

Abstract
The Src homology‐2 domain‐containing tyrosine phosphatase 2 (SHP‐2) regulates many cellular processes, including proliferation, differentiation and survival. Polymorphisms in the gene encoding SHP‐2 are associated with an increased susceptibility to develop ulcerative colitis. We recently reported that intestinal epithelial cell (IEC)‐specific deletion of Shp‐2 in mice (Shp‐2 IEC‐KO) leads to chronic colitis and colitis‐associated cancer. This suggests that SHP‐2‐dependent signaling protects the colonic epithelium against inflammation and colitis‐associated cancer development. To verify this hypothesis, we generated mice expressing the Shp‐2 E76K activated form specifically in IEC. Our results showed that sustained Shp‐2 activation in IEC increased intestine and crypt length, correlating with increased cell proliferation and migration. Crypt regeneration capacity was also markedly enhanced, as revealed by ex vivo organoid culture. Shp‐2 activation alters the secretory cell lineage, as evidenced by increased goblet cell numbers and mucus secretion. Notably, these mice also demonstrated elevated ERK signaling in IEC and exhibited resistance against both chemical‐ and Citrobacter rodentium‐induced colitis. In contrast, mice with IEC‐specific Shp‐2 deletion displayed reduced ERK signaling and rapidly developed chronic colitis. Remarkably, expression of an activated form of Braf in Shp‐2‐deficient mice restored ERK activation, goblet cell production and prevented colitis. Altogether, our results indicate that chronic activation of Shp‐2/ERK signaling in the colonic epithelium confers resistance to mucosal erosion and colitis. © 2018 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: Shp‐2, colitis, intestinal barrier, goblet cells, epithelial regeneration, wound healing, ERK/MAPK
Introduction
Inflammatory bowel diseases (IBD) include Crohn's disease (CD) and ulcerative colitis (UC). Both are characterized by inflammation of the lining of part of the digestive tract. In CD, inflammation occurs throughout the digestive system, from the mouth to the anus, mostly in the intestine, whereas in UC, inflammation is localized to the rectum and colon 1. Both genetic and environmental factors are associated with increased IBD risks. Genome‐wide association studies have identified more than 230 IBD susceptibility genes 2, 3, 4. Thus, the etiology of IBD involves a complex interaction between genetic and environmental factors that trigger an inappropriate immune response 5, 6, 7, 8, 9, 10.
Recently, tyrosine phosphatase (PTP) variants in the genes PTPN22, PTPN2 and PTPN11 have been associated with IBD onset 11. However, only a few studies have addressed their functional role in intestinal inflammation. PTP regulate fundamental signaling processes by modulating the activity of their substrates through tyrosine residue dephosphorylation 12. For example, PTPN22 controls inflammatory signaling such as NFκB, in lymphocytes and mononuclear cells, resulting in aberrant cytokine secretion and autophagosome formation. PTPN22 deficiency in vivo increases colitis symptoms, demonstrating the importance of PTPN22 to maintain intestinal homeostasis 11, 13. PTPN2 (or T‐cell phosphatase) regulates intestinal barrier function as well as innate and adaptive immune responses 14, 15. PTPN2 dysfunction in intestinal epithelial cells (IEC) also results in defective formation of autophagosomes with impaired handling of invading bacteria 16, suggesting that CD‐associated PTPN2 variants in IEC could contribute to the onset of inflammation in the intestine. Finally, polymorphisms in the PTPN11 gene encoding SHP‐2 have been described in UC patients 17. Interestingly, we 18 and others 19, 20 recently demonstrated that mice with an IEC‐specific deletion of Shp‐2 (Shp‐2 IEC‐KO) rapidly develop colonic inflammation. Intestinal mucosa disruption with crypt abscesses and immune cell infiltration indicate that the Shp‐2 IEC‐KO phenotype is similar to the phenotype observed in UC patients as opposed to CD patients 18. Importantly, a marked reduction in goblet cell numbers is observed before the inflammation onset 21. Hence, the decrease in goblet cell numbers associated with reduced secretion of the protective mucus layer could explain the spontaneous colitis developed by Shp‐2 IEC‐KO mice 18.
These findings prompted us to investigate whether sustained Shp‐2 activation in IEC could protect the mucosa against injuries. We therefore generated a conditional knock‐in mouse model expressing an activated form of Shp‐2 specifically in IEC (Shp‐2 IEC‐E76K). We show that Shp‐2 IEC‐E76K mice are resistant to dextran sulfate sodium (DSS)‐induced colitis and Citrobacter rodentium infection. We also demonstrate that, by activating the ERK pathway, Shp‐2 promotes IEC proliferation and regeneration, as well as wound healing and goblet cell differentiation, all crucial cellular processes for maintenance of the intestinal epithelial barrier and homeostasis.
Materials and methods
The antibodies used are described in supplementary material, Supplementary materials and methods. All other materials were from Sigma‐Aldrich (Oakville, ON, Canada), unless stated otherwise.
Conditional knock‐in and knock‐out mice
The complete mating information is available in supplementary material, Supplementary materials and methods. In brief, to express an active Shp‐2 protein in IEC, knock‐in Ptpn11 E76KNeo/+ mice 22 were crossed with Villin‐Cre mice 23 to generate double heterozygous experimental mice (Shp‐2 IEC‐E76K) and control littermates 24. The Shp‐2 deletion model was previously described (Shp‐2 IEC‐KO) 18. Stat3 fx/fx mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA) and crossed with Shp‐2 fx/fx mice and Villin‐Cre mice to generate double IEC‐specific knock‐out mice (Shp‐2;Stat3 IEC‐KO) and their control littermates. Finally, we crossed Braf V600E/V600E mice (provided by Dr D. Danfort from McGill University and Dr M. McMahon from University of Utah Health Sciences) described previously 25 with Shp‐2 fx/fx and Villin‐Cre to generate the experimental Shp‐2 IEC‐KO ;Braf V600E/+ mice. All experiments were approved by the Animal Research Ethics Committee of the Faculty of Medicine and Health Sciences of the Université de Sherbrooke.
Histological staining
Colons and ileums were fixed, sectioned and stained as described previously 24, 26. Immunohistochemistry was performed using a DAKO EnVision+ System kit (Lexington, MA, USA). Slides were scanned using a Nanozoomer apparatus from Hamamatsu (Shizuoka Prefecture, Japan). Mucus was visualized using Alcian blue (Polysciences, Warrington, PA, USA) with staining carried out on distal colon tissues after Carnoy's fixation.
In vivo migration assay
To determine colonic epithelial cell migration, mice were injected with 5‐Bromo‐2′‐deoxyuridine (BrdU) (10 mg/kg) (Invitrogen, Burlington, ON, Canada). Tissues were collected 18 h after injection and fixed with 4% Paraformaldehyde (PFA) prior to immunostaining sections for BrdU.
Colitis induction with DSS and clinical evaluation
Fourteen‐week‐old co‐housed Shp‐2 IEC‐E76K mice and littermates were administered 2.5% DSS (colitis grade; MP Biomedical, Solon, OH, USA) in their drinking water ad libitum for 7 days. Clinical parameters such as weight loss, rectal bleeding and diarrhea were monitored every day. The disease activity index was measured at day 7 according to Cooper et al 27 (see supplementary material, Table S1). Histological damage scoring was assessed on hematoxylin and eosin‐stained sections (see supplementary material, Table S2). All clinical scorings were performed in a blinded manner.
Citrobacter rodentium infection and bacterial counting
Co‐housed Shp‐2 IEC‐E76K mice and control littermates (10–14 weeks old) were infected by oral gavage with ∼2.5 × 108 colony‐forming units (CFU) of streptomycin‐resistant C. rodentium DBS100 28 from an overnight culture. Stools were collected every day for 10 days and the fecal bacterial load was counted (see supplementary material, Supplementary materials and methods for details). Histological damage scoring and Alcian blue staining were carried out as described above.
