

Abstract
Objectives
N‐acetylcysteine (NAC) is a clinically approved thiol‐containing redox modulatory compound currently in trials for many neurological and psychiatric disorders. Although generically labeled as an “antioxidant,” poor understanding of its site(s) of action is a barrier to its use in neurological practice. Here, we examined the efficacy and mechanism of action of NAC in rodent models of hemorrhagic stroke.
Methods
Hemin was used to model ferroptosis and hemorrhagic stroke in cultured neurons. Striatal infusion of collagenase was used to model intracerebral hemorrhage (ICH) in mice and rats. Chemical biology, targeted lipidomics, arachidonate 5‐lipoxygenase (ALOX5) knockout mice, and viral‐gene transfer were used to gain insight into the pharmacological targets and mechanism of action of NAC.
Results
NAC prevented hemin‐induced ferroptosis by neutralizing toxic lipids generated by arachidonate‐dependent ALOX5 activity. NAC efficacy required increases in glutathione and is correlated with suppression of reactive lipids by glutathione‐dependent enzymes such as glutathione S‐transferase. Accordingly, its protective effects were mimicked by chemical or molecular lipid peroxidation inhibitors. NAC delivered postinjury reduced neuronal death and improved functional recovery at least 7 days following ICH in mice and can synergize with clinically approved prostaglandin E2 (PGE2).
Interpretation
NAC is a promising, protective therapy for ICH, which acted to inhibit toxic arachidonic acid products of nuclear ALOX5 that synergized with exogenously delivered protective PGE2 in vitro and in vivo. The findings provide novel insight into a target for NAC, beyond the generic characterization as an antioxidant, resulting in neuroprotection and offer a feasible combinatorial strategy to optimize efficacy and safety in dosing of NAC for treatment of neurological disorders involving ferroptosis such as ICH. Ann Neurol 2018;84:854–872
Hemorrhagic stroke, defined as bleeding within the brain parenchyma, accounts for 13% to 15% of all stroke cases. Nearly half of patients afflicted die, and for those that who survive, long‐term disability is common.1, 2 Identification of novel targets to treat this disease is an important unmet public need.
N‐acetylcysteine (NAC) is an US Food and Drug Administration–approved cysteine prodrug, established for its role in treating acetaminophen‐induced liver failure3 and other non‐CNS (central nervous system) indications.4, 5 Considerable preclinical data have accumulated to support the use of NAC in the CNS in acute and chronic neurological conditions as well as neuropsychiatric disorders.6, 7, 8, 9 Recent studies show that NAC inhibits ferroptosis in vitro,10 and because ferroptosis appears to be a mechanism of cell loss in hemorrhagic stroke in vivo in rodent models,10, 11 we considered its use for this stroke sub‐type.
Among the challenges in moving NAC forward as a therapeutic for CNS disorders are its many putative targets of action. Indeed, NAC has been thought to have broad salutary effects in diverse animal models by its ability to affect multiple targets. For example, NAC increases levels of cellular cysteine. Cysteine is the rate‐limiting precursor for glutathione synthesis. Glutathione acts as a co‐factor with a number of distinct antioxidant and detoxifying enzymes.12
NAC can also enhance CNS function through increases in glutamatergic neurotransmission. It accomplishes this by driving activity of the transporter, a cystine/glutamate antiporter, which pumps glutamate out of cells in exchange for NAC‐derived cyste(i)ne.13 NAC acts on immune cells to inhibit proinflammatory cytokines tumor necrosis factor alpha and interleukin‐1β in preclinical ischemic models14; or to improve mitochondrial dysfunction in a model of Huntington's disease15 or traumatic brain injury in rats.16 Also, high‐dose NAC has been shown to inhibit neurotropic Sindbis virus–induced apoptosis, in part by suppressing prodeath, nuclear factor kappa B signaling.17 Finally, NAC was recently shown to have a thrombolytic effect on occlusive arterial clot.18
Because NAC has many putative targets, it has been assumed that its salutary effects cannot be correlated with any single target. Accordingly, deciding on an optimal dose of NAC to use in the CNS has been challenging. Because CNS penetration of NAC is also poor,8 in the absence of a bona‐fide target, it is difficult to know whether current systemic administration is adequate to achieve therapeutic concentrations in the CNS. The need to identify a therapeutic target is further underscored by other factors, which constrain the clinical applications of NAC. Although NAC is believed to be generally safe, it has been reported to dose dependently cause nausea and vomiting, induce bronchospasm, slow blood clotting, and induce neurotoxicity, which could be problematic for patients with stroke, especially of the hemorrhagic subtype.
Therefore, the goal of this study was to understand whether NAC is effective in models of intracerebral hemorrhage (ICH); if so, to identify the target of its action as a first step toward defining the optimal dose in vivo for functional recovery. Here, we show that NAC acts therapeutically as a precursor for glutathione which in turn together with glutathione dependent enzymes (eg, glutathione S‐transferases [GSTs]) targets lipid‐derived reactive electrophilic species derived from increased arachidonate 5‐lipoxygenase (ALOX5). We also show that simultaneous, exogenous infusion of a clinically approved, protective lipid species, prostaglandin E2 (PGE2), can reduce the concentration of NAC required to stimulate protection and functional recovery in vitro or in vivo in mice.
Materials and Methods
Animals
C57BL/6 and pregnant CD1 mice of 10 to 12 weeks were purchased from Charles River Laboratories (Wilmington, MA). Homozygous ALOX5 knockout (ALOX‐5 –/–) mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Animals were housed in a pathogen‐free facility on a 12‐hour light/dark cycle and, unless otherwise stated, provided ad libitum access to food and water. All mice and procedures were approved by the Institutional Animal Care and Use Committee of the Weill Cornell Medical College (New York, NY) and were in accord with the guidelines established by National Institutes of Health. Rat experiments were approved and conducted at the University of Alberta (Edmonton, Alberta, Canada) according to published methods.19, 20 Briefly, 219 adult male Sprague–Dawley rats (∼350–400g; Charles River Laboratories, Montreal, Quebec, Canada) were used in several studies to examine the effects of NAC (40 or 75mg/kg given intraperitoneally [IP] 2 hours after collagenase infusion and thereafter as in the mice efficacy studies, versus saline control). We assessed body temperature, edema, eicosanoids, cerebral bleeding, behavior, and lesion volume (out to a 28‐day survival). Animals were randomly assigned to groups and data were analyzed blinded.
Primary Cortical Neuronal Cultures
Primary cortical neurons were prepared as previously described.21 Briefly, cortices were dissected from embryonic (E15.5) CD1 mice, homogenized, papain digested, and cells were plated in minimum essential medium containing 10% fetal bovine serum, 5% horse serum, and 1% penicillin/streptomycin in 96‐well plates, six‐well plates, or 10‐cm dishes. Neurons were maintained at 37°C with 5% CO2. All experiments were started at 24 hours after plating.
In Vitro ICH Model
Blood breakdown product, hemin, was used to induce cell death in primary cortical neurons. For the protective studies, cells were treated with hemin (100 μM) in the presence of NAC (0.01–1.00mM), Trolox (0.1–100.0 μM), α‐lipoic acid (0.01–2.00mM), U 73122 (0.1–100.0 μM), β carotene (0.1–100.0 μM), MS‐PPOH (0.1–100.0 μM), aspirin (0.1–100.0 μM), celecoxib (0.1–100.0 μM), Indomethacin (0.1–100.0 μM), Zileuton (0.1–100.0 μM), BW B70 (0.1–100.0 μM), BW A4C (0.1–100.0 μM), NCTT‐956 (0.1–100.0 μM), PD146176 (0.1–100.0 μM), MK 561 (0.1–100.0 μM), glutathione ethyl ester (1–10mM), L‐oxothiazolidine‐4‐carboxylate (1–10mM), cystamine (0.1–10.0 μM), and nordihydroguaiaretic acid (0.1–10.0 μM). Cell viability was analyzed 24 hours after treatment. Cells were rinsed with warm phosphate‐buffered saline (PBS) and assessed by methyl thiazolyl tetrazolium (MTT) assay. The fidelity of MTT assays in measuring viability was verified by calcein‐AM/ethidium homodimer‐1 staining (Live/Dead assay; Molecular Probes, Eugene, OR), following the manufacturer's instructions.
Collagenase‐Induced Mouse Model of ICH
Male C57BL/6 mice (8–12 weeks of age; Charles River Laboratories, Wilmington, MA) were anesthetized with isoflurane (2–5%) and placed on a stereotaxic frame. During the procedure, the animal's body temperature was maintained at 37°C with a homeothermic blanket. With a nanomite syringe pump (Harvard Apparatus, Holliston, MA) and a Hamilton syringe, 1 μl of collagenase (0.075IU; Sigma‐Aldrich, St. Louis, MO) was infused into the right striatum at a flow rate of 0.120 μl/min. Relative to the bregma point, the stereotaxic coordinates of the injection were as follows: lateral, −0.20; anteroposterior, 0.62; and dorsoventral, −0.40. In control animals, 1 μl of saline was infused. The treatment group received NAC (75 and 300mg/kg dissolved in vehicle [normal saline] and administered IP) once a day for 7 days starting at 2 hours after collagenase infusion. For combinatorial studies, NAC (40mg/kg, IP) and PGE2 (10 μM; intracerebroventricular [ICV]) was administered 2 hours after collagenase injection and then NAC (40mg/kg) alone was given daily for 7 days. The control groups received vehicle (normal saline) alone. Animals were randomized to sham or ICH groups. Identity of mice that received vehicle or NAC was masked to surgeons who performed the ICH. Identity was revealed after the data were collected. Proper postoperative care was taken until animals recovered completely. Similar surgical procedures were used for rat experiments where 0.14U (0.7 μl) of collagenase was infused into striatum (0.5mm anterior, 3.5mm lateral, and 6.5mm deep) under isoflurane anesthesia. Randomization and blinding were also used.
Behavioral Analysis
The corner task assessed the integrated sensorimotor function in both stimulation of vibrissae (sensory/neglect) and rearing (motor response). Briefly, mice were placed between two cardboard pieces forming a corner with a 30‐degree angle. While maintaining the 30‐degree angle, the boards were gradually moved toward the mouse until the mouse approached the corner, reared upward, and turned 180 degrees to face the open end. The direction (left or right) in which the mouse turned around was recorded for each trial. Ten trials were performed for each mouse. For the sensory neglect task (adhesive tape removal task), adhesive tape was placed on the planter region of the forward paw (right and left) of mice. The time from which the tape was applied to when the mouse successfully removed it was recorded for each paw. A maximum of 300 seconds for each paw was allowed.22 In the rat studies, a neurological deficit scale, the corner turn, tape removal, and staircase reaching tasks were used.
In Vivo Metal Distribution Imaging Analysis by x‐Ray Fluorescence Microscopy
Seven days following collagenase‐induced ICH, mice were euthanized and perfused with trace metal‐free PBS. Brains were removed and flash frozen with Freeze'it. Tissue was cut into 20‐μm‐thick sections and deposited on 4‐μm‐thick ultralene. Iron and zinc content was imaged in the same samples using x‐ray fluorescence microscopy at beamline X27A at the National Synchrotron Light Source. x‐ray fluorescence spectra were collected using an x‐ray excitation energy of 11keV and a beam size of 9 μm (vertical) × 17 μm (horizontal) in 15‐μm steps, with an integration time of 7 sec/pixel. Intensity for each metal was quantified by integrating the area under the curve for the respective peak in the XRF spectrum (iron Kα = 6,405eV and zinc Kα = 8,637eV). National Institute of Standards and Technology thin film standard reference materials 1832 and 1833 were used to calculate concentration and normalize for any differences between the multiple beam time runs required to collect the data. Molar concentrations were determined by dividing the μm/cm2 values by the product of the volume of x‐ray beam on the sample (area × thickness of the sample), density of tissue (estimated to be 0.9 g/cm3), and molecular weight of the element.
Fluoro‐Jade B Staining
Neurodegeneration was assessed in mice with collagenase‐induced ICH by analyzing staining with Fluoro‐Jade B (Millipore, Burlington, MA) carried out following the supplier's protocol. Briefly, 40‐μM brain sections were mounted on gelatin‐coated slides and dried at room temperature overnight. Sections were immersed in a graded series of alcohol solutions before being immersed in a 0.06% potassium permanganate (KMnO4) solution for 15 minutes. Sections were washed with water before being immersed in a 0.001% Fluoro‐Jade staining solution for 30 minutes with gentle shaking in the dark. Sections were then washed with water and then dried overnight at room temperature in the dark before being dehydrated and coverslipped with DPX (Sigma‐Aldrich). Fluoro‐Jade B staining was examined within perihematoma regions using a fluorescence microscope (Zeiss Axiovert; Carl Zeiss, Jena, Germany). Quantitation of Fluoro‐Jade B staining was performed from three brain sections (anterior to posterior of hematoma) using Metamorph analysis (Molecular Devices, LLC, San Jose, CA). In rats, total lesion volume was determined at 14 or 28 days post‐ICH.
Measurement of Eicosanoids
Analysis of oxidation products of arachidonic acid and other polyunsaturated fatty acids, such as leukotrienes, prostaglandins, and F2‐IsoP, were carried out at the Eicosanoid Core Laboratory at Vanderbilt University Medical Center (Nashville, TN). Briefly, lipid species were measured in control and ICH brains using gas chromatography/negative ion chemical ionization mass spectrometry (GC/MS). The methods used have been published in detail previously.41
Transcriptomic Analysis
Weighted Gene Coexpression Network Analysis (WGCNA) was performed using the R package (R Foundation for Statistical Computing, Vienna, Austria), as previously described.23, 24 Briefly, correlation coefficients were constructed between expression levels of genes, and a connectivity measure (topological overlap; TO) was calculated for each gene by summing the connection strength with other genes. Genes were then clustered based on their TO, and groups of coexpressed genes (modules) were identified. Each module was assigned a color, and the first principal component (eigengene) of a module was extracted from the module and considered to be representative of the gene expression profiles in a module. The phenotypic trait of interest was then regressed on the eigengene to examine whether there was a significant relationship between the module and trait. For modules that showed a statistically significant relationship with a phenotypic trait of interest (presence of ICH), gene ontology and pathway analyses were conducted. Enrichment of genes for specific ontologies and pathways was analyzed using Enrichr (23586463), and corrected p values were used. An independent list of coexpressed genes was obtained from COEXPRESdb (version 6.0; 25392420).
Quantitative Real‐Time PCR
Total RNA from cells and tissues were isolated using the NucleoSpin RNA II kit (MACHEREY‐NAGEL GmbH & Co. KG, Düren, Germany), according to the manufacturer's protocol. Duplex real‐time PCR reactions were performed with gene expression assays using 6‐carboxyfluorescein–labeled probes (Applied Biosystems, Foster City, CA) for ALOX5 (Mm 01182747_m1) and ALOX5 AP (Mm 01218551_m1). Expression levels were normalized to β‐actin gene expression levels, which were determined with a VIC‐labeled probe (Applied Biosystems). All experiments were performed using a 7500 Real‐Time PCR System (Applied Biosystems).
Adenoviral Overexpression of Antioxidant Enzymes
Adenoviral constructs for managenese‐containing superoxide dismutase (MnSOD), catalase, and respective green fluorescent protein (GFP) control were obtained from ViraQuest, Inc. (North liberty, IA), and adenoviral constructs for GSTA4, peroxiredoxin 3, and respective GFP control were purchased from Vector BioLabs (Malvern, PA). Primary neuronal cultures were transduced with all adenoviruses at a multiplicity of infection of 100 in serum‐free media for 1 hour at 370C and then were incubated in complete plating media for 48 hours. Then, these neurons were treated with hemin (100 μM) for 16 hours, and thereafter cell survival was determined by MTT assay.
Immunoblot Analyses
Nuclear and cytoplasmic proteins were obtained using the NE‐PER Nuclear and Cytoplasmic Extraction Reagents (Pierce Biotechnology, Rockford, IL) in the presence of protease inhibitors (phenylmethane sulfonylfluoride, MG132), according to the manufacturer's protocol. For determination of catalase and MnSOD levels, cells were lysed in RIPA‐B (1% Triton X‐100, 1% sodium dodecyl sulfate, 50mM of Tris‐Cl [pH 7.4], 500mM of NaCl and 1mM of ethylenediaminetetraacetic acid) buffer before being diluted with Laemmli buffer and boiled. All samples and were electrophoresed under reducing conditions on NuPAGE Novex 4% to 12% Bis‐Tris Gel polyacrylamide gels (Invitrogen, Carlsbad, CA) before being transferred to a nitrocellulose membrane (Bio‐Rad Laboratories, Hercules, CA). For detection of specific proteins, the following primary antibodies and dilutions were used: ALOX‐5 (610694; BD Biosciences, San Jose, CA), MnSOD (HPA001814; Sigma‐Aldrich), catalase (C0979; Sigma‐Aldrich), and β‐actin (A2066 and A5316; Sigma‐Aldrich) were diluted in Odyssey blocking buffer, and membranes were incubated overnight at 4°C. After three washes with Tris‐buffered saline with Tween‐20, membranes were incubated in fluorophore‐conjugated Odyssey IRDye‐680 or IRDye‐800 secondary antibody (LI‐COR Biosciences, Lincoln, NE) for 1 hour at room temperature. Finally, proteins were detected using an Odyssey infrared imaging system (LI‐COR Biosciences).
Lipoxygenase UV‐Vis‐Based Half Maximal Inhibitory Concentration IC50 Assay
Inhibition potencies were determined by following the formation of the conjugated diene products, 5‐HpETE [ε = 27,000M–1 cm–1 for arachidonic acid [AA] turnover) at 234nm with a PerkinElmer Lambda 40 UV/Vis spectrophotometer (PerkinElmer, Waltham, MA), as described previously.25 All reaction mixtures were 2ml in volume and constantly stirred using a magnetic stir bar at room temperature (23°C). All reactions were carried out in Buffer A containing 200 μM of adenosine triphosphate and 10 μM of AA. AA concentration was verified by full turnover with soybean‐1 lipoxygenase and quantitating product concentration. Inhibitors were stored in dimethyl sulfoxide (DMSO) at –20˚C. Enzymatic reactions were initiated by the addition of approximately 100 to 300nM of ammonium sulfate–precipitated wild‐type enzyme. Half maximal inhibitory concentration values were obtained by determining the enzymatic rate at a minimum of five inhibitor concentrations and plotting them against inhibitor concentration, followed by a hyperbolic saturation curve fit. Inhibitor concentrations were assayed in duplicate or triplicate, depending on the quality of the data.
Statistical Analyses
Results are expressed as means ± standard error of the mean (SEM) of multiple individual experiments, each carried out in triplicate. Unless stated otherwise, the statistical analysis was carried out with GraphPad Prism software (version 5; GraphPad Software Inc., La Jolla, CA. One‐way analysis of variance (ANOVA) with Bonferroni's or Dunnett's multiple comparisons post‐hoc test was used if more than two groups were compared, and a two‐way ANOVA with Bonferroni's post‐hoc test if two independent variables were compared.
Results
NAC Abrogated Hemin Toxicity In Vitro, Reduced Neuronal Death, and Improved Functional Recovery in an in vivo Model of ICH
To investigate the effects of NAC on hemorrhagic stroke, we first examined its ability to protect neurons from hemin‐induced toxicity. Hemin is the oxidized form of heme, which is released from hemoglobin following erythrocyte lysis. It has been implicated in secondary injury following ICH. Treatment of primary cortical neurons with 100 μM of hemin induced cell death, as quantitated with MTT and confirmed by qualitative live (green) and dead (red) staining (Fig 1A) through a ferroptotic mechanism.10 Treatment with NAC abrogated hemin‐induced ferroptosis (half maximal effective concentration [EC50] = 500 μM; Fig 1B), and NAC could be delivered 2 hours following hemin exposure and protect all cells. To determine whether NAC (300mg/kg) is effective for treatment of ICH in mice, we injected collagenase into the mediolateral striatum to break down the basal lamina and induce striatal brain bleeding. Collagenase injection simulates the spontaneous rupture of an intraparenchymal vessel with bleeding into the tissue over several hours.26 We assessed neuronal loss from sections of mice treated with vehicle or NAC (300mg/kg, IP administration) initially delivered 2 hours following experimental ICH and daily up to 7 days following the ictus using a nonspecific marker of neurodegeneration, Fluoro‐Jade (Fig 1C). As expected, NAC (300mg/kg) reduced neuronal degeneration in perihematomal regions of the mouse brain (Fig 1D,E). Mice with striatal hemorrhage showed a preference for ipsilateral turns because of deficits in the weight‐balancing movements of the limbs contralateral to the injury, as well as spatial neglect. These deficits were corrected by 300mg/kg of NAC, but not 75mg/kg of NAC, as measured by the corner turn task (Fig 1F). Moreover, analysis of the tape removal task (a test of sensory neglect) also improved significantly in NAC‐treated mice 1, 3, and 7 days after ICH, but again only at the highest dose (300mg/kg; Fig 1G). Together, these findings demonstrate that NAC (300mg/kg) reduces cell death in vitro and in vivo and improves behavior after ICH in mice. Examination of NAC in a collagenase ICH model in rats showed that 40mg/kg was the highest dose tolerated without toxicity. Higher doses that were effective in mice (300mg/kg) caused significant toxicity in rats, including paralytic ileus and/or death (not shown). Of note, like mice, NAC delivered at 40 or 75mg/kg in rats 2 hours post‐ICH was not effective in reducing lesion volume or improving functional recovery, suggesting that not only efficacy, but also safety of NAC will be important in translating positive findings from mice to humans.
Figure 1.
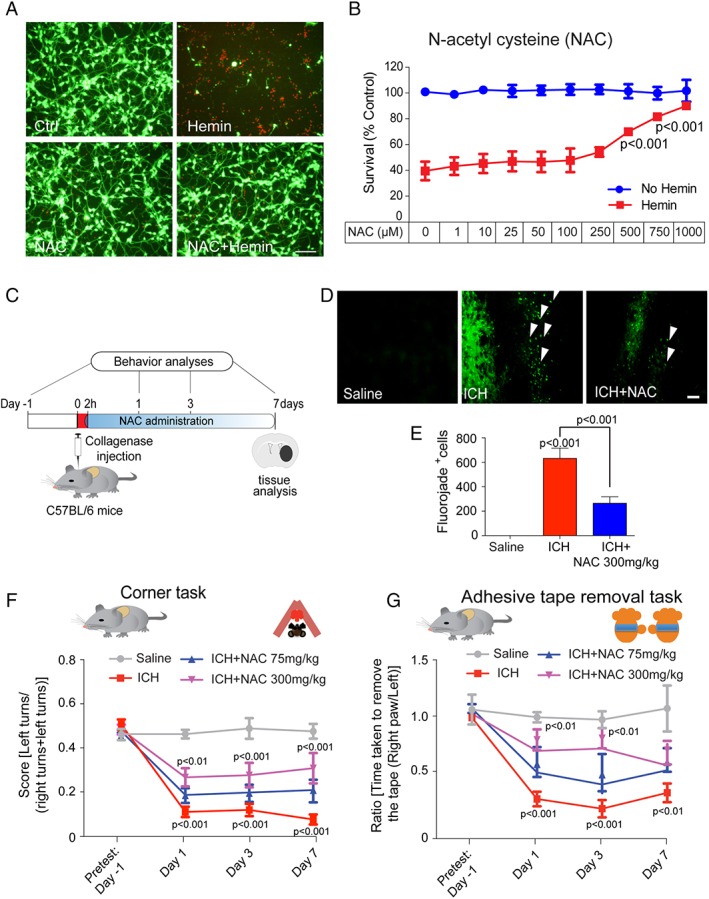
N‐acetylcysteine (NAC) abrogated hemin‐induced ferroptosis in primary neurons in vitro and reduced cell death and enhanced functional recovery in a collagenase ICH model in mice. (A) Representative images of primary cortical neurons 24 hours after treatment with saline, NAC (1mM), hemin (100 μM) and hemin (100 μM) + NAC (1mM). LIVE/DEAD assay; green fluorescent cells labeled with calcein‐AM are alive; red fluorescent cells labeled with ethidium homodimer are dead. Scale bars, 100 μm. (B) NAC protected primary cortical neurons from hemin‐induced ferroptosis in a concentration‐dependent manner. Cell death was analyzed 24 hours after hemin treatment with or without NAC by monitoring MTT reduction, a population measure of cell viability. (C) Experimental design for delivery of NAC post‐ICH in mice. NAC (75mg and 300mg/kg; intraperitoneal) was delivered 2 hours after collagenase injection and then daily for 7 days. Behavior was assessed using the corner task (spatial neglect) and adhesive tape removal task (sensory neglect) and was assessed on days 1, 3, and 7 after ICH. (D) NAC reduced neuronal degeneration was monitored by Fluoro‐Jade (FJ) staining (green) in the perihematomal regions of the mouse brain. Representative images show increased numbers of degenerating neurons (white arrows) in the ICH‐treated group; this was reduced by NAC treatment. Scale bar, 100 μm. (E) Quantification of FJ staining of neurons, a nonspecific marker of degeneration, after NAC treatment in ICH mice brains. (F) NAC (300mg/kg) significantly reduces spatial neglect associated with ICH. (G) NAC (300mg/kg) reduces sensory neglect (adhesive tape removal task) induced by ICH (n = 11). NAC (75mg/kg) had no significant effect in mice or rats (not shown). Significance was determined by two‐way ANOVA and Bonferroni's post‐hoc test. All graphs are mean ± SEM. ANOVA = analysis of variance; ICH = intracerebral hemorrhage; NAC = N‐acetylcysteine; MTT = methyl thiazolyl tetrazolium; SEM = standard error of the mean.
NAC Did Not Influence Collagenase Enzyme Activity or Bulk Iron Chelation
To address the mechanism of NAC protection in hemorrhagic stroke in mice, we first verified that NAC's protective effects do not result from its influence on collagenase enzyme activity, which was used to induce brain hemorrhage. Accordingly, we treated mice 2 hours post‐ICH with NAC and sacrificed them after 24 hours and assessed hematoma volume (Fig 2A). Measurements of hematoma size 24 hours after collagenase injection verified that NAC (75 or 300mg/kg) does not inhibit collagenase activity in mice (Fig 2B–D), demonstrating that the significant behavioral benefits cannot be attributed to suppression of collagenase activity by NAC.
Figure 2.
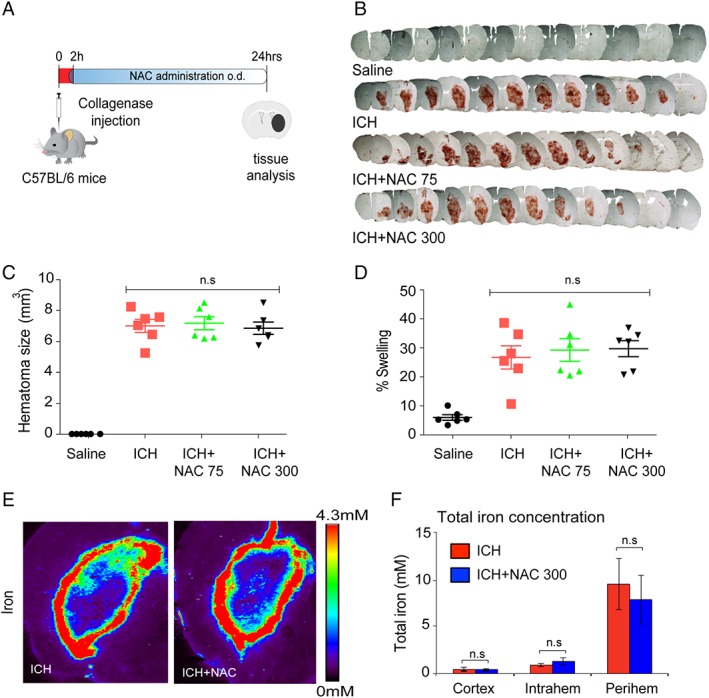
N‐acetylcysteine (NAC) enhanced functional recovery following intracerebral hemorrhage (ICH) without influencing collagenase activity in vivo or iron levels or iron distribution in the brain after ICH. (A) Schematic illustration of NAC treatment, post‐ICH. (B) Serial brain sections from saline‐, ICH‐, and ICH + NAC–treated groups. (C,D) Quantification of hematoma size and brain edema by light microscopy revealed no significant difference between control and NAC‐treated groups in mice (n = 6). (E) x‐ray fluorescence microscopy analysis of pseudo‐colored images from coronal sections of collagenase alone or NAC treatment in collagenase‐induced ICH in mice after 7 days. Quantification in cortex, hematomal, and perihematomal regions revealed that total iron does not change in the brain following NAC treatment (F). Significance determined by one‐way ANOVA followed by Dunnett's comparison test, for vehicle treatment ICH or NAC treatment (C,D). All graphs are mean ± SEM. ANOVA = analysis of variance; ICH = intracerebral hemorrhage; NAC = N‐acetylcysteine; ns = not significant; SEM = standard error of the mean.
NAC has been shown to be able to chelate metals, including iron, and it is established that small molecules with iron‐binding capacity can improve outcomes in ICH.22, 27, 28 Accordingly, we asked whether NAC affects total iron levels in the CNS. We measured total iron in the brain sections from saline and NAC‐treated ICH mice at 7 days by x‐ray fluorescence microscopy. As expected, ICH dramatically increased iron levels; however, NAC had no effect on iron levels or distribution following ICH (Fig 2E,F). These findings suggest NAC acts to protect from ICH independent of its effects on total iron levels or distribution in the CNS.
NAC Failed to Synergize With Other “antioxidants” as Part of a Previously Validated Cocktail
One strategy for identifying putative targets of NAC is to assess its ability to synergize with antioxidants with established, single target of action. A lack of synergy would suggest target congruence between the antioxidant with a known target of action and NAC. Trolox is a water soluble analog of vitamin E and a scavenger of lipid peroxyl radicals, whereas α‐lipoic acid (LA) is a known thiol active compound that acts as a cofactor for many mitochondrial enzymes, thus reducing mitochondrial superoxide production. We selected these for initial investigation because a previous study by Pujol et al established that a cocktail of antioxidants (NAC, trolox, and α‐lipoic acid) diminished disease progression and reversed axonal damage in a mouse model of X‐linked adrenoleukodystrophy.29 To investigate the effects of NAC, trolox, and LA individually and in combination, we studied their effects in mouse cortical neurons exposed to hemin. Treatment of NAC or trolox, individually, dose dependently abrogated hemin‐induced toxicity (Fig 3A). However, LA failed to prevent hemin toxicity at any dose examined (Fig 3A). To examine synergy, we tested subthreshold concentrations of the antioxidants (eg, NAC and trolox) together in hemin model. Interestingly, all combinations of the cocktail of antioxidants failed to provide any synergistic protection against hemin toxicity in primary cortical neurons, which would be consistent with a convergent mechanism at the level of lipid peroxidation, an established target of the vitamin E analog, trolox (Fig 3B).
Figure 3.
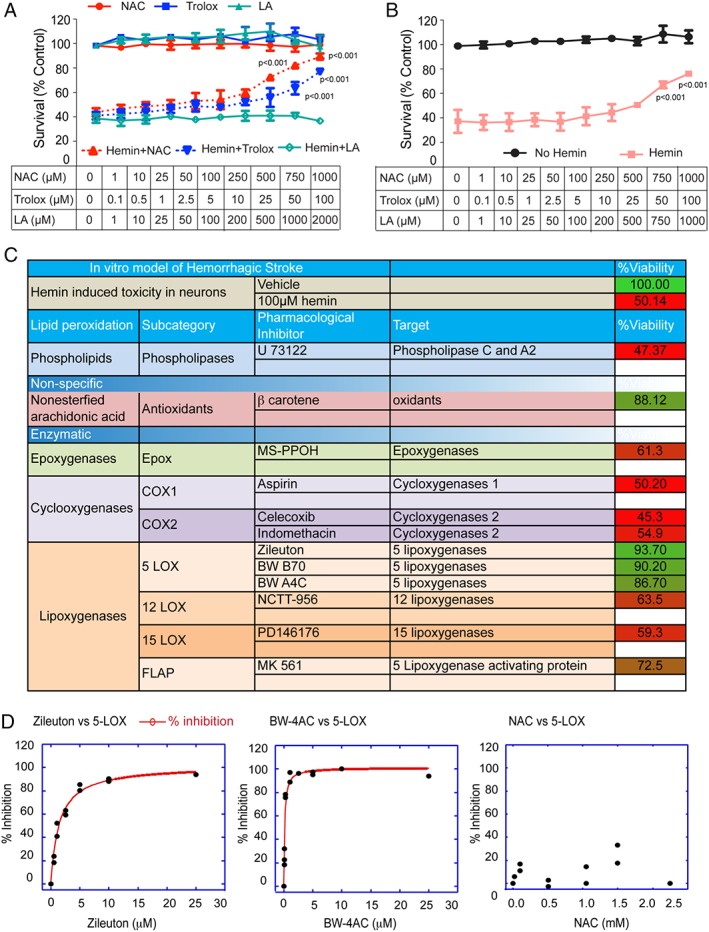
Systematic pharmacological characterization of arachidonic acid metabolizing enzymes in hemin‐induced ferroptosis in primary cortical neurons identified 5‐Lipoxygenase (ALOX5) as a target. (A) N‐acetylcysteine (NAC), a cysteine prodrug, or Trolox (TRO) dose dependently prevented hemin‐induced ferroptosis in primary cortical neurons as measured by MTT assay. Lipoic acid (LA) failed to prevent hemin‐induced ferroptosis in primary cortical neurons. (B) Combinations of nonprotective concentrations of NAC, TRO, or LA failed to synergize in preventing hemin‐induced ferroptosis in primary cortical neurons. (C) Table illustrates that structurally diverse inhibitors of 5‐lipoxygenase, but not inhibitors of other arachidonate metabolizing enzymes, prevent hemin‐induced ferroptosis in primary cortical neurons. (D) Assay of recombinant ALOX5 activity in vitro (test tube) showed that putative ALOX5 inhibitors are potently effective in inhibiting the enzyme, whereas NAC is not. Significance was determined by two‐way ANOVA and Bonferroni's post‐hoc test. ANOVA = analysis of variance; MTT = methyl thiazolyl tetrazolium.
NAC Prevented Hemin‐Induced Death by Neutralizing Toxic Lipids Generated by Nuclear ALOX5
Reactive lipid oxidants can be produced enzymatically by specific enzymes or nonenzymatically by direct oxidant modification. Because NAC appeared to be targeting oxidized lipids, we used structurally diverse, but well‐characterized, pharmacological tools to further understand how hemin kills neurons. Neuronal membranes are composed of phospholipid bilayers in which AA is esterified into phosphatidylcholine, phosphatidylserine, and phosphatidylinositol. Following brain injury, AA is liberated by increases in calcium‐dependent, phospholipase A2 or phospholipase C activity.30 Released AA can be oxidized nonenzymatically by oxidants or enzymatically (by cyclooxygenase [COX], lipoxygenase, or epoxygenase enzymes) to produce bioactive lipid mediators (isoprostanes, hydroxynonenol, malondialdehye, hydroxyl‐PUFAs [HETEs], epoxy‐PUFAs [EETs], prostaglandins, and leukotrienes) that regulate a diverse set of homeostatic and inflammatory processes.31 We performed a systematic characterization of AA metabolizing enzymes involved in hemin‐induced toxicity by using a cassette of known, but diverse, chemical inhibitors. Selective inhibitors of epoxygenases (MS‐PPOH), COX‐1 (aspirin), COX‐2 (celecoxib; indomethacin), 12‐LOX (NCTT‐956), or 15‐LOX (PD146176) failed to protect against hemin toxicity (Fig 3C). However, structurally diverse ALOX5 inhibitors (Zileuton, EC50 = 7 μM; BW B70, EC50 = 4 μM, and BW A4C, EC50 = 5 μM) significantly reduced hemin‐induced toxicity in primary cortical neurons (Fig 3C). Test‐tube assays involving recombinant ALOX5 verified that each of these inhibitors inhibits ALOX5 activity with high potency, whereas NAC (Fig 3D) and β‐carotene (not shown), which were also protective, did not (Fig 3D). These data suggest that 5‐lipoxygenase–derived lipid metabolites are necessary for hemin‐induced ferroptosis in vitro. They also raise the possibility that NAC, trolox, and β‐carotene are acting to neutralize the toxic metabolic products of ALOX5 activity rather than ALOX5 activity itself. These findings predicted that inhibitors of arachidonate‐5‐lipoxygenase activating protein (FLAP), an integral membrane protein within the nuclear envelope, which serves to recruit ALOX5 to the membrane, should also be protective. As expected, a chemical FLAP inhibitor (MK 561) also significantly reduced hemin‐induced death (Fig 3C).
ICH Induced Nuclear ALOX5 Protein and ALOX5‐Derived Gene Expression in Rodents and Humans
Our pharmacological data support the hypothesis that ALOX5 mediates death in vitro ICH by localizing to the nuclear envelope by interactions with FLAP. Previous studies have shown that ALOX5 protein levels increase at the nuclear envelope to release leukotrienes and attract inflammatory cells.32 Accordingly, we wanted to determine whether ICH induces ALOX5 levels in the cytoplasm or nucleus and to assay whether ALOX5‐derived AA species are increased following ICH (Fig 4A). To address this question, we first analyzed protein levels of ALOX5 in the nucleus after ICH. Immunoblot analysis of the cytosolic and nuclear fraction after ICH revealed that ALOX5 significantly increases in the nucleus (Fig 4B). ALOX5 metabolizes AA to produce 5‐hydroperoxyeicosatetraenoic acid (5‐HPETE), which can then form inflammatory mediators leukotriene B4 (LTB4) and cysteinyl leukotrienes (CysLTs; including LTC4, LTD4, and LTE4).33 To assess whether ALOX5 is activated in a rodent model of ICH, we monitored its metabolites by GC/MS. GC/MS analysis of ICH striatum showed a significant time‐dependent increase in the ALOX5 products, 5‐HETE, LTB4, and LTE4 (Fig 4C–E). These findings are consistent with previous transcriptomic analyses of brain tissues from human ICH patients that identified an increased expression of mRNAs encoding ALOX5 and 5‐LO–activating protein FLAP (ALOX5 AP).34
Figure 4.
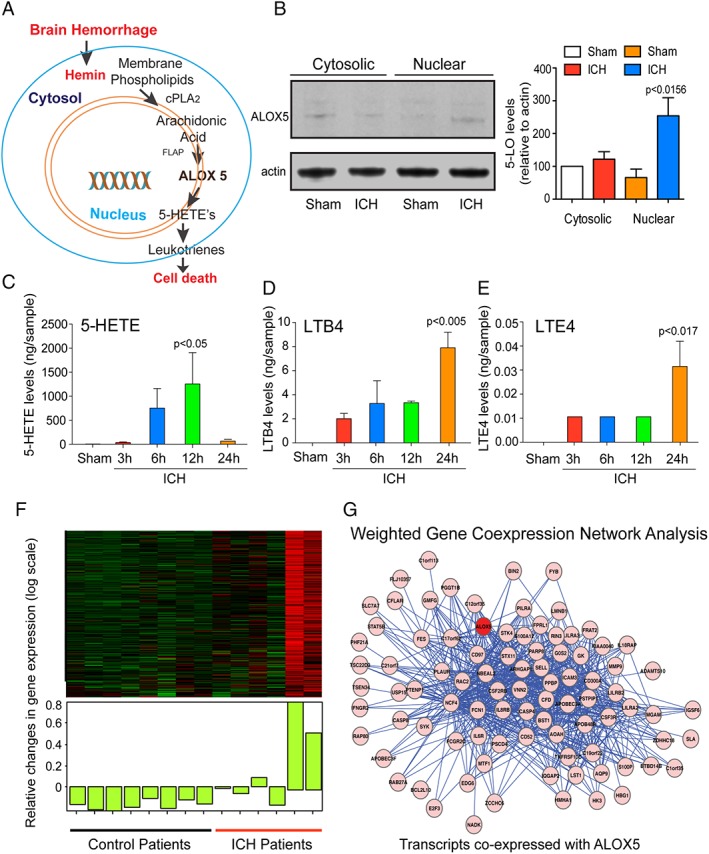
Intracerebral hemorrhage (ICH)‐induced ALOX5‐derived oxidized lipids and gene expression in mice and humans. (A) Schematic model of ALOX5 pathway activation in ICH. (B) ICH increased ALOX5 protein levels in the nuclear fraction as verified by immunoblot analysis. Gas chromatography/mass spectrometry analysis revealed a significant increase in ALOX5‐derived lipid species after ICH in rats (n = 4) compared to sham, including 5‐hydroxyeicosatetraenoic acid (5‐HETE) (C), Leukotriene B4 (LTB4) (D), and Leukotriene E4 (E). Data from sham control brains from each time point were pooled for the analysis. (F) Transcriptomic analysis of brain tissues from control (n = 8) and ICH (n = 6) patients. (G) Weighted gene coexpression network analysis (WGCNA) revealed transcripts most closely coregulated with ALOX5. Significance was determined by one‐way ANOVA and Dunnet's multiple‐comparison test. All graphs are mean ± SEM. ANOVA = analysis of variance; ICH = intracerebral hemorrhage; SEM = standard error of the mean.
To further explore the network of transcripts coexpressed with ALOX5, we reanalyzed the human gene expression data from Carmichael et al (including eight control samples and six samples from patients with ICH), using WGCNA, a systems biology approach used to identify networks of coexpressed genes in relation to phenotypic data.23, 24 WGCNA identified 21 groups of coexpressed genes, or modules, and ALOX5 was included in the greenyellow module, a group of 302 transcripts overall upregulated in ICH (Fig 4F). We queried a large transcriptional database (http://coxpresdb.jp/cgi-bin/coex_list.cgi?gene=240&sp=Hsa) and extracted the top 100 transcripts correlated with ALOX5 (Fig 4G). Functional annotation of this module revealed over‐representation of genes involved in inflammation in general and neutrophil degranulation specifically. Functional annotation of these transcripts revealed similar ontology terms. Fifteen of these 100 transcripts were included in the WGCNA greenyellow module, a significant overlap (p = 8 × 10E‐9, hypergeometric test) providing independent validation of the WGCNA analysis (not shown). These findings suggest ICH induces accumulation of ALOX5 in the nucleus to increase ALOX5‐derived lipid species, which can trigger cell death (Fig 4) and/or represent a signal sent from the nuclear envelope of dead cells to trigger inflammation.32, 35
Germline Deletion of ALOX5 or Pharmacological Reduction of Its Expression by NAC Leads to Neuroprotection and Improved Behavioral Recovery
Our model suggests that NAC should neutralize ALOX5‐derived lipid species. Because hemin leads to increases in ALOX5 protein in the nucleus, we examined whether hemin also induced increases in ALOX5 and ALOX AP mRNA levels and whether this was inhibited by NAC. Consistent with our protein data (Fig 4B), hemin time dependently induced ALOX AP and ALOX5 mRNA. Unexpectedly, NAC significantly suppressed these increases in vitro as well as in vivo in mice (Fig 5A–D). If ALOX5 and its products are targets for NAC treatment, then ALOX5 deletion should be sufficient for neuroprotection and functional recovery following ICH.
