

Abstract
Gap junctions (GJs) coupling oligodendrocytes to astrocytes and to other oligodendrocytes are formed mainly by connexin47 (Cx47) and a smaller portion by connexin32 (Cx32). Mutations in both connexins cause inherited demyelinating disorders, but their expression is also disrupted in multiple sclerosis (MS). To clarify whether the loss of either Cx47 or Cx32 could modify the outcome of inflammation and myelin loss, we induced experimental autoimmune encephalomyelitis (EAE) in fully backcrossed Cx32 knockout (KO) and Cx47KO mice and compared their outcome with wild type (WT, C57BI/6 N) mice. Cx47KO EAE mice developed the most severe phenotype assessed by clinical scores and behavioral testing, followed by Cx32KO and WT mice. Cx47KO more than Cx32KO EAE mice developed more microglial activation, myelin, and axonal loss than did WT mice. Oligodendrocyte apoptosis and precursor proliferation was also higher in Cx47KO than in Cx32KO or WT EAE mice. Similarly, blood‐spinal cord barrier (BSCB) disruption and inflammatory infiltrates of macrophages, T‐ and B‐cells were more severe in Cx47KO than either Cx32KO or WT EAE groups. Finally, expression profiling revealed that several proinflammatory cytokines were higher at the peak of inflammation in the Cx47KO mice and persisted at later stages of EAE in contrast to reduction of their levels in WT EAE mice. Thus, loss of oligodendrocyte GJs aggravates BSCB disruption and inflammatory myelin loss, likely due to dysregulation of proinflammatory cytokines. This mechanism may play an important role in MS brain with reduced connexin expression, as well as in patients with inherited mutations in oligodendrocyte connexins and secondary inflammation.
Keywords: Connexins, experimental autoimmune encephalomyelitis, multiple sclerosis, myelin, oligodendrocytes
Main Points
We induced EAE in WT, Cx32KO and Cx47KO mice.
EAE scores and pathological findings were exacerbated in Cx47KO more than in Cx32KO mice with dysregulation of several cytokines.
Loss of oligodendrocyte connexins has pro‐inflammatory effects.
1. INTRODUCTION
Multiple sclerosis (MS) is a chronic progressive neurological disease of the central nervous system (CNS) characterized by inflammation and destruction of myelin, tissue remodeling and microglial activation resulting in axonal degeneration (Lublin & Reingold, 1996; Weiner, 2009). The pathological correlate of relapses is a disruption of the blood–brain barrier (BBB) and inflammatory cell infiltration into the CNS (Correale & Villa, 2007; Larochelle, Alvarez, & Prat, 2011). Glia cells play important roles in brain function and are directly relevant to MS pathogenesis. Oligodendrocytes myelinate axons, facilitating rapid impulse propagation, but they also provide trophic and metabolic support to axons that is required for normal axonal transport and long‐term survival (Nave, 2010). Astrocytes also serve important homeostatic roles in the CNS, regulating metabolic and synaptic balance, as well as inflammatory responses (Alvarez, Katayama, & Prat, 2013; Luissint, Artus, Glacial, Ganeshamoorthy, & Couraud, 2012). Moreover, they participate in the formation of the BBB (Miljkovic & Spasojevic, 2013).
Both oligodendrocytes and astrocytes are extensively connected via intercellular gap junctions (GJs). These include oligodendrocyte:oligodendrocyte (O:O) oligodendrocyte:astrocyte (O:A), and astrocyte:astrocyte (A:A) GJs. Most O:O junctions are formed by connexin47 (Cx47) and a lesser portion by connexin32 (Cx32) (Altevogt & Paul, 2004; Kamasawa et al., 2005; Kleopa, Orthmann, Enriquez, Paul, & Scherer, 2004; Nagy, Ionescu, Lynn, & Rash, 2003a; Rash, Yasumura, Dudek, & Nagy, 2001; Wasseff & Scherer, 2011). Cx47 is expressed in the perikarya and proximal processes of oligodendrocytes and couples with astrocytic Cx43 (Orthmann‐Murphy, Freidin, Fischer, Scherer, & Abrams, 2007) to form most O:A GJs (Altevogt & Paul, 2004; Kamasawa et al., 2005). Cx32 forms some intercellular GJs with astrocytic Cx30 (Altevogt & Paul, 2004; Nagy et al., 2003a; Nagy, Ionescu, Lynn, & Rash, 2003b) but is mainly found along large myelinated fibers of the white matter (WM) (Altevogt, Kleopa, Postma, Scherer, & Paul, 2002; Kleopa et al., 2004) forming mostly reflexive intracellular channels in the myelin sheath (Kamasawa et al., 2005). The functional role of oligodendrocyte GJs has been highlighted by human disorders caused by mutations in the respective connexin genes. Recessive GJC2/Cx47 mutations cause hypomyelinating leukodystrophy type 2 (Uhlenberg et al., 2004), while GJB1/Cx32 mutations cause in addition to peripheral neuropathy acute transient encephalopathy syndromes, often induced by conditions of metabolic stress (Altevogt et al., 2002; Paulson et al., 2002; Taylor, Simon, Marks, & Scherer, 2003). Mouse models including the Cx32 knockout (KO) and Cx47KO are available but show little baseline CNS pathology; however mice lacking both Cx32 and Cx47 (Menichella, Goodenough, Sirkowski, Scherer, & Paul, 2003; Odermatt et al., 2003) confirmed that loss of oligodendrocyte GJ connectivity causes severe CNS demyelination.
The strong inflammatory reaction in the CNS of these connexin mutants (Georgiou et al., 2017; Schiza et al., 2015; Wasseff & Scherer, 2015) suggests that oligodendrocyte GJs may play a role in acquired neuroinflammatory disorders. Furthermore, analysis of postmortem human MS brain has demonstrated extensive alterations of glial GJ formation in MS, both within WM lesions as well as in the normal‐appearing WM (Markoullis et al., 2012; Masaki et al., 2012) and cortex (Markoullis et al., 2014). These alterations have also been reproduced in experimental autoimmune encephalomyelitis (EAE) mouse model of the disease (Markoullis et al., 2012). This widespread disruption of oligodendrocyte GJs in MS brain and EAE model are predicted to contribute to further demyelination, failed remyelination, and axonal loss, accelerating disease progression and associated disability (Markoullis et al., 2014). However, it remains unclear whether the loss of oligodendrocyte GJs is sufficient to drive the pathological process in inflammatory demyelination.
The aims of this study were to examine the hypothesis that loss of either of the two GJ‐forming connexins in oligodendrocytes would lead to increased severity of induced EAE and to clarify the underlying mechanisms. We show that clinical and pathological outcomes were exacerbated in mutant mice with loss of either Cx32 or Cx47 GJs, with more severe outcomes in mice lacking Cx47 than in mice lacking Cx32. Increased myelin loss and oligodendrocytes apoptosis in GJ deficient mice was associated with a more severe disruption of the blood‐spinal cord barrier (BSCB), as well as stronger and more persistent elevation of inflammatory cytokines. These results confirm that oligodendrocyte GJs may play a regulatory role in inflammatory conditions and their loss has a proinflammatory effect, a mechanism that may be directly relevant for MS pathogenesis and progression.
2. MATERIALS AND METHODS
2.1. Experimental animals
Breeding pairs of C57BL/6 N mice (wild type [WT]), as well as Gjb1‐null/Cx32 KO and Gjc2/Cx47KO mice fully backrossed (at least 10 generations) onto a C57BL/6 N background from the same breeding colony used as controls and were housed in the pathogen‐free mouse facility of the Cyprus Institute of Neurology and Genetics. All mice were genotyped prior to use. Animals were kept under controlled conditions of temperature (~21°C), humidity, air exchange and bright cycle (12/12‐h dark/light) and provided with standardized mouse diet and drinking water ad libitum. All animal procedures were approved by the Government's Chief Veterinary Officer according to EU guidelines (License Nr. CY/EXP/PR.L2/2010/2014) and by the SUNY Downstate Medical Center IACUC. “Animal research: reporting in vivo experiments (ARRIVE)” guidelines were also followed (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010).
2.2. Induction and assessment of EAE
The immunogen, myelin oligodendrocyte glycoprotein (MOG) peptide 35–55 (NH2–MEVGWYRSPFSRVVHLYRNGK–OH) was synthesized in the peptide synthesis laboratory at Johns Hopkins University lab (Baltimore, USA). For disease induction, mice were immunized subcutaneously (s.c.) at the base of the tail with 200 μl of an emulsion containing Complete Freund's Adjuvant (CFA) and MOG35–55 peptide as previously described (Jones et al., 2008), with minor modifications. Briefly, the peptide was dissolved in saline at 2 mg/ml, mixed at a 1:1 ratio with complete adjuvant, supplemented with 5 mg/ml heat‐inactivated Mycobacterium tuberculosis and mixed until a stable emulsion was formed. On the day of immunization and 2 days later mice were also injected intraperitoneally (i.p.) with 200 ng of pertussis toxin in 200 μl PBS. Control animals of each genotype were immunized with an equivalent volume of CFA emulsion without MOG peptide (CFA control groups). The mice were observed daily and were scored on a scale of 0–5 with gradations of 0.5 for intermediate scores (Jones et al., 2008): 0, no clinical signs; 1, loss of tail tone; 2, wobbly gait; 3, hind limb paralysis; 4, moribund; and 5, death. The results presented in this work are in accordance with the guidelines suggested for EAE publications (Baker & Amor, 2012).
2.3. Behavioral analysis
2.3.1. Rotarod test
For evaluation of motor abnormalities, the fixed‐speed rotarod test was used. Mice were placed on a rotarod (diameter, 3.2 cm; Ugo Basile, Comerio, Italy), while it was turning at a fixed speed (12 and 20 rpm). Retention time on the rod was recorded in seconds, up to a maximum of 1,800. Mice were trained before immunization on three consecutive days by performing three trials on each day.
2.3.2. Grip strength
The strength of both the fore‐ and hind‐limbs of immunized mice was measured using the grip strength meter (Ugo Basile, Comerio, Italy) according to manufacturer's instructions. The peak force (in grams; g) was measured. All force measurements were repeated five consecutive times and the mean value was calculated.
2.4. Immunostaining
For immunofluorescence staining, mice were deeply anesthetized and subsequently transcardially perfused with ice‐cold saline, followed by 4% paraformaldehyde (PFA) solution in 0.1 M phosphate buffer (pH = 7.2). Spinal cord tissues were removed, post‐fixed in 4% PFA for 2 h and then transferred to 20% sucrose (in 0.1 M phosphate buffer, pH = 7.2). The spinal cord tissues were cut into six blocks corresponding to cervical, lower cervical‐upper thoracic, mid thoracic, lower thoracic‐upper lumbar, lower lumbar and sacral segments. Subsequently, tissues were embedded in OCT compound (Sakura Finetek, USA) and stored at −80°C. Cryosections were mounted onto glass slides, permeabilized in cold acetone (−20°C for 10 min), incubated at room temperature (RT) with blocking solution (5% bovine serum albumin [BSA] containing 0.5% Triton X‐100) for 1 h and then incubated overnight at 4°C with primary antibodies: Ms anti‐neurofilament antibody RT97 (Developmental Studies Hybridoma Bank, 1:1,000), Rt‐proteolipid protein (PLP) (gift of Prof. Richard Reynolds; 1:10), Rb‐Iba1 (Biocare Medical 1:500), Ms‐CC1 (Calbiochem 1:50), Ms‐Glut1 (Abcam; 1:200), activated Rb‐caspase 3 (Abcam 1:100), Rt‐CD68 (Serotec 1:100), Rb‐CD3 (Abcam 1:100), Gt‐CD20 (Santa Cruz 1:100), Ms‐Cx43 (Millipore 1:50), Ms‐Cx32 (Invitrogen 1:50), Rb‐Cx32 (Invitrogen 1:50), Ms‐Cx47 (Invitrogen 1:300) Rb‐Cx30 (Invitrogen 1:300). Sections were washed in PBS and incubated with appropriate Dylight fluorescein or rhodamine conjugated AffiniPure F(ab’)2 secondary antibodies (Jackson ImmunoResearch Laboratories) for 1 h at RT. Cell nuclei were visualized with 4′,6′‐diamidino‐2‐phenylindole (DAPI) (Sigma‐Aldrich). Images were obtained under a Zeiss fluorescence microscope with a digital camera and comparable exposure times using the Zeiss Axiovision software (Carl Zeiss MicroImaging).
For assessment of peripheral immune reaction, spleen sections were cut from PFA fixed tissue blocks (12 μm), thaw‐mounted onto glass slides, permeabilized for 10 min in cold (−20°C) acetone and blocked (5% BSA in PBS containing 0.5% Triton‐X 100) for 1 h at RT. Subsequently, sections were incubated overnight at 4°C with the following primary antibodies diluted in blocking solution: rabbit antisera against Cx47 (clone 935, 1:800) (Orthmann‐Murphy et al., 2007), Cx32 (Invitrogen 1:50), CD20 (M‐20) (Santa Cruz 1:100), CD68‐Alexa488 (Serotec 1:50), CD3 (Invitrogen 1:100). Sections were washed, incubated with appropriate secondary antibodies and imaged as above.
2.5. BSCB permeability assay
BSCB leakiness was assessed in vivo by the Evans Blue (EB) dye method (Sigma, St. Louis, MO, USA) and by staining for endogenous fibronectin and fibrinogen in WT, Cx32KO, and Cx47KO mice. Briefly, 5 μl/g of body weight of a 4% EB stain solution (in PBS) was injected intravenously into each mouse 30 min before they were sacrificed. Slides containing spinal cord sections were directly visualized using a fluorescence microscope. Cryosections from lumbar spinal cord were immunostained for fibronectin (DAKO, Glostrup, Denmark, 1:100) and FITC‐labeled polyclonal fibrinogen antibody (DAKO, 1:500) to assess serum protein leakage into the parenchyma.
2.6. Quantification of EAE pathology
All aspects of EAE pathology were quantified by an observer blinded to genotype using the Image Pro Analysis 6.3 software (Media Cybernetics) on whole immunostained lumbar spinal cord cross sections imaged in an automated fashion and captured sequentially with the 20× objective as described previously (Markoullis, Sargiannidou, Gardner, et al., 2012). Areas showing loss of myelin lacking PLP immunoreactivity but with axonal preservation were outlined, measured, and expressed as a percentage of total lumbar spinal cord WM area delineated by the myelin staining. Axonal loss was evaluated in sections stained with a mouse monoclonal antibody against RT97 labeling neurofilament heavy subunits. We counted the total number of axons in the WM of a lumbar spinal cord hemisection in each of the EAE mice.
2.7. TUNEL assay
For the TUNEL assay, sections were processed using the in situ BrdU‐Red DNA fragmentation (TUNEL) assay kit (Abcam) and counterstained with DAPI. Spinal cord sections cut from PFA fixed tissue blocks (10–12 μm) were dried, immersed in 10 mM Tris‐HCI (pH = 7.4) at 37°C for 5 min, and then incubated in 20 μg/ml Proteinase K solution (2 μl Proteinase K 10 mg/ml + 998 μl 10 mM Tris‐HCl pH = 8.0 containing 50 mM Ethylenediaminetetraacetic acid [EDTA]) for 5 min, washed and incubated in PFA for 5 min. Tissues were covered with DNA labelling solution and incubated 1 h in the dark in a humidified 37°C incubator. Sections were then washed and incubated with anti‐BrdU‐Red antibody for 30 min, then incubated with 7‐AAD/RNase A Staining Buffer for optional counterstain for total DNA for 30 min. Cell nuclei were visualized with DAPI (Sigma‐Aldrich). Microscopy and image acquisition were performed as above. The total number of TUNEL‐positive cells per lumbar spinal cord section was counted in four different sections, averaged and compared between genotypes.
2.8. BrdU staining
Mice were injected i.p. with BrdU (50 μg/g of body weight) 6 h before sacrificing (Cameron, 2006). Spinal cord sections cut from PFA fixed tissue blocks were denatured with 1 N HCI and incubated at 37°C for 10 min and subsequently washed and neutralized with PBS for 5 min. Tissues were then thaw‐mounted onto glass slides, permeabilized for 10 min in cold (−20°C) acetone and blocked (5% BSA in PBS containing 0.5% Triton‐X 100) for 1 h at RT. Sections were then incubated overnight at 4°C with the anti‐BrdU antibody (Abcam 1:100) in blocking solution. Finally, they were washed and incubated with appropriate secondary antibodies before microscopy as above.
2.9. Cytokine expression analysis
Protein lysates were obtained by homogenization of lumbar spinal cords in RIPA buffer (10 mM sodium phosphate pH = 7.0, 150 mM NaCl, 2 mM EDTA, 50 mM sodium fluoride, 1% NP‐40, 1% sodium deoxycholate, and 0.1% sodium dodecyl sulphate [SDS]) containing a protease‐inhibitor cocktail (Roche; Cat. Nr. 11,836,153 00). Tissues were sonicated and protein concentrations were measured on a NanoDrop. The expression level of various cytokines was assessed using an array membrane (RayBio® C‐Series Mouse Cytokine Antibody Array C3) incubated overnight at 4°C, using the protocol recommended for the CNS. After washes, the membranes were incubated with the biotin‐conjugated antibodies overnight at 4 °C and processed according to the manufacturer's instructions. Following imaging and densitometry, OD signals from each blot were subjected to background subtraction and intra‐blot normalization. Fold change for each group was calculated and Log2 transformed for further statistical testing by multiple comparisons with corrections using the two‐stage step‐up method of Benjamini, Krieger and Yekutieli (false discovery rate, FDR = 0.1 or 0.15).
2.10. Immunoblot analysis
Fresh tissue samples were collected from lumbar spinal cord and lysed in ice‐cold RIPA buffer (as above). Tissues were sonicated and protein concentrations were measured on a NanoDrop. Proteins (150 μg) from each tissue lysate were fractioned on a 12% SDS‐polyacrylamide gel electrophoresis (PAGE) gel and transferred to a Hybond‐C extra membrane (GE Healthcare Bio‐Sciences), using a semi‐dry transfer unit. Nonspecific sites on the membrane were blocked with 5% nonfat milk in Tris‐buffered saline containing 0.1% Tween 20 (TBS‐T) for 1 h at RT. Immunoblots were then incubated with a rabbit antiserum against Cx43 (Millipore 1:500) at 4°C. After washing, blots were incubated with or anti‐mouse horseradish peroxidase‐conjugated secondary antiserum (Jackson ImmunoResearch Laboratories, 1:3,000) in 5% milk‐TBS‐T, for 1 h. Membranes were then reblotted with GAPDH (Santa Cruz, 1:4,000) antibody as a loading control. The bound antibody was visualized by enhanced chemiluminescence system (ECL Plus, GE Healthcare Bio‐Sciences, Amersham). To compare protein levels between samples, band intensity was measured using TinaScan software version 2.07d.
2.11. Statistical analysis
GraphPad Prism 6.0 (San Diego, CA) was used for statistical analysis. Errors are reported as SEM. EAE scores were tabulated as daily treatment group averages starting on the day of immunization with MOG35–55. Animals that died during the course of this study (i.e., a score of 5) were excluded from group average scores beyond that day. The Kruskal–Wallis test was used for assessing the differences in EAE clinical scores among the three groups while analysis of variances (ANOVAs) plus post hoc Tukey's honest significant difference test was used to compare the results of rotarod and grip strength tests. In both tests, p < .05 was considered significant. Differences between genotypes in EAE pathological features and blood spinal cord permeability assays were examined with the two‐tailed Student's t‐test and Bonferroni correction. Since three groups were compared in each experiment, the statistical threshold was set at 0.0167 and thus p‐values < .0167 were considered significant in all comparisons.
3. RESULTS
3.1. Clinical and behavioral analysis of EAE in connexin mutants
We have previously demonstrated that Cx32KO mice have a more severe clinical pathology compared with the WT mice when subjected to EAE (Markoullis, Sargiannidou, Gardner, et al., 2012). To further understand the role of oligodendrocyte connexins in the development and outcome of EAE, age matched (6‐ to 8‐week‐old) female fully back‐crossed Cx32KO or Cx47KO and WT mice were immunized with MOG33–55 peptide in order to induce EAE. The clinical course of EAE showed significant differences when comparing WT mice with connexin deficient mice (Figure 1a,c). Specifically, mice lacking Cx47 developed clinical signs of EAE earlier (mean day of disease onset 9 ± 1) than mice lacking Cx32 or WT mice (mean day of disease onset 11 ± 1 and 12 ± 1, respectively). Overall, EAE induced in Cx47 KO mice caused more severe mean clinical score (MCS) on a 0–5 scale compared with Cx32KO and WT mice. The MCS of Cx47KO group at the peak of the disease (20–24 days postimmunization‐dpi) was significantly higher (MCS = 3.3 ± 0.4) than that of Cx32KO (MCS = 2.8 ± 0.2) and WT (MCS = 2.0 ± 0.3) mice. Three mice from the Cx47KO group were fully paralyzed (EAE score was equal to 4.5) at 21–23 dpi and therefore they were sacrificed before the end of the experiment. A score of 4.5 was included in the calculation of daily MCS on day of death and in the calculation of MCS until the end of the experiment. Thus, EAE started earlier and was more severe in mice lacking Cx47 than in mice lacking Cx32 or in the WT group. EAE was induced in mice of the three genotypes a total of three times (n = 6–9 per group and experiment) with similar outcomes in each experiment (Supporting Information Figure S1).
Figure 1.

Clinical scores and motor performance of WT, Cx32KO and Cx47KO EAE mice. (a–c) Representative comparison of MCSs ± SEM between EAE mice of the three genotypes through day 24 dpi. Arrows indicate deaths in EAE groups (one Cx32KO and one Cx47KO mouse were found dead at 18 and 19 dpi, respectively). Death was scored as 5 on the day the animal found dead and was not included in subsequent calculations of MCS. Comparison of MCS at each time point starting from 10 dpi showed significantly higher scores in Cx32KO (a) and Cx47KO (b) compared with WT EAE groups. Cx47KO mice showed higher scores than Cx32KO (c) only at the peak of the disease (20–24 dpi) but these differences were not statistically significant. Rotarod test at both 12 (d) and 20 (e) rotations per minute (rpm) showed significantly worse performance for both Cx32KO and Cx47KO compared with WT EAE groups, as well as for Cx47KO compared with Cx32KO groups. EAE scores correlated with time to fall in all genotypes (f–h). Grip strength evaluation of fore‐ (i) and hind‐limbs (j) revealed significantly lower strength in Cx32KO as well as Cx47KO compared with WT EAE groups, while Cx47KO performed more poorly than Cx32KO mice in hind‐ but not in forelimb strength evaluation. Grip strength correlated with EAE scores in all EAE groups (k–m). Data in (a–c) represent mean ± SEM; Kruskal–Wallis test: *p < .005; **p < .01; ***p < .001; n.s.: Non‐significant. Data in d, e, i, and j represent mean ±SEM; one‐way ANOVA with Tukey's Post Hoc Test: #p < .05; ##p < .01; ###p < .001 [Color figure can be viewed at wileyonlinelibrary.com]
In addition to the classical method of scoring EAE we performed two other behavioral measures to assess motor performance, the rotatod test (n = 8–9 mice per group) and the grip‐strength test (n = 8–9 mice per group). We tested groups of WT, Cx32KO, and Cx47KO EAE and CFA‐control mice for each of the three genotypes and determined their relationship to the classical method of scoring EAE mice using the five‐step scale. The fixed‐speed rotarod test was performed only up to 18 dpi since animals with EAE score > 2.5 fell off the rotor immediately after it began turning. Cx47KO EAE treated mice spent less time on the rotarod compared with Cx32KO and WT mice at both 12 rpm (Figure 1d) and 20 rpm (Figure 1e). Rotarod performance correlated strongly with EAE score in all genotypes (Figure 1f–h).
Furthermore, we measured the strength of both the fore‐ and hind‐limbs of immunized mice using a mouse grip strength meter. Mice with an EAE score ≥ 3 were unable to perform this test because they failed to hold the wire of the grip‐strength meter. Thus, we could perform this test up to 18 dpi. Grip strength of the fore‐limbs started decreasing after the onset of EAE and continuously deteriorated up to 18 dpi (Figure 1i). A 7% loss in forelimb strength was observed for WT group while an approximately 15% loss of initial forelimb was recorded for Cx32KO and Cx47KO groups. Statistical analysis revealed that the decrease in forelimb strength of Cx32KO and Cx47KO groups was significantly higher when compared with WT group. However, no significant differences in the strength of forelimbs were observed between Cx32KO and Cx47KO EAE mice. In contrast, hind limb grip strength in the WT EAE group decreased by approximately 25% at 18 dpi, whereas in Cx32KO and Cx47KO the drops were 40 and 60%, respectively (Figure 1j). In addition, the decrease of both fore‐ and hind‐limb strength correlated with EAE scores in all genotypes (Figure 1k–m). Grip‐strength of CFA‐control Cx32KO and Cx47KO groups did not change significantly over time (data not shown). Thus, EAE mice show marked motor deficits and these are most severe in the Cx47KO groups, followed by the Cx32KO and then by the WT mice.
3.2. Pathological features of EAE in connexin mutant mice
All aspects of EAE pathology were compared among EAE mice of all three genotypes. Immunization of WT, Cx32KO, and Cx47KO mice with the MOG35–55 peptide caused inflammation and loss of myelin in the spinal cord (Figure 2a–c). Quantification of myelin loss in whole lumbar spinal cord sections (% WM area with lack of myelin immunoreactivity) showed that Cx47KO EAE mice were more affected than WT mice, whereas the difference in the degree of myelin loss observed between Cx47KO and Cx32KO EAE mice as well as between Cx32KO and WT EAE mice did not reach significance (Figure 2j). We further examined how the lack of either Cx47 of Cx32 relates to the loss of axons in EAE. Axon loss, which is often observed in EAE animal models (Markoullis, Sargiannidou, Gardner, et al., 2012) was detected in lumbar spinal cord sections from all three groups (Figure 2d–f). Axon counts in the WM quadrant of lumbar spinal cord sections from EAE mice at 24 dpi (Figure 2k) revealed a trend for more severe reduction in Cx47KO mice compared with Cx32KO and WT EAE groups, but these differences did not reach statistical significance.
Figure 2.
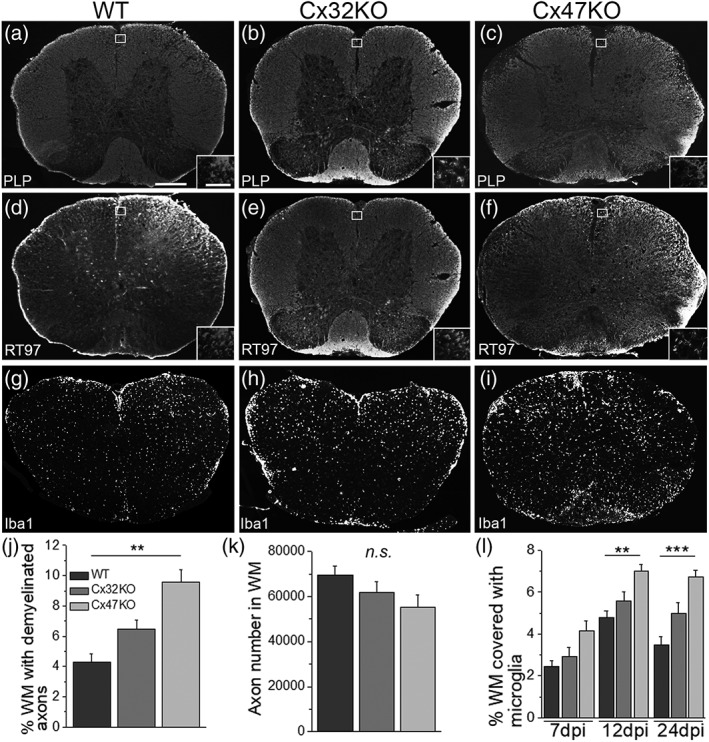
Pathological features of EAE in connexin mutants. These are representative images of whole lumbar spinal cord sections from EAE mice at 24 dpi, stained as indicated with antibodies to myelin marker PLP (a–c) to quantify myelin loss and with axonal neurofilament marker RT97 to quantify axonal loss (d,e), or for microglial marker Iba‐1 (g–i). Areas exhibiting loss of myelin as well as diffuse microglia activation appear more extensive in connexin deficient mice, especially in the Cx47KO. Insets show areas near the lesions at higher magnification. (j) Quantification of myelin loss (% of WM with lack of PLP immunoreactivity) (n = 4 mice per genotype at 24 dpi) showed significantly more myelin loss in Cx47KO compared with WT group, but not between WT and Cx32KO or between Cx32KO and Cx47KO groups. (k): Counts of axons (total number of axons in WM; n = 4 mice per group) show a trend for more severe loss in Cx47KO than in Cx32KO than in WT EAE mice, but these differences were not statistically significant. (l) Quantification of % WM area covered with Iba1 immunofluorescence at 7, 12, and 24 dpi shows more activated microglia in the Cx47KO compared with either the WT or Cx32KO EAE groups at all time points; these differences reach statistical significance at 12 and 24 dpi, but not at 7 dpi (data in j–l data represent mean ± SEM); Student's t‐test with Bonferroni correction and statistical threshold adjusted to *p < .0167; **p < .01; ***p < .001; only statistically significant differences are shown). Scale bars: In a = 100 μm; in inset (a) =30 μm
To examine whether the amount of activated microglia/macrophages differs between WT and Cx32‐ or Cx47‐deficient EAE mice, whole lumbar spinal cord sections of EAE mice from the three genotypes at 7, 12, and 24 dpi were immunolabeled with antibody to Iba‐1, a microglial marker. Extensive microglia/macrophages activation with increased Iba‐1 immunoreactivity was observed in all EAE groups, but this inflammatory activity was significantly greater in Cx47KO EAE mice (Figure 2g–i). Quantification of inflammation (% WM area covered with Iba1 immunoreactivity) at 7, 12, and 24 dpi (Figure 2l) confirmed that mice lacking Cx47 exhibited more activated microglia compared with Cx32KO or WT EAE mice. At 7 dpi there was a trend for higher Iba‐1 immunoreactivity in Cx47KO EAE than in Cx32KO or WT mice, but these differences were not statistically significant. At 12 dpi, Iba‐1 immunoreactivity increased in all EAE groups reaching its maximum value with Cx47KO mice showing more inflammation than WT mice (p < .01). The differences between Cx47KO and Cx32KO as well as between Cx32KO and WT mice were not statistically significant. At 24 dpi, Iba‐1 immunoreactivity remained high in Cx47KO, decreased slightly in Cx32KO EAE mice and decreased much more in the WT group. At this time point the WM area covered by Iba‐1 positive cells was significantly larger in Cx47KO compared with WT (p < .001) but did not differ significantly between Cx32KO and WT EAE or between Cx32KO and Cx47KO mice.
3.3. Cx47 deficient EAE mice show increased disruption of the BSCB
The BBB/BSCB is a major contributor to the maintenance of the CNS homeostasis by regulating soluble factor and cellular exchange between the CNS and the blood (Banks, 1999). BSCB breakdown is an underlying event in the pathogenesis of EAE (Fabis, Scott, Kean, Koprowski, & Hooper, 2007) and therefore an important determinant of pathology and clinical phenotype. Given the observed differences in EAE manifestations among connexin deficient mice, we compared the extent of BSCB permeability changes in connexin KOs and WT EAE mice at 12 dpi and at 24 dpi. We quantified the extravasation of intravenously injected EB dye into the lumbar spinal cord parenchyma. In addition, we examined lumbar spinal cord tissue obtained from EAE and CFA‐control mice for extravasation of fibrinogen and fibronectin into the parenchyma (Figure 3).
Figure 3.
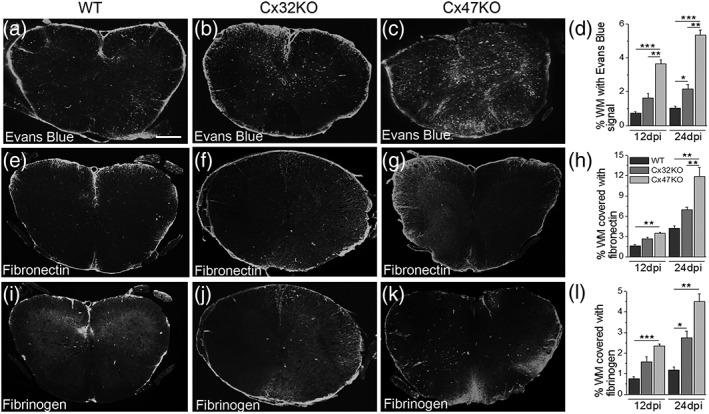
Evaluation of BSBC disruption. (a–c) Representative images of whole lumbar spinal cord sections from 24 dpi EAE mice following intravenous administration of EB dye show increased parenchymal leakage in the Cx47KO group compared with WT and Cx32KO groups. Quantification of total fluorescence for EB (n = 4 mice per genotype) at 12 and 24 dpi (d) confirms the more severe disruption of BSCB in Cx47KO group compared with the WT and Cx32KO groups. BSCB disruption was also assessed by immunofluorescence with antibodies to fibronectin (e–g) and fibrinogen (i–k). Quantification of total immunofluorescence for fibronectin (h) and fibrinogen (l) at 12 and 24 dpi (n = 4 mice per group) verified the increased BSCB leakage in the Cx47KO group compared with the WT and Cx32KO groups. (data in d, h, and l represent mean ± SEM. Student's t‐test with Bonferroni correction: *p < .0167; **p < .01; ***p < .001; only statistically significant differences are shown). Scale bars (in a) = 100 μm
Overall, BSCB leakiness increased from 12 to 24 dpi in all EAE groups. Significantly more extensive penetration of EB dye was observed in spinal cord sections of Cx47KO EAE mice than in sections from the Cx32KO or WT mice at 24 dpi (Figure 3a–c). We quantified the density of EB signal in the WM of whole lumbar spinal cord sections (n = 4 mice per genotype and time point) at both 12 and 24 dpi (Figure 3d). At 12 dpi, EB signal density reflecting BSCB leakage was significantly higher in Cx47KO EAE spinal cord compared with both Cx32KO (p < .01) and WT (p < .001) groups, whereas Cx32KO and WT EAE groups did not differ significantly. At 24 dpi, penetration of EB dye in spinal cord sections was further increased by approximately 35–45% in all EAE groups. Quantification at 24 dpi confirmed significantly higher EB density in Cx47KO EAE compared with either Cx32KO (p < .01) or WT EAE groups (p < .001), and BSCB leakage in Cx32KO EAE mice was also higher compared with the WT EAE group (p < .0167).
To further assess the effect of either Cx32 or Cx47 deficiency on the leakage of BSCB in EAE and to confirm the loss of BSCB integrity, we subsequently immunostained whole lumbar spinal cord sections from CFA‐control and EAE mice of all genotypes with antibodies to fibronectin and fibrinogen at both 12 and 24 dpi. Fibrinogen, plasminogen, and thrombin are blood proteins that have vital functions in the maintenance of integral vascular homeostatic processes involved in blood clotting. However, under pathological conditions associated with BBB and BSCB disruption, these proteins extravasate into brain and spinal cord parenchyma (Ryu & McLarnon, 2009). Moreover, fibrinogen/fibrin deposition is often seen in tissue from MS patients (Yang, Tian, Wu, Huang, & Wu, 2011). Our results revealed significantly increased leakiness for both fibronectin (Figure 3e–g) and fibrinogen (Figure 3i–k) in spinal cord sections of Cx47KO EAE mice compared with Cx32KO and WT groups; quantitative analysis of both the fibronectin (Figure 3h) and fibrinogen (Figure 3l) signals at 12 and 24 dpi confirmed this. In addition, the leakage of BSCB was significantly higher for EB and fibrinogen in Cx32KO compared with WT EAE mice at 24 dpi. As expected, immunostaining of spinal cord sections from CFA‐control mice from all genotypes did not show any BCSB leakage (data not shown). Overall, these results suggest that the differences in disease onset and severity between the WT, Cx32KO, and Cx47KO EAE mice may be related to the differences in the extent of BSCB leakage in each genotype.
3.4. Higher oligodendrocyte apoptosis and oligodendrocyte precursor cell proliferation rates are seen in EAE mice lacking Cx47
To assess the degree of oligodendrocyte apoptosis, whole lumbar spinal cord sections from EAE mice at 24 dpi were double immunolabeled with antibodies to activated caspase‐3 and oligodendrocyte marker CC1 (Figure 4a–c). Although apoptotic oligodendrocytes immunoreactive to activated caspase‐3/CC1 were seen in all EAE groups, their numbers per lumbar spinal cord section were significantly higher in Cx47KO compared with WT mice (p < .01); comparisons between WT and Cx32KO and between Cx32KO and Cx47KO EAE mice showed no significant differences (Figure 4d) although there was a trend towards higher numbers of caspase‐3 positive cells in Cx32KO compared with WT mice. No immunoreactivity for activated caspase‐3 was detected in CFA control mice of all groups (data not shown). In order to confirm these findings, we further examined the extent of oligodendrocyte apoptosis by TUNEL assay in lumbar spinal cord sections of CFA‐control and EAE mice from the WT, Cx32KO, and Cx47KO at 24 dpi. In EAE mice of all genotypes, TUNEL positive nuclei were detected in the form of small clusters (Figure 4e–g). Quantification of TUNEL positive cells (Figure 4h) revealed that the number of apoptotic cells was higher in Cx47KO (13.74 ± 1.22 cells per section) compared with both Cx32KO (10 ± 0.34) (p < .0167) and WT EAE mice (6.82 ± 0.42) (p < .001). Cx32KO also showed significantly more TUNEL positive cells than WT EAE mice (p < .001). In contrast, no nuclear TUNEL staining was detected in CFA control mice from all genotypes (data not shown).
Figure 4.
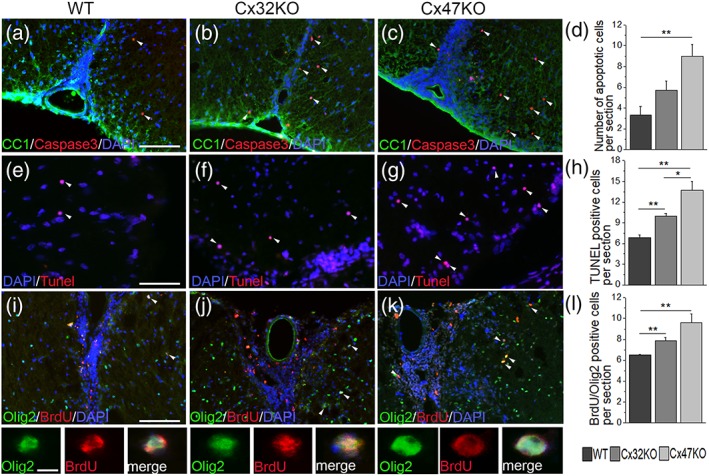
Assessment of apoptosis and proliferation of oligodendrocytes lineage cells in EAE. (a–c) Double staining of 24 dpi EAE lumbar spinal cord sections with apoptosis marker (activated) caspase‐3 (red) and oligodendrocyte marker CC1 (green) reveals the presence of apoptotic oligodendrocytes in all genotypes, that appear more numerous in Cx47KO compared with WT and Cx32KO spinal cord WM. Cell nuclei are labeled with DAPI (blue) throughout. D. Counts of caspase‐3 immunoreactive oligodendrocytes (n = 4 mice per genotype) confirm a significantly higher number in Cx47KO compared with WT, while differences between WT and Cx32KO or Cx32KO and Cx47KO groups are not significant. (e–g) Representative images of lumbar spinal cord sections from 24 dpi EAE mice, visualized with TUNEL (red) to demonstrate apoptosis, reveal several apoptotic cells (white arrowheads) in the WM, with apparently higher numbers in connexin deficient mice. TUNEL positives cells display round homogeneous nuclei typical for oligodendrocytes. (h) Quantitative analysis (n = 4 per group) confirmed that the numbers of TUNEL positive cells are significantly higher in Cx32KO and Cx47KO compared with the WT group. Significant differences in the mean number of apoptotic cell were also observed between the Cx32KO and Cx47KO EAE mice. (i–k) Double staining (merged overview images and higher magnification images with separated channels below) with antibodies to BrdU (red) and Olig2 (green) revealed more extensive OPC proliferation in Cx47KO as well as in Cx32KO compared with WT EAE mice. Counts of BrdU/Olig2 double positive cells (n = 4 mice per group) confirmed these findings (L). (in D, H, and L data represent mean ±SEM; Student's t‐test with Bonferroni correction; *p < .0167; **p < .01; ***p < .001; only statistically significant differences are shown). Scale bars: In a, E and I = 20 μm; in insets (I) = 10 μm
The effect of the loss of either Cx32 or Cx47 on oligodendrocyte precursor cell (OPC) proliferation in EAE was assessed by double immunolabeling with antibodies to Olig2 and BrdU. Increased numbers of BrdU/Olig2 double positive cells were detected in Cx47KO and Cx32KO EAE mice compared with WT EAE mice (Figure 4i–k). Counts of Olig2/BrdU positive cells in the WM of whole lumbar spinal cord cross sections (n = 4 per genotype) confirmed that they were higher in Cx47KO mice (9.63 ± 0.83) compared with WT mice (6.50 ± 0.07) (p < .01) as well as in Cx32KO (7.88 ± 0.31) compared with WT mice (p < .01) but did not differ significantly between Cx32KO and Cx47KO mice (Figure 4l).
3.5. Assessment of inflammatory cells in EAE groups
Lumbar spinal cord sections of EAE mice from all three genotypes at 24 dpi (n = 4 mice per EAE group) were immunostained for markers of immune cells including antibodies to CD68 (macrophage marker), CD3 (T‐cell marker) and CD20 (B‐cell marker). Double labeling for macrophage marker CD68 and T‐cell marker CD3 (Figure 5a–c) showed the highest numbers of CD68+ and CD3+ cells in Cx47KO EAE mice followed by Cx32KO mice. Quantification of CD68 immunoreactivity confirmed that macrophage infiltrates were more extensive in Cx47KO compared with either Cx32KO (p < .0167) or WT (p < .001) EAE groups, as well as in Cx32KO compared with the WT group (p < .001) (Figure 5d). Likewise, CD3+ T‐cells were significantly more numerous in sections of Cx47KO (30.5 ± 2.25 CD3+ cells per section) compared with WT (19.5 ± 1.35 CD3+ cells) EAE group (p < .05) (Figure 5e). However, the differences in CD3+ cell counts between Cx47KO and Cx32KO (26.5 ± 2.1 CD3+ cells) or between Cx32KO and WT EAE mice were not significant. Finally, immunostaining at 24 dpi for B‐cell marker CD20 (Figure 5f–h) revealed numerous CD20+ B‐cells in all genotypes. Whereas the WM area covered by CD20+ cells was similar in WT and Cx32KO mice, it was significantly (over two‐fold) higher in Cx47KO EAE mice (Figure 5i).
Figure 5.
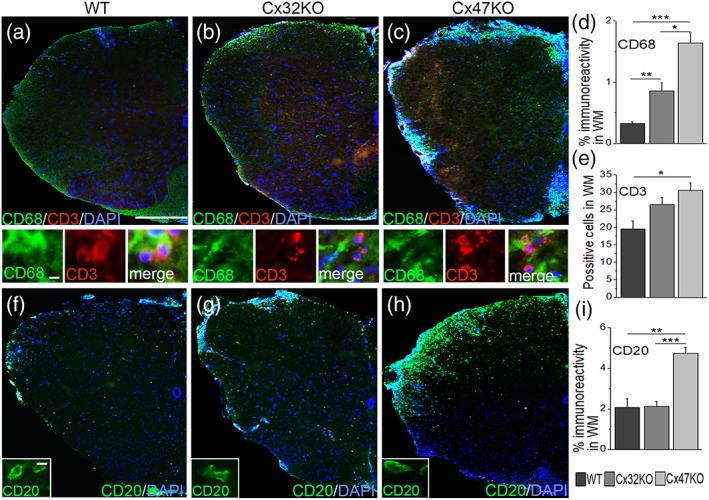
Assessment of immune cells in WM of connexin mutant EAE mice. Representative images of lumbar spinal cord hemisections from 24 dpi WT, Cx32KO, and Cx47KO EAE mice as indicated (merged overview images and higher magnification images with separate channels underneath) double stained with macrophage marker CD68 (green) and T‐cell marker CD3 (red) (a–c), or stained with B‐cell marker CD20 (green) (f–h). Cell nuclei are visualized with DAPI (blue). Inflammatory cell infiltrates appear more pronounced in Cx47KO mice. D. Quantitative analysis reveals that macrophage immunoreactivity (% WM with CD68 immunoreactivity) is higher in mice lacking either Cx32 or Cx47 compared with the WT group, as well as in Cx47KO compared with Cx32KO mice. e. The mean number of CD3+ positive cells (n = 3 mice per group) was also higher in Cx47KO than in WT mice. (i) Quantification of CD20+ cells (% WM with CD20 immunoreactivity) revealed that B‐cell infiltrates were more extensive in Cx47KO compared with WT mice (n = 3 per group) data in (d), (e), and (i) represent mean ± SEM; Student's t‐test with Bonferroni correction and statistical threshold adjusted to 0.0167; *p < .025; **p < .01; ***p < .001; only statistically significant differences are shown). Scale bars: In a = 100 μm; in insets (a, f) =5 μm
3.6. Evaluation of peripheral immune reaction in EAE connexin mutant mice
To explore the possibility that either Cx32 or Cx47 plays a role in peripheral immune cells and that their differential activation in EAE might explain the observed differences in EAE manifestations, we immunostained fixed spleen sections from 24 dpi EAE mice from all three genotypes for Cx32 and Cx47 in combination with macrophage marker CD68, T‐cell marker CD3 and B‐cells marker CD20. This analysis showed that neither Cx32 nor Cx47 were expressed in immune cells of the peripheral lymphatic tissue. Furthermore, immune cell activation, including macrophages, T‐cells and B‐cells was similar in the spleens of Cx32KO, Cx47KO and WT 24 dpi EAE mice (Supporting Information Figure S2). Finally, double staining for Cx47 and endothelial marker Glut1, which is normally expressed in microvessels with blood‐tissue barrier function, showed no evidence of expression of Cx47 in capillaries or venules in WT mouse brain (Supporting Information Figure S3).
3.7. Assessment of astrocyte connexin expression in EAE mice lacking oligodendrocyte connexins
Astrocytic Cx43 is the main partner of Cx47 at A:O GJs (Orthmann‐Murphy et al., 2007), and Cx43 GJ plaques normally show a concentration around oligodendrocytes, especially in the WM. Cx30, the other major astrocytic connexin is mostly expressed in the grey matter and primarily forms A:O GJs with Cx32 (Kleopa et al., 2004; Nagy et al., 2003b). In order to examine the distribution of expression of these two astrocyte connexins in Cx47KO, Cx32KO, and WT mice, we performed immunostaining for Cx43 and Cx30 on lumbar spinal cord sections from CFA controls as well as 12 dpi and 24 dpi EAE mice from all three genotypes.
This analysis showed that both astrocytic connexins show expression patterns and intensity that are similar among the three EAE groups. In particular, we observed similar baseline expression of Cx30 and Cx43 in CFA control tissues in all three genotypes (Supporting Information Figure S4a–c), as well as a reduction of Cx43 immunoreactivity in the WM of EAE mice from all genotypes at 12 dpi, the peak of inflammation, (Supporting Information Figure S4d–f) as previously described for WT and Cx32KO mice (Markoullis, Sargiannidou, Gardner, et al., 2012). At 24 dpi, Cx43 expression in EAE mice from all genotypes showed recovery to levels similar to CFA controls (Supporting Information Figure S4g–l). Concordant with the immunostaining results, immunoblot analysis of Cx43 levels in EAE lumbar spinal cord lysates from all three genotypes revealed no statistically significant differences at 12 or at 24 dpi (Supporting Information Figure S4m–p).
3.8. Cytokine expression profile in EAE mice lacking oligodendrocyte connexins
Our results indicate that increased EAE severity seen in the Cx47KO and Cx32KO mice is associated with increased neuroinflammation and more severe BSCB disruption in connexin KO mice. To further clarify the underlying mechanism, we performed a detailed expression analysis of 62 cytokines relevant to CNS inflammation using the RayBio® C‐Series Mouse Cytokine Antibody Array C3 in spinal cord tissue lysates from 7 and 12 dpi EAE mice from WT, Cx32 KO and Cx47 KO groups (Figure 6a–f). Expression analysis at 7 dpi revealed increased normalized expression levels of several cytokines including the Axl, BLC (CXCL 13), CD30 Ligand, CRG‐2, CTACK (CCL27), CXCL16, Eotaxin‐1, Fas Ligand (Fas‐L), Fractalkine, GCSF, GM‐CSF, IL‐2, IL‐3, IL‐3 R beta (IL‐3 R β), IL‐4, IL‐10, IL‐12 p40/p70, KC (CXCL1), MCP‐1 (CCL2), MIP‐1 alpha (MIP‐1 α) MIP‐1 gamma (MIP‐1 γ), and MCP‐3 beta (MCP‐3 β). At 12 dpi most of these cytokines, except IL‐4, IL‐10, and KC showed decreasing levels in WT EAE mice (Figure 6g), whereas in Cx32 KO EAE mice 14 of 22 cytokines persisted at similar high levels (Figure 6h) and in Cx47KO EAE mice there was further increase in the expression of CRG‐2, CTACK, Eotaxin‐1, IL‐2, IL‐3, IL‐3Rβ, KC, MIP‐1 α, MIP‐1 γ, and MCP‐3β while the others persisted at similar levels (Figure 6f).
Figure 6.
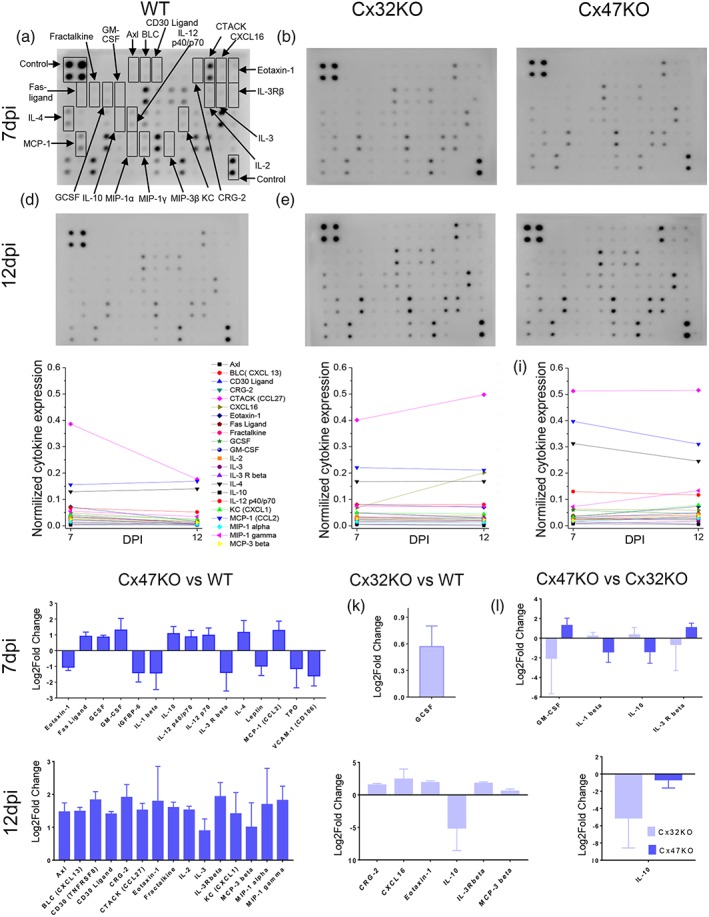
Cytokine expression analysis. Cytokine antibody array membranes were used to compare the expression levels of pro‐inflammatory cytokines related to CNS in EAE mice of the three genotypes at both 7 dpi (a–c) and at 12 dpi (d–f). Cytokine expression levels were normalized to internal positive controls. Among the 64 cytokines tested, several cytokines were upregulated as indicated by arrows in (a). In WT EAE mice all cytokines except IL‐4, IL‐10, and KC decreased from 7 to 12 dpi (g), whereas in Cx32KO the expression levels of the majority (14 of 22) of tested cytokines at 12 dpi were as high as at 7 dpi (h) and in Cx47KO EAE mice several (10 of 22) of the tested cytokines including CRG‐2, CTACK, Eotaxin‐1, IL‐2, IL‐3, IL‐3 R beta, KC, MIP‐1α, MIP‐1γ, and MCP‐3β showed a further increase in expression levels at 12 dpi compared with 7 dpi (i). Direct comparison between the three genotypes at 7 dpi (j–l) and 12 dpi (m–o) showed significant differences in the expression levels of several cytokines between Cx47KO and WT groups as indicated (data in j–o were tested by multiple comparisons with corrections using the two‐stage step‐up method of Benjamini, Krieger, and Yekutieli; false discovery rate, FDR = 0.1 or 0.15); (n = 3 mice examined per genotype and time point) [Color figure can be viewed at wileyonlinelibrary.com]
Direct comparison between genotypes at 7 dpi showed that several cytokines were upregulated in Cx47KO compared with WT EAE mice, including the FasL, GCSF, GM‐CSF, IL‐10, IL‐12 p40/p70, IL‐4, and MCP‐1 (CCL2) (Figure 6j). By comparison, in Cx32KO EAE mice only GCSF was upregulated at 7 dpi (Figure 6k). In addition, the cytokines GM‐CSF and IL‐3R beta were upregulated in Cx47KO compared with Cx32KO EAE mice (Figure 6l). At 12 dpi, several cytokines showed significantly higher expression levels in Cx47KO mice compared with WT EAE, including Axl, CXCL13, CD30 ligand, CRG‐2, CTACK (CCL27), Eotaxin‐1, Fraktalkine (CX2CL1), IL‐2, IL‐3, IL‐3Rbeta, CXCL1, MCP‐3beta (CCL19), MIP‐1alpha (CCL3), and MIP‐1 gamma (Figure 6m). Some cytokines were also upregulated in the Cx32KO compared with WT EAE at 12 dpi, including CRG‐2, CXCL16, Eotaxin‐1, IL‐3Rbeta and MCP‐3beta (Figure 6n). On the contrary, the expression level of IL‐10 was downregulated in both Cx32KO and Cx47 KO mice (Figure 6o). Overall, EAE resulted in a greater number of significantly differentially regulated cytokines in Cx47KO than in Cx32KO at both time points examined. This correlates with the increased clinical severity observed in Cx47KO following EAE and suggests an enhanced proinflammatory response in mice lacking Cx47 relative to Cx32KO and WT mice.
4. DISCUSSION
The aim of this study was to investigate whether the loss of either Cx32 or Cx47 affects the clinical course and pathological changes in mice subjected to EAE. Although extensive previous studies in our (Markoullis et al., 2014;Markoullis, Sargiannidou, Gardner, et al., 2012 ; Markoullis, Sargiannidou, Schiza, et al., 2012) and other laboratories (Brand‐Schieber et al., 2005; Masaki et al., 2012; Roscoe, Kidder, & Karlik, 2007; Sharma et al., 2010) have highlighted the disruption of glia GJs as a result of neuroinflammation and demyelination in MS brain and in EAE models, a direct link between oligodendrocyte GJ deficiency, especially as it pertains to Cx47, and increased susceptibility to EAE mediated dysfunction has not been directly demonstrated .
Several differences were demonstrated between WT EAE mice and two different mutants, each lacking one of the two major oligodendrocyte GJ proteins, Cx32 or Cx47. First, we found that connexin mutants differ from the WT group in susceptibly to EAE. In particular, Cx47KO mice developed EAE more rapidly compared with the Cx32KO and WT mice. Furthermore, we observed a gradient of severity in clinical scores and motor performance deficits. EAE phenotype was more severe in Cx47KO mice compared with Cx32KO mice and in turn, both connexin mutants showed more severe manifestations compared with WT mice. Associated with these findings, pathological analysis revealed that Cx47KO mice developed more myelin and axonal loss than did WT and Cx32KO mice, while the increased myelin loss in Cx32KO compared with WT EAE mice was not statistically significant. Finally, further experiments confirmed that several parameters of EAE pathogenesis, including the breakdown of the BSCB, the infiltration of spinal cord tissue by inflammatory cells, and oligodendrocyte apoptosis were more severe in Cx47KO mice than in Cx32KO and WT mice, while differences between Cx32KO and WT were generally smaller and only statistically significant for some of these parameters.
Interestingly, EAE outcome was worse in Cx47KO compared with Cx32KO mice. Cx47 is normally expressed in all oligodendrocytes in the CNS (Kleopa et al., 2004; Menichella et al., 2003; Odermatt et al., 2003), while Cx32 is expressed only in a subset of oligodendrocytes myelinating larger diameter fibers (Altevogt et al., 2002; Kleopa et al., 2004). Thus, a smaller population of oligodendrocytes might be affected by loss of Cx32 than by loss of Cx47. The more important functional role of Cx47 in the CNS compared with that of Cx32 is also highlighted by the fact that patients with mutations in Cx47 develop a severe hypomyelinating leukodystrophy (Uhlenberg et al., 2004), while patients with Cx32 mutations develop only subclinical or transient encephalopathy under conditions of additional metabolic stress (Sargiannidou, Kim, Kyriakoudi, Eun, & Kleopa, 2015; Taylor et al., 2003). Moreover, Cx47 forms the majority of both homotypic O:O and heterotypic O:A GJs in the CNS, coupling with the astrocytic partner Cx43 (Altevogt & Paul, 2004; Kamasawa et al., 2005). Reduced expression of Cx43 by astrocytes and loss of Cx43 GJs occurs early in EAE (Markoullis, Sargiannidou, Gardner, et al., 2012) as well as in other CNS injury models (Karpuk, Burkovetskaya, Fritz, Angle, & Kielian, 2011). O:A GJs formed by Cx30 and Cx32 are less likely to play an important role in EAE outcome because Cx30, the astrocytic partner of Cx32, is expressed mostly in the gray matter and at much lower levels in the WM (Markoullis, Sargiannidou, Gardner, et al., 2012; Markoullis, Sargiannidou, Schiza, et al., 2012) where EAE pathology mainly develops. Nevertheless, loss of Cx32 may also promote inflammation and increase vulnerability of myelinated fibers, as shown previously in Cx32KO mice following EAE induction (Markoullis, Sargiannidou, Gardner, et al., 2012) and confirmed by this study.
A major finding in our study was the much more severe disruption of the BSCB in connexin mutants, especially the Cx47KO, than in WT EAE mice. How the loss of O:A and O:O GJs affects the development and outcome of EAE and degree of BBB/BSCB disruption remains speculative. Infiltration of the CNS by inflammatory leukocytes and the disruption of BBB/BSCB are histopathological hallmarks of MS and EAE (Errede et al., 2012). Disruption of the BBB/BSCB is also a clinically relevant factor as it represents the initial stage of MS lesion formation (Frischer et al., 2009; Weiner, 2009). Both BBB and BSCB are responsible for the correct functioning and homeostasis of the CNS by regulating cellular and molecular trafficking between blood and brain/spinal cord parenchyma (Banerjee & Bhat, 2007). Astrocytes play a pivotal role in the formation and maintenance of the BBB/BSCB and in regulation of blood flow and neurovascular coupling by formation of dynamic tight junctions at the glia limitans, by releasing growth factors, and by facilitating cell to cell interactions (Horng et al., 2017; Miljkovic & Spasojevic, 2013). OPCs have also been implicated in the regulation of BBB/BSCB through the release of soluble factors such as TGF‐β (Seo et al., 2013; Seo et al., 2014). Furthermore, they are known to express both Cx47 (Markoullis, Sargiannidou, Schiza, et al., 2012) and Cx32 (Melanson‐Drapeau et al., 2003), thus contributing to GJ network integration and differentiation. Loss of O:A GJs could dysregulate both oligodendrocytes and OPCs as well as astrocytes directly.