Western blotting and RT‐qPCR
Protein and RNA extractions, reverse transcription (RT) and western blot analyses were performed as described 24. Quantitative polymerase chain reaction (qPCR) was performed using the RNomics Platform at the Université de Sherbrooke. All primer sequences and cycling conditions are described in supplementary material, Supplementary materials and methods.
Organoids
For enteroids, crypts were isolated from the jejunum of 8‐week‐old mice with EDTA, as described previously29. Organoids were cultured and passaged as described by Sato et al 30. Culture conditions and the crypt isolation method for colonoids are detailed in supplementary material, Supplementary materials and methods. Phase‐contrast images were taken using a Zeiss Axiovert 200 M inverted microscope (Toronto, ON, Canada). Proliferation was evaluated in organoid culture using the Click‐it 5‐Ethynyl‐2′‐deoxyuridine (EdU) Alexa Fluor 555 imaging kit (Thermo Fisher Scientific, Chelmsford, MA, USA) according to the manufacturer's protocol and visualized by confocal microscopy (LSM Olympus FV1000 apparatus and FV10‐ASW 3.1 software; Olympus; Center Valley, PA, USA).
Details of the methods used for western blotting, cell culture, cell monolayer wounding assays, antibiotic pretreatments and RT‐qPCR are provided in supplementary material, Supplementary materials and methods.
Statistical analyses and figure assembly
All assays were carried out in at least triplicate. Typical results shown are representative of three independent experiments if not stated otherwise. A Student's t‐test was used for populations that followed a normal distribution curve and a Mann–Whitney test was performed for other cases. Data were expressed as mean ± standard error of the mean (SEM). Results were considered statistically significant at p ≤ 0.05. Graphs and statistics were generated using GraphPad Prism (GraphPad Software, La Jolla, CA, USA) and figures were constructed using Adobe Photoshop software (Adobe Systems, San Jose, CA, USA). Mucus layer thickness and wound healing migration were measured using ImageJ 1.45 s software (National Institutes of Health, Bethesda, MD, USA; https://imagej.nih.gov/ij/).
Results
Shp‐2 activation promotes IEC proliferation and migration
To analyze the impact of Shp‐2 activation on intestinal homeostasis, we used the Ptpn11 E76KNeo/+;Villin‐Cre transgenic mouse model (Shp‐2 IEC‐E76K). The Villin (Vil1) promoter drives Cre expression specifically in IEC 23, leading to DNA recombination and removal of transcriptional stop and Neo elements in these cells (Figure 1A). This recombination allows the expression of the gain‐of‐function Shp‐2E76K allele under control of Shp‐2 endogenous regulatory elements 22. Shp‐2 IEC‐E76K mice were born at the expected Mendelian ratio with normal body weight (data not shown). Three months after birth, Shp‐2 IEC‐E76K mice exhibited a significant increase in small and large intestine lengths in comparison with control littermates (Figure 1B). Mutant mice also exhibited deeper crypts associated with an increased number of Ki67‐labeled proliferative cells both in the small intestine (data not shown) and the colon (Figure 1C,D). Finally, epithelial cell migration rate along the colonic crypt axis, as assessed by BrdU labeling, was increased in Shp‐2 IEC‐E76K mice as opposed to controls (Figure 1E).
Figure 1.
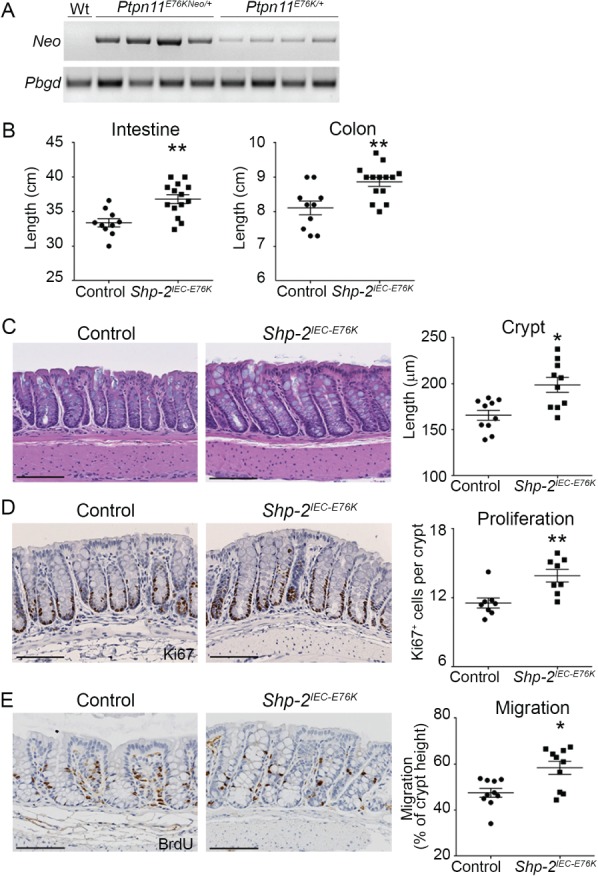
Shp‐2 activation in mouse IEC leads to hyperplasia. (A) Expression of the mutant Shp‐2E76K in enriched colonic mucosal extracts was validated by comparing Neo cassette excision from genomic DNA of Ptpn11 E76KNeo/+ and Ptpn11 E76K/+ (Shp‐2 IEC‐E76K) mice by PCR and Pbgd was used as a reference gene. (B) Small intestine and colon length was measured on both Shp‐2 IEC‐E76K mice and control littermates at 3 months of age. n ≥ 10. (C) Colon tissue architecture was visualized with hematoxylin and eosin staining and crypt length was measured. n = 10. (D) Immunohistochemistry for Ki67 was performed to measure proliferation. The number of positive cells per crypt was counted. n = 15 crypts per mouse. Graph presents mean value of n = 8 mice. (E) In vivo migration was assessed with an 18 h BrdU pulse prior to sacrifice, followed by immunohistochemistry on colonic sections. The migration front distance was measured and reported relative to total crypt length. n = 15 crypts per mouse. Graph shows mean value of n = 10 mice. Graphs present mean ± SEM. Mann–Whitney *p ≤ 0.05, **p ≤ 0.01. Scale bars = 100 μm.
Intestinal epithelial Shp‐2 activation perturbs secretory cell maturation
We have previously shown that IEC‐specific Shp‐2 ablation leads to lower goblet cell numbers while promoting higher Paneth cell numbers in the small intestine 18, 21. We therefore analyzed differentiated secretory cell types in the contrasting Shp‐2 activation model. Interestingly, Shp‐2 activation promoted goblet cell expansion (Figure 2A) but decreased Paneth cell numbers (Figure 2B,C) in the small intestine, as well as Reg3γ (Reg3g) and Angiogenin‐4 (Ang4) antimicrobial peptide gene expression (Figure 2D). Goblet cell number was also increased in the colon of Shp‐2 IEC‐E76K mice in comparison with control littermates (Figure 2E), which correlates with a thicker inner mucus layer (Figure 2F), suggesting an increase in goblet cell secretory function upon Shp‐2 activation 31. Proper mucus maturation was observed in both Shp‐2 IEC‐E76K and control colons, as indicated by Ulex europaeus‐I agglutinin (UEA‐I) lectin staining of fucosylated mucins 32 (Figure 2G). Tff3 and Muc5ac gene expression was also enhanced following Shp‐2 activation in mouse colon (Figure 2H). As previously suggested 33, the expression of goblet cell factors is not evenly distributed, indicating the existence of different goblet cell subsets. For example, strong Tff3 staining was mostly localized on goblet cells closer to the crypt tips, suggesting that its synthesis and accumulation occurred in the more mature subset of cells. No major difference in Tff3 localization was observed between the two groups of mice (see supplementary material, Figure S1A). Furthermore, a sparse pattern for Relm‐beta expression was observed on isolated goblet cells, which was maintained in the Shp‐2 IEC‐E76K mice (see supplementary material, Figure S1B). In addition, western blotting revealed that, although Tff3 protein synthesis was increased in Shp‐2 IEC‐E76K mice, there were no significant changes in Relm‐beta protein levels (see supplementary material, Figure S1C,D). Therefore, sustained IEC‐specific Shp‐2 activation promotes the expansion of some, but not all, goblet cell subsets.
