. Biol. Chem. 1994, 269, 25095–25105. [PubMed] [Google Scholar]
- 71. Shin K.-S., Lee Y.-J., Arch. Biochem. Biophys. 2000, 384, 109–115. [DOI] [PubMed] [Google Scholar]
- 72. Kwiatkowski L. D., Kosman D. J., Biochem. Biophys. Res. Commun. 1973, 53, 715–721. [DOI] [PubMed] [Google Scholar]
- 73. Hartmans S., de Vries H. T., Beijer P., Brady R. L., Hofbauer M., Haandrikman A. J. in Hemicelluloses: Science and Technology (Eds.: P. Gatenholm, M. Tenkanen), American Chemical Society, Washington, DC, 2003, pp. 360–371. [Google Scholar]
- 74. Sygmund C., Kittl R., Volc J., Halada P., Kubátová E., Haltrich D., Peterbauer C. K., J. Biotechnol. 2008, 133, 334–342. [DOI] [PubMed] [Google Scholar]
- 75. Sherif M., Waung D., Korbeci B., Mavisakalyan V., Flick R., Brown G., Abou-Zaid M., Yakunin A. F., Master E. R., Microb. Biotechnol. 2013, 6, 588–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ludwig R., Ozga M., Zámocky M., Peterbauer C., Kulbe K. D., Haltrich D., Biocatal. Biotransform. 2004, 22, 97–104. [Google Scholar]
- 77. Suri J. T., Mitsumori S., Albertshofer K., Tanaka F., Barbas C. F., J. Org. Chem. 2006, 71, 3822–3828. [DOI] [PubMed] [Google Scholar]
- 78. Krieger E., Vriend G., Bioinformatics 2014, 30, 2981–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Krieger E., Vriend G., J. Comput. Chem. 2015, 36, 996–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Manta B., Cassimjee K. E., Himo F., ACS Omega 2017, 2, 890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Steffen-Munsberg F., Vickers C., Thontowi A., Schätzle S., Meinhardt T., Humble M. S., Land H., Berglund P., Bornscheuer U. T., Höhne M., ChemCatChem 2013, 5, 154–157. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary