

Abstract
Phenotyping, ie, identification of patients possessing a characteristic of interest, is a fundamental task for research conducted using electronic health records. However, challenges to this task include imperfect sensitivity and specificity of clinical codes and inconsistent availability of more detailed data such as laboratory test results. Despite these challenges, most existing electronic health records–derived phenotypes are rule‐based, consisting of a series of Boolean arguments informed by expert knowledge of the disease of interest and its coding. The objective of this paper is to introduce a Bayesian latent phenotyping approach that accounts for imperfect data elements and missing not at random missingness patterns that can be used when no gold‐standard data are available. We conducted simulation studies to compare alternative phenotyping methods under different patterns of missingness and applied these approaches to a cohort of 68 265 children at elevated risk for type 2 diabetes mellitus (T2DM). In simulation studies, the latent class approach had similar sensitivity to a rule‐based approach (95.9% vs 91.9%) while substantially improving specificity (99.7% vs 90.8%). In the PEDSnet cohort, we found that biomarkers and clinical codes were strongly associated with latent T2DM status. The latent T2DM class was also strongly predictive of missingness in biomarkers. Glucose was missing in 83.4% of patients (odds ratio for latent T2DM status = 0.52) while hemoglobin A1c was missing in 91.2% (odds ratio for latent T2DM status = 0.03 ), suggesting missing not at random missingness. The latent phenotype approach may substantially improve on rule‐based phenotyping.
Keywords: Bayesian, electronic health records, latent class, missing data, phenotype
1. INTRODUCTION
Electronic health records (EHR) have emerged as an important data resource for conducting observational studies of health care and outcomes. However, analyses using these data must account for the imperfect and inconsistent quality of EHR data in order to avoid erroneous inference.1, 2 One important data quality issue for EHR‐based research is high levels of missing data that arise through a complex process.3, 4, 5, 6 Missing data in this context are often missing not at random (MNAR) because patients who are sicker tend to have more complete and extensive EHR data available due to their greater frequency of interaction with the health care system. Despite the near ubiquity of this complex missingness mechanism, analyses of EHR data often do not explicitly and rigorously account for missing data.
Patient phenotyping, ie, characterizing patients with respect to the presence or absence of characteristics such as diseases of interest, is a fundamental task that must be undertaken in order to make use of EHR data for research. Although EHR data contain many potentially informative data elements, they often do not include gold‐standard phenotypes, and the most informative data elements, such as results of diagnostic tests, are often missing for the majority of patients. The most commonly used approach to phenotyping using EHR data is rule‐based phenotyping, in which an algorithm based on clinical knowledge of the disease process and coding practices for the disease is prespecified.7 For instance, the Phenotype Knowledgebase (http://phekb.org) provides several dozen rule‐based phenotypes for a variety of conditions of interest. Approaches to handling missing data included in a rule‐based phenotype include treating lack of information as indicative of the absence of disease, excluding the patient from further analysis, or using more formal approaches to handling missing data such as multiple imputation (MI). However, the implications of these alternative approaches for bias and efficiency of phenotypes under different missing data mechanisms have not been explored in this setting.
Complete case analysis, ie, exclusion of all individuals with any missing data elements included in the phenotype definition, is inefficient, potentially resulting in substantial losses of data, and can lead to bias if individuals with missing information represent a nonrandom subset of the population. In the context of multisite research networks where some data elements may be missing for all patients at a given site, excluding individuals with missing data elements is particularly problematic since it would amount to the elimination of those sites from the analysis. Such exclusions are likely to bias results by systematically eliminating subgroups of patients that may differ in important respects from patients who have no missing data. One approach to handling this challenge is to use a phenotype that only includes data available for all individuals. For instance, in the case of diabetes, biomarker measurements are likely to be missing for many patients. A phenotype might therefore be based only on the presence or absence of clinical codes, which would be available for all patients. While using a simplified phenotype obviates the need to address missing data in a potentially large subgroup of individuals, it does so at the expense of discarding potentially more informative predictors that have been measured among people without missing data.
An alternative to complete case analysis is to impute missing data elements and then apply the phenotype to the imputed data. Multiple imputation via chained equations8 is a frequently used approach to imputing missing data because it allows for multiple variable types and can be implemented easily. This flexibility is convenient for use in the context of EHR data where multiple variable types are likely to be encountered. However, MI methods rely on an assumption of missing at random (MAR) missingness, which is likely to be violated in EHR data. In cases in which receipt of a diagnostic test is related to the phenotype of interest, which is unobserved, the assumption of MAR missingness is violated. Including measures of comorbidity and healthcare utilization in the imputation model may help to mitigate bias.9 However, it is unlikely that the missing data mechanism can be completely specified using available data elements.
Latent variable methods have recently been suggested as an alternative approach to phenotyping10 that can handle incomplete or inconsistently assessed patient characteristics. While MI approaches attempt to impute missing values for each measurement based on the joint distribution of the measurements, latent variable models posit the existence of an underlying characteristic through which the observed data are related. Several prior studies have used latent variables to address challenges arising from inconsistently available administrative data. For instance, He et al11 combined data from chart abstraction and Medicare claims in which both data sources were considered imperfect and incomplete. They proposed a Bayesian MI method via data augmentation for their latent variable model and estimated associations between the underlying characteristic of interest and outcomes, even in the presence of substantial missingness. A similar latent variable model was proposed by Chen and Zhou,12 who considered the additional complicating factor of complex correlation structures due to nesting of observations within individuals and clinics. However, their estimation strategy focused on marginal modeling, integrating over unobserved patient characteristics rather than explicitly estimating them. Coley et al13 used EHR data to predict a latent prostate cancer recurrence phenotype using Bayesian estimation methods. In their setting, this phenotype was explicitly observed for a subset of patients but selection into this subset was assumed to be informative. Their model thus accommodated both inconsistently available data across individuals and MNAR missingness for some data elements. These prior approaches demonstrate the feasibility of a latent variables approach in the analysis of EHR data.
In this paper, we propose a Bayesian latent class model for EHR‐based phenotyping that addresses heterogeneity in available data elements across patients and possibly MNAR missingness. The Bayesian approach bridges rule‐based phenotypes, which are based entirely on expert knowledge and opinions, and data‐driven approaches, which are derived solely from information contained in the data. In comparison to commonly used rule‐based phenotyping approaches, the Bayesian approach facilitates incorporation of expert knowledge via prior distributions while allowing for estimation of relationships among variables included in the model based on the observed data. In contrast to the approach of Coley et al,13 we consider an unsupervised context in which gold‐standard phenotype information is not available for any subjects. This is a common setting in EHR‐based research where many studies are conducted without access to validation data.
The structure of this paper is as follows. Section 2 introduces data from the PEDSnet consortium on a cohort of pediatric patients at elevated risk of type 2 diabetes mellitus (T2DM), which we use to illustrate challenges arising due to missing data and motivate parameter choices for our simulation studies. We then describe the proposed Bayesian latent class model and alternative rule‐based approaches to phenotyping. In Section 3, we present results of simulation studies comparing alternative phenotyping approaches and an analysis of the PEDSnet data on T2DM. Finally, we conclude in Section 4 with a summary and discussion of methodological alternatives to addressing the challenges of EHR‐based phenotyping.
2. METHODS
2.1. Pediatric T2DM
Our investigation of alternative methods for EHR‐based phenotyping was motivated by the case of pediatric T2DM. Unlike T2DM in adults which has high prevalence, T2DM in children is rare, with estimated prevalence of <0.1%.14 Data on pediatric T2DM came from one of eight sites in the Patient Centered Outcomes Research Institute‐funded PEDSnet consortium, the Children's Hospital of Philadelphia (CHOP). Included children were 9‐18 years old, had at least two outpatient clinical encounters captured in the CHOP EHR in 2001‐2017, and had at least one body mass index (BMI) z‐score in excess of the 95th percentile for their age and sex. While biomarkers including hemoglobin A1c (HbA1c) and serum glucose have good operating characteristics for diagnosing T2DM,15 biomarker data are not available for the majority of children. In this sample, <9% of children had HbA1c data available and <17% had glucose data. In contrast, information on presence or absence of diagnostic codes, comorbidities, healthcare utilization, and prescription medications is available for all children. However, these data elements may not have good sensitivity or specificity with respect to underlying disease status. For this analysis, we included data from all in‐person clinical encounters occurring within 2 years of the first clinical encounter at which a patient met study inclusion criteria, which was defined as the baseline visit. Data elements derived from the EHR and included in our analysis as potential predictors of pediatric T2DM were age, sex, race/ethnicity, visit to an endocrinologist, metformin prescription, insulin prescription, age and sex standardized BMI z‐score, average glucose value, average HbA1c value, and diagnosis codes for type 1 diabetes mellitus (T1DM) and T2DM.