Figure 2.
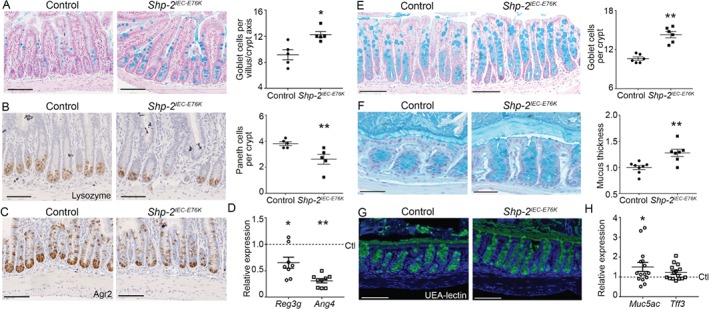
Shp‐2 activation in mouse IEC leads to Paneth cell depletion but promotes goblet cell differentiation. Alcian blue staining (A) and lysozyme and Agr2 immunohistochemistry (B, C) were performed on ileal tissues from control and Shp‐2 IEC‐E76K mice to visualize goblet and Paneth cells. Goblet cells were counted per villus–crypt axis and Paneth cells per crypt. Approximately 15 villus–crypt axes per animal were counted. Graph represents mean value of n = 5. (D) Reg3γ (Reg3g) and Angiogenin‐4 (Ang4) expression were measured by RT‐qPCR in ileal mucosal samples. Expression was normalized to the reference transcripts Psmc4, Pum1 and Tbp and results were normalized to the control. n = 8. (E) Alcian blue staining was performed on colon sections from control and Shp‐2 IEC‐E76K mice to visualize goblet cells. The number of goblet cells per crypt was counted. n = 15 crypts per mouse. Graph represents mean value of n = 6 mice. (F) Inner mucus layer thickness was measured after Carnoy's fixation followed by Alcian blue staining on distal colon from Shp‐2 IEC‐E76K and control littermates using ImageJ software. n ≥ 7. (G) Mucus fucosylation was visualized by immunofluorescence with a UEA‐lectin‐FITC reagent. (H) Muc5ac and Tff3 (p = 0.0581) expression were measured by RT‐qPCR in colonic mucosal samples. Expression was normalized to the reference transcript Psmc4, Pum1 and Tbp and results were normalized to the control. n = 15. Graphs present mean ± SEM. Mann–Whitney *p ≤ 0.05, **p ≤ 0.01. (A) Scale bars = 150 μm. (B–D) Scale bars = 100 μm. (F) Scale bars = 50 μm.
Intestinal epithelial Shp‐2 activation protects against chemical colitis
Based on the homeostatic changes observed in Shp‐2 IEC‐E76K murine intestines, particularly in the colon, and on our previous observation of increased colonic inflammation after IEC‐specific Shp‐2 deletion 18, we postulated that activation of Shp‐2 would protect against the onset of colitis. We employed the widely used DSS‐induced colitis model to analyze acute colonic inflammation 27 by giving both control and mutant mice 2.5% DSS in drinking water for 7 days. While control mice lost nearly 20% of their body weight (Figure 3A) and exhibited severe signs of diarrhea and rectal bleeding resulting in a high disease activity index and histological scoring (Figure 3B,C), Shp‐2 IEC‐E76K mice exhibited less weight loss, diarrhea and bloody stools after DSS treatment as well as milder epithelial alterations at the microscopic level (Figure 3A–C). These findings suggest a protective effect of sustained Shp‐2 activity upon DSS‐induced acute injury.
Figure 3.
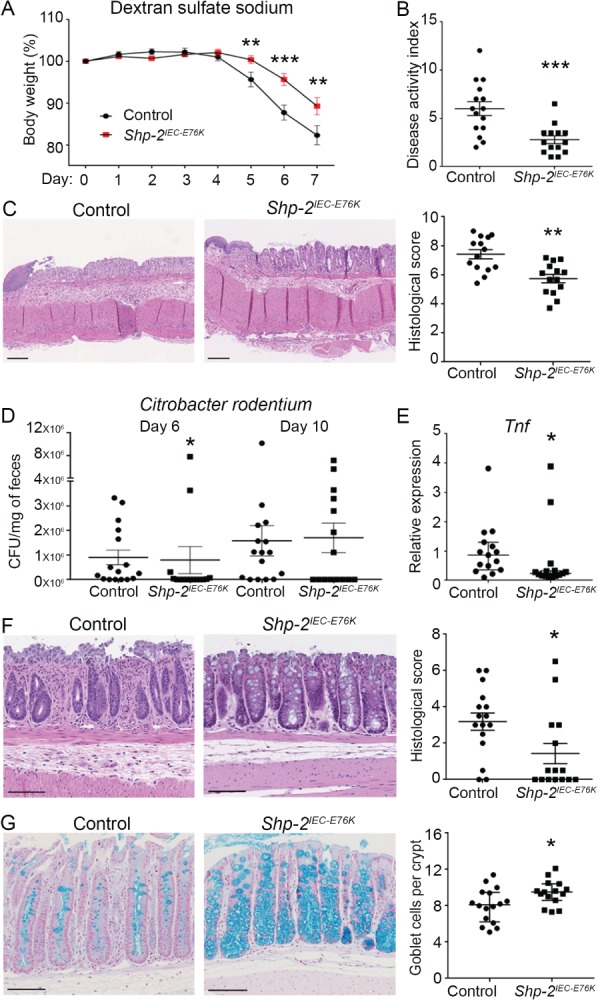
Shp‐2 activation protects against DSS and C. rodentium‐induced colitis in mice. (A) Shp‐2 IEC‐E76K mice and control littermates were treated with 2.5% DSS for 7 days to induce acute colitis. Weight was measured every day during treatment. Graph presents weight loss as a percentage of initial weight. n ≥ 14. (B) Disease activity index of Shp‐2 IEC‐E76K mice and control littermates was calculated by scoring stool softness, occult fecal blood, rectal bleeding and colon rigidity at sacrifice. n ≥ 14. (C) Hematoxylin and eosin staining of distal colon tissue from control and Shp‐2 IEC‐E76K mice was used to score the mucosal damage after DSS treatment, according to the presence of mucosal architecture destruction, immune cell infiltration, muscle thickening, goblet cell depletion and crypt abscesses. n ≥ 14. (D) Shp‐2 IEC‐E76K mice and littermate controls were gavaged with 2.5 × 108 C. rodentium and fecal bacterial load (CFU/mg feces) was measured on days 6 and 10. n ≥ 14. (E) Tnf expression was measured by RT‐qPCR from RNA isolated from total colonic tissues 10 days postinfection. Expression was normalized to the reference transcripts Psmc4, Pum1 and Tbp. n ≥ 14. (F) Histological scoring was performed on the distal colon from Shp‐2 IEC‐E76K mice and their littermate controls after hematoxylin and eosin staining. n ≥ 14. (G) Alcian blue was used to stain goblet cells in the colon of Shp‐2 IEC‐E76K mice and their control littermates. The number of Alcian blue‐positive cells was counted in 15 crypts per animal. Graph presents mean value of n ≥ 14. Scale bars = 100 μm. Graphs present mean ± SEM. Mann–Whitney *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Rapid and efficient mucosal wound healing after epithelial inflammatory damages is essential for colitis remission 34. To determine whether Shp‐2 signaling may regulate IEC restitution after wounding, we infected Caco‐2/15 cells with recombinant lentiviruses encoding anti‐SHP‐2 short hairpin RNA (shRNA) to stably suppress SHP‐2 expression. Notably, SHP‐2 silencing inhibited Caco‐2/15 cell monolayer migration after wounding (see supplementary material, Figure S2). Thus, SHP‐2 induces restitution processes in IEC.