Although our cytokine array analysis represents only an initial screening of possible variations in immune responses caused by loss of oligodendrocyte connexins, there is clear evidence of dysregulated cytokine expression. Cx47KO and to a lesser degree Cx32KO mice showed persistent elevation of GM‐CSF, associated with increased inflammatory activity in MS and in acute EAE (Lovett‐Racke, Bittner, Cross, Carlino, & Racke, 1998; Rust, Kuhle, Kappos, & Derfuss, 2016) and of G‐CSF (Xiao, Lu, & Link, 2007). Both can be produced by astrocytes (Choi, Lee, Lim, Satoh, & Kim, 2014; Rothhammer & Quintana, 2015) and both are regulated at the posttranscriptional level (Frei, Nohava, Malipiero, Schwerdel, & Fontana, 1992). Cx47KO mice showed also higher expression of FasL, which regulates T‐cell functions, activates the caspase cascade and is present on glial cells in chronic MS plaques (Dowling et al., 1996; Volpe, Sambucci, Battistini, & Borsellino, 2016). Moreover, IL‐2, IL‐3, and IL3‐beta, involved in MS pathogenesis (Renner et al., 2016; Zandian et al., 2011), showed persistent elevation at 12 dpi in Cx47KO in contrast to WT EAE mice. Finally, CCL2 produced by activated astrocytes (Rothhammer & Quintana, 2015) is significantly upregulated in Cx47KO EAE mice at 7 dpi, before the disruption of BBB/BSCB (Aubé et al., 2014). CCL2 regulates overall BBB integrity by destabilizing endothelial tight junctions and ultimately enables leukocyte penetration into the CNS (Paul et al., 2014). Thus, upregulated CCL2 could play a role in the increased severity of BBB/BSCB disruption in Cx47KO mice. Interestingly, astrocytic CCL2 expression is controlled by NF‐κB (Giraud, Caron, Pham‐Dinh, Kitabgi, & Nicot, 2010) and by activation of stat1 (Wang et al., 2013), while Cx43, the major partner of Cx47 at O:A GJs (Orthmann‐Murphy et al., 2007), mediates NF‐κB signaling activation (Xie et al., 2013). Thus, loss of Cx47 and the potential dysfunction of Cx43 (Markoullis, Sargiannidou, Gardner, et al., 2012) may directly affect the regulation of the astrocyte activation cascade.
Overall, several cytokines orchestrating BBB/BSCB disruption and inflammatory cell activation in MS and EAE are significantly more elevated in Cx47KO and to a lesser degree in Cx32KO mice and remain elevated at later stages when their downregulation has already taken place in WT EAE mice. A major question raised by these studies, and requiring further investigation, is whether the connexin loss leads to a direct reduction of BBB/BSCB integrity, which in turn promotes an increased inflammatory response, or whether loss of connexins promotes a more vigorous intrinsic brain immune response, as indicated by the abnormal cytokine responses in GJ deficient mice, which secondarily causes BBB/BSCB disruption. Previous studies in brains of mice deficient in oligodendrocyte GJs demonstrated activation of immune cells (Georgiou et al., 2017; Markoullis, Sargiannidou, Gardner, et al., 2012; Olympiou et al., 2016; Schiza et al., 2015; Tress et al., 2011) and upregulation of pro‐inflammatory genes expressed by both microglia and astrocytes (Wasseff & Scherer, 2015). Some evidence for a pro‐inflammatory environment in the CNS resulting from loss of oligodendrocyte connexins has been shown in Cx32/Cx47 double KO mice (Georgiou et al., 2017; Schiza et al., 2015; Wasseff & Scherer, 2015). Similar proinflammatory effects of GJ deficiency have also been found in peripheral nerves of Cx32 mutant mice (Freidin, Asche‐Godin, & Abrams, 2015; Groh et al., 2012; Kobsar et al., 2003; Kobsar, Maurer, Ott, & Martini, 2002) and the cytokine colony stimulating factor‐1 (CSF‐1) has been implicated (Groh et al., 2015; Groh, Basu, Stanley, & Martini, 2016).
The mechanisms underlying the increased inflammatory responses associated with exacerbated clinical outcomes in mice with connexin deficient oligodendrocytes remain unclear. Both primary and secondary effects on BBB/BSCB permeability, or intrinsically increased non‐specific oligodendrocyte susceptibility to injury may be involved. Loss of oligodendrocyte connexins could directly render the BBB/BSCB more permeable or more prone to disruption. However, we found no evidence of Cx47 expression in CNS endothelial cells. Thus, a direct role of endothelial cell dysfunction in the increased susceptibility to EAE seems less likely. Nevertheless, further studies of how the loss of Cx47 in particular affects the BBB/BSCB at baseline and at earlier stages of EAE, as well as in non‐EAE models of neuroinflammation would help to clarify this question.
Although we found no evidence for a direct involvement of Cx47 in the peripheral cellular immune response, Cx47 expression in lymphatic epithelium (Meens et al., 2017; Munger, Davis, & Simon, 2017) may suggest a possible role in peripheral immune reactions. This could be relevant for the heightened CNS B cell response in Cx47KO EAE brains, since the pathway from brain lymphatics to draining cervical lymph nodes may modulate the CNS immune response (Louveau, Da Mesquita, & Kipnis, 2016). Interestingly, at least one study reported that in MS patients’ brains, B cells mature in the cervical lymph nodes which collect from the brain lymphatics (Stern et al., 2014). In any case, the possibility of non‐oligodendrocyte expression of Cx47 as a contributor to increased neuroinflammation should be directly addressed in future studies, for example by generating and studying conditional Cx47KO mice with oligodendrocyte‐specific loss of Cx47.
Our results provide further evidence for the crucial role of glial GJ channels in the regulation of homeostasis in the CNS including a timely and controlled response to inflammation and loss of myelin. The panglial GJ network appears to influence cytokine expression and BBB integrity. Thus, disruption of glial GJs may be both a consequence of acquired MS and EAE pathology (Markoullis et al., 2014; Markoullis, Sargiannidou, Gardner, et al., 2012; Markoullis, Sargiannidou, Schiza, et al., 2012; Masaki et al., 2012) and also a causative factor, driving lesion formation, expansion, and finally disease progression, representing a vicious circle. Further studies to better understand the relation between glial GJ connectivity and control of inflammation are warranted and may provide novel therapeutic targets for MS.
Supporting information
Supporting Information FIGURE S1 Clinical Scores of WT, Cx32KO, and Cx47KO EAE mice in three different experiments. Comparison of MCSs between EAE mice of the three genotypes through day 24 postimmunization (dpi) obtained from three different experiments (i: A–C; ii: D–F; and iii: G–I), performed at different time points with sample sizes as indicated. Arrows indicate deaths in EAE groups (a and b). In all experiments, there is a similar exacerbation of EAE scores in Cx47KO more than in Cx32KO compared with the corresponding WT groups. Data in A‐I represent mean MCS ± SEM; Kruskal–Wallis test: *p < .005; **p < .01; ***p < .001; n.s.: nonsignificant
Supporting Information FIGURE S2 Lack of Cx32 or Cx47 expression in the spleen and similar patterns of immune cell activation in 24 dpi EAE mice from all genotypes. Representative images (low magnification overview and higher magnification insets in separate channels underneath) of spleen sections double stained with macrophage marker CD68 (green) and Cx32 (red) (a–c); or with B‐cell marker CD20 (green) and Cx47 (red) (d–f); or with T‐cell marker CD3 (green) and Cx47 (red) (g–i). Cell nuclei are visualized with DAPI (blue). There is no specific Cx32 or Cx47 immunoreactivity in the spleen and in particular in the immune cells labelled. Furthermore, expression patterns for macrophages, T‐ and B‐cells in Cx32KO and Cx47KO mice are similar to WT mice following EAE induction. Scale bars: in A = 50 μm; in insets (A) =10 μm
Supporting Information FIGURE S3 Cx47 is not expressed in endothelial cells of microvessels in the CNS that are important for BBB function. Double immunostaining of sections through the basal ganglia area of WT mouse brain for Cx47 (red) and endothelial marker erythrocyte‐type glucose transporter protein Glut1 (green) shows no expression of Cx47 in Glut1 positive endothelial cells of capillaries (marked with *) (a) or venules (marked with **) (b). Cell nuclei are visualized with DAPI (blue). Cx47 is only expressed in perikarya and proximal process of oligodendrocytes forming the characteristic GJ‐like plaques (open arrowheads), but is not found in microvessels. Overview images and higher magnification insets are shown. Scale bars: 10 μm
Supporting Information Figure S4 Expression of astrocyte connexins in EAE mice. a‐l: Representative images of lumbar spinal cord sections (merged overview images and higher magnification images with separated channels shown as insets) from 12 and 24 dpi CFA (a–c and g–i) and EAE (d–f and j–l) mice, as indicated, double stained for Cx43 (green) and Cx30 (red). Cell nuclei are visualized with DAPI (blue). There is a reduction of Cx43 immunoreactivity in spinal cord WM in 12 dpi tissues of EAE mice of all three genotypes, with beginning recovery at 24 dpi, whereas Cx30 expression that is largely restricted to the gray matter shows no apparent change in EAE. However, there is no obvious difference in either Cx43 or Cx30 expression patterns among the three genotypes. Immunoblot analysis of Cx43 expression at 12 and 24 dpi EAE groups (WT, Cx32KO and Cx47KO) as indicated. Representative images of the blots in (m) and (o) with GAPDH for loading control shown under each connexin blot. Quantitative analysis of Cx43 expression levels (average normalized band intensity) at 12 (n) and 24 (p) dpi as indicated shows no significant difference between the three genotypes. Scale bars: in A = 25 μm; in insets (A) =10 μm
ACKNOWLEDGMENT
This study has been funded by the National Multiple Sclerosis Society (RG459A1/2 grant to CKA and KAK).
Papaneophytou CP, Georgiou E, Karaiskos C, et al. Regulatory role of oligodendrocyte gap junctions in inflammatory demyelination. Glia. 2018;66:2589–2603. 10.1002/glia.23513
Funding information National Multiple Sclerosis Society, Grant/ Award Number: RG459A1/2
Christos P. Papaneophytou and Elena Georgiou contributed equally to this study.