Figure 5.
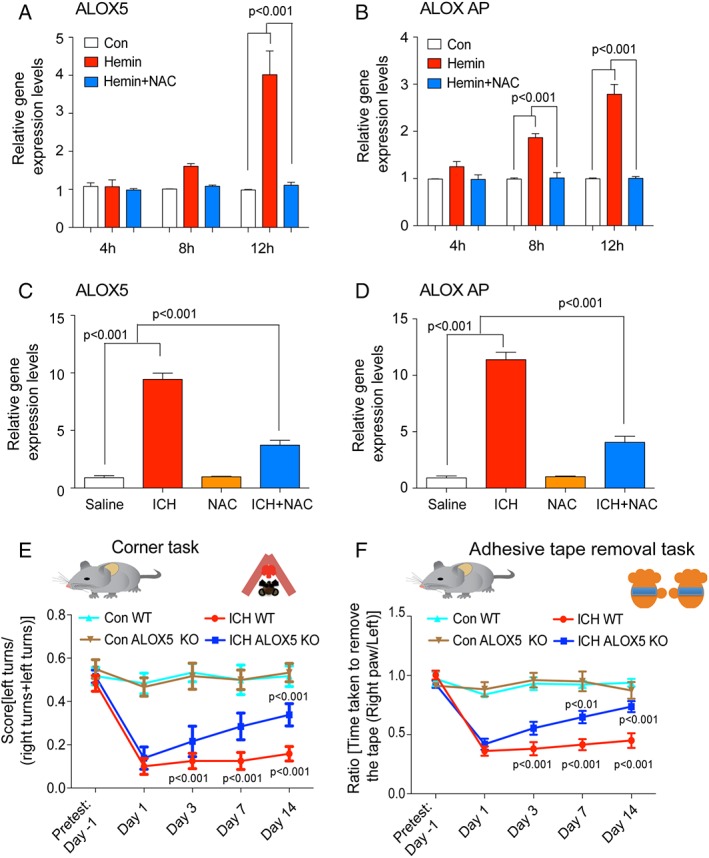
N‐acetylcysteine (NAC) reduced ICH‐induced ALOX5 gene expression and molecular knockdown of ALOX5 improves ICH‐induced behavioral deficits. RT‐PCR analysis revealed that toxic levels of hemin time dependently (4, 8, and 12 hours) increased ALOX5 and ALOX activating protein (AP) levels in primary neurons. Protective doses of NAC (1mM) blocked this expression in primary cortical neurons (A,B). In mice, NAC significantly blocked ICH‐induced ALOX5 (C) and ALOX AP levels (D). Molecular knockdown of ALOX5 improved spatial neglect behavioral deficits and sensory neglect (adhesive tape removal task) behavioral deficits associated with ICH compared to WT controls. Significance was determined by two‐way ANOVA and Bonferroni's post‐hoc test. All graphs are mean ± SEM. ANOVA = analysis of variance; ICH = intracerebral hemorrhage; NAC = N‐acetylcysteine; SEM = standard error of the mean; WT, wild type.
We therefore induced ICH in wild‐type and ALOX5 knockout mice. As expected, molecular knockdown of ALOX5 significantly improved somatosensory function at 3 and 7 days following ICH (Fig 5E,F). Collectively, these findings are consistent with a model of injury in ICH where hemin, by as yet unclear mechanisms, induces ALOX5 activity at the nuclear envelope. In a feed‐forward manner, these metabolites increase ALOX5 and FLAP transcription and activity, generating adequate reactive lipid species to mediate ferroptotic cell death with features of necrosis. Necrotic cells could then release leukotrienes from the nuclear envelope to activate the inflammatory response.32 Our human data suggested that transcriptional induction of ALOX5 occurs temporally in concert with neutrophil degranulation, possibly coupling cell‐death mechanisms with the inflammatory response (Fig 4G). Converging data suggest that ICH‐induced ALOX5 triggers cell death and impairment following ICH in mice.
NAC Reduced the Lipid Protein Adducts and Efficacy Required Increased Levels or Flux in Glutathione and Its Protection Is Mimicked by GSTA4
Given the unexpected effects of NAC on ALOX5 transcription, it is formally possible that NAC acts up‐ and/or downstream of ALOX5 transcriptional induction. Whatever the site(s) of action, the model further predicts that NAC, trolox, and more‐selective ALOX5 inhibitors should inhibit incorporation of ALOX5‐derived reactive lipids into proteins. Electrophilic attack by ALOX5 products could alter cellular protein‐mediated signaling to trigger cell death. To detect covalent lipid protein adducts, we used AA tagged with biotin (Bt‐AA) followed by western blots of lysates probed with streptavidin/horseradish peroxidase, as previously described.37 As expected, Bt‐AA incorporation into protein increased with increasing concentrations of hemin (Fig 6A). Nonprotective doses of NAC (0.1mM), vitamin E (1 μM), or zileuton (ALOX5 inhibitor, 1 μM) did not decrease AA‐protein adduct formation. However, protective doses of NAC (1mM), vitamin E (10 μM), or zileuton (10 μM) decreased lipid protein adduction formation (Fig 6B; see arrow). Quantification of the intensity of the experimental bands revealed that NAC, Vit E, and zileuton (an ALOX5 inhibitor) all inhibit lipid protein adduct formation (Fig 6C). The bands that are evident in the control wells are the endogenous biotin containing carboxylases and are an additional confirmation of equal gel loading between groups.38 These results suggest that reactive lipids incorporated into proteins are abrogated by ALOX5 inhibitors, NAC, or vitamin E and are consistent with the ability of ALOX5 metabolites to incorporate into proteins to induce changes in signaling or dysfunction and induce cell death. The catalytic action of ALOX5 involves the formation of site‐specific alkyl and lipid peroxyl radicals, which are generally not released from the enzyme and not then accessible to lipid radical scavenging agents, such as vitamin E, which cannot access the catalytic site of the enzyme. However, low levels of the lipid radicals can exit the active site and, if this occurs, are capable of initiating lipid peroxidation and in this case can be quenched by selective ALOX5 inhibitors or chain‐breaking antioxidants such as vitamin E or NAC (Fig 6C). Cell death can be neutralized by specific ALOX5 inhibition or by NAC, which neutralizes lipid species downstream of ALOX5 activity and prevents the feed‐forward increases in ALOX5 and FLAP expression and ferroptotic cell death (Fig 1) and, although not directly explored here, possibly inflammation.32
Figure 6.
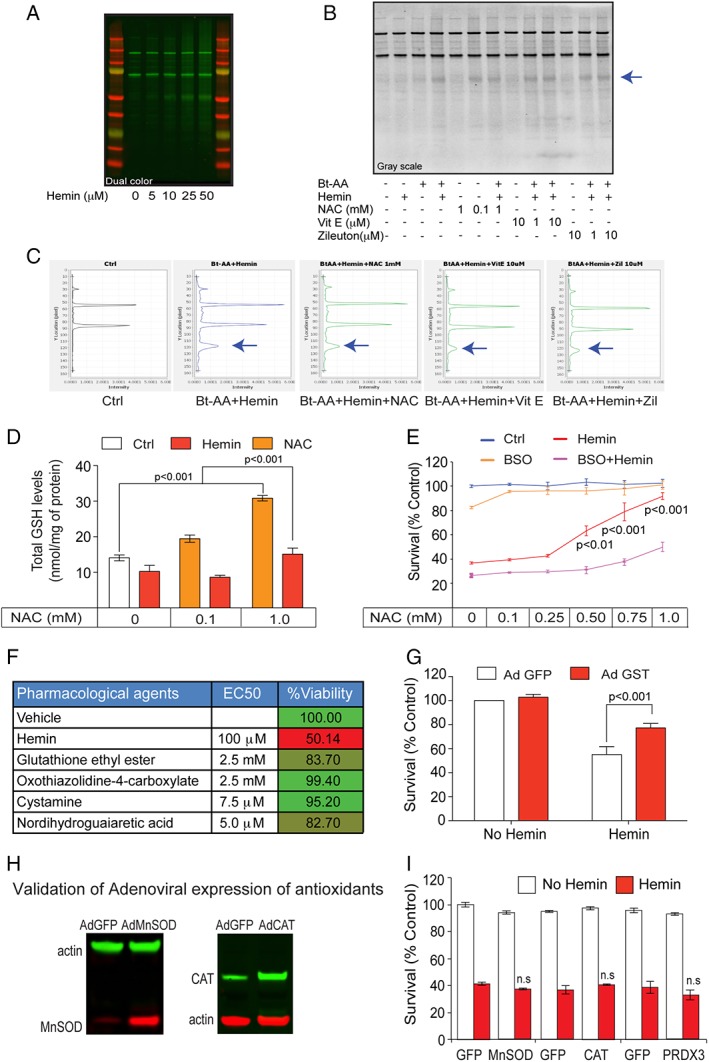
Protective NAC reduced lipid protein adducts and required glutathione (GSH) synthesis for neuroprotection. (A) Primary cortical neurons were treated with progressively higher concentrations of hemin. Lipid protein adducts (arachidonic acid‐biotin) were detected by western blots probed with streptavidin‐HRP (horseradish peroxidase). (B) NAC (1mM), α‐tocopherol (10 μM), and Zileuton (10 μM) attenuated hemin‐induced oxidized lipid protein adducts. Grayscale image of lipid protein adducts probed with streptavidin‐HRP. Representative blot from replicates of three experiments. (C) Quantification of bands reveals a significant reduction in oxidized lipid protein adducts after NAC, Zileuton, and vitamin E. (D) High‐performance liquid chromatography analysis of total GSH revealed that NAC increases GSH levels in control and hemin‐treated cortical neurons. (E) Pharmacological inhibition of γ‐glutamylcysteine synthetase, the rate‐limiting enzyme in GSH synthesis, by BSO blocked the ability of NAC to prevent hemin‐induced ferroptosis in primary cortical neurons. (F) As predicted from results in (B), table shows that multiple established strategies to increase GSH levels in neurons or glia prevented hemin‐induced ferroptosis. GSH ethyl ester (a membrane permeant form of GSH); L‐oxothiazolidine‐4‐carboxylate (OTC), a cysteine donor; Nrf2 activators cystamine and NDGA, which increase GSH synthesizing and GSH‐dependent detoxification enzymes, abrogate hemin‐induced ferroptosis in primary cortical neurons as measured by MTT assay. (G) Adenoviral overexpression of GSTA4, a GSH enzyme known to neutralize toxic lipids, protects against hemin‐induced ferroptosis. (H) Representative immunoblot for forced expression of antioxidants. (I) Overexpression of MnSOD, catalase, and peroxiredoxin 3 failed to protect against hemin‐induced toxicity. Significance was determined by two‐way ANOVA followed by Bonferroni's comparison test. All graphs are mean ± SEM. Ad GFP = adenoviral green fluorescent protein; ANOVA = analysis of variance; BSO = buthionine sulfoximine; EC50 = half maximal effective concentration; MTT = methyl thiazolyl tetrazolium; NAC = N‐acetylcysteine; ns = not significant; SEM = standard error of the mean.
In this scheme, NAC could interact directly with reactive lipids, or, alternatively, it could act by increasing concentrations and/or flux of cysteine, the rate‐limiting precursor for glutathione, a versatile, tripeptide antioxidant.12, 21 To determine whether NAC‐induced protection from hemin requires glutathione, we inhibited glutathione synthesis using buthionine sulfoximine (BSO), a selective inhibitor of the enzyme, γ‐glutamylcysteine synthetase, the rate‐limiting step in glutathione synthesis,21 and determined whether NAC‐induced protection could be abrogated. As expected, NAC dose dependently increased total glutathione levels in the presence or absence of hemin, although in the presence of hemin absolute levels were reduced (Fig 6D). Consistent with the notion that glutathione is required for NAC protection from hemin‐induced ferroptosis, we found that BSO significantly reduced NAC's beneficial effects (Fig 6E). The results predicted that other strategies that also lead to increased glutathione levels in neuronal cultures should also abrogate hemin‐induced ferroptosis. As expected, increasing glutathione with a membrane permeable form of glutathione, glutathione ethyl ester, by adding the cysteine prodrug, L‐oxothiazolidine‐4‐carboxylate (OTC39), or by addition of the established transcriptional activators of nuclear factor erythroid 2–related factor 2 (Nrf2) and glutathione synthesis, cystamine, or nordihydroguaiaretic acid (NDGA40) prevented hemin‐induced ferroptosis as effectively as NAC in vitro (Fig 6F). Together, these results suggest that NAC acts to increase glutathione to prevent ALOX5 induced ferroptosis.
Glutathione can act by several glutathione‐dependent enzymes to neutralize reactive lipids, including GSTs. One GST isoform, GSTA4, has been specifically implicated in this role.41, 42 Accordingly, adenoviral overexpression of GSTA4 in primary cortical neuronal cultures protected against hemin‐induced toxicity (Fig 6G). As controls for specificity of this effect, we overexpressed other “glutathione‐independent” antioxidants, such as MnSOD, catalase, and peroxiredoxin, which target superoxide (MnSOD) or hydrogen peroxide (catalase, peroxredoxin) in the cytosol or mitochondria, As expected, we did not find that forced expression of any of these enzymes could protect against hemin‐induced toxicity. Although our results do not establish whether chemical or molecular agents are acting in glia or neurons or both, these findings are consistent with a model in which NAC increases reduced glutathione and the activity of glutathione‐dependent antioxidant enzymes (eg, GSTA4) to nullify toxic reactive lipid species.
Targeted Lipidomics Identified Expected Increases in ALOX5 Metabolites in ICH and a Prosurvival Prostaglandin, PGE2, Which Synergized With NAC in ICH
As described above, we observed expected increases of ALOX5 metabolites in vivo in rats following ICH (Fig 4). Our mass spectrometric analysis from rats also revealed that ICH‐induced COX‐dependent lipid species (Fig 7A), including PGE2, prostaglandin D2 (PGD2), prostaglandin F2 (PGF2), and 6‐keto‐prostaglandin F2 (6keto‐PGF2), are increased following ICH (Fig 7C–F). Interestingly, F2‐isoprostanes, which are a marker of nonenzymatic lipid peroxidation, were not increased (Fig 7B). Collectively, these data suggest that AA metabolites downstream of ALOX5 and COX are increased following ICH, whereas a canonical nonenzymatic lipid species, F2 isoprostanes, are not. The findings are consistent with previous observations from several labs that showed that COX‐2 and downstream metabolic enzymes are induced following ICH.43, 44
Figure 7.
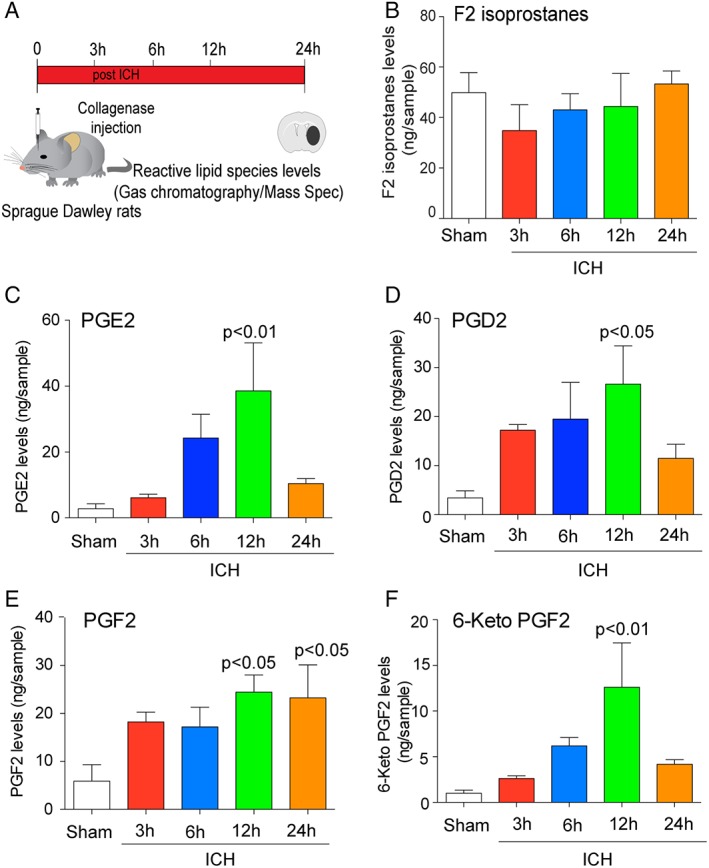
Intracerebral hemorrhage (ICH) in rodents induced COX‐derived oxidized lipids. (A) Experimental design for analyzing eicosanoid levels post‐ICH in mice. (B) GC/MS analysis revealed that levels of F2 isoprostanes, established markers of nonenzymatic lipid peroxidation, were not altered after ICH (n = 4). COX‐derived species PGE2 (C), PGD2 (D), PGF2 (E), and 6‐keto PGF2 (F) were time dependently increased post‐ICH (n = 4). Data from sham control brains from each time points were pooled for the analysis. Significance was determined by one‐way ANOVA and Dunnet's multiple‐comparison test. All graphs are mean ± SEM. ANOVA = analysis of variance; COX = cyclooxygenase; GC/MS = gas chromatography/mass spectrometry; ICH = intracerebral hemorrhage; PGD2 = prostaglandin D2; PGE2 = prostaglandin E2; PGF2 = prostaglandin F2; SEM = standard error of the mean.
Although a number of prostaglandin species downstream of COX‐2 activity were induced following ICH, we focused our attention on PGE2. Indeed, a zebrafish model was used to show that PGE2 combined with NAC synergistically prevented cell death from acetaminophen‐induced toxicity in liver. Acetominophen, like hemin, triggers death by insufficient glutathione levels or flux (Fig 6; see a previous work45. Accordingly, we asked whether PGE2 would synergize with NAC protection against hemin‐induced ferroptosis in vitro. Despite modest protection from hemin‐induced ferroptosis by 16,16 dimethyl PGE2 (dmPGE2, a long‐acting analog of PGE2) in cortical neurons, treatment of NAC (100 μM) and dmPGE2 (10 μM) provided synergistic protection (Fig 8A). These results showed that combinatorial treatment of dmPGE2 and NAC could reduce the concentration of NAC required for protection by 10‐fold in vitro. To determine whether NAC and dmPGE2 would synergize to induce functional recovery after ICH in vivo, NAC (40mg/kg; IP, a dose ineffective in mice and rats) and dmPGE2 10 μM (ICV) was delivered 2 hours postinjury in mice, and then NAC 40mg/kg was administered IP once‐daily for 7 days (Fig 8B). Whereas the combination of NAC and dmPGE2 significantly reduced neuronal degeneration, as monitored by Fluoro‐Jade staining (green) in the perihematomal regions of the mouse brain after ICH (Fig 8C,D), NAC or dmPGE2 alone had no significant effect. Behavioral studies following ICH showed that NAC plus dmPGE2 combination significantly improves behavioral deficits induced by ICH (Fig 8E,F), whereas either agent alone had no effect. Of note, we attempted to evaluate the combination of PGE2 plus NAC in rats, but again we found critical species differences, in that rats, not mice, revealed large changes in body temperature to ∼40°C (and probably high blood pressure), leading to a significantly enlarged hematoma size, which likely explains the significantly worsened lesion size with this PGE2/NAC combination in rats. Together, these findings demonstrate that combinatorial administration of NAC and PGE2 synergistically protects the brain, and reduces the concentrations of NAC required to improve functional recovery following ICH in mice, whereas side effects limit its utility in rats.
Figure 8.
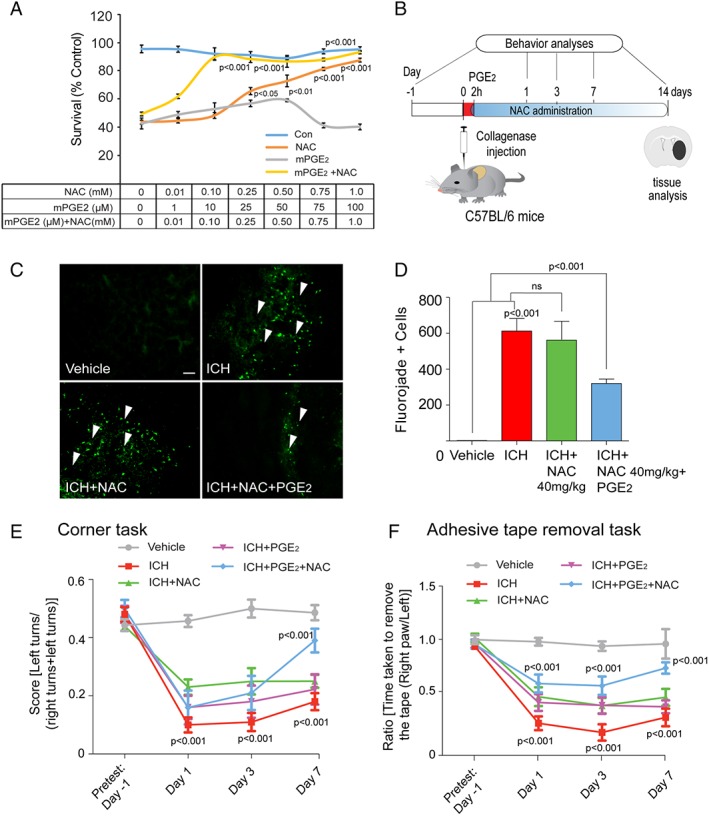
Prostaglandin (PGE2) synergized with NAC to prevent hemin‐induced ferroptosis in vitro and in improving functional recovery following ICH in mice in vivo. (A) NAC in combination with PGE2 synergized against hemin‐induced ferroptosis in primary cortical neurons. (B) Schematic illustration of the combinatorial delivery of NAC (40mg/kg, intraperitoneal) and PGE2 (10 μM, intracerebroventricular) after ICH in mice. The corner task (spatial neglect) and adhesive tape removal task (sensory neglect) were assessed on days 1, 3, and 7 after ICH to assess behavioral improvement. (C) The NAC (40 mg/kg, intraperitoneal) and intracerebroventricular (10 μM) PGE2 combination reduced neuronal degeneration as monitored by Fluoro‐Jade staining (green) in the perihematomal regions of the mouse brain after ICH. (D,E) Intraperitoneal NAC (40mg/kg) plus intracerebroventricular PGE2 reduces spatial neglect and sensory neglect induced by ICH, whereas each agent alone does not (n = 10). Significance was determined by two‐way ANOVA and Bonferroni's post hoc test. All graphs are mean ± SEM. ANOVA = analysis of variance; ICH = intracerebral hemorrhage; NAC = N‐acetylcysteine; ns = not significant; PGE2 = prostaglandin E2; SEM = standard error of the mean.