Intestinal epithelial Shp‐2 activation promotes intestinal colonization resistance to C. rodentium
Our data showed that IEC‐specific Shp‐2 activation protected against chemically induced colitis. To determine whether Shp‐2 activation could also protect against bacterial infections, we used the C. rodentium infection model to study host immune responses against bacterial pathogens in the intestine 35, 36, 37. Normally, upon infection with C. rodentium, mice display modest and transient weight loss and diarrhea with colonic crypt elongation, immune cell infiltration and goblet cell depletion 36. Interestingly, 6 days after C. rodentium infection, Shp‐2 IEC‐E76K mice displayed reduced bacterial titers in their stools compared with their control littermates (Figure 3D). This apparent resistance to C. rodentium colonization was consistently observed in Shp‐2 IEC‐E76K mice, regardless of ancestry (breeders), gender or housing. Consistent with reduced bacterial colonization, Shp‐2 IEC‐E76K‐infected mice displayed reduced proinflammatory cytokine Tnf expression in the distal colon (Figure 3E), in contrast to infected wild‐type mice. In addition, Shp‐2 IEC‐E76K‐infected mice were protected against inflammatory lesions (Figure 3F) and goblet cell depletion (Figure 3G), phenotypes usually induced by C. rodentium 36, 37. These findings suggest that activation of IEC Shp‐2 plays a protective role against bacterial‐induced colitis.
To verify whether the apparent resistance of Shp‐2 IEC‐E76K mice to Citrobacter infection may reflect changes in the microbiota, we tested the susceptibility of these mice to C. rodentium infection following a streptomycin pretreatment. As shown in supplementary material, Figure S3, Shp‐2 IEC‐E76K mice exhibited similar bacterial burdens, goblet cell depletion and sensitivity to C. rodentium infection as control mice after pretreatment with streptomycin. These results clearly suggest that the microbiota indeed plays a key role in the resistance to infection observed in Shp‐2 IEC‐E76K mice.
Activation of Braf/ERK signaling promotes goblet cell expansion and represses colitis development in Shp‐2 IEC‐KO mice
We previously reported that IEC‐specific Shp‐2 deletion in mice (Shp‐2 IEC‐KO mice) triggers spontaneous colitis associated with barrier function impairment. Importantly, before the appearance of inflammation, Stat3 transcription factor was hyperphosphorylated while ERK1/2 phosphorylation was reduced in the colonic epithelium of Shp‐2 IEC‐KO mice 18. We therefore analyzed the contribution of Stat3 and ERK1/2 signaling in colitis development observed in Shp‐2‐deficient mice. Interestingly, epithelial Stat3 deletion modestly but significantly attenuated colitis severity in 2‐week‐old Shp‐2 IEC‐KO mice, as illustrated by the partial restoration of body weight and reduction of histological damages (see supplementary material, Figure S4). To determine if ERK inhibition contributes to colitis development in these mice, we crossed Shp‐2‐deficient mice with Braf V600E mice carrying a Cre‐activated allele of the murine Braf gene 25. Braf V600E and Shp‐2 IEC‐KO ;Braf V600E mice were born at normal Mendelian frequency and were apparently healthy (data not shown). As expected, phosphorylation levels of MEK1/2, the direct substrates of Braf, were clearly enhanced in Braf V600E murine colons, whereas they were barely detectable in Shp‐2 IEC‐KO colons (Figure 4A). Interestingly, MEK phosphorylation levels observed in Shp‐2 IEC‐KO ;Braf V600E mice were quite similar to the levels observed in Braf V600E mice, hence indicating that Braf activation prevented the downregulation of MEK1/2 phosphorylation induced by Shp‐2 deletion (Figure 4A). Of further importance, none of the Shp‐2 IEC‐KO ;Braf V600E mice developed spontaneous colitis, in contrast to Shp‐2 IEC‐KO mice, as demonstrated by histological analysis and score (Figure 4B,C). Alcian blue staining revealed a marked increase in goblet cell numbers in Braf V600E murine colons as opposed to controls and the goblet cell reduction observed in Shp‐2 IEC‐KO mice was rescued in Shp‐2 IEC‐KO ;Braf V600E mice (Figure 4D). Additionally, we did not find lysozyme‐positive cells in Shp‐2 IEC‐KO ;Braf V600E colons, in contrast to Shp‐2 IEC‐KO colons (Figure 4E, see arrows) 21. Of importance, we found increased phosphorylated ERK1/2 MAPK in colonic epithelial enrichments from Shp‐2 IEC‐E76K mice (Figure 4F). Taken together, these data suggest that Shp‐2 may primarily protect against colonic infl ammation by most likely activating the Braf/MEK/ERK signaling, which promotes goblet cell expansion.
Figure 4.
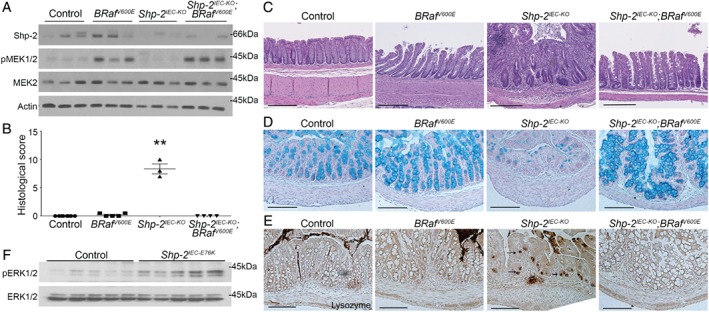
Shp‐2 controls the intestinal epithelial secretory cell fate through the Raf/MAPK pathway. (A) Western blotting of enriched colonic mucosal protein extracts to measure MEK (S217/S221) activation in Shp‐2 IEC‐KO, Braf V600E‐expressing and dual Shp‐2 IEC‐KO; Braf V600E‐expressing mice. Total MEK2 and β‐actin were used as loading controls. n = 3. (B,C) Colon tissue architecture was visualized following hematoxylin and eosin staining and colonic inflammation observed in Shp‐2 IEC‐KO mice was prevented by Braf activation. Histological score was calculated for each 1‐month‐old mouse. n ≥ 3. (D) Alcian blue staining was used to visualize goblet cells in the proximal colon of control, Braf V600E, Shp‐2 IEC‐KO and Shp‐2 IEC‐KO ;Braf V600E mice. n ≥ 3. (E) Immunohistochemistry for lysozyme was performed on proximal colonic tissues to confirm Paneth cell metaplasia in Shp‐2 IEC‐KO mice (arrow) and its absence in Shp‐2 IEC‐KO ;Braf V600E mice. n ≥ 3. (F) Western blotting was performed on enriched colonic protein extracts to measure ERK (T202/Y204) activation in Shp‐2‐activated mucosa. Total ERK1/2 was used as a loading control. n = 5. Scale bars = 100 μm. Graph presents mean ± SEM. Mann–Whitney **p ≤ 0.01.