REFERENCES
- Altevogt, B. M. , Kleopa, K. A. , Postma, F. R. , Scherer, S. S. , & Paul, D. L. (2002). Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. The Journal of Neuroscience, 22, 6458–6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altevogt, B. M. , & Paul, D. L. (2004). Four classes of intercellular channels between glial cells in the CNS. The Journal of Neuroscience, 24, 4313–4323. 10.1523/JNEUROSCI.3303-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez, J. I. , Katayama, T. , & Prat, A. (2013). Glial influence on the blood brain barrier. Glia, 61, 1939–1958. 10.1002/glia.22575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubé, B. , Lévesque, S.A. , Paré, A. , Chamma, É. , Kébir, H. , Gorina, R. , … Lacroix, S. (2014). Neutrophils mediate blood‐spinal cord barrier disruption in demyelinating neuroinflammatory diseases. Journal of Immunology, 193, 2438–2454. 10.4049/jimmunol.1400401 [DOI] [PubMed] [Google Scholar]
- Baker, D. , & Amor, S. (2012). Publication guidelines for refereeing and reporting on animal use in experimental autoimmune encephalomyelitis. Journal of Neuroimmunology, 242, 78–83. 10.1016/j.jneuroim.2011.11.003 [DOI] [PubMed] [Google Scholar]
- Banerjee, S. , & Bhat, M. A. (2007). Neuron‐glial interactions in blood‐brain barrier formation. Annual Review of Neuroscience, 30, 235–258. 10.1146/annurev.neuro.30.051606.094345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks, W. A. (1999). Physiology and pathology of the blood‐brain barrier: Implications for microbial pathogenesis, drug delivery and neurodegenerative disorders. Journal of Neurovirology, 5, 538–555. [DOI] [PubMed] [Google Scholar]
- Brand‐Schieber, E. , Werner, P. , Iacobas, D. A. , Iacobas, S. , Beelitz, M. , Lowery, S. L. , … Scemes, E. (2005). Connexin43, the major gap junction protein of astrocytes, is down‐regulated in inflamed white matter in an animal model of multiple sclerosis. Journal of Neuroscience Research, 80, 798–808. 10.1002/jnr.20474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron, H. A. (2006). Quantitative analysis of in vivo cell proliferation. Current Protocols in Neuroscience, Chapter 3 . Unitas, 3, 9–3.9.15. 10.1002/N0471142301.ns0309s37 [DOI] [PubMed] [Google Scholar]
- Choi, S. S. , Lee, H. J. , Lim, I. , Satoh, J.‐i. , & Kim, S. U. (2014). Human astrocytes: Secretome profiles of cytokines and chemokines. PLoS One, 9, e92325 10.1371/journal.pone.0092325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correale, J. , & Villa, A. (2007). The blood‐brain‐barrier in multiple sclerosis: Functional roles and therapeutic targeting. Autoimmunity, 40, 148–160. 10.1080/08916930601183522 [DOI] [PubMed] [Google Scholar]
- Dowling, P. , Shang, G. , Raval, S. , Menonna, J. , Cook, S. , & Husar, W. (1996). Involvement of the CD95 (APO‐1/Fas) receptor/ligand system in multiple sclerosis brain. The Journal of Experimental Medicine, 184, 1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errede, M. , Girolamo, F. , Ferrara, G. , Strippoli, M. , Morando, S. , Boldrin, V. , … Virgintino, D. (2012). Blood‐brain barrier alterations in the cerebral cortex in experimental autoimmune encephalomyelitis. Journal of Neuropathology and Experimental Neurology, 71, 840–854. 10.1097/NEN.0b013e31826ac110 [DOI] [PubMed] [Google Scholar]
- Fabis, M. J. , Scott, G. S. , Kean, R. B. , Koprowski, H. , & Hooper, D. C. (2007). Loss of blood‐brain barrier integrity in the spinal cord is common to experimental allergic encephalomyelitis in knockout mouse models. Proceedings of the National Academy of Sciences of The United States of America, 104, 5656–5661. 10.1073/pnas.0701252104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei, K. , Nohava, K. , Malipiero, U. V. , Schwerdel, C. , & Fontana, A. (1992). Production of macrophage colony‐stimulating factor by astrocytes and brain macrophages. Journal of Neuroimmunology, 40, 189–195. [DOI] [PubMed] [Google Scholar]
- Freidin, M. , Asche‐Godin, S. , & Abrams, C. K. (2015). Gene expression profiling studies in regenerating nerves in a mouse model for CMT1X: Uninjured Cx32‐knockout peripheral nerves display expression profile of injured wild type nerves. Experimental Neurology, 263, 339–349. 10.1016/j.expneurol.2014.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischer, J. M. , Bramow, S. , Dal‐Bianco, A. , Lucchinetti, C. F. , Rauschka, H. , Schmidbauer, M. , … Lassmann, H. (2009). The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain, 132, 1175–1189. 10.1093/brain/awp070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiou, E. , Sidiropoulou, K. , Richter, J. , Papaneophytou, C. , Sargiannidou, I. , Kagiava, A. , … Kleopa, K. A. (2017). Gene therapy targeting oligodendrocytes provides therapeutic benefit in a leukodystrophy model. Brain , 140, 599–616. doi: 10.1093/brain/aww351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraud, S. N. , Caron, C. M. , Pham‐Dinh, D. , Kitabgi, P. , & Nicot, A. B. (2010). Estradiol inhibits ongoing autoimmune neuroinflammation and NFkappaB‐dependent CCL2 expression in reactive astrocytes. Proceedings of the National Academy of Sciences of the United States of America, 107, 8416–8421. 10.1073/pnas.0910627107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh, J. , Basu, R. , Stanley, E. R. , & Martini, R. (2016). Cell‐surface and secreted isoforms of CSF‐1 exert opposing roles in macrophage‐mediated neural damage in Cx32‐deficient mice. The Journal of Neuroscience, 36, 1890–1901. 10.1523/JNEUROSCI.3427-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh, J. , Klein, I. , Hollmann, C. , Wettmarshausen, J. , Klein, D. , & Martini, R. (2015). CSF‐1‐activated macrophages are target‐directed and essential mediators of Schwann cell dedifferentiation and dysfunction in Cx32‐deficient mice. Glia, 63, 977–986. 10.1002/glia.22796 [DOI] [PubMed] [Google Scholar]
- Groh, J. , Weis, J. , Zieger, H. , Stanley, E. R. , Heuer, H. , & Martini, R. (2012). Colony‐stimulating factor‐1 mediates macrophage‐related neural damage in a model for Charcot–Marie–tooth disease type 1X. Brain, 135, 88–104. 10.1093/brain/awr283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horng, S. , Therattil, A. , Moyon, S. , Gordon, A. , Kim, K. , Argaw, A. T. , … John, G. R. (2017). Astrocytic tight junctions control inflammatory CNS lesion pathogenesis. The Journal of Clinical Investigation, 127, 3136–3151. 10.1172/jci91301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, M. V. , Nguyen, T. T. , Deboy, C. A. , Griffin, J. W. , Whartenby, K. A. , Kerr, D. A. , & Calabresi, P. A. (2008). Behavioral and pathological outcomes in MOG 35‐55 experimental autoimmune encephalomyelitis. Journal of Neuroimmunology, 199, 83–93. 10.1016/j.jneuroim.2008.05.013 [DOI] [PubMed] [Google Scholar]
- Kamasawa, N. , Sik, A. , Morita, M. , Yasumura, T. , Davidson, K. G. , Nagy, J. I. , & Rash, J. E. (2005). Connexin‐47 and connexin‐32 in gap junctions of oligodendrocyte somata, myelin sheaths, paranodal loops and Schmidt‐Lanterman incisures: Implications for ionic homeostasis and potassium siphoning. Neuroscience, 136, 65–86. 10.1016/j.neuroscience.2005.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpuk, N. , Burkovetskaya, M. , Fritz, T. , Angle, A. , & Kielian, T. (2011). Neuroinflammation leads to region‐dependent alterations in astrocyte gap junction communication and hemichannel activity. The Journal of Neuroscience, 31, 414–425. 10.1523/jneurosci.5247-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleopa, K. A. , Orthmann, J. L. , Enriquez, A. , Paul, D. L. , & Scherer, S. S. (2004). Unique distributions of the gap junction proteins connexin29, connexin32, and connexin47 in oligodendrocytes. Glia, 47, 346–357. 10.1002/glia.20043 [DOI] [PubMed] [Google Scholar]
- Kobsar, I. , Berghoff, M. , Samsam, M. , Wessig, C. , Maurer, M. , Toyka, K. V. , & Martini, R. (2003). Preserved myelin integrity and reduced axonopathy in connexin32‐deficient mice lacking the recombination activating gene‐1. Brain, 126, 804–813. [DOI] [PubMed] [Google Scholar]
- Kobsar, I. , Maurer, M. , Ott, T. , & Martini, R. (2002). Macrophage‐related demyelination in peripheral nerves of mice deficient in the gap junction protein connexin 32. Neuroscience Letters, 320, 17–20. [DOI] [PubMed] [Google Scholar]
- Larochelle, C. , Alvarez, J. I. , & Prat, A. (2011). How do immune cells overcome the blood‐brain barrier in multiple sclerosis? FEBS Letters, 585, 3770–3780. 10.1016/j.febslet.2011.04.066 [DOI] [PubMed] [Google Scholar]
- Louveau, A. , Da Mesquita, S. , & Kipnis, J. (2016). Lymphatics in neurological disorders: A Neuro‐Lympho‐vascular component of multiple sclerosis and Alzheimer's disease? Neuron, 91, 957–973. 10.1016/j.neuron.2016.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett‐Racke, A. E. , Bittner, P. , Cross, A. H. , Carlino, J. A. , & Racke, M. K. (1998). Regulation of experimental autoimmune encephalomyelitis with insulin‐like growth factor (IGF‐1) and IGF‐1/IGF‐binding protein‐3 complex (IGF‐1/IGFBP3). The Journal of Clinical Investigation, 101, 1797–1804. 10.1172/jci1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lublin, F. D. , & Reingold, S. C. (1996). Defining the clinical course of multiple sclerosis: Results of an international survey. National Multiple Sclerosis Society (USA) advisory committee on clinical trials of new agents in multiple sclerosis. Neurology, 46, 907–911. [DOI] [PubMed] [Google Scholar]
- Luissint, A.‐C. , Artus, C. , Glacial, F. , Ganeshamoorthy, K. , & Couraud, P.‐O. (2012). Tight junctions at the blood brain barrier: Physiological architecture and disease‐associated dysregulation. Fluids and Barriers of the CNS, 9, 23–23. 10.1186/2045-8118-9-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markoullis, K. , Sargiannidou, I. , Gardner, C. , Hadjisavvas, A. , Reynolds, R. , & Kleopa, K. A. (2012). Disruption of oligodendrocyte gap junctions in experimental autoimmune encephalomyelitis. Glia, 60, 1053–1066. 10.1002/glia.22334 [DOI] [PubMed] [Google Scholar]
- Markoullis, K. , Sargiannidou, I. , Schiza, N. , Hadjisavvas, A. , Roncaroli, F. , Reynolds, R. , & Kleopa, K. A. (2012). Gap junction pathology in multiple sclerosis lesions and normal‐appearing white matter. Acta Neuropathologica, 123, 873–886. 10.1007/s00401-012-0978-4 [DOI] [PubMed] [Google Scholar]
- Markoullis, K. , Sargiannidou, I. , Schiza, N. , Roncaroli, F. , Reynolds, R. , & Kleopa, K. A. (2014). Oligodendrocyte gap junction loss and disconnection from reactive astrocytes in multiple sclerosis gray matter. Journal of Neuropathology and Experimental Neurology, 73, 865–879. 10.1097/NEN.0000000000000106 [DOI] [PubMed] [Google Scholar]
- Masaki, K. , Suzuki, S. O. , Matsushita, T. , Yonekawa, T. , Matsuoka, T. , Isobe, N. , … Kira, J. (2012). Extensive loss of connexins in Balo's disease: Evidence for an auto‐antibody‐independent astrocytopathy via impaired astrocyte‐oligodendrocyte/myelin interaction. Acta Neuropathologica, 123, 887–900. 10.1007/s00401-012-0972-x [DOI] [PubMed] [Google Scholar]
- Meens, M. J. , Kutkut, I. , Rochemont, V. , Dubrot, J. , Kaladji, F. R. , Sabine, A. , … Kwak, B. R. (2017). Cx47 fine‐tunes the handling of serum lipids but is dispensable for lymphatic vascular function. PLoS One, 12, e0181476 10.1371/journal.pone.0181476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melanson‐Drapeau, L. , Beyko, S. , Dave, S. , Hebb, A. L. , Franks, D. J. , Sellitto, C. , … Bennett, S. A. (2003). Oligodendrocyte progenitor enrichment in the connexin32 null‐mutant mouse. The Journal of Neuroscience, 23, 1759–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menichella, D. M. , Goodenough, D. A. , Sirkowski, E. , Scherer, S. S. , & Paul, D. L. (2003). Connexins are critical for normal myelination in the CNS. The Journal of Neuroscience, 23, 5963–5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miljkovic, D. , & Spasojevic, I. (2013). Multiple sclerosis: Molecular mechanisms and therapeutic opportunities. Antioxidants & Redox Signaling, 19, 2286–2334. 10.1089/ars.2012.5068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger, S. J. , Davis, M. J. , & Simon, A. M. (2017). Defective lymphatic valve development and chylothorax in mice with a lymphatic‐specific deletion of Connexin43. Developmental Biology, 421, 204–218. 10.1016/j.ydbio.2016.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy, J. , Ionescu, A. V. , Lynn, B. D. , & Rash, J. E. (2003a). Connexin29 and connexin32 at oligodendrocyte and astrocyte gap junctions and in myelin of the mouse central nervous system. The Journal of Comparative Neurology, 464, 356–370. 10.1002/cne.10797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy, J. I. , Ionescu, A. V. , Lynn, B. D. , & Rash, J. E. (2003b). Coupling of astrocyte connexins Cx26, Cx30, Cx43 to oligodendrocyte Cx29, Cx32, Cx47: Implications from normal and connexin32 knockout mice. Glia, 44, 205–218. 10.1002/glia.10278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave, K. A. (2010). Myelination and the trophic support of long axons. Nature Reviews. Neuroscience, 11, 275–283. 10.1038/nrn2797 [DOI] [PubMed] [Google Scholar]