Discussion
ICH, a stroke subtype, remains a significant cause of mortality and morbidity around the world.2 Accordingly, there is a need to identify therapeutic agents with known safety in humans such as NAC to expand options for treating ICH. Whereas previous studies have suggested that NAC has many potential targets in the CNS for preventing cell death or enhancing functional recovery, our data point to nuclear ALOX5‐derived reactive lipid species as the target of action for NAC in mediating neuroprotective effects in an in vitro and an in vivo mouse model of ICH. First, NAC failed to synergize with established lipid peroxidation inhibitors, including trolox (a water‐soluble form of vitamin E; Fig 3B), vitamin E (not shown), or β‐carotene (not shown), consistent with them working on a convergent pathway centered in lipid peroxidation and its products. Second, NAC protection in an in vitro model of ICH (ferroptosis) was mimicked by structurally diverse inhibitors of ALOX5, an AA‐metabolizing enzyme that produces signaling, reactive lipids (Fig 3C). Similarly, molecular deletion of ALOX5 in mice improved functional recovery after ICH (Fig 5E,F). Third, AA reacted with protein following in vitro ICH, and the interaction of proteins and reactive lipids was blocked by chemical ALOX5 inhibition, a nonselective lipid peroxidation inhibitor (vitamin E), or NAC (Fig 6B,C). Fourth, NAC protection in vitro was dependent on glutathione levels or flux, indicating that direct scavenging or reaction with electrophiles is not required for NAC's beneficial effects (Fig 6). In further support of this finding, we found that the effects of NAC were mimicked by other agents known to increase cysteine (rate‐limiting precursor of glutathione), such as OTC, or increase glutathione (glutathione ethyl ester; Fig 6F). Moreover, Nrf2 activators (cystamine, NDGA),40 which induce glutathione synthesizing and glutathione detoxification enzymes, transcriptionally also prevent hemin toxicity (Fig 6F). Fifth, forced expression of GST isoform (GSTA4), known to neutralize reactive lipids in a glutathione‐dependent manner, also prevented hemin‐induced toxicity in vitro (Fig 6G). Sixth, increases in ALOX5 metabolites were observed following ICH in mice and rats, suggesting that toxic factors downstream of hemorrhage in vivo also produce reactive lipids (Fig 4). Seventh, increases in ALOX5 and ALOX5 AP message are observed in mice, rats, and humans; NAC treatment decreased hemin‐induced ALOX5 gene expression and hemin‐induced oxidized lipid species (Fig 5). Methods for detecting incorporation of reactive lipids downstream of ALOX5 in vivo are not currently available, suggesting that other biomarkers will need to be developed as a proxy for NAC's ability to decrease ALOX5‐derived reactive lipids. Nevertheless, previous studies have shown that zileuton can improve functional recovery following ICH in mice.36
Stoichiometric antioxidants, such as vitamin E or cerovive (NXY‐059), have been disappointing in the clinic for stroke treatment, likely because the concentrations required for these agents to be effective are relatively high compared to therapeutic agents that work catalytically. NAC also appears to act stoichiometrically to increase glutathione and is used at relatively high concentrations in vitro and in vivo (Fig 1). For stoichiometric antioxidants, one molecule of antioxidant is required to neutralize one oxidant. Accordingly, if the concentration of antioxidant drug that reaches the CNS is below the level of injury‐induced oxidant production, then the agent will be ineffective. Given that previous studies have not attempted to establish a relevant target for antioxidants in the required cell type(s), it has been difficult to establish the optimal dose of stoichiometric antioxidants to use in a clinical trial. Our studies suggest that following ICH, 5‐lipoxygenase–derived lipids are important for cell death, and thus provide a starting point for future studies aimed at establishing target engagement for NAC in ICH in humans. Indeed, previous transcriptomic analyses of brain tissues from human ICH patients identified an increased expression of mRNAs encoding ALOX5 and 5‐LOX–activating protein FLAP (ALOX5 AP).34 Additionally, a recent study identified a polymorphism in the promoter for FLAP (ALOX5 AP) that is associated with ICH in a Korean population.46 The ability of NAC to abrogate increases in ALOX5 and ALOX5 AP in hemin‐induced ferroptosis suggests a feed‐forward model in which ALOX5 products further augment levels of ALOX5 and ALOX5AP. This provides a strong argument for getting NAC to patients as early as possible at the appropriate dose after ICH to confer maximal effect against cell death as well as inflammation.
Targeted lipidomics of eicosanoids (signaling molecules resulting from oxidation of 20 carbon arachidonic acids) revealed that both products of ALOX5 and COX‐2 are increased following ICH in vivo. We decided to focus on the effects of PGE2, one product of COX‐2 metabolism, because of a previous study in zebrafish that showed that PGE2 and NAC could synergize to improve the therapeutic window and efficacy in preventing acetaminophen toxicity.45 Similar to hemin‐induced toxicity of neurons in vitro, NAC‐induced protection from acetaminophen toxicity requires increases in levels of flux of glutathione, so we considered the possibility that results from those studies might inform our investigations as well. Remarkably, we found synergy between PGE2 and NAC in preventing hemin‐induced ferroptosis in vitro, and improving recovery following collagenase‐induced ICH in vivo in mice. Of note, in vitro, we did not find that COX inhibitors (which would be expected to diminish PGE2) worsened hemin‐induced death in mixed neuronal‐glia cultures (not shown). These findings raise the possibility that a cell type other than neurons or glia is relevant for producing PGE2 in ICH, and it is formally possible that PGE2 is produced by microglia or macrophages, which are not present in our in vitro cultures. Whatever the source of PGE2 in ICH, our results are the first to demonstrate synergy between NAC and PGE2 in protecting the brain. With the advent of multiple strategies to deliver drugs to the hematoma site, including minimally invasive surgery47 or BrainPath,48 a combinatorial neuroprotective treatment delivered right to the hematoma site is now feasible.
We do not understand the basis for synergy between NAC and PGE2. Unexpectedly, we did not find that ALOX5 inhibitors synergized with PGE2 (not shown), raising the possibility that NAC may alter PGE2 signaling directly rather than by its effects on ALOX5‐derived products. Previous studies suggest that PGE2 affords neuroprotection against Aβ‐induced toxicity in primary neurons through EP2 or EP4 receptors, by cAMP/protein kinase A–dependent pathway.44, 49 Indeed, activation of EP2 receptor by PGE2 or agonist butaprost blocked hemin‐induced neuronal death.50 These findings contrast with other studies that have shown that PGE2 activation of EP3 receptors can worsen outcomes following ICH in mice, raising the possibility that NAC enhances PGE2‐EP2/EP4 binding while diminishing PGE2‐EP3 interactions. Of note, in mice or rats low doses of NAC (40mg/kg) were ineffective (Figs 1 and 8). We evaluated the PGE2/NAC combination in rats, but unlike mice, rats showed much greater increases in temperature (and probably blood pressure) in response to ICV infusion of 190ng of PGE2. These effects lead to an increase in ICH hematoma size in rats, which we did not observe in mice. Until this lack of species translation is understood, NAC and/or PGE2 should not be considered for treatment of ICH in humans. Indeed, future studies will focus on the mechanism by which NAC and PGE2 synergize in vitro and in vivo in mice as a strategy for moving this effective combination safely into rats and humans.
We could not examine doses of NAC in rats (300mg/kg) that were effective in mice because of dose‐limiting toxicity. Doses in rats that were well tolerated (40mg/kg) were ineffective. Similar doses (40–75mg/kg) in mice were also ineffective (Fig. 8C,D). Although it is formally possible that the unexpected toxicity of NAC in rats relates to the intraperitoneal delivery strategy we used (NAC is delivered intravenously or orally in humans), our results suggest that NAC may not have as wide a therapeutic window as previously considered. Our results in mice highlight that replication experiments in a different species can fail not only because of lack of efficacy, but also because of species differences in toxicity (Fig 2).
A survey of trials involving NAC in neurological disorders on clinical trials.gov reveals trials in process or completed in a host of neurological conditions, including subarachnoid hemorrhage, Parkinson's disease, Alzheimer's disease, traumatic brain injury, autism, and schizophrenia. Almost all studies chose doses of NAC that are safe, but without clear elucidation of the dose required to induce a therapeutic effect. Some studies are using MR spectroscopy to monitor increases in glutathione or glutamate with NAC, but, again, these studies and ours do not establish a clear target for how much of an increase in glutathione or glutamate is required for behavioral improvement. Cerebrospinal fluid measurements could improve fidelity of glutathione measurements, but a threshold level of increase of glutathione has yet to be elucidated in any paradigm.51 As we plan to try to avoid these enormous challenges in moving our studies of NAC forward for ICH, we will need to develop a good biomarker for NAC's therapeutic effect in humans. Given the ability of PGE2 to synergize with NAC in mice to induce functional recovery, it is our hypothesis that understanding precisely how this combination is effective will provide the best chances of neuroprotection with the widest therapeutic window in humans, once a good biomarker for NAC efficacy (possibly an ALOX5 metabolite) is identified.
Author Contributions
S.S.K., F.C., and R.R.R. conceived and designed the study, drafted the figures, and wrote the paper. S.S.K., L.A., Y.C., D.B., M.W.B., K.D., C.M.W., C.A.N., A.K., S.P., J.T.P., and S.S. conducted, acquired, and analyzed the data. S.S.K., G.L.M., V.D.‐U., D.P., T.R.H., S.T.C., G.C., F.C., and R.R.R. analyzed data and contributed to the manuscript preparation. All the authors reviewed and approved the final manuscript.
Potential Conflicts of Interest
R.R.R. and S.S.K. are co‐inventors on a patent related to the use of N‐acetylcysteine and/or PGE2 in neurological disorders. These patents have been licensed by Neuronasal, Inc., an early‐stage biotechnology company. R.R.R. is on the SAB for Neuronasal and has a small equity interest in the company along with receiving occasional consulting fees.
