

Abstract
Chronic inflammation of the colon (colitis) is a risk factor for colorectal cancer (CRC). Hormone‐replacement therapy reduces CRC incidences, and the estrogen receptor beta (ERβ/ESR2) has been implicated in this protection. Gut microbiota is altered in both colitis and CRC and may influence the severity of both. Here we test the hypothesis that intestinal ERβ impacts the gut microbiota. Mice with and without intestine‐specific deletion of ERβ (ERβKOVil) were generated using the Cre‐LoxP system. Colitis and CRC were induced with a single intraperitoneal injection of azoxymethane (AOM) followed by administration of three cycles of dextran sulfate sodium (DSS) in drinking water. The microbiota population were characterized by high‐throughput 16S rRNA gene sequencing of DNA extracted from fecal samples (N = 39). Differences in the microbiota due to AOM/DSS and absence of ERβ were identified through bioinformatic analyses of the 16S‐Seq data, and the distribution of bacterial species was corroborated using qPCR. We demonstrate that colitis‐induced CRC reduced the gut microbiota diversity and that loss of ERβ enhanced this process. Further, the Bacteroidetes genus Prevotellaceae_UCG_001 was overrepresented in AOM/DSS mice compared to untreated controls (3.5‐fold, p = 0.004), and this was enhanced in females and in ERβKOVil mice. Overall, AOM/DSS enriched for microbiota impacting immune system diseases and metabolic functions, and lack of ERβ in combination with AOM/DSS enriched for microbiota impacting carbohydrate metabolism and cell motility, while reducing those impacting the endocrine system. Our data support that intestinal ERβ contributes to a more favorable microbiome that could attenuate CRC development.
Keywords: microbiota, colitis‐associated colon cancer, AOM/DSS model, estrogen receptor beta
Short abstract
What's new?
Chronic inflammation of the colon is a risk factor for colorectal cancer (CRC). Hormone‐replacement therapy reduces CRC incidence, and the estrogen receptor beta (ERβ/ESR2) has been implicated in this protection. The microbiota of the gut is altered in both colitis and CRC, but whether intestinal ERβ affects gut microbiota remains to be investigated. Here, the authors demonstrate, in a mouse model, that colitis‐induced CRC reduces the gut microbiota diversity and that loss of ERβ enhances this process. The findings could enable novel therapeutic or preventive approaches toward a more favorable microbiome in inflammatory bowel disease and/or colon cancer development.
Abbreviations
- AOM
azoxymethane
- CRC
colorectal cancer
- DSS
dextran sulfate sodium
- ERβ
estrogen receptor beta
- KO
knockout
- LEfSe
Linear discriminant analysis effect size
- NMDS
nonmetric dimensional scaling
- OTU
operational taxonomic unit
- PERMANOVA
permutational analysis of variance
- PICRUSt
Phylogenetic investigation of communities by reconstruction of unobserved states
- qPCR
quantitative polymerase chain reaction
- WT
wild type
Introduction
Colorectal cancer (CRC) is the second leading cause of cancer death worldwide.1 Inflammation of the gut (colitis) is considered an important risk factor.2 Colitis is thought to contribute to cancer development through several mechanisms, including release of cytokines that activate pro‐survival NFκB and STAT3 signaling3 and alter the immune system,4 and by increasing risk of gene mutations5 and epigenetic changes.6 Colitis can also change the dynamics of the gut microbiota which can lead to dysbiosis and contribute to cancer development (reviewed in [7]). Dysbiosis is characterized by a reduction of beneficial bacteria (anaerobic species) and an increase of facultative anaerobic bacteria, such as pathogenic Enterobacteriaceae.8 In attempts to identify specific microbial species that may contribute to colorectal carcinogenesis, studies have characterized microbiota diversity in individuals with colorectal cancer.9, 10 These studies have, however, not yet reached consensus. A number of factors make these analyses challenging, including large variations regarding biological, environmental, dietary, inter‐individual, and technical parameters such as sample origin (fecal or mucosal samples) and time of sample collection.11, 12, 13 Genetically homogenous mice, housed in controlled environment and fed identical diet, provide a less variable setting. A mouse model that combines one azoxymethane (AOM, a pro‐carcinogenic agent) injection with dextran sulfate sodium (DSS, a chemical colitogen) in drinking water, causes epithelial injury and induces wound‐healing response, that mimic human colitis‐induced CRC.14 Some studies have found changes in the richness and diversity of the microbiota composition due to AOM and/or DSS exposure.15, 16, 17, 18
Further, estrogen through estrogen receptor β (ERβ/ESR2) appears to reduce the risk of CRC development (reviewed in [19]). Our previous cell‐based analyses have shown that re‐introduction of ERβ into CRC cells exhibits anti‐proliferative functions, represses oncogenes, and mediates anti‐inflammatory signaling.20, 21, 22 Studies in mice have generated support in vivo for such a preventive role.23, 24 Application of the AOM/DSS model on full‐body ERβ knockout (BERKO) mice demonstrated an increase in number and size of polyps or tumors compared to wild type (WT) mice,24 and similar results were found using the APCmin/+ model.25 However, whether intestinal ERβ affects the microbiota of the gut has not been investigated. To explore the effect of ERβ in the intestines, we have generated intestine‐specific knockout of ERβ (referred to as ERβKOVil or simply KO in the remainder of the text).20 Here, we characterize how the microbiota composition is affected by AOM/DSS‐driven colitis and CRC development in mice with and without intestinal ERβ.
Materials and Methods
Animals
The animal study was performed at University of Houston and approved by the Institutional Animal Care and Use Committee (12–026 and 13–012). Mice with floxed ERβ (B6.129X1‐Esr2tmGust) were crossed with mice expressing Cre‐recombinase under effect of the Villin promoter (B6.SJL‐Tg(Vilcre)997 Gum/J), generating intestine‐specific ERβ knockout mice (ERβKOVil or KO). ERβKOVil (n = 21) and corresponding WT littermates (n = 18) were distributed into two major experiments, 9 weeks (n = 20) and 16 weeks (n = 19) after AOM injection, and respective controls (no AOM/DSS). The successful deletion of ERβ expression in the intestinal epithelium of these mice has been previously demonstrated,20 and ERβ expression in its main organs (primarily the ovary 26) is maintained (data not shown) and the mice are fully fertile (full KO of ERβ generate sub‐fertile females with nearly no offspring). The layout of the experiment is illustrated in (Fig. 1). WT and ERβKOVil littermates were housed in the same cages, separated only by sex and AOM/DSS treatment, thereby minimizing cage, strain, and age‐dependent effects in terms of microbiota. At age 8–11 weeks and an average weight of 27 g for males and 21 g for females, mice were injected intraperitoneally (IP) with 10 mg azoxymethane (AOM, Sigma‐Aldrich) per kg bodyweight, followed by three 1‐week cycles of 2.5% dextran sulfate sodium (DSS, MP Biomedical) in drinking water. The DSS cycles were separated by 2 weeks of regular drinking water. Stool pellets were collected 9 weeks [WT (n = 9) and ERβKOVil (n = 11)], and 16 weeks [WT (n = 9) and ERβKOVil (n = 10)] after AOM injection, and stored at −20 °C.
Figure 1.
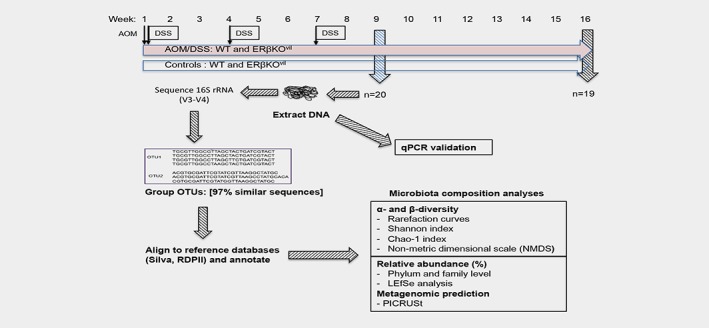
Schematic workflow of experimental design and analysis. Male and female mice on the same mixed C57BL/6 J background, with and without deletion of intestinal ERβ, were either injected with AOM (10 mg/kg bw, IP) at time point 0 and subjected to DSS (2.5%, drinking water) at weeks 1, 4 and 7, or maintained as parallel untreated controls. Stool pellets were collected from all mice at 9 weeks and 16 weeks. DNA was extracted, and genomic 16S rRNA gene was sequenced, OTUs were aligned to reference databases, annotated and analyzed for alpha and beta diversity, relative abundance of taxa at phyla and family level, differential abundance (LEfSe) and metagenomic prediction (PICRUSt). The sequencing results were validated with qPCR. [Color figure can be viewed at wileyonlinelibrary.com]
DNA extraction, library construction and 16S‐sequencing
The microbiota composition was determined by 16S‐Seq of the variable V3‐V4 genomic regions of bacterial 16S rRNA. Libraries for sequencing were prepared from DNA extracted using QIAamp DNA stool mini kit (Qiagen, Sweden), according to the manufacturer's recommendations. Briefly, each fecal pellet was weighed, homogenized by pellet pestles in lysis buffer, vortexed and centrifuged. The supernatant was treated with InhibitEX tablets to adsorb PCR inhibitors, and with proteinase K to remove proteins. DNA was precipitated in ethanol (99%), purified using spin columns, and eluted in 200 μL elution buffer. Purity and concentration of DNA were measured using Nanodrop 2000 spectrophotometer (Thermo Scientific). Libraries were generated for all samples at the same time: Ten nanograms of DNA per sample was PCR amplified with Phusion master mix (20 cycles of 95 °C for 30 s, 55 °C for 30 s, 72 °C for 30 s) and degenerated forward (5’‐TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG CCT ACG GGN GGC WGC AG‐3′) and reverse (5’‐GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GGA CTA CHV GGG TAT CTA ATC C‐3′) primers. These primers target the variable V3‐V4 genomic regions of bacterial 16S rRNA,27 and the 5′ ends include Illumina adapter sequences. The expected amplicon size was approximately 550 bp, and unbound primers and primer‐dimer fragments were removed using AMPure XP beads according to the manufacturer's protocol. Next, dual indices were attached to the Illumina sequencing adapters using the Nextera XT index kit and a second PCR was performed to complete the library (13 cycles of 95 °C for 30 s, 55 °C for 30 s, 72 °C for 30 s). The resulting product (expected size approximately 630 bp) was purified using AMPure XP beads, and the quality and size of end products was assessed with a DNA Bioanalyzer 1000 broad range kit. DNA concentration was assessed with the QuantIt kit, and adjusted to 4 nM. Ten microliters of each library was pooled, denatured with NaOH to generate single‐stranded DNA, and sequenced in one run using Illumina MiSeq V3 kit with 5% PhiX library as spike‐in internal control. In total, 3,397,153 reads were generated for these samples.
Bioinformatic pipeline and statistical analyses
A software designed to remove adapter sequences from reads, Cutadapt,28 was used to trim away the primer sequences as well as 3′‐bases with a Phred quality score below 15. Read pairs where one read was shorter than 120 bp were discarded. Remaining read pairs were merged using Uparse,29 and pairs that did not overlap or were predicted to have more than 3 errors over the full sequence length were discarded. Overall, 640,965 reads were kept. They were dereplicated and denoised with Unoise2,30 producing 2000 corrected sequences with abundance >1. All remaining reads were mapped back to these corrected sequences for quantification. We ensured that the samples of different groups were sequenced to approximately the same depths: The final 16S‐Seq analysis generated 126,551 approved read pairs for the 9‐week experiment (n = 20) with an average of 7723 reads per sample (range: 4140‐10,837), and 178,519 read pairs for the 16‐week experiment (n = 19) with an average of 6437 (range: 4236‐9832) reads per sample. There was no significant difference of read counts between AOM/DSS and control mice, or between WT mice and KO mice within the experiments. This supports that the removal of potential PCR contaminants (such as DSS) with InhibitEX was successful during the preparation protocol. Taxonomic assignment was based on the SILVA database31 using the strategy described by Hugerth et al.32 All unique operational taxonomic units (OTUs) and their 16S rRNA sequences, as well as the codes and scripts used in our study, are attached as Additional file 1 and Additional file 2, respectively. Our data is submitted to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) as BioProject [PRJNA475738] and BioSamples [SAMN09405040‐SAMN09405078].
Bioinformatic microbiota analyses
Alterations of the microbiota were analyzed bioinformatically through several measures, including rarefaction curves, alpha‐diversity, beta diversity, Bray‐Curtis divergence, and linear discriminant analysis (LDA) effect size (LEfSe). Rarefaction curves were used to confirm that sufficient sequencing coverage had been achieved, and to identify differences of species diversity between groups. Significant differences between rarefaction curves of different groups were calculated as described by Cayuela et al. 33 using the function EcoTest.sample from R package rareNMtests v1.1. Alpha diversity is a measure of microbiota abundance and how evenly these are distributed (evenness), and was estimated through Shannon's diversity index. Another estimate of alpha diversity is the richness of the microbiota, which was estimated through the Chao1 index. Beta diversity is the ratio of the diversity between groups versus individuals within each group, and we used the Bray‐Curtis divergence to quantify this compositional dissimilarity between different groups. Statistical testing was performed using permutational multivariate analysis of variance (PERMANOVA). All these calculations were performed in R v. 3.3.1 using packages Vegan v. 2.4–5, Fossil v. 0.3.7,34 and Vioplot v. 0.2.35 LEfSe36 was used to discover enriched features applying an LDA threshold of 2.0 and a significance cut‐off less than 0.05. AOM/DSS was used as main category (Classes) while the genotype or sex were analyzed as subcategories and plotted individually. Other comparisons with different classes and subclasses as input parameters were also conducted, as specified in the text and figures. Finally, to predict the functionality of the metagenomic content in each sample, phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt) software was used, based on the abundance of corresponding OTUs. 37
Quantitative PCR
qPCR was used for validation of 16S‐Seq data. Specific primers were designed for Prevotellaceae_UCG_001 and Parasutterella, or adopted for Bacteroidetes, Firmicutes, Gammaproteobacteria, Bifidobacteria, Cyanobacteria, Tenericutes, Verrucomicrobia, and total bacteria (primer sequences in Supporting Information Table S1). The designed primer pairs were confirmed for specificity of the intended target group by alignment to the ribosomal database project (RDPII) using the probe match tool. Ten nanograms of fecal DNA was amplified in a total volume of 10 μL using iTaq Universal SYBR Green Supermix (Bio‐Rad Laboratories) according to the manufacturer's recommendations (5 min at 95 °C, 40 cycles of 30 s at 95 °C, 30 s at 60 °C, 30 s at 72 °C), and followed by disassociation curve analysis to ensure amplification of a single amplicon. Each sample was analyzed in duplicates, and relative expression calculated using the 2‐ΔΔCt method normalized to total bacteria (total genomic 16S rRNA). The WT controls were used as calibrator samples in each experiment. Differences were tested with Welch's t‐test, and a p‐value equal or less than 0.05 was considered significant.
Results
To reduce the impact of normal variation of microbiota we compared co‐housed (shared cages) and age‐matched KO and WT siblings. All mice (males and females, WT and KO) were on the same mixed C57BL/6 J background. An overview of the experiment is presented in Figure 1.
Identification of baseline microbiota compositions in WT controls
We first compared the microbiota diversity of the two different WT control groups (the untreated 9‐week and 16‐week mice) in order to investigate the general variability of the microbiota and to establish a baseline microbiota composition and species diversity. We also corroborated the relative abundance of phyla in the WT controls of both time points using qPCR. We did not observe significant differences between the controls of the two different time points, except for two phyla (Firmicutes and Tenericutes) which increased at the later time point (Supporting Information Fig. S1). The ages differ by an average of 7 weeks between the two groups (19 versus 26 weeks old at end of experiments), but both experimental groups are categorized as young mice according to the frailty index scores of origin strains (C57BL/6).38 We conclude that, overall, the microbiota does not differ significantly between the two different groups of WT control mice.
AOM/DSS reduces gut microbiota diversity in WT animals
In order to identify the general impact that colitis‐induced CRC development has on the composition of microbiota, we compared the species diversity between AOM/DSS and untreated WT animals through several measures. First, we assessed species richness per sampling by plotting the number of detected species as a function of number of sequences analyzed. The resulting rarefaction curves leave the exponential phase around 2000 reads per sample, and signs of saturation are noted after around 5000 reads, confirming that sequencing coverage was sufficient (Fig. 2 a, b). These curves also show that AOM/DSS exposure reduced the number detected species (diversity) with more than 100 OTUs (Fig. 2 a, b) at both the 9‐week (p = 0.025) and 16‐week (p = 0.035) time point. Next, we investigated the abundance of the microbiota and how evenly these are distributed (evenness) using Shannon's alpha‐diversity index, and their richness using the Chao‐1 alpha‐diversity index (Fig. 2 c, d). We find that AOM/DSS in WT mice decreased the Shannon index from 4.9 to 4.2 (not significant) and the Chao‐1 index from 940 to 630 (p = 0.036) at 9 weeks. A decrease was noted also at 16 weeks, where Shannon index was reduced from 5.15 to 4.85 (p = 0.009) and Chao‐1 index from 910 to 750 (p = 0.08). We also explored the compositional dissimilarity of the species diversity between AOM/DSS and control groups by calculating beta diversity using Bray–Curtis dissimilarity. We found a significant dissimilarity between AOM/DSS‐treated mice and their controls at both time points (p = 0.001, Fig. 2 e, f, Supporting Information Table S2). Pooling of WT control mice from both time points (n = 16), maintained significant separation from the AOM/DSS cohorts (p = 0.001, Supporting Information Fig. S2A‐B, Supporting Information Table S3). We conclude that AOM/DSS‐mediated colitis and subsequent CRC significantly reduce the microbiota diversity in WT mice.
Figure 2.
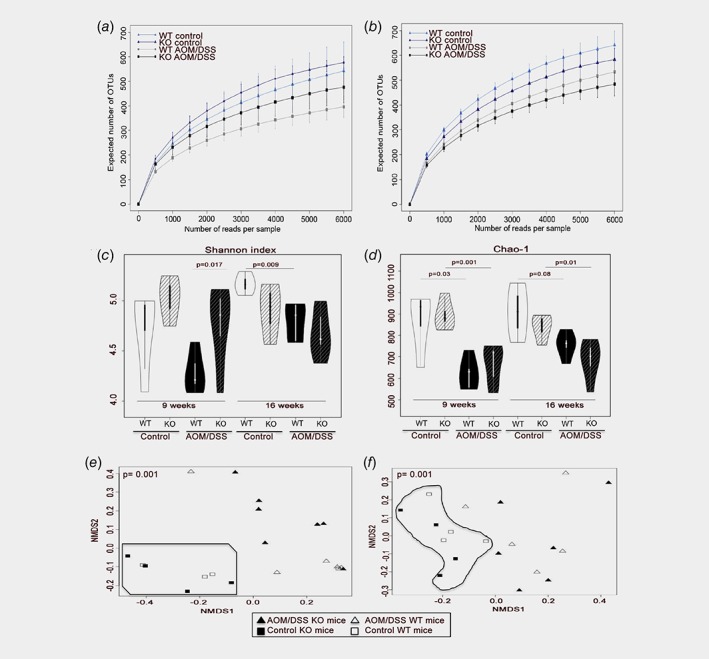
Microbiota diversity is reduced by treatment and lack of intestinal ERβ. (a, b) Rarefaction curves (mean +/− SD) show that sufficient sequencing depth was reached, and that a reduced number of species were detected in WT mice 9 weeks after treatment (p = 0.025, (a) and in both WT and KO mice after 16 weeks (WT: p = 0.035, KO: p = 0.030, B). (c, d) Diversity and richness measured with Shannon's and Chao1, respectively. AOM/DSS significantly reduces the diversity, and after 16 weeks, the lowest diversity was found in treated KO mice. (e, f) Beta diversity visualized with non‐metric dimensional scattering plot (NMDS), is significantly separated by AOM/DSS treatment at both 9 weeks (n = 20, p = 0.001, panel e) and 16 weeks (n = 19, p = 0.001, panel f). Significance calculated by PERMANOVA test, statistical comparisons are summarized in Supplemental Table 2. [Color figure can be viewed at wileyonlinelibrary.com]
AOM/DSS in WT animals alters the abundance of species related to metabolism
Next, we plotted the relative abundance of microbiota at the phyla level as bar charts (Fig. 3 a, b, with values of relative abundance at both time points summarized in Supporting Information Tables S4‐S5). Relative abundance at the family levels is presented in Supporting Information Figure S2C‐D with corresponding values in Supporting Information Tables S6‐S7. The phyla Bacteroidetes and Firmicutes were the most prevalent taxa in all groups and both time points, followed by Proteobacteria, Verrucomicrobia and Cyanobacteria. A transient AOM/DSS‐mediated increase of several phyla (Bacteroidetes, Firmicutes, Verrucomicrobia, and Cyanobacteria) after 9 weeks was corroborated using qPCR, but none of these changes remained after 16 weeks (Fig. 3 c–f). To discover significant alteration of taxa abundance in response to AOM/DSS in a systematic manner, we conducted LEfSe. This approach revealed 18 OTUs as significantly affected 9 weeks after initiation of AOM/DSS treatment, and 12 OTUs after 16 weeks (Fig. 4 a). Parasutterella was the most induced species (5.5‐fold enrichment) by AOM/DSS at 9 weeks (Fig. 4 a, left panel, and 4b) and Prevotellaceae_UCG_001 (phylum Bacteroidetes, family Prevotellaceae) the most reduced species (4.5‐fold reduced, Fig. 4 a and c) in WT mice. This was supported by qPCR (Fig. 4 d, e). At 16 weeks, Prevotellaceae_UCG_001 was enriched by AOM/DSS treatment, along with the taxa Odoribacter, Bacteroides and Parabacteroides (Fig. 4 a, right panel). We corroborated also this impact on Prevotellaceae_UCG_001 using qPCR (3.3‐fold enrichment, p = 0.004, Fig. 4 e). Finally, in order to make a prediction of functional effects resulting from the altered microbiota community, we used PICRUSt software. PICRUSt analyses can predict the functional KEGG pathways that are related to the composition of a metagenome, and has been demonstrated to provide a good representation of metagenomic prediction.37 Applying LEfSe onto this analysis revealed that several predicted KEGG pathways were enriched due to AOM/DSS treatment. After 9 weeks, AOM/DSS significantly increased bacteria associated with metabolism and immune system diseases (KEGG level 2, LDA >2, Fig. 4 f). More specifically (KEGG level 3), we find that AOM/DSS enhanced bacteria that metabolize butanoate, cofactors and vitamins, while reducing those that metabolize cyanoaminoacid, starch, and sucrose (Fig. 4 g). We also find that AOM/DSS increased bacteria associated with phosphotransferase system (PTS), fatty acid biosynthesis, and transcription machinery (Fig. 4g). We conclude that AOM/DSS treatment and subsequent colitis and CRC significantly alter specific microbial species in the colon of WT mice, and that this could impact metabolism, transcription, and the immune system of the gut.
Figure 3.
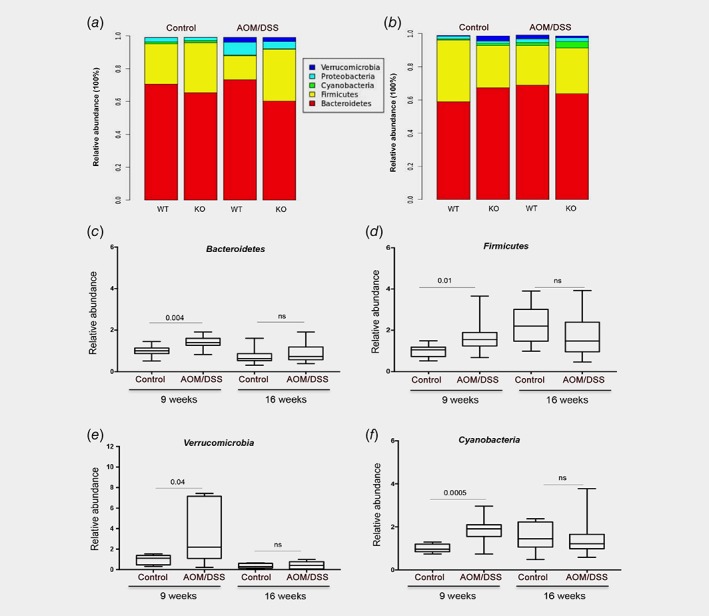
Relative abundance of microbiota. (a, b) Microbiota distribution at the phyla level at 9 weeks (a) and 16 weeks (b), with corresponding values of relative abundances summarized in Supplemental Tables 4–7. (c–f) Relative abundance measured with qPCR for Bacteroidetes (c), Firmicutes (d), Verrucomicrobia (e), and Cyanobacteria (f) are statistically significant (Welch's t‐test, WT and KO combined) at 9 weeks. [Color figure can be viewed at wileyonlinelibrary.com]
Figure 4.
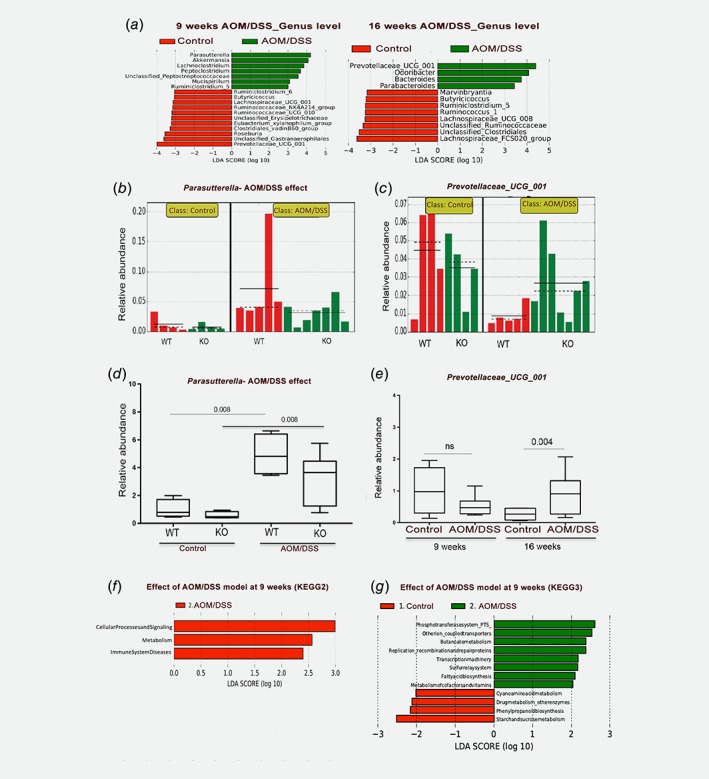
Differential abundance of microbiota demonstrated after AOM/DSS treatment. (a) LDA score indicate differentially abundant taxa (genus level) due to AOM/DSS at 9 weeks (left) and 16 weeks (right), per LEfSe analysis (p < 0.05 for all). (b) Histogram of the relative distribution of Parasutterella at 9 weeks, which shows an enrichment in WT mice after AOM/DSS treatment. (c) Histogram of the relative distribution of Prevotellaceae_UCG_001 at 9 weeks, which shows the abundance in control WT and KO. (d, e) qPCR supports 16S‐Seq identified alterations of Parasutterella (d) and Prevotellaceae_UCG_001 (e). (f, g) Functional classification of the predicted metagenome (PICRUSt) in response to AOM/DSS at 9 weeks using KEGG level 2 (f) and level 3 (g) pathways and visualized with LEfSe. [Color figure can be viewed at wileyonlinelibrary.com]
Microbiota diversity and specific species are influenced by sex
To investigate whether the microbiota diversity is different between male and female mice, we explored the impact of this parameter on the beta diversity using Bray–Curtis dissimilarity and PERMANOVA testing. We found that sex alone impacted the diversity at both time points (9 W: p = 0.048 and 16 W: p = 0.014, Supporting Information Table S2). This separation remained also when controls from 9 weeks (n = 8) and 16 weeks (n = 8) were pooled (9 W: p = 0.066 and 16 W: p = 0.007, Supporting Information Table S3). LEfSe further revealed specific species differences between the sexes in the untreated controls (Fig. 5 a, b). Bifidobacterium and Turicibacter were enriched in females at both time points, while males had higher levels of the genera Mucispirillum (both time points) and Anaeroplasma (at 9 weeks). qPCR analysis for the phyla Tenericutes (genus Anaeroplasma) and Bifidobacteria (genus Bifidobacterium) confirmed sex differences (Fig. 5 c, d). Males (untreated) had 5‐fold higher levels of Tenericutes (p = 0.02 at 9 weeks), and females (untreated) had 11‐fold higher levels of Bifidobacteria (p = 0.03 at 16 weeks). Further, the phylum Verrucomicrobia was 2.4‐fold higher in males (p = 0.009 at 9‐weeks, Supporting Information Fig. S3A), and Gammaproteobacteria higher in females (p = 0.05 at 16‐weeks, Supporting Information Fig. S3C). PICRUSt and LEfSe analysis further proposed that sex‐related microbiota differences impacted metabolism and the endocrine system in females, and cell motility and membrane transport functions in males (LDA >3, Fig. 5 h).
Figure 5.
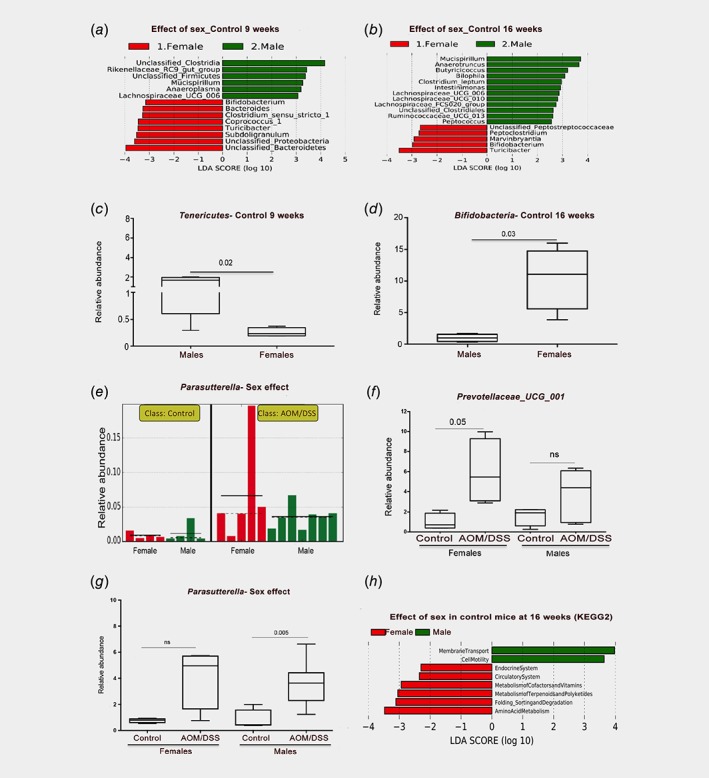
Microbiota abundance are sex‐dependent. (a, b) LEfSe analysis of differentially abundant taxa (genus level) indicates sex differences in control mice at 9 weeks (a) and 16 weeks (b). (c, d) qPCR corroborated differences identified in a‐b for phylum Tenericutes (genus Anaeroplasma) and phylum Bifidobacteria (genus Bifidobacterium). Welch's t‐test was used for significance testing. (e) Relative distribution of Parasutterella in individual mice after 9 weeks, per LEfSe analysis with AOM/DSS model as class and sex as subclass (corroborated by qPCR in panel g). (f) Sex‐dependent enrichment of Prevotellaceae_UCG_001 in AOM/DSS‐treated females at 16 weeks. (g) Parasutterella is significantly enriched in AOM/DSS‐treated males at 9 weeks. (h) Metagenomic prediction (PICRUSt) of sex differences using KEGG pathways and visualized with LEfSe, for untreated mice at 16 weeks. [Color figure can be viewed at wileyonlinelibrary.com]
After treatment, the same sex differences did not remain or were not significant (exemplified in Supporting Information Fig. S3B and D). However, we note that the induction of Prevotellaceae_UCG_001 by AOM/DSS at 16 weeks (Fig. 4 e), was 6‐fold enriched in females (p = 0.05) while not significantly enriched in males, as corroborated by qPCR (Fig. 5 f). Further, the AOM/DSS‐induced Parasutterella in WT mice (p = 0.008, Fig. 4 d) was significant only in males (p = 0.005, Fig. 5 g). Thus, our analyses demonstrated clear sex differences in the microbiota, which affect specific clades associated with colitis and dysbiosis.
Intestinal ERβ deletion modulates colitis‐induced microbiota diversity
Finally, we explored whether ERβ in the intestinal epithelium impacts the microbiota diversity. First, we investigated untreated animals, where rarefaction curves indicated a slight increase in the number of OTUs in KO mice compared to WT mice (about 50 OTUs) at the 9‐week time point (Fig. 2 a) and a slight decrease (85 OTUs) at the 16‐week time point (Fig. 2 b), but these changes were not significant. Similarly, both the Shannon and Chao‐1 indices indicated a slight increased diversity at 9 weeks, and decreased diversity at 16 weeks compared to WT mice, but neither reached statistical significance (Fig. 2 c, d). Treatment with AOM/DSS reduced the number of OTUs detected per rarefaction curves in KO animals with about 100 OTUs compared to untreated KO animals at both time points (Fig. 2 a, b, p = 0.01 at 9 weeks, p = 0.03 at 16 weeks), but the difference between KO and WT after AOM/DSS was not significant. There was, however, a significant increase of diversity in KO mice compared to WT after treatment per Shannon index (p = 0.017) at 9 weeks. Further, at the 16‐week time point, all three measurements (rarefaction curves, Shannon and Chao‐1 indices) indicated that AOM/DSS ERβKOVil mice exhibited the lowest microbiota alpha diversity. The statistical significance of the AOM/DSS‐induced reduction of microbiota richness (Chao‐1 index) was also more robust in KO mice (9w: p = 0.001, 16w: p = 0.01) than in WT mice (9w: p = 0.03, 16w: p = 0.08) for both time points (Fig. 2 d). Analysis of beta diversity using Bray–Curtis dissimilarity revealed a separation due to genotype (ERβ) (p = 0.028), and genotype in combination with sex (p = 0.011) at 9 weeks (Supporting Information Table S2), but no significant separation at 16 weeks. Overall, our analyses indicate that absence of intestinal ERβ reduces the microbiota diversity in conjunction with AOM/DSS treatment and CRC.
ERβ affects specific microbiota species during induced colitis
In order to explore whether intestinal ERβ KO impacts the abundance of specific species, we conducted further analysis with LEfSe. Comparing KO and WT control mice, we found that deletion of ERβ impacted few genera (Fig. 6 a–c). A reduction of unclassified Firmicutes were indicated in control KO mice at 16 weeks (Fig. 6 c, d). This was supported by qPCR with generic Firmicutes primers (p = 0.07, Fig. 6 e). After AOM/DSS treatment (9 weeks), more OTUs were enriched in KO animals compared to WT (Fig. 6 f). Interestingly, this included butyrate‐producing bacteria members of the phylum Firmicutes, such as Lachnospiraceae and Intestinimonas (Fig. 6 f–h). Further, the AOM/DSS‐mediated reduction of Prevotellaceae_UCG_001 at 9 weeks (Fig. 4 a and e), was almost exclusively resulting from reduction in the WT mice, while the decrease in KO mice were minimal (Fig. 4 c). We also note that while Parasutterella was significantly induced in both WT and KO animals (p = 0.008, Fig. 4 d), its levels appear lower in KO both before and after treatment (Fig. 4 b and d). Applying PICRUSt followed by LEfSe analysis, indicated that lack of ERβ in combination with treatment (9 weeks) enriched for microbiota with functions in membrane transport, cell motility, and carbohydrate metabolism, and reduced functions related to endocrine systems and other types of energy metabolism (Fig. 6 i). Some differences at the 16‐week time point included enrichment of Rikenellaceae_RC9 and exclusive presence of Lachnospiracae_UCG_010 in AOM/DSS‐treated KO (Fig. 6 j–l). Overall, this may suggest that intestinal ERβ impacts the microbiota diversity and specific microbial species, especially during early onset of inflammatory events, which we hypothesize could contribute to progression of chronic inflammation and/or CRC development.
Figure 6.
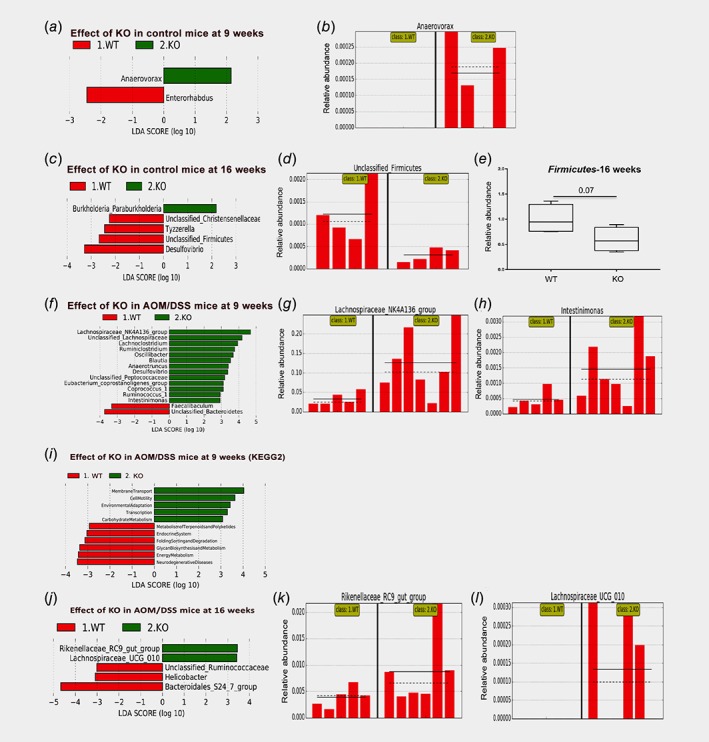
Specific taxa are differentially enriched in response to ERβ knockout. (a) LEFsE analysis indicates increase of the genus Anaerovorax (phylum Firmicutes) in KO untreated mice compared to WT, at 9 weeks, and (b) corresponding relative distribution of Anaerovorax in individual mice. (c) LEfSe analysis indicates decrease of unclassified Firmicutes in untreated KO mice at 16 weeks, and (d) corresponding relative distribution of unclassified Firmicutes in individual mice, which (e) is supported by qPCR of phylum Firmicutes (Welch's t‐test for significance testing). (f) LEFsE analysis indicates differential abundance between AOM/DSS‐treated WT and KO at 9 weeks, with (g, h) corresponding histograms illustrating increase in relative abundances of Lachnospiraceae_NK4A136 group and genus Intestinimonas in KO mice. (i, j) Metagenomic prediction (PICRUSt) of alterations in response to KO in AOM/DSS‐treated mice at 9 weeks (panel i) and 16 weeks (panel j) using KEGG pathways and visualized with LEfSe. (k, l) The histogram of the increased abundances of Rikenellaceae_RC9_gut_group and genus Lachnospiraceae_UCG_010 in KO‐treated mice at 16 weeks (corresponding to data in panel j). [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
In our study, we have characterized how colitis‐associated CRC driven by AOM/DSS impacts the microbiota diversity in mice with and without intestinal ERβ. In particular, we have identified novel ERβ and sex‐dependent effects.
Although our study was conducted in mice on mixed C57BL/6 J background, the identified AOM/DSS‐induced alterations of the microbiota corroborates several previous reports using AOM and/or DSS in other mice strains. Such studies include analysis of the dynamic diversity of microbiota in fecal contents of BALB/c mice after AOM injection and 4 cycles of DSS (3%) for 7 days,17 male A/J mice treated with AOM and DSS (2.5%) while on high‐fat diet at 77 days18, C57BL/6 mice after DSS only (5%) for 3 and 14 days, respectively39, and C57Bl/10 treated with DSS only (3.5%) for 7 days.15 Although different mice strains and different protocols of AOM and/or DSS treatment were used, all are in accordance with our study in terms of a decreased overall abundance and reduced beta diversity of the gut microbiota community after AOM and/or DSS treatment. However, there are also notable differences regarding the structural composition of the microbiota community, including the baseline composition of microbiota species.
The patterns we find are also in agreement with studies describing impact of colitis in the genetically modified IL10−/− colitis animal model,8 and in patients diagnosed with inflammatory bowel disease (IBD).40 For example, we found that the family Lachnospiraceae, was among the most decreased genera in AOM/DSS WT mice after 9 and 16 weeks (Fig. 4 a, b). This family was also reduced in the IL10−/− colitis model41 and in human IBD and CRC patients.42, 43 Other microbial species were transiently enriched by AOM/DSS (at 9 weeks), including Verrucomicrobia, Firmicutes, Bacteroidetes, and Cyanobacteria. Some members of those phyla are responsible for the degradation of host glycans and mucous layers which they use as nutritional and energy source, leading to disturbance of epithelial barriers.44 Our data showed that pathogenic gram‐negative bacteria Parasutterella (Fig. 4 a, b and d) and host glycan‐degradation bacteria Akkermansia (Fig. 4 a) were enriched after AOM/DSS treatment. This can lead to gut barrier disruption, damaging the mucus layer and the epithelium and eventually induction of immune responses that may result in chronic inflammation.4, 45 This agrees with our PICRUSt metagenomic prediction, which indicated that AOM/DSS enriched for microbiota affecting functions involved in immune system diseases and metabolic functions. Moreover, phosphotransferase system (PTS) was highly enriched after AOM/DSS treatment. This function allows a selective uptake and phosphorylation of sugar molecules by the bacteria, which inhibits the efflux of sugar molecules back across the bacterial membrane.46
Our data are also in agreement with studies showing that DSS leads to a shift of the microbiota toward abundance of proinflammatory gram‐negative bacteria. For example, we find enrichment of Bacteroides and Odoribacter in tumor‐bearing mice which is in agreement with studies carried out by Zackular et al.16 Further, Parasutterella, which we found increased by AOM/DSS (Fig. 4 d and 5 g), has been found to be higher in IBD patients with Crohn's disease,47 and was one of only five genera significantly enriched in CRC patients compared to healthy volunteers.43 Zackular et al. also found Parasutterella and Akkermansia to be enriched 7 days after DSS in C57BL/6 mice.16 Several of the AOM/DSS affected species, including the enrichment of Mucispirillum, have the genetic arsenal to degrade the mucous layer,48 or secrete pathogenic molecules such as lipopolysaccharides, that signal through Toll‐like receptors (TLRs)49, and could therefore contribute to colitis and cancer development. The agreement of our study with other reports of the impact of AOM or DSS on the microbiota, support the accuracy of observations and methodological considerations of our study. We also identify novel changes due to AOM/DSS‐induced colitis, such as the emergence and enrichment of the genus Prevotellaceae_UCG_001 in AOM/DSS‐treated mice at 16 weeks (Fig. 4 a and c), especially in females (Fig. 5 f). Members of the family Prevotellaceae have previously been shown to increase the severity of DSS‐induced colitis in mice.50
Our study is the first study investigating the impact of intestinal epithelial ERβ on microbiota. A previous limited study used T‐RFLP and Sanger's sequencing to characterize 16S rRNA gene variants in fecal pellets from body‐wide ERβ knockout mice (BERKO).12 That study found indications that the diet impacted microbiota differently in BERKO mice, especially the phyla Bacteroidetes, Firmicutes and Proteobacteria.12 Our analysis, using 16S‐Seq, found a lower abundance of Firmicutes in KO mice (Fig. 6 c–e). Importantly, we showed that the reduced alpha diversity that resulted 16 weeks after AOM/DSS in WT animals was further reduced in KO mice. Additionally, the beta diversity was significantly impacted by loss of ERβ, and this was enhanced by sex (Supporting Information Tables S2 and S3), at 9 weeks after initiation of AOM/DSS treatment. A number of taxa were differently affected in AOM/DSS‐treated KO mice compared to WT mice, including Prevotellaceae_UCG_001 which was transiently reduced 5.6‐fold in WT mice but barely reduced (1.2‐fold) in KO mice at 9 weeks. This genus could be of particular interest to consider for future studies aimed at exploring how estrogen and ERβ impact gut inflammation and cancer via microbiota. Further, Lachnospiraceae_UCG_001 was enriched in treated KO mice at 16 weeks (Fig. 6 j and l). This is important because it has been shown that Lachnospiraceae_UCG_001 serves as an energy source for epithelial cells via production of short chain fatty acids, which maintain the epithelial barrier integrity.42, 43 Fatty acid biosynthesis was induced by AOM/DSS, and membrane transport and carbohydrate metabolism functions were specifically enriched in KO mice exposed to AOM/DSS, per PICRUSt analysis. Lachnospiraceae_UCG_001 represents a source of energy for proliferation of the epithelial cells and tumor progression at 16 weeks, and it would be of interest to explore the role of this enrichment in future studies, and to explore whether knockout of intestinal ERβ also impacts the short chain fatty acids in the gut. ERβ in the colon has been proposed to maintain the integrity of epithelial barrier,51 and our results support a role for ERβ in maintenance of the eubiosis status in the gut.
The effect of knockout was, however, not significant for all parameters analyzed. It is possible that more ERβ‐related differences, including those that are specific for each gender, would be identified in a larger study. It is also possible that genotype‐affected microbiota changes were diminished in our study due to our choice to co‐host WT and KO littermates in the same cages. This was done in order to avoid cage effects, but may result in a partial sharing of microbiota. Finally, we did not explore the mucosal‐associated microbiota profiles nor the impact of different diets on these mice. Further studies, exploring luminal microbiota, metabolic interactions with the host, a variety of diets including a purified diet, will yield further information.
In our study, we observed significant segregation of microbiota beta‐diversity depending on sex after both 9 weeks and 16 weeks (Supporting Information Tables S2 and S3). We also found differences in the relative abundance of e.g. Tenericutes, Bifidobacter Verrucomicrobia and Gammaproteobacteria between males and females (Fig. 5 c and Supporting Information Figure S3), although the difference was diminished after AOM/DSS treatment. Men have a higher incidence of CRC compared to women and it is possible that sex differences impact the microbiota diversity and hence susceptibility to microbiota‐associated diseases. For example, ICR (Swiss albino) males have been reported to be more vulnerable to microbiota infections and development of inflammation than corresponding females.52 A few animal studies have, indeed, demonstrated a correlation between microbiota diversity and sex53, and in response to different nutrients.54 Human studies have also indicated a difference in overall microbiota communities in the gut or of specific bacteria related to gender.55 In addition, studies have shown that microbiota composition can influence the levels of sex hormones and also regulate autoimmune diseases.56 However, other reports claim that there is no strong correlation between sex and microbiota diversity.57 We have shown here that intestinal ERβ has an impact on the microbiota, and we have identified sex‐dependent differences of the microbiota. It is possible that the sex‐dependent differences are related to differential activation of ERβ in males and females. In order to investigate the combination of sex‐differences, ERβ, and colitis and CRC, larger studies should be conducted in a time‐dependent manner and in more mice strains, to assess the dynamic shifts of microbiota.
Conclusions
We identify that induced colitis and CRC reduce the microbiota diversity, enrich gram‐negative bacteria, and strongly impact several phyla. The genus Prevotellaceae_UCG_001 is increased when tumors are established, especially in females, and Parasutterella is strongly increased in AOM/DSS‐induced colitis but decreased once CRC is established. Furthermore, for the first time, we identify an effect of intestinal ERβ on the modulation of microbiota diversity.
Supporting information
Supplemental Figure 1 Relative abundance of selected taxa in WT control mice per qPCR analysis. A‐F) qPCR was used to compare the relative abundance of Bacteroidetes (A), Cyanobacteria (B), Firmicutes (C), Tenericutes (D), Verrucomicrobia (E), and Bifidobacteria (F). Only Firmicutes and Tenericutes were significantly changed between the 9‐ and 16‐week WT control groups. Significance was tested with Welch's t‐test, with p < 0.05 considered as significant.
Supplemental Figure 2 Beta diversity of microbiota visualized with non‐metric dimensional scattering plot (NMDS). A‐B) Beta diversity of pooled 9‐week and 16‐week untreated controls (n = 16) compared to AOM/DSS treated samples at 9‐week (n = 12, panel A) or 16‐week (n = 11, panel B) time points. The significance was calculated by PERMANOVA test and all values provided in Supplemental Table S3. AOM/DSS treatment (9 week: p = 0.001; 16 week p = 0.001), sex (9 week p = 0.066; 16 week p = 0.007) and combination of genotype and sex (9 week p = 0.040) changed beta diversity. C‐D). Microbiota distribution at the family level at 9 weeks (C) and 16 weeks (D). Corresponding values of the relative abundances are summarized in Supplemental Tables 6–7.
Supplemental Figure 3 Sex‐dependent microbiota changes before and after initiation of AOM/DSS model corroborated by qPCR. A‐B) Relative abundance of Verrucomicrobia in untreated (A) males (n = 4) and females (n = 4), and AOM/DSS treated males (n = 7) and females (n = 5), at 9 weeks. C‐D) Relative abundance of Gammaproteobacteria in untreated (C) males (n = 4) and females (n = 4), and AOM/DSS treated (D) males (n = 7) and females (n = 4), at 16 weeks. Significance values were calculated with Welch's t test, WT and KO combined, with p < 0.05 considered as significant.
Supplemental Table S1 Primer sequences, designed or adopted. The sequences were aligned against Ribosomal database project (RDP release 11) to confirm matching against selected species.
Supplemental Table S2. Statistical PERMANOVA tests significance values for beta diversity analyses related to Figure 2E and F
Supplemental Table S3. Statistical PERMANOVA tests significance values for beta diversity analyses related to Supporting Information Figure S2A and B.
Supplemental Table S4. Taxonomy ratios of phyla at 9 weeks related to Figure S3A and B.
Supplemental Table S5. Taxonomy ratios of phyla at 16 weeks related to Figure S3A and B
Supplemental Table S6. Taxonomy ratios of families at 9 weeks relating to Supporting Information Figure S2C and D
Supplemental Table S7. Taxonomy ratios of families at 16 weeks relating to Supporting Information Figure S2C and D.
Acknowledgements
We would like to thank Dr. Trang Vu‐Nguyen (University of Houston) for assistance with animals, and Dr. Sanghyuk Chung (University of Houston) for advice. The authors acknowledge support from the Clinical Genomics facility and National Genomics Infrastructure in Stockholm (funded by Science for Life Laboratory, the Knut and Alice Wallenberg Foundation and the Swedish Research Council) for assistance with massively parallel sequencing, and SNIC/Uppsala Multidisciplinary Center for Advanced Computational Science and UPPMAX for access to computational infrastructure. This work was partially supported by the Ragnar Söderberg's Foundation (to LE), Robert A. Welch Foundation (E‐0004, to JÅG), National Cancer Institute at the National Institutes of Health (R01CA172437), Marie Curie Actions FP7‐PEOPLE‐2011‐COFUND (GROWTH 291795) via the VINNOVA programme Mobility for Growth, the Swedish Cancer Society (CAN2015/591, CAN 2018/596), Swedish Research Council (2017‐01658), and Stockholm County Council (all to CW).
References
- 1. Ferlay J, Colombet M, Soerjomataram I, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer 2018. [DOI] [PubMed] [Google Scholar]
- 2. Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta‐analysis. Gut 2001;48:526–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF‐kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev 2010;21:11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shahanavaj K, Gil‐Bazo I, Castiglia M, et al. Cancer and the microbiome: potential applications as new tumor biomarker. Expert Rev Anticancer Ther 2015;15:317–30. [DOI] [PubMed] [Google Scholar]
- 5. Westbrook AM, Wei B, Braun J, et al. Intestinal mucosal inflammation leads to systemic genotoxicity in mice. Cancer Res 2009;69:4827–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Colotta F, Allavena P, Sica A, et al. Cancer‐related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 2009;30:1073–81. [DOI] [PubMed] [Google Scholar]
- 7. Zitvogel L, Ma Y, Raoult D, et al. The microbiome in cancer immunotherapy: diagnostic tools and therapeutic strategies. Science 2018;359:1366–70. [DOI] [PubMed] [Google Scholar]
- 8. Lupp C, Robertson ML, Wickham ME, et al. Host‐mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2007;2:119–29. [DOI] [PubMed] [Google Scholar]
- 9. Zhu Q, Gao R, Wu W, et al. The role of gut microbiota in the pathogenesis of colorectal cancer. Tumour Biol 2013;34:1285–300. [DOI] [PubMed] [Google Scholar]
- 10. Arthur JC, Jobin C. The complex interplay between inflammation, the microbiota and colorectal cancer. Gut Microbes 2013;4:253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Benson AK, Kelly SA, Legge R, et al. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci U S A 2010;107:18933–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Menon R, Watson SE, Thomas LN, et al. Diet complexity and estrogen receptor beta status affect the composition of the murine intestinal microbiota. Appl Environ Microbiol 2013;79:5763–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hoy YE, Bik EM, Lawley TD, et al. Variation in taxonomic composition of the fecal microbiota in an inbred mouse strain across individuals and time. PLoS One 2015;10:e0142825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuraishy A, Karin M, Grivennikov SI. Tumor promotion via injury‐ and death‐induced inflammation. Immunity 2011;35:467–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heimesaat MM, Fischer A, Siegmund B, et al. Shift towards pro‐inflammatory intestinal bacteria aggravates acute murine colitis via toll‐like receptors 2 and 4. PLoS One 2007;2:e662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zackular JP, Baxter NT, Iverson KD, et al. The gut microbiome modulates colon tumorigenesis. MBio 2013;4:e00692–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Klimesova K, Kverka M, Zakostelska Z, et al. Altered gut microbiota promotes colitis‐associated cancer in IL‐1 receptor‐associated kinase M‐deficient mice. Inflamm Bowel Dis 2013;19:1266–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang CZ, Huang WH, Zhang CF, et al. Role of intestinal microbiome in American ginseng‐mediated colon cancer protection in high fat diet‐fed AOM/DSS mice. Clin Transl Oncol 2017;20:302–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Williams C, Dileo A, Niv Y, et al. Estrogen receptor beta as target for colorectal cancer prevention. Cancer Lett 2016;372:48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nguyen‐Vu T, Wang J, Mesmar F, et al. Estrogen receptor beta reduces colon cancer metastasis through a novel miR‐205 ‐ PROX1 mechanism. Oncotarget 2016;7:42159–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Edvardsson K, Nguyen‐Vu T, Kalasekar SM, et al. Estrogen receptor beta expression induces changes in the microRNA pool in human colon cancer cells. Carcinogenesis 2013;34:1431–41. [DOI] [PubMed] [Google Scholar]
- 22. Edvardsson K, Strom A, Jonsson P, et al. Estrogen receptor beta induces antiinflammatory and antitumorigenic networks in colon cancer cells. Mol Endocrinol 2011;25:969–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hartman J, Edvardsson K, Lindberg K, et al. Tumor repressive functions of estrogen receptor beta in SW480 colon cancer cells. Cancer Res 2009;69:6100–6. [DOI] [PubMed] [Google Scholar]
- 24. Saleiro D, Murillo G, Benya RV, et al. Estrogen receptor‐beta protects against colitis‐associated neoplasia in mice. Int J Cancer 2012;131:2553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cleveland AG, Oikarinen SI, Bynote KK, et al. Disruption of estrogen receptor signaling enhances intestinal neoplasia in Apc(min/+) mice. Carcinogenesis 2009;30:1581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Andersson S, Sundberg M, Pristovsek N, et al. Insufficient antibody validation challenges oestrogen receptor beta research. Nat Commun 2017;8:15840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Klindworth A, Pruesse E, Schweer T, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next‐generation sequencing‐based diversity studies. Nucleic Acids Res 2013;41:e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martin M. Cutadapt removes adapter sequences from high‐throughput sequencing reads. Bioinform Action 2012;17:10–2. [Google Scholar]
- 29. Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 2013;10:996–8. [DOI] [PubMed] [Google Scholar]
- 30. Edgar RC. UNOISE2: improved error‐correction for Illumina 16S and ITS amplicon sequencing. BioRxiv:081257 2016.
- 31. Quast C, Pruesse E, Yilmaz P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web‐based tools. Nucleic Acids Res 2013;41:D590–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hugerth LW SM, Pennhag AA, Du J, et al. A comprehensive automated pipeline for human microbiome sampling, 16S rRNA gene sequencing and bioinformatics processing. BioRxiv:286526 2018.
- 33. Cayuela L, Gotelli NJ, Colwell RK. Ecological and biogeographic null hypotheses for comparing rarefaction curves. Ecol Monogr 2015;85:437–55. [Google Scholar]
- 34. Vavrek MJ. fossil: Palaeoecological and palaeogeographical analysis tools. Palaentol Electron 2011;141T:16p. [Google Scholar]
- 35. Vioplot D A.: Violin Plot. R Package version 0.2. http://wsopuppenkiste.wiso.ni-goettingen.de/daldler 2005.
- 36. Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol 2011;12:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Langille MG, Zaneveld J, Caporaso JG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013;31:814–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Langille MG, Meehan CJ, Koenig JE, et al. Microbial shifts in the aging mouse gut. Microbiome 2014;2:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nagalingam NA, Kao JY, Young VB. Microbial ecology of the murine gut associated with the development of dextran sodium sulfate‐induced colitis. Inflamm Bowel Dis 2011;17:917–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hansen J, Gulati A, Sartor RB. The role of mucosal immunity and host genetics in defining intestinal commensal bacteria. Curr Opin Gastroenterol 2010;26:564–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ye J, Lee JW, Presley LL, et al. Bacteria and bacterial rRNA genes associated with the development of colitis in IL‐10(−/−) mice. Inflamm Bowel Dis 2008;14:1041–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Frank DN, St Amand AL, Feldman RA, et al. Molecular‐phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 2007;104:13780–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang T, Cai G, Qiu Y, et al. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J 2012;6:320–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Derrien M, Vaughan EE, Plugge CM, et al. Akkermansia muciniphila gen. Nov., sp. nov., a human intestinal mucin‐degrading bacterium. Int J Syst Evol Microbiol 2004;54:1469–76. [DOI] [PubMed] [Google Scholar]
- 45. Davenport M, Poles J, Leung JM, et al. Metabolic alterations to the mucosal microbiota in inflammatory bowel disease. Inflamm Bowel Dis 2014;20:723–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McCoy JG, Levin EJ, Zhou M. Structural insight into the PTS sugar transporter EIIC. Biochim Biophys Acta 2015;1850:577–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ricanek P, Lothe SM, Frye SA, et al. Gut bacterial profile in patients newly diagnosed with treatment‐naive Crohn's disease. Clin Exp Gastroenterol 2012;5:173–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Derrien M, Van Baarlen P, Hooiveld G, et al. Modulation of mucosal immune response, tolerance, and proliferation in mice colonized by the Mucin‐degrader Akkermansia muciniphila. Front Microbiol 2011;2:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zarember KA, Godowski PJ. Tissue expression of human toll‐like receptors and differential regulation of toll‐like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol 2002;168:554–61. [DOI] [PubMed] [Google Scholar]
- 50. Elinav E, Strowig T, Kau AL, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011;145:745–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wada‐Hiraike O, Imamov O, Hiraike H, et al. Role of estrogen receptor beta in colonic epithelium. Proc Natl Acad Sci U S A 2006;103:2959–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee SM, Kim N, Son HJ, et al. The effect of sex on the Azoxymethane/dextran sulfate sodium‐treated mice model of colon cancer. J Cancer Prev 2016;21:271–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Org E, Mehrabian M, Parks BW, et al. Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes 2016;7:313–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shastri P, McCarville J, Kalmokoff M, et al. Sex differences in gut fermentation and immune parameters in rats fed an oligofructose‐supplemented diet. Biol Sex Differ 2015;6:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dominianni C, Sinha R, Goedert JJ, et al. Sex, body mass index, and dietary fiber intake influence the human gut microbiome. PLoS One 2015;10:e0124599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Markle JG, Frank DN, Mortin‐Toth S, et al. Sex differences in the gut microbiome drive hormone‐dependent regulation of autoimmunity. Science 2013;339:1084–8. [DOI] [PubMed] [Google Scholar]
- 57. Kovacs A, Ben‐Jacob N, Tayem H, et al. Genotype is a stronger determinant than sex of the mouse gut microbiota. Microb Ecol 2011;61:423–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1 Relative abundance of selected taxa in WT control mice per qPCR analysis. A‐F) qPCR was used to compare the relative abundance of Bacteroidetes (A), Cyanobacteria (B), Firmicutes (C), Tenericutes (D), Verrucomicrobia (E), and Bifidobacteria (F). Only Firmicutes and Tenericutes were significantly changed between the 9‐ and 16‐week WT control groups. Significance was tested with Welch's t‐test, with p < 0.05 considered as significant.
Supplemental Figure 2 Beta diversity of microbiota visualized with non‐metric dimensional scattering plot (NMDS). A‐B) Beta diversity of pooled 9‐week and 16‐week untreated controls (n = 16) compared to AOM/DSS treated samples at 9‐week (n = 12, panel A) or 16‐week (n = 11, panel B) time points. The significance was calculated by PERMANOVA test and all values provided in Supplemental Table S3. AOM/DSS treatment (9 week: p = 0.001; 16 week p = 0.001), sex (9 week p = 0.066; 16 week p = 0.007) and combination of genotype and sex (9 week p = 0.040) changed beta diversity. C‐D). Microbiota distribution at the family level at 9 weeks (C) and 16 weeks (D). Corresponding values of the relative abundances are summarized in Supplemental Tables 6–7.
Supplemental Figure 3 Sex‐dependent microbiota changes before and after initiation of AOM/DSS model corroborated by qPCR. A‐B) Relative abundance of Verrucomicrobia in untreated (A) males (n = 4) and females (n = 4), and AOM/DSS treated males (n = 7) and females (n = 5), at 9 weeks. C‐D) Relative abundance of Gammaproteobacteria in untreated (C) males (n = 4) and females (n = 4), and AOM/DSS treated (D) males (n = 7) and females (n = 4), at 16 weeks. Significance values were calculated with Welch's t test, WT and KO combined, with p < 0.05 considered as significant.
Supplemental Table S1 Primer sequences, designed or adopted. The sequences were aligned against Ribosomal database project (RDP release 11) to confirm matching against selected species.
Supplemental Table S2. Statistical PERMANOVA tests significance values for beta diversity analyses related to Figure 2E and F
Supplemental Table S3. Statistical PERMANOVA tests significance values for beta diversity analyses related to Supporting Information Figure S2A and B.
Supplemental Table S4. Taxonomy ratios of phyla at 9 weeks related to Figure S3A and B.
Supplemental Table S5. Taxonomy ratios of phyla at 16 weeks related to Figure S3A and B
Supplemental Table S6. Taxonomy ratios of families at 9 weeks relating to Supporting Information Figure S2C and D
Supplemental Table S7. Taxonomy ratios of families at 16 weeks relating to Supporting Information Figure S2C and D.