ERK signaling promotes differentiation of goblet cells by inhibiting the NOTCH pathway
To determine the cell‐intrinsic effect of ERK signaling on goblet cell function, we used LS174T cells, which are a representative model of secretory progenitor cells committed to differentiate into goblet cells 38, 39. As shown in Figure 5, inhibition of ERK signaling with either an ERK inhibitor (SCH772984) or MEK inhibitor (CI‐1040) significantly reduced levels of MUC2 as well as Alcian blue staining in LS174T cells. As expected, γ‐secretase inhibition increased MUC2 expression and Alcian blue staining (Figure 5B,C). Because NOTCH signaling governs the differentiation of secretory lineage specification and MUC2 expression 39, 40, 41, we evaluated whether MEK/ERK signaling modulates NOTCH pathway activation in LS174T cells. As shown in Figure 5D, pharmacological inhibition of the MEK/ERK pathway promoted NOTCH signaling, as evidenced by increased protein expression of the NOTCH intracellular domain (NICD) and the NOTCH target HES1. Concomitant NOTCH pathway inhibition with DBZ and MEK/ERK inhibition prevented NICD and HES1 activation, indicating a NOTCH receptor‐dependent mechanism (Figure 5D). As these data suggest that MEK/ERK inhibition activates NOTCH signaling, we verified mRNA expression of NOTCH receptors and ligands. Notably, MEK and ERK inhibition significantly increased DLL1 and DLL4 gene expression (Figure 5E). NOTCH1 and NOTCH3 transcript levels were also modestly enhanced following MEK/ERK inhibition, but not significantly. Taken together, these results suggest that MEK/ERK signaling promotes goblet cell progenitor differentiation by reducing activation of the NOTCH signaling pathway.
Figure 5.
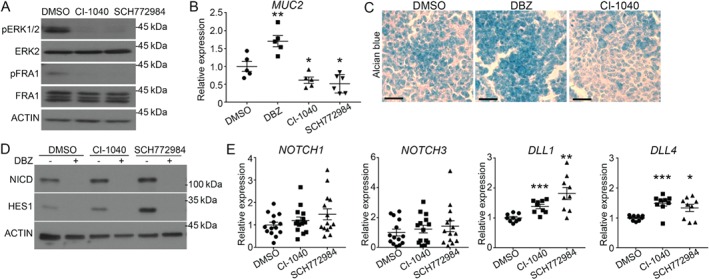
ERK signaling promotes differentiation of LS174T goblet cells by inhibiting the NOTCH pathway. (A) Western blotting for the level of ERK phosphorylation (T202/Y204) and phosphorylation of ERK substrate FRA1 (S265) in LS174T cells treated for 3 h with inhibitors of either MEK (CI‐1040, 2 μm) or ERK (SCH772984, 1 μm). Total ERK2, FRA1 and β‐actin were used as loading controls. n = 3. (B) LS174T cells were treated with DMSO, 1 μm DBZ (γ‐secretase inhibitor), 2 μm CI‐1040 (MEK inhibitor) or 1 μm SCH772984 (ERK1/2 inhibitor) for 72 h before measuring MUC2 expression by RT‐qPCR. Expression was normalized to the reference transcripts PUM1, MRPL19 and RPL13A. n ≥ 5. (C) Alcian blue staining was performed on EtOH fixed cells following 72 h of treatment with DMSO, DBZ or CI‐1040. n = 3. (D) Western blotting to measure the level of HES1 expression and Notch cleavage (NICD) in LS174T cells treated with either DBZ or MEK/ERK inhibitors, with β‐actin used as a loading control. n = 3. (E) NOTCH1, NOTCH3 as well as DLL1 and DLL4 mRNA expression were measured by RT‐qPCR in LS174T cells treated with CI‐1040 and SCH772984. Expression was normalized to the reference transcripts PUM1, MRPL19 and RPL13A. n ≥ 9 for each treatment. Graphs present mean ± SEM. Student's t‐test *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Shp‐2 signaling induces intestinal crypt regeneration through the activation of MEK/ERK signaling pathway
We then examined the effect of Shp‐2 activation on IEC regeneration by using the ex vivo organoid culture system 42, 43. As shown in Figure 6A,B, organoids derived from both Shp‐2 IEC‐E76K small intestine and colon developed much more rapidly in comparison with organoids derived from control mice. Furthermore, activated Shp‐2 promoted de novo crypt formation in enteroids, as demonstrated by increased numbers of protrusions compared with controls after 5 days (Figure 6C). Proliferation was markedly increased in Shp‐2 IEC‐E76K ‐derived enteroids and colonoids, as determined by EdU incorporation (Figure 6D,E).
Figure 6.
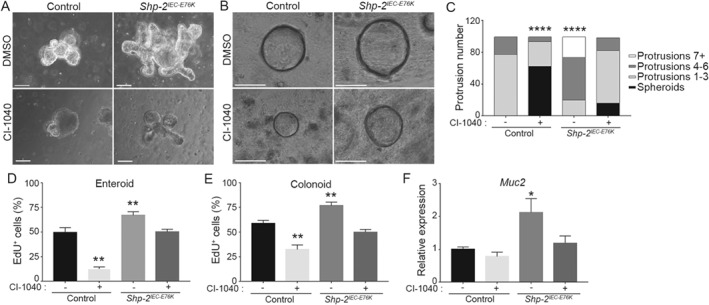
Shp‐2 drives crypt regeneration through ERK/MAPK pathway. Intestinal crypt enteroids and colonoids from Shp‐2 IEC‐E76K or control mice were cultured in Matrigel for 12 h before treatment with the MEK inhibitor CI‐1040 (8 μm) or DMSO. Images present (A) enteroids and (B) colonoids cultured for 4 days with or without MEK inhibitor. Scale bars = 50 μm. (C) At day 4, protrusion numbers from 100 control or Shp‐2 IEC‐E76K enteroids were counted for each condition. The graph presents the percentage of organoids with a certain number of protrusions. A representative experiment is shown. n = 5. EdU incorporation staining was used to measure proliferation in control and Shp‐2 IEC‐E76K enteroids (D) and colonoids (E), treated with or without CI‐1040 at 8 μm for 4 days. For enteroids, EdU‐positive cells per protrusion were counted. For colonoids, EdU‐positive cells were counted per whole organoid. Approximatively 100 organoids were analyzed per condition and the experiments were conducted with n = 5 mice. (F) Muc2 gene expression by RT‐qPCR in control and Shp‐2 IEC‐E76K enteroids with or without MEK inhibition. Expression was normalized to the reference transcripts Psmc4, Pum1 and Tbp. n = 5. Graphs present mean ± SEM. Student's t‐test *p ≤ 0.05, **p ≤ 0.01, ****p ≤ 0.0001.
To determine whether elevated ERK signaling contributes to these phenotypic alterations, we treated organoids with the CI‐1040 inhibitor. As expected, MEK inhibition resulted in a reduction in the number of protrusions and EdU‐positive cells in control organoids (Figure 6C–E). However, MEK inhibition in Shp‐2 IEC‐E76K‐derived organoids reduced protrusion numbers and proliferation to levels observed in control organoids under untreated conditions (Figure 6C–E). Notably, Shp‐2 activation in enteroids also significantly enhanced Muc2 transcription in a MEK‐dependent manner (Figure 6F). Taken together, these results indicate that Shp‐2 promotes intestinal crypt self‐renewing capacity by activating the ERK pathway.