Acknowledgment
This work was supported by the Sperling Center for Hemorrhagic Stroke Recovery at Burke Neurological Institute, the Burke Foundation, Dr. Miriam and Sheldon G Adelson Medical Research Foundation grant to R.R.R., and the National Institutes of Health (Grant P01 NIA AG014930, Project 1 to R.R.R.).
We thank Soah J. Khim for providing technical assistance. Research support for the rat experiments was provided to F.C. from the Heart and Stroke Foundation of Canada. Analysis of eicosanoids (including specific compounds such as isoprostanes, prostaglandins, or leukotrienes) were performed in the Vanderbilt University Eicosanoid Core Laboratory.
REFERENCES
- 1. Qureshi AI, Suri MF, Georgiadis AL, et al. Intra‐arterial recanalization techniques for patients 80 years or older with acute ischemic stroke: pooled analysis from 4 prospective studies. AJNR Am J Neuroradiol 2009;30:1184–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. van Asch CJ, Oudendijk JF, Rinkel GJ, Klijn CJ. Early intracerebral hematoma expansion after aneurysmal rupture. Stroke 2010;41:2592–2595. [DOI] [PubMed] [Google Scholar]
- 3. Green JL, Heard KJ, Reynolds KM, Albert D. Oral and intravenous acetylcysteine for treatment of acetaminophen toxicity: a systematic review and meta‐analysis. West J Emerg Med 2013;14:218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dekhuijzen PN, van Beurden WJ. The role for N‐acetylcysteine in the management of COPD. Int J Chron Obstruct Pulmon Dis 2006;1:99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. De Rosa SC, Zaretsky MD, Dubs JG, et al. N‐acetylcysteine replenishes glutathione in HIV infection. Eur J Clin Invest 2000;30:915–929. [DOI] [PubMed] [Google Scholar]
- 6. Adair JC, Knoefel JE, Morgan N. Controlled trial of N‐acetylcysteine for patients with probable Alzheimer's disease. Neurology 2001;57:1515–1517. [DOI] [PubMed] [Google Scholar]
- 7. Berk M, Malhi GS, Gray LJ, Dean OM. The promise of N‐acetylcysteine in neuropsychiatry. Trends Pharmacol Sci 2013;34:167–177. [DOI] [PubMed] [Google Scholar]
- 8. Clark J, Clore EL, Zheng K, et al. Oral N‐acetyl‐cysteine attenuates loss of dopaminergic terminals in alpha‐synuclein overexpressing mice. PLoS One 2010;5:e12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Monti DA, Zabrecky G, Kremens D, et al. N‐acetyl cysteine may support dopamine neurons in Parkinson's disease: preliminary clinical and cell line data. PLoS One 2016;11:e0157602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zille M, Karuppagounder SS, Chen Y, et al. Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke 2017;48:1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li Q, Han X, Lan X, et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2017;2:e90777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress‐induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci 1994;14:4385–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baker DA, Xi ZX, Shen H, et al. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci 2002;22:9134–9141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Khan M, Sekhon B, Jatana M, et al. Administration of N‐acetylcysteine after focal cerebral ischemia protects brain and reduces inflammation in a rat model of experimental stroke. J Neurosci Res 2004;76:519–527. [DOI] [PubMed] [Google Scholar]
- 15. Wright DJ, Renoir T, Smith ZM, et al. N‐Acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington's disease. Transl. Psychiatry 2015;5:e492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xiong Y, Peterson PL, Lee CP. Effect of N‐acetylcysteine on mitochondrial function following traumatic brain injury in rats. J. Neurotrauma 1999;16:1067–1082. [DOI] [PubMed] [Google Scholar]
- 17. Lin KI, Lee SH, Narayanan R, et al. Thiol agents and Bcl‐2 identify an alphavirus‐induced apoptotic pathway that requires activation of the transcription factor NF‐kappa B. J Cell Biol 1995;131:1149–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martinez de Lizarrondo S, Gakuba C, Herbig BA, et al. Potent thrombolytic effect of N‐acetylcysteine on arterial thrombi. Circulation 2017;136:646–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. MacLellan CL, Davies LM, Fingas MS, Colbourne F. The influence of hypothermia on outcome after intracerebral hemorrhage in rats. Stroke 2006;37:1266–1270. [DOI] [PubMed] [Google Scholar]
- 20. Williamson MR, Dietrich K, Hackett MJ, et al. Rehabilitation augments hematoma clearance and attenuates oxidative injury and ion dyshomeostasis after brain hemorrhage. Stroke 2017;48:195–203. [DOI] [PubMed] [Google Scholar]
- 21. Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem 1994;62:376–379. [DOI] [PubMed] [Google Scholar]
- 22. Karuppagounder SS, Alim I, Khim SJ, et al. Therapeutic targeting of oxygen‐sensing prolyl hydroxylases abrogates ATF4‐dependent neuronal death and improves outcomes after brain hemorrhage in several rodent models. Sci Transl Med 2016;8:328ra329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang B, Horvath S. A general framework for weighted gene co‐expression network analysis. Stat Appl Genet Mol Biol 2005;4: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 24. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Robinson SJ, Hoobler EK, Riener M, et al. Using enzyme assays to evaluate the structure and bioactivity of sponge‐derived meroterpenes. J Nat Prod 2009;72:1857–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grossetete M, Rosenberg GA. Matrix metalloproteinase inhibition facilitates cell death in intracerebral hemorrhage in mouse. J Cereb Blood Flow Metab 2008;28:752–763. [DOI] [PubMed] [Google Scholar]
- 27. Lin KI, DiDonato JA, Hoffmann A, et al. Suppression of steady‐state, but not stimulus‐induced NF‐κB activity inhibits alphavirus‐induced apoptosis. J Cell Biol 1998;141:1479–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hatakeyama T, Okauchi M, Hua Y, et al. Deferoxamine reduces neuronal death and hematoma lysis after intracerebral hemorrhage in aged rats. Transl Stroke Res 2013;4:546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lopez‐Erauskin J, Fourcade S, Galino J, et al. Antioxidants halt axonal degeneration in a mouse model of X‐adrenoleukodystrophy. Ann Neurol 2011;70:84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chuang DY, Simonyi A, Cui J, et al. Botanical polyphenols mitigate microglial activation and microglia‐induced neurotoxicity: role of cytosolic phospholipase A2. Neuromolecular Med 2016;18:415–425. [DOI] [PubMed] [Google Scholar]
- 31. Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 2001;294:1871–1875. [DOI] [PubMed] [Google Scholar]
- 32. Enyedi B, Jelcic M, Niethammer P. The cell nucleus serves as a mechanotransducer of tissue damage‐induced inflammation. Cell 2016;165:1160–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Samuelsson B. The discovery of the leukotrienes. Am J Respir Crit Care Med 2000;161:S2–S6. [DOI] [PubMed] [Google Scholar]
- 34. Carmichael ST, Vespa PM, Saver JL, et al. Genomic profiles of damage and protection in human intracerebral hemorrhage. J Cereb Blood Flow Metab 2008;28:1860–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chang W, Gundersen GG. Swollen nuclei signal from the grave. Cell 2016;165:1051–1052. [DOI] [PubMed] [Google Scholar]
- 36. Hijioka M, Anan J, Ishibashi H, et al. Inhibition of leukotriene B4 action mitigates intracerebral hemorrhage‐associated pathological events in mice. J Pharmacol Exp Ther 2017;360:399–408. [DOI] [PubMed] [Google Scholar]
- 37. Higdon AN, Dranka BP, Hill BG, et al. Methods for imaging and detecting modification of proteins by reactive lipid species. Free Radic Biol Med 2009;47:201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Higdon AN, Benavides GA, Chacko BK, et al. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: the protective role of autophagy. Am J Physiol Heart Circ Physiol 2012;302:H1394–H1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Porta P, Aebi S, Summer K, Lauterburg BH. L‐2‐oxothiazolidine‐4‐carboxylic acid, a cysteine prodrug: pharmacokinetics and effects on thiols in plasma and lymphocytes in human. J Pharmacol Exp Ther 1991;257:331–334. [PubMed] [Google Scholar]
- 40. Smirnova NA, Haskew‐Layton RE, Basso M, et al. Development of Neh2‐luciferase reporter and its application for high throughput screening and real‐time monitoring of Nrf2 activators. Chem Biol 2011;18:752–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Milne GL, Zanoni G, Porta A, et al. The cyclopentenone product of lipid peroxidation, 15‐A2t‐isoprostane, is efficiently metabolized by HepG2 cells via conjugation with glutathione. Chem Res Toxicol 2004;17:17–25. [DOI] [PubMed] [Google Scholar]
- 42. Benes H, Vuong MK, Boerma M, et al. Protection from oxidative and electrophilic stress in the Gsta4‐null mouse heart. Cardiovasc Toxicol 2013;13:347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gong C, Ennis SR, Hoff JT, Keep RF. Inducible cyclooxygenase‐2 expression after experimental intracerebral hemorrhage. Brain Res 2001;901:38–46. [DOI] [PubMed] [Google Scholar]
- 44. Echeverria V, Greenberg DL, Dore S. Expression of prostaglandin E2 synthases in mouse postnatal cortical neurons. Ann N Y Acad Sci 2005;1053:460–471. [DOI] [PubMed] [Google Scholar]
- 45. North TE, Babu IR, Vedder LM, et al. PGE2‐regulated wnt signaling and N‐acetylcysteine are synergistically hepatoprotective in zebrafish acetaminophen injury. Proc Natl Acad Sci U S A 2010;107:17315–17320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim DH, Ahn WY, Kim DK, et al. A Promoter polymorphism (rs17222919, ‐1316T/G) of ALOX5AP is associated with intracerebral hemorrhage in Korean population. Prostaglandins Leukot Essent Fatty Acids 2011;85:115–120. [DOI] [PubMed] [Google Scholar]
- 47. Hanley DF, Thompson RE, Muschelli J, et al. Safety and efficacy of minimally invasive surgery plus alteplase in intracerebral haemorrhage evacuation (MISTIE): a randomised, controlled, open‐label, phase 2 trial. Lancet Neurol 2016;15:1228–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Labib MA, Shah M, Kassam AB, et al. The safety and feasibility of image‐guided brainpath‐mediated transsulcul hematoma evacuation: a multicenter study. Neurosurgery 2017;80:515–524. [DOI] [PubMed] [Google Scholar]
- 49. McCullough L, Wu L, Haughey N, et al. Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J Neurosci 2004;24:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mohan S, Narumiya S, Dore S. Neuroprotective role of prostaglandin PGE2 EP2 receptor in hemin‐mediated toxicity. Neurotoxicology 2015;46:53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Reyes RC, Cittolin‐Santos GF, Kim JE, et al. Neuronal glutathione content and antioxidant capacity can be normalized in situ by N‐acetyl cysteine concentrations attained in human cerebrospinal fluid. Neurotherapeutics 2016;13:217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]