The University of Pennsylvania and CHOP Institutional Review Boards determined that this project did not constitute human subjects research because it used only existing de‐identified data resources.
2.2. Bayesian latent phenotype model
To address the missing data issues described in Section 1, we developed a Bayesian latent class model applicable to EHR data that features variation in the number and type of observations available across individuals, where true disease status may influence the type of data available for an individual, and in which the true disease status is assumed unobserved for all patients. We allow for covariate dependence of the latent phenotype as well as the observed data elements, conditional on the latent phenotype. Subsequently, we describe our approach to model specification and estimation.
Let D i represent a phenotype for i = 1,…n patients, which is assumed unobserved. Available data for estimating D i include vectors of J biomarkers (Y i = (Y i1,…,Y iJ)), K clinical codes (W i = (W i1,…,W iK)), L prescription medications (P i = (P i1,…,P iL)), and M patient covariates such as demographics (X i = (X i1,…,X iM)). Table 1 summarizes the elements of the proposed Bayesian model for latent phenotypes, using T2DM as an illustrative example. Although our development is motivated by pediatric T2DM, the general classes of EHR‐derived variables used in this example are common to many phenotypes, allowing the latent phenotyping approach to be used beyond this specific context. Biomarkers are assumed doubly informative in that not only the biomarker value but the availability of a biomarker measurement (R i = (R i1,…,R iJ)) may be indicative of the underlying phenotype. Availability of biomarkers, biomarker measurements, clinical codes, and prescription medications are assumed conditionally independent given the underlying disease status, D i, and patient characteristics.
Table 1.
Model specification for Bayesian latent variable model for EHR‐derived phenotypes for the ith patient.
| Latent Phenotype | Availability of | Biomarkers | Clinical Codes | Prescription | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Biomarkers | Medications | |||||||||
| Example | Type 2 Diabetes | Availability of glucose | Glucose or | Diabetes ICD‐9 code; | Diabetes | |||||
| or HbA1c data | HbA1c values | Endocrinologist visits | medication | |||||||
| Variable | D i | R i j, j = 1,…,J | Y i j, j = 1,…,J | W ik, k = 1,…,K | P il, l = 1,…,L | |||||
| Model |
|
|
|
|
|
|||||
| Priors | β D∼MVN(0,ΣD) |
|
|
|
|
|||||
| η i∼Unif(a,b) | ||||||||||
|
|
Abbreviations: N, normal; Bern, Bernoulli; MVN, multivariate normal; Unif, uniform; InvGamma, inverse gamma, HbA1c, Hemoglobin A1c.
Without loss of generality, we assume that binary indicators of the presence of clinical codes and prescription medications are available for all patients while biomarkers are only available for a subset. The likelihood for the ith patient is given by
where β D denotes the association between patient characteristics and the phenotype of interest; η i denotes a subject‐specific random effect for the phenotype; and , , , and, denote the association between patient characteristics and the underlying phenotype and availability of biomarkers, biomarker values, clinical codes, and medications, respectively. Using the specifications in Table 1 yields a model in which mean biomarker levels are shifted by a quantity for patients with the phenotype of interest compared to those who do not possess this characteristic. Similarly, sensitivity and specificity of binary indicators for clinical codes, medications, and presence of biomarkers are given by combinations of regression parameters. For instance, in a model with no patient covariates, specificity of the kth code is given by while sensitivity is given by , where . Additionally, the proposed model allows for a unique combination of available data elements for each patient. The likelihood for each individual consists of the product of the likelihood contributions for all available measurements for that individual for each variable type. If the jth biomarker is missing, R i j = 0, which results in exclusion of the likelihood contribution for Y i j.
Prior knowledge about the classification accuracy of biomarkers and codes can be encoded through suitable choice of priors for these parameters. In the case of normally distributed biomarkers, the proposed model implies a binormal receiver operating characteristic (ROC) model. In general, for a normally distributed biomarker with mean and variance given by μ 1 and in diseased individuals and μ 0 and in controls, the area under the curve (AUC) is given by , where Φ(·) represents the standard normal cumulative distribution function.16 In our specific case in which we assume a common variance for cases and controls and a difference in means for the jth biomarker of , the AUC is given by . If prior information on AUC for a given biomarker is available, this can be used to inform selection of a prior for .
We recommend the use of a uniform prior, Uniform(a,b), for to constrain prevalence of the phenotype of interest within some plausible range. For instance, in the case of pediatric T2DM, past studies suggest that prevalence is very low.14 Thus, setting a < b < 0 is appropriate in this case. Constraining η i via a prior with finite range is useful to prevent “label switching,” which can otherwise present a problem for model identifiability17 and Bayesian latent variable models.18
Estimation can be carried out using Markov Chain Monte Carlo (MCMC) methods. Specifically, in simulation studies and analyses of PEDSnet data, we used JAGS via the R package runjags to obtain samples from the posterior distribution of model parameters. Samples from the posterior distribution of , the patient‐specific probability of membership in the D i = 1 class, were used to describe a patient's latent phenotype. Example R and JAGS code used for the analysis of PEDSnet data are provided in our GitHub repository (https://github.com/rhubb/Latent-phenotype/).
2.3. Clinical decision rules for T2DM
We compared the performance of the above described latent phenotype approach to three classification rules based on biomarkers and clinical codes. These comparator approaches were motivated by a prior study that explored data elements used in T2DM phenotypes for adults19 as well as existing clinical decision rules for T2DM.20 Three alternative classification rules were considered in which a patient was classified as having T2DM (ie, ) if he or she had:
biomarker in the abnormal range, ie, glucose ≥ 200 mg/dl or HbA1c ≥ 6.5%;
presence of clinical codes or prescriptions related to T2DM;
biomarker in the abnormal range OR presence of clinical codes or prescriptions related to T2DM.
In typical applications of T2DM rule‐based phenotyping, if glucose or HbA1c are missing they are assumed to be in the normal range. In addition to this approach, we also explored the use of MI for missing biomarkers. We used MI via chained equations to impute missing biomarkers conditional on patient characteristics, diagnosis codes, and prescription medications. In simulation studies and analyses of PEDSnet data, five imputed data sets were generated, biomarker values were averaged across imputations, and mean imputed biomarker values were then included in the rule‐based approaches above. In application of rule‐based phenotypes to PEDSnet data, we additionally required no occurrence of T1DM codes to account for the much higher prevalence of T1DM in pediatric cohorts compared to T2DM.
2.4. Simulation study
We evaluated the performance of alternative phenotyping approaches using a series of simulation studies. The objective of our simulations was to produce simulated data sets that resembled real EHR data on pediatric patients in the PEDSnet cohort in terms of the types, distributions, and missingness patterns of variables. In our simulated example motivated by the context of pediatric T2DM, we simulated fully observed patient covariates age, race, BMI z‐score; binary indicators for presence of clinical codes for T2DM and making a visit to an endocrinologist; and a binary indicator for presence of a medication prescription for metformin. We also simulated two biomarkers, glucose and HbA1c. Data were simulated for 1000 patients according to the distributions provided in Table 2. Standard deviations for the two biomarkers were selected to correspond to an AUC of approximately 0.95 for each biomarker.
Table 2.
Distributional assumptions and parameter values used in simulation studies. Normal distributions are parameterized using mean and standard deviation.
| Variable | Distribution |
|---|---|
| Age (X i1) | Uniform (9,18) |
| White race (X i2) | Bernoulli (0.524) |
| T2DM (D i | X i1,X i2) | Bernoulli (expit(η i + 0.01X i1 − 0.07X i2)),η i∼Normal( − 2.64,0.04) |
| BMI percentile (X i3 | D i) | Truncated Normal (2.2D i + 2.0(1 − D i),0.3, lowerbound = 1.645) |
| T2DM code (W i1| D i) | Bernoulli (0.8D i + 0.004(1 − D i)) |
| Endocrinologist visit code (W i2 | D i) | Bernoulli (0.5D i + 0.078(1 − D i)) |
| Metformin code (P i1| D i) | Bernoulli (0.2D i + 0.0112(1 − D i)) |
| Glucose (Y i1| D i) | Normal (90.6 + 42D i,16.93) |
| HbA1c (Y i2| D i) | Normal (5.4 + 1.00D i,0.45) |
Abbreviations: BMI, body mass index; T2DM, type 2 diabetes mellitus.
We next introduced missingness in biomarkers according to two alternative missingness mechanisms. Under MAR missingness, missignness was simulated according to a Bernoulli distribution with patient‐specific missingness probabilities that varied according to age, race, and BMI. Under MNAR missingness, these missingness probabilities were additionally allowed to depend on T2DM status. Availability of data on the jth biomarker was simulated according to a Bernoulli distribution with probability, . Across all simulations, we set and , for k = 1,2,3. These values were selected to represent a moderately strong effect of patient risk factors on biomarker missingness, with less frequent missingness in glucose (Y i1) and more frequent missingness in HbA1c (Y i2). Specifically, patients with clinical characteristics associated with T2DM were more likely to have available biomarker data, with the strength of this risk factor effect greater for HbA1c (Y i2) than for glucose (Y i1). We then investigated four different scenarios for missing data:
Low MAR missingness: , ;
High MAR missingness: , ;
Low MNAR missingness: , ;
High MNAR missingness: , .
Setting in MNAR scenarios represents a strong dependence of biomarker availability on underlying presence of T2DM. We also note that, while we refer to these scenarios as “low” and “high” missingness, these terms are applied relatively. Missingness was high in an absolute sense across all simulations with, on average, 25% missingness in Y i1 and 48% missingness in Y i2 in the “low missingness” scenarios and 81% missingness in Y i1 and 95% missingness in Y i2 in the “high missingness” scenarios. In data from PEDsnet, missingness in biomarkers was more similar to the high missingness scenario than the low missingness scenario.
For the Bayesian latent phenotype approach, we used normal priors with variance 100 for parameters in biomarker models and normal priors with variance 10 for parameters in logistic models for binary indicators. We placed a Uniform(−5,−1) prior on η i. For each simulated data set, we drew 1000 samples from the posterior distribution for for each patient after 1100 burn‐in iterations. Based on this posterior sample, we computed the posterior mean for each patient and classified the patient according to diabetes status.
A cutpoint for dichotomous classification as to T2DM status was chosen such that patients with posterior mean probability of T2DM in the top 5% of the distribution were classified as diabetic and all others were classified as nondiabetic. This cutpoint was chosen because the prevalence of T2DM in simulated data was approximately 5%. This represents the type of dichotomization that could be made in a latent phenotype if an approximate population prevalence was known. This dichotomous classification for the latent phenotype approach was compared to classification based on (1) biomarkers only, (2) codes only, (3) biomarkers and codes, (4) biomarkers with missing values replaced via MI, and (5) biomarkers and codes with missing biomarker values replaced via MI.
We characterized the performance of alternative methods applied to simulated data in terms of their predictive accuracy relative to underlying true T2DM status. Sensitivity, specificity, and proportion of patients misclassified were calculated for each method relative to underlying true T2DM status. In addition, the Bayesian latent phenotype model and biomarkers provided continuous measures of disease risk. Discrimination for these measures was additionally evaluated using an ROC analysis and the mean area under the ROC curve (AUC) was calculated across simulations. In ROC analyses, individuals with missing biomarkers were assigned a biomarker value of zero in order to ensure that they were classified as nondiseased across all biomarker thresholds.
We conducted a series of sensitivity analyses to evaluate the robustness of our results to (1) the prevalence of the phenotype of interest, (2) the choice of cutpoint for the Bayesian latent phenotyping approach, and (3) the violation of the assumption of equal variance of the biomarkers in cases and controls. To address (1), we repeated the simulation study using the parameter values and distributional assumptions described above but increasing the prevalence of the T2DM phenotype in the simulated data to ∼20%. For (2), we used the simulation settings described above but, rather than dichotomizing posterior probabilities at the known population prevalence, we dichotomized the posterior probabilities at the estimated population prevalence based on the mean of the posterior means, , where is the posterior mean probability of T2DM for the ith subject. Finally, to investigate (3), we used the simulation settings described above but increased the biomarker variance by a factor of four in cases only. In this scenario, we continued to carry out estimation using the Bayesian model assuming a common variance for cases and controls in order to investigate the effect of model misspecification on our results. All sensitivity analyses were conducted in the setting of MAR missingness.
Each simulation scenario was repeated 100 times and results were summarized across simulations.
2.5. Analysis of PEDSnet data
We used the alternative methods described above to identify patients with T2DM using the PEDSnet data described in Section 2.1. Clinical codes included in our analysis were T2DM and an endocrinologist visit. We selected these variables to illustrate two types of information available in EHR data (diagnosis codes and utilization information). Two prescription medications, metformin and insulin, were included in the latent phenotype model. Continuous biomarkers included in the analysis were glucose and HbA1c. For binary measures, we evaluated all medical records within two years of a patient's baseline visit to PEDSnet for presence or absence of corresponding codes. For continuous biomarkers, we took the average of available measures within two years of baseline, if any. To account for possible MNAR missingness in biomarkers, we also included binary indicators of presence of any glucose or HbA1c measurement, modeled separately, within two years of baseline. Patient characteristics, ie, age, nonwhite race or hispanic ethnicity, and BMI, were included in the model for D i, the latent T2DM status and in models for missingness in biomarkers.
To explore the value of MI in this setting, we estimated patient phenotypes using the two biomarker‐based approaches with and without missing values imputed via MI. Variables included in our MI model were patient age, year, gender, race, ethnicity, number of endocrinologist visits, number of metformin prescriptions, number of insulin prescriptions, whether metformin was prescribed prior to insulin, BMI, height, weight, cholesterol, glucose, HbA1c, number of T1D codes, and number of T2D codes. We imputed missing data five times and calculated the average glucose and HbA1c across imputations. These average biomarker levels were then included in the two phenotyping approaches.
For parameters in logistic regression models for binary measures, we used Normal (0,100) priors. For biomarkers, we used Normal(0,100) priors for the mean biomarker value in nondiabetics. To incorporate known information about the predictive accuracy of glucose and HbA1c, we used informative priors for the additive difference in the biomarker for diabetic patients compared to nondiabetics. The value for this mean shift was selected to correspond to an AUC of 0.95 under the binormal model. For glucose, we used a Normal(75.6, 10) prior and for HbA1c a Normal(2.9, 1) prior. We discarded the first 2000 iterations of the MCMC sampler and based all results on a subsequent sample of 5000 draws from the posterior distribution of the model parameters. Posterior probability of T2DM was characterized based on the posterior mean of for each child. The sum of the posterior means was used to estimate the number of children expected to have T2DM and the mean of the posterior means provided an estimated prevalence.
3. RESULTS
3.1. Simulation results
Under MAR missingness, the latent phenotype, codes only, and codes or biomarkers approaches all achieved high sensitivity with mean sensitivity across simulations in excess of 90% for all three approaches (Figure 1). Imputing missing biomarkers had little effect on sensitivity of the codes or biomarkers approach. In contrast, the biomarkers only approach had poor sensitivity. Sensitivity of biomarker‐based classification is strongly influenced by missingness. Therefore, unsurprisingly, sensitivity of this approach is poor under high levels of missingness and decreases with increasing missingness. Imputing missing biomarkers had little effect on the mean sensitivity of biomarkers in the low missingness scenario. In the high missingness scenario, imputing missing biomarkers improved mean sensitivity from 1.8% to 23.6%. As expected, specificity was inversely related to sensitivity with the codes only, codes or biomarkers, and codes or biomarkers with MI approaches achieving fair specificity while the biomarkers and biomarkers MI strategies had near perfect specificity (>99%). The latent phenotype approach achieved very high specificity (99.7% in the high missingness scenario) in addition to its good sensitivity. Because our simulation scenarios investigated a rare condition (average prevalence of 5%), overall classification accuracy was more highly influenced by specificity than sensitivity. As a result, the Bayesian approach outperformed all other methods on this measure. Under the low missingness scenario, mean classification accuracy of