Discussion
Polymorphisms in the gene PTPN11 encoding SHP‐2 are associated with UC susceptibility 17. However, how SHP‐2 and SHP‐2 variants contribute to disease susceptibility is still unknown. We and others have demonstrated that IEC‐specific Shp‐2‐deleted mice (Shp‐2 IEC‐KO) rapidly and spontaneously develop severe colonic inflammation 18, 19, 20. The gut microbiota may play an important role as antibiotic treatment remarkably impaired the colitis development in Shp‐2 IEC‐KO mice 18. Importantly, SHP‐2 gene expression is reduced in mucosae from patients suffering from colitis 18, hence emphasizing the inverse relationship between SHP‐2 expression and the inflammatory phenotype. The above observations therefore prompted us to speculate that sustained Shp‐2 activation would protect the colonic mucosa against injury and inflammation. Indeed, we have shown that conditional knock‐in mice, specifically expressing the constitutively active Shp‐2E76K mutant in IEC, developed attenuated colitis symptoms following DSS treatment and C. rodentium infection. Thus, Shp‐2 activation exerts protective actions against mucosal damage and pathogen infection.
The most important question raised by our data is how IEC‐specific Shp‐2 mediates such protective action. Being on the front line of defense, the intestinal epithelium continuously sustains injuries. After mucosal damage, wound healing needs to be rapid and efficient in order to limit microbial contamination of surrounding tissues. Hence, through a sequence of events involving IEC restitution or migration, proliferation and differentiation, the wound is resealed and homeostasis is re‐established 34. In the present study, we provide several lines of evidence indicating that Shp‐2 plays a significant role in mediating key steps of mucosal repair after damage. First, colonic crypt cell proliferation and migration rates are both increased in Shp‐2 IEC‐E76K mice. Second, goblet cell expansion associated with a thicker mucus layer and increased Tff3 expression are observed in Shp‐2 IEC‐E76K murine colons, suggesting a reinforcement of barrier integrity. Indeed, Tff3 stabilizes the mucus layer and contributes to epithelial healing 44. Third, SHP‐2 silencing in IEC monolayers reduces migration after wounding. Fourth, sustained Shp‐2 activity strongly increases intestinal crypt self‐renewing and differentiating capacity, as demonstrated in ex vivo cultured organoids. Taken together, these data suggest that Shp‐2 actively confers resistance to colonic mucosae against various insults by promoting mucosal healing, goblet cell function and epithelium regeneration.
It remains unclear how activation of Shp‐2 leads to lower C. rodentium burden during infection of Shp‐2 IEC‐E76K mice, although it would be fair to assume that such an effect is associated with the increased goblet cell number and function. A thicker and more stable mucus layer would be more effective at eliminating the bacteria given that the mucus actively helps ‘to flush’ attaching/effacing pathogens such as C. rodentium 33. Alternatively, the observed changes in goblet and Paneth cells in Shp‐2 IEC‐E76K mice might be expected to alter the intestinal microbiome 45, which is now recognized as an important factor influencing susceptibility to C. rodentium infection 37, 46. Accordingly, we found that, after streptomycin pretreatment, Shp‐2 IEC‐E76K mice exhibit similar bacterial burdens and sensitivity to C. rodentium infection as control mice. Although we did not examine microbial changes, these data reveal that the microflora does protect Shp‐2 IEC‐E76K mice against C. rodentium infection. By which pathways the microbiota of Shp‐2 IEC‐E76K mice mediates such protection remains to be investigated. Ghosh et al 46 previously demonstrated that the colonic microbiota can induce proinflammatory and pro‐oxidant responses that control pathogen load. Among other pathways of protection, we noticed that Shp‐2 IEC‐E76K mice were resistant to goblet cell depletion usually induced by C. rodentium. However, goblet cell depletion was observed if mice were pretreated with streptomycin before infection. Hence, one might speculate that microbiota changes occurring in Shp‐2 IEC‐E76K mice contribute to the observed goblet cell expansion. In this regard, a recent study has shown that microbiota promotes secretory cell fate determination by inhibiting host Notch signaling in the intestinal epithelium 47.
Several signaling pathways, including Jak/Stat, NF‐kB, PI3K/Akt and RhoA pathways, have been shown to be regulated directly or indirectly by SHP‐2 in cellulo 48, 49, 50. Among these pathways, Stat3 may be particularly relevant because it can be inactivated by SHP‐2 in IECs and it is an important regulator of mucosal wound healing in the intestine 51, 52. Unexpectedly, we observed that IEC‐specific Stat3 deletion attenuated colitis severity in Shp‐2 IEC‐KO mice, suggesting that sustained activation of Stat3 in IECs may rather contribute to inflammation. In line with this, increases in serum interleukin‐6 concentrations and Stat3 hyperactivation were observed in IBD patients, as well as in several experimental colitis models 53, 54.
Another pathway that is tightly regulated by SHP‐2 is the ERK MAPK pathway. Indeed, many studies have demonstrated that SHP‐2 is required for normal ERK activation in multiple cellular contexts, including IECs 55, 56. Accordingly, our data show increased ERK1/2 phosphorylated levels in conditional knock‐in mice expressing the IEC‐specific Shp‐2 E76K activating mutation. Conversely, MEK/ERK signaling is reduced in Shp‐2‐deficient intestinal epithelium (18 and herein). These results emphasize the positive role of Shp‐2 in mediating the activation of IEC‐specific ERK signaling. Notably, expression of an activated Braf mutant in Shp‐2 IEC‐KO mice restores MEK phosphorylation and goblet cell numbers and prevents spontaneous colitis development. Hence, inhibition of ERK signaling may be crucial in the deregulation of goblet cell differentiation and colitis development observed upon Shp‐2 inactivation.
Heuberger et al 19 recently reported that Shp‐2‐dependent ERK signaling controls the choice between goblet and Paneth cell fates in the small intestine. Although the molecular mechanisms involved still remain elusive, the authors hypothesize that Shp‐2/MAPK suppression in intestinal crypts may increase Wnt/β‐catenin signaling, which in turn promotes stem cell function and Paneth cell differentiation. However, it remains unclear why goblet cell differentiation is so markedly modulated following Shp‐2 deletion and ERK inhibition 18, 19. One of the key pathways controlling differentiation of secretory cell lineages, including intestinal goblet cells, is the Notch pathway 40, 41, 57, 58. For example, simultaneous inactivation of both Notch1 and Notch2 receptors resulted in complete conversion of progenitor cells in postmitotic goblet cells 59. Interestingly, the Notch signaling was stimulated following MEK or ERK inhibition in LS174T cells, which are committed to differentiate into goblet cells 38, 39. Notch signaling activation after MEK/ERK inhibition may be a consequence of increased expression of Dll1 and Dll4, two potent ligands of Notch receptors in progenitor cells. Indeed, binding of these ligands to Notch receptors induces a conformational change exposing a cleavage site for ADAM family proteases. This allows cleavage by γ‐secretase, therefore resulting in the production of NICD, the cleaved form of Notch 58. Our data showing that inhibition of the γ‐secretase complex by DBZ prevents NICD and HES1 accumulation induced by ERK inhibition confirm this NOTCH‐dependent mechanism.
To our knowledge, this is the first study to report that Shp‐2 activation protects the colonic mucosa against chemical or bacterial insults. In part through the activation of ERK signaling, the Shp‐2 phosphatase regulates multiple aspects of colon epithelial homeostasis, including proliferation, differentiation, barrier function and wound closure, to optimize intestinal homeostatic responses to various injuries. Importantly, polymorphisms in the PTPN11 gene coding for SHP‐2 were previously associated with UC susceptibility. However, the impact of these polymorphisms on SHP‐2 expression or activity remains known. One could speculate that PTPN11 SNPs reduce SHP‐2 expression. Experiments are currently in progress in specimens from IBD patients to test this hypothesis.
Author contributions statement
JG‐S performed most of the research shown in Figures 1, 2, 3, 4, 5, 6 and in supplementary material, Figure S2. GC performed the experiments shown in supplementary material, Figure S1, while AL performed experiments shown in some panels of Figures 4 and 5 and in supplementary material, Figure S2. VV‐L and M‐JL helped with the handling of mice and cell lines. M‐JL also contributed to the organoid experiments shown in Figure 6. CKQ provided Ptpn11 E76KNeo/+ mice and revised the manuscript. ST and AM designed the experiments shown in Figure 3D–G (C. rodentium infection) and revised the manuscript. JG‐S, M‐JL and NR wrote the paper. NR was in charge of overall direction and planning. All authors read and approved the final manuscript.
SUPPLEMENTARY MATERIAL ONLINE.
Supplementary materials and methods
Supplementary figure legends
Figure S1. Shp‐2 activation enhances a goblet cell subset producing Tff3 but not Resistin‐like beta
Figure S2. Shp‐2 is required for epithelial wound healing
Figure S3. Intestinal microbiota is involved in Shp‐2‐induced protection against C. rodentium infection
Figure S4. Stat3 activation enhanced colitis burden in Shp‐2IEC‐KO mice
Table S1. Disease activity index
Table S2. Histological damage scoring
Table S3. Primer sequences for RT‐qPCR
Supporting information
Supplementary materials and methods
Supplementary figure legends
Figure S1. Shp‐2 activation enhances a goblet cell subset producing Tff3 but not Resistin‐like beta
Figure S2. Shp‐2 is required for epithelial wound healing
Figure S3. Intestinal microbiota is involved in Shp‐2‐induced protection against C. rodentium infection
Figure S4. Stat3 activation enhanced colitis burden in Shp‐2 IEC‐KO mice
Table S1. Disease activity index
Table S2. Histological damage scoring
Table S3. Primer sequences for RT‐qPCR
Acknowledgements
We thank Pr Claude Asselin for critical reading of the manuscript. We thank the RNomics and Electron Microscopy & Histology Platforms of the FMSS at the Université de Sherbrooke for services. Special thanks to Drs David Dankort (McGill University) and Martin McMahon (University of Utah Health Sciences) for providing Braf V600E mice and to Dr Daniel Podolsky (UT Southwestern Medical Center) for sharing a Tff3 antibody. This research was supported by a grant from Canadian Institutes of Health Research (CIHR) to Nathalie Rivard. Jessica Gagné‐Sansfaçon is a FRQS student scholar. Nathalie Rivard and Alfredo Menendez are members of the FRQS‐Funded Centre de Recherche du CHUS. Nathalie Rivard is a recipient of a Canadian Research Chair in colorectal cancer and inflammatory cell signaling.
No conflicts of interest were declared.
References
*Cited only in supplementary material.
- 1. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol 2010; 28: 573–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cho JH, Brant SR. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology 2011; 140: 1704–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Loddo I, Romano C. Inflammatory bowel disease: genetics, epigenetics, and pathogenesis. Front Immunol 2015; 6: 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu JZ, van Sommeren S, Huang H, et al Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 2015; 47: 979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ogura Y, Bonen DK, Inohara N, et al A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 2001; 411: 603–606. [DOI] [PubMed] [Google Scholar]
- 6. Hugot JP, Chamaillard M, Zouali H, et al Association of NOD2 leucine‐rich repeat variants with susceptibility to Crohn's disease. Nature 2001; 411: 599–603. [DOI] [PubMed] [Google Scholar]
- 7. Jostins L, Ripke S, Weersma RK, et al Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491: 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ek WE, D'Amato M, Halfvarson J. The history of genetics in inflammatory bowel disease. Ann Gastroenterol 2014; 27: 294–303. [PMC free article] [PubMed] [Google Scholar]
- 9. McCole DF. IBD candidate genes and intestinal barrier regulation. Inflamm Bowel Dis 2014; 20: 1829–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Laukoetter MG, Nava P, Nusrat A. Role of the intestinal barrier in inflammatory bowel disease. World J Gastroenterol 2008; 14: 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Spalinger MR, McCole DF, Rogler G, et al Role of protein tyrosine phosphatases in regulating the immune system: implications for chronic intestinal inflammation. Inflamm Bowel Dis 2015; 21: 645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tiganis T, Bennett AM. Protein tyrosine phosphatase function: the substrate perspective. Biochem J 2007; 402: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Glas J, Wagner J, Seiderer J, et al PTPN2 gene variants are associated with susceptibility to both Crohn's disease and ulcerative colitis supporting a common genetic disease background. PLoS One 2012; 7: e33682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Spalinger MR, Kasper S, Chassard C, et al PTPN2 controls differentiation of CD4+ T cells and limits intestinal inflammation and intestinal dysbiosis. Mucosal Immunol 2015; 8: 918–929. [DOI] [PubMed] [Google Scholar]
- 15. Scharl M, Paul G, Weber A, et al Protection of epithelial barrier function by the Crohn's disease associated gene protein tyrosine phosphatase n2. Gastroenterology 2009; 137: 2030–2040.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hendriks WJAJ, Pulido R. Protein tyrosine phosphatase variants in human hereditary disorders and disease susceptibilities. Biochim Biophys Acta 2013; 1832: 1673–1696. [DOI] [PubMed] [Google Scholar]
- 17. Narumi Y, Isomoto H, Shiota M, et al Polymorphisms of PTPN11 coding SHP‐2 as biomarkers for ulcerative colitis susceptibility in the Japanese population. J Clin Immunol 2009; 29: 303–310. [DOI] [PubMed] [Google Scholar]
- 18. Coulombe G, Leblanc C, Cagnol S, et al Epithelial tyrosine phosphatase SHP‐2 protects against intestinal inflammation in mice. Mol Cell Biol 2013; 33: 2275–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Heuberger J, Kosel F, Qi J, et al Shp2/MAPK signaling controls goblet/paneth cell fate decisions in the intestine. Proc Natl Acad Sci U S A 2014; 111: 3472–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamashita H, Kotani T, Park J‐H, et al Role of the protein tyrosine phosphatase Shp2 in homeostasis of the intestinal epithelium. PLoS One 2014; 9: e92904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Coulombe G, Langlois A, De Palma G, et al SHP‐2 phosphatase prevents colonic inflammation by controlling secretory cell differentiation and maintaining host‐microbiota homeostasis. J Cell Physiol 2016; 231: 2529–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu D, Liu X, Yu W‐M, et al Non‐lineage/stage‐restricted effects of a gain‐of‐function mutation in tyrosine phosphatase Ptpn11 (Shp2) on malignant transformation of hematopoietic cells. J Exp Med 2011; 208: 1977–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Madison BB, Dunbar L, Qiao XT, et al Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem 2002; 277: 33275–33283. [DOI] [PubMed] [Google Scholar]
- 24. Gagné‐Sansfaçon J, Coulombe G, Langlois M‐J, et al SHP‐2 phosphatase contributes to KRAS‐driven intestinal oncogenesis but prevents colitis‐associated cancer development. Oncotarget 2016; 7: 65676–65695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dankort D, Filenova E, Collado M, et al A new mouse model to explore the initiation, progression, and therapy of BRAFV600E‐induced lung tumors. Genes Dev 2007; 21: 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leblanc C, Langlois M‐J, Coulombe G, et al Epithelial Src homology region 2 domain‐containing phosphatase‐1 restrains intestinal growth, secretory cell differentiation, and tumorigenesis. FASEB J 2017; 31: 3512–3526. [DOI] [PubMed] [Google Scholar]