- Odermatt, B. , Wellershaus, K. , Wallraff, A. , Seifert, G. , Degen, J. , Euwens, C. , … Willecke, K. (2003). Connexin 47 (Cx47)‐deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. The Journal of Neuroscience, 23, 4549–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olympiou, M. , Sargiannidou, I. , Markoullis, K. , Karaiskos, C. , Kagiava, A. , Kyriakoudi, S. , … Kleopa, K. A. (2016). Systemic inflammation disrupts oligodendrocyte gap junctions and induces ER stress in a model of CNS manifestations of X‐linked Charcot‐Marie‐tooth disease. Acta Neuropathologica Communications, 4, 95 10.1186/s40478-016-0369-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthmann‐Murphy, J. L. , Freidin, M. , Fischer, E. , Scherer, S. S. , & Abrams, C. K. (2007). Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins. The Journal of Neuroscience, 27, 13949–13957. 10.1523/jneurosci.3395-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul, D. , Ge, S. , Lemire, Y. , Jellison, E. R. , Serwanski, D. R. , Ruddle, N. H. , & Pachter, J. S. (2014). Cell‐selective knockout and 3D confocal image analysis reveals separate roles for astrocyte‐and endothelial‐derived CCL2 in neuroinflammation. Journal of Neuroinflammation, 11, 10 10.1186/1742-2094-11-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson, H. L. , Garbern, J. Y. , Hoban, T. F. , Krajewski, K. M. , Lewis, R. A. , Fischbeck, K. H. , … Shy, M. E. (2002). Transient central nervous system white matter abnormality in X‐linked Charcot‐Marie‐tooth disease. Annals of Neurology, 52, 429–434. 10.1002/ana.10305 [DOI] [PubMed] [Google Scholar]
- Rash, J. E. , Yasumura, T. , Dudek, F. E. , & Nagy, J. I. (2001). Cell‐specific expression of connexins and evidence of restricted gap junctional coupling between glial cells and between neurons. The Journal of Neuroscience, 21, 1983–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner, K. , Hellerbrand, S. , Hermann, F. , Riedhammer, C. , Talke, Y. , Schiechl, G. , … Mack, M. (2016). IL‐3 promotes the development of experimental autoimmune encephalitis. JCI Insight, 1, e87157 10.1172/jci.insight.87157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscoe, W. A. , Kidder, G. M. , & Karlik, S. J. (2007). Experimental allergic encephalomyelitis in connexin 43–heterozygous mice. Cell Communication and Adhesion, 14, 57–73. 10.1080/15419060701459569 [DOI] [PubMed] [Google Scholar]
- Rothhammer, V. , & Quintana, F. J. (2015). Control of autoimmune CNS inflammation by astrocytes. Seminars in Immunopathology, 37, 625–638. 10.1007/s00281-015-0515-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust, H. , Kuhle, J. , Kappos, L. , & Derfuss, T. (2016). Severe exacerbation of relapsing‐remitting multiple sclerosis after G‐CSF therapy. Neurology Neuroimmunology and Neuroinflammation, 3, e215 10.1212/NXI.0000000000000215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu, J. K. , & McLarnon, J. G. (2009). A leaky blood‐brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer's disease brain. Journal of Cellular and Molecular Medicine, 13, 2911–2925. 10.1111/j.1582-4934.2008.00434.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargiannidou, I. , Kim, G. H. , Kyriakoudi, S. , Eun, B. L. , & Kleopa, K. A. (2015). A start codon CMT1X mutation associated with transient encephalomyelitis causes complete loss of Cx32. Neurogenetics, 16, 193–200. 10.1007/s10048-015-0442-4 [DOI] [PubMed] [Google Scholar]
- Schiza, N. , Sargiannidou, I. , Kagiava, A. , Karaiskos, C. , Nearchou, M. , & Kleopa, K. A. (2015). Transgenic replacement of Cx32 in gap junction‐deficient oligodendrocytes rescues the phenotype of a hypomyelinating leukodystrophy model. Human Molecular Genetics, 24, 2049–2064. 10.1093/hmg/ddu725 [DOI] [PubMed] [Google Scholar]
- Seo, J. H. , Maki, T. , Maeda, M. , Miyamoto, N. , Liang, A. C. , Hayakawa, K. , … Arai, K. (2014). Oligodendrocyte precursor cells support blood‐brain barrier integrity via TGF‐β Signaling. PLoS One, 9, e103174 10.1371/journal.pone.0103174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo, J. H. , Miyamoto, N. , Hayakawa, K. , Pham, L. D. , Maki, T. , Ayata, C. , … Arai, K. (2013). Oligodendrocyte precursors induce early blood‐brain barrier opening after white matter injury. The Journal of Clinical Investigation, 123, 782–786. 10.1172/jci65863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, R. , Fischer, M. T. , Bauer, J. , Felts, P. A. , Smith, K. J. , Misu, T. , … Lassmann, H. (2010). Inflammation induced by innate immunity in the central nervous system leads to primary astrocyte dysfunction followed by demyelination. Acta Neuropathologica, 120, 223–236. 10.1007/s00401-010-0704-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern, J. N. , Yaari, G. , Vander Heiden, J. A. , Church, G. , Donahue, W. F. , Hintzen, R. Q. , … O'Connor, K. C. (2014). B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Science Translational Medicine, 6, 248ra107 10.1126/scitranslmed.3008879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, R. A. , Simon, E. M. , Marks, H. G. , & Scherer, S. S. (2003). The CNS phenotype of X‐linked Charcot‐Marie‐tooth disease: More than a peripheral problem. Neurology, 61, 1475–1478. [DOI] [PubMed] [Google Scholar]
- Tress, O. , Maglione, M. , Zlomuzica, A. , May, D. , Dicke, N. , Degen, J. , … Willecke, K. (2011). Pathologic and phenotypic alterations in a mouse expressing a connexin47 missense mutation that causes Pelizaeus‐Merzbacher‐like disease in humans. PLoS Genetics, 7(7), e1002146 10.1371/journal.pgen.1002146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlenberg, B. , Schuelke, M. , Ruschendorf, F. , Ruf, N. , Kaindl, A. M. , Henneke, M. , … Gartner, J. (2004). Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus‐Merzbacher‐like disease. American Journal of Human Genetics, 75, 251–260. 10.1086/422763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe, E. , Sambucci, M. , Battistini, L. , & Borsellino, G. (2016). Fas–Fas ligand: Checkpoint of T cell functions in multiple sclerosis. Frontiers in Immunology, 7, 382 10.3389/fimmu.2016.00382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Deckert, M. , Xuan, N. T. , Nishanth, G. , Just, S. , Waisman, A. , … Schluter, D. (2013). Astrocytic A20 ameliorates experimental autoimmune encephalomyelitis by inhibiting NF‐kappaB‐ and STAT1‐dependent chemokine production in astrocytes. Acta Neuropathologica, 126, 711–724. 10.1007/s00401-013-1183-9 [DOI] [PubMed] [Google Scholar]
- Wasseff, S. K. , & Scherer, S. S. (2011). Cx32 and Cx47 mediate oligodendrocyte:Astrocyte and oligodendrocyte:Oligodendrocyte gap junction coupling. Neurobiology of Disease, 42, 506–513. 10.1016/j.nbd.2011.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasseff, S. K. , & Scherer, S. S. (2015). Activated immune response in an inherited leukodystrophy disease caused by the loss of oligodendrocyte gap junctions. Neurobiology of Disease, 82, 86–98. 10.1016/j.nbd.2015.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner, H. L. (2009). The challenge of multiple sclerosis: How do we cure a chronic heterogeneous disease? Annals of Neurology, 65, 239–248. 10.1002/ana.21640 [DOI] [PubMed] [Google Scholar]
- Xiao, B. G. , Lu, C. Z. , & Link, H. (2007). Cell biology and clinical promise of G‐CSF: Immunomodulation and neuroprotection. Journal of Cellular and Molecular Medicine, 11, 1272–1290. 10.1111/j.1582-4934.2007.00101.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, X. , Lan, T. , Chang, X. , Huang, K. , Huang, J. , Wang, S. , … Huang, H. (2013). Connexin43 mediates NF‐κB signalling activation induced by high glucose in GMCs: Involvement of c‐Src. Cell Communication and Signaling, 11, 38–38. 10.1186/1478-811X-11-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Tian, S. J. , Wu, L. , Huang, D. H. , & Wu, W. P. (2011). Fibrinogen depleting agent batroxobin has a beneficial effect on experimental autoimmune encephalomyelitis. Cellular and Molecular Neurobiology, 31, 437–448. 10.1007/s10571-010-9637-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandian, M. , Mott, K. R. , Allen, S. J. , Dumitrascu, O. , Kuo, J. Z. , & Ghiasi, H. (2011). Use of cytokine immunotherapy to block CNS demyelination induced by a recombinant HSV‐1 expressing IL‐2. Gene Therapy, 18, 734–742. 10.1038/gt.2011.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information FIGURE S1 Clinical Scores of WT, Cx32KO, and Cx47KO EAE mice in three different experiments. Comparison of MCSs between EAE mice of the three genotypes through day 24 postimmunization (dpi) obtained from three different experiments (i: A–C; ii: D–F; and iii: G–I), performed at different time points with sample sizes as indicated. Arrows indicate deaths in EAE groups (a and b). In all experiments, there is a similar exacerbation of EAE scores in Cx47KO more than in Cx32KO compared with the corresponding WT groups. Data in A‐I represent mean MCS ± SEM; Kruskal–Wallis test: *p < .005; **p < .01; ***p < .001; n.s.: nonsignificant
Supporting Information FIGURE S2 Lack of Cx32 or Cx47 expression in the spleen and similar patterns of immune cell activation in 24 dpi EAE mice from all genotypes. Representative images (low magnification overview and higher magnification insets in separate channels underneath) of spleen sections double stained with macrophage marker CD68 (green) and Cx32 (red) (a–c); or with B‐cell marker CD20 (green) and Cx47 (red) (d–f); or with T‐cell marker CD3 (green) and Cx47 (red) (g–i). Cell nuclei are visualized with DAPI (blue). There is no specific Cx32 or Cx47 immunoreactivity in the spleen and in particular in the immune cells labelled. Furthermore, expression patterns for macrophages, T‐ and B‐cells in Cx32KO and Cx47KO mice are similar to WT mice following EAE induction. Scale bars: in A = 50 μm; in insets (A) =10 μm
Supporting Information FIGURE S3 Cx47 is not expressed in endothelial cells of microvessels in the CNS that are important for BBB function. Double immunostaining of sections through the basal ganglia area of WT mouse brain for Cx47 (red) and endothelial marker erythrocyte‐type glucose transporter protein Glut1 (green) shows no expression of Cx47 in Glut1 positive endothelial cells of capillaries (marked with *) (a) or venules (marked with **) (b). Cell nuclei are visualized with DAPI (blue). Cx47 is only expressed in perikarya and proximal process of oligodendrocytes forming the characteristic GJ‐like plaques (open arrowheads), but is not found in microvessels. Overview images and higher magnification insets are shown. Scale bars: 10 μm
Supporting Information Figure S4 Expression of astrocyte connexins in EAE mice. a‐l: Representative images of lumbar spinal cord sections (merged overview images and higher magnification images with separated channels shown as insets) from 12 and 24 dpi CFA (a–c and g–i) and EAE (d–f and j–l) mice, as indicated, double stained for Cx43 (green) and Cx30 (red). Cell nuclei are visualized with DAPI (blue). There is a reduction of Cx43 immunoreactivity in spinal cord WM in 12 dpi tissues of EAE mice of all three genotypes, with beginning recovery at 24 dpi, whereas Cx30 expression that is largely restricted to the gray matter shows no apparent change in EAE. However, there is no obvious difference in either Cx43 or Cx30 expression patterns among the three genotypes. Immunoblot analysis of Cx43 expression at 12 and 24 dpi EAE groups (WT, Cx32KO and Cx47KO) as indicated. Representative images of the blots in (m) and (o) with GAPDH for loading control shown under each connexin blot. Quantitative analysis of Cx43 expression levels (average normalized band intensity) at 12 (n) and 24 (p) dpi as indicated shows no significant difference between the three genotypes. Scale bars: in A = 25 μm; in insets (A) =10 μm