- 27. Cooper HS, Murthy SN, Shah RS, et al Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest 1993; 69: 238–249. [PubMed] [Google Scholar]
- 28. Tremblay S, Romain G, Roux M, et al Bile acid administration elicits an intestinal antimicrobial program and reduces the bacterial burden in two mouse models of enteric infection. Infect Immun 2017; 85: e00942–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gonneaud A, Jones C, Turgeon N, et al A SILAC‐based method for quantitative proteomic analysis of intestinal organoids. Sci Rep 2016; 6: 38195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sato T, Stange DE, Ferrante M, et al Long‐term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology 2011; 141: 1762–1772. [DOI] [PubMed] [Google Scholar]
- 31. Birchenough GMH, Johansson MEV, Gustafsson JK, et al New developments in goblet cell mucus secretion and function. Mucosal Immunol 2015; 8: 712–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Holmén Larsson JM, Thomsson KA, Rodríguez‐Piñeiro AM, et al Studies of mucus in mouse stomach, small intestine, and colon. III. Gastrointestinal Muc5ac and Muc2 mucin O‐glycan patterns reveal a regiospecific distribution. Am J Physiol Gastrointest Liver Physiol 2013; 305: G357–G363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Allaire JM, Morampudi V, Crowley SM, et al Frontline defenders: goblet cell mediators dictate host–microbe interactions in the intestinal tract during health and disease. Am J Physiol Gastrointest Liver Physiol 2018; 314: G360–G377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sturm A, Dignass AU. Epithelial restitution and wound healing in inflammatory bowel disease. World J Gastroenterol 2008; 14: 348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Garmendia J, Frankel G, Crepin VF. Enteropathogenic and enterohemorrhagic Escherichia coli infections: translocation, translocation, translocation. Infect Immun 2005; 73: 2573–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bhinder G, Sham HP, Chan JM, et al The Citrobacter rodentium mouse model: studying pathogen and host contributions to infectious colitis. J Vis Exp 2013; 72: e50222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Collins JW, Keeney KM, Crepin VF, et al Citrobacter rodentium: infection, inflammation and the microbiota. Nat Rev Microbiol 2014; 12: 612–623. [DOI] [PubMed] [Google Scholar]
- 38. Bu X‐D, Li N, Tian X‐Q, et al Caco‐2 and LS174T cell lines provide different models for studying mucin expression in colon cancer. Tissue Cell 2011; 43: 201–206. [DOI] [PubMed] [Google Scholar]
- 39. van Klinken BJ, Oussoren E, Weenink JJ, et al The human intestinal cell lines Caco‐2 and LS174T as models to study cell‐type specific mucin expression. Glycoconj J 1996; 13: 757–768. [DOI] [PubMed] [Google Scholar]
- 40. Tian H, Biehs B, Chiu C, et al Opposing activities of Notch and Wnt signaling regulate intestinal stem cells and gut homeostasis. Cell Rep 2015; 11: 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Van Dussen KL, Carulli AJ, Keeley TM, et al Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development 2012; 139: 488–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Merker SR, Weitz J, Stange DE. Gastrointestinal organoids: how they gut it out. Dev Biol 2016; 420: 239–250. [DOI] [PubMed] [Google Scholar]
- 43. Drost J, Artegiani B, Clevers H. The generation of organoids for studying Wnt signaling. Methods Mol Biol 2016; 1481: 141–159. [DOI] [PubMed] [Google Scholar]
- 44. Taupin D, Podolsky DK. Trefoil factors: initiators of mucosal healing. Nat Rev Mol Cell Biol 2003; 4: 721–732. [DOI] [PubMed] [Google Scholar]
- 45. Salzman NH, Bevins CL. Dysbiosis – a consequence of Paneth cell dysfunction. Semin Immunol 2013; 25: 334–341. [DOI] [PubMed] [Google Scholar]
- 46. Ghosh S, Dai C, Brown K, et al Colonic microbiota alters host susceptibility to infectious colitis by modulating inflammation, redox status, and ion transporter gene expression. Am J Physiol Gastrointest Liver Physiol 2011; 301: G39–G49. [DOI] [PubMed] [Google Scholar]
- 47. Troll JV, Hamilton MK, Abel ML, et al Microbiota promote secretory cell determination in the intestinal epithelium by modulating host Notch signaling. Development 2018; 145: dev155317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Qu CK. The SHP‐2 tyrosine phosphatase: signaling mechanisms and biological functions. Cell Res 2000; 10: 279–288. [DOI] [PubMed] [Google Scholar]
- 49. You M, Flick LM, Yu D, et al Modulation of the nuclear factor kappa B pathway by Shp‐2 tyrosine phosphatase in mediating the induction of interleukin (IL)‐6 by IL‐1 or tumor necrosis factor. J Exp Med 2001; 193: 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schoenwaelder SM, Petch LA, Williamson D, et al The protein tyrosine phosphatase Shp‐2 regulates RhoA activity. Curr Biol 2000; 10: 1523–1526. [DOI] [PubMed] [Google Scholar]
- 51. Xu D, Qu C‐K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front Biosci 2008; 13: 4925–4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang W, Chan RJ, Chen H, et al Negative regulation of Stat3 by activating PTPN11 mutants contributes to the pathogenesis of Noonan syndrome and juvenile myelomonocytic leukemia. J Biol Chem 2009; 284: 22353–22363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Suzuki A, Hanada T, Mitsuyama K, et al Cis3/Socs3/Ssi3 plays a negative regulatory role in Stat3 activation and intestinal inflammation. J Exp Med 2001; 193: 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang K, Yuan C‐P, Wang W, et al Expression of interleukin 6 in brain and colon of rats with TNBS‐induced colitis. World J Gastroenterol 2010; 16: 2252–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Araki T, Nawa H, Neel BG. Tyrosyl phosphorylation of Shp2 is required for normal ERK activation in response to some, but not all, growth factors. J Biol Chem 2003; 278: 41677–41684. [DOI] [PubMed] [Google Scholar]
- 56. Cunnick JM, Meng S, Ren Y, et al Regulation of the mitogen‐activated protein kinase signaling pathway by SHP2. J Biol Chem 2002; 277: 9498–9504. [DOI] [PubMed] [Google Scholar]
- 57. Milano J, McKay J, Dagenais C, et al Modulation of notch processing by gamma‐secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci 2004; 82: 341–358. [DOI] [PubMed] [Google Scholar]
- 58. Vooijs M, Liu Z, Kopan R. Notch: architect, landscaper, and guardian of the intestine. Gastroenterology 2011; 141: 448–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Riccio O, van Gijn ME, Bezdek AC, et al Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27Kip1 and p57Kip2. EMBO Rep 2008; 9: 377–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *. Wlodarska M, Willing B, Keeney KM, et al Antibiotic treatment alters the colonic mucus layer and predisposes the host to exacerbated Citrobacter rodentium‐induced colitis. Infect Immun 2011; 79: 1536–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *. Hellemans J, Mortier G, De Paepe A, et al qBase relative quantification framework and software for management and automated analysis of real‐time quantitative PCR data. Genome Biol 2007; 8: R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *. Brosseau J‐P, Lucier J‐F, Lapointe E, et al High‐throughput quantification of splicing isoforms. RNA 2010; 16: 442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Supplementary figure legends
Figure S1. Shp‐2 activation enhances a goblet cell subset producing Tff3 but not Resistin‐like beta
Figure S2. Shp‐2 is required for epithelial wound healing
Figure S3. Intestinal microbiota is involved in Shp‐2‐induced protection against C. rodentium infection
Figure S4. Stat3 activation enhanced colitis burden in Shp‐2 IEC‐KO mice
Table S1. Disease activity index
Table S2. Histological damage scoring
Table S3. Primer sequences for RT‐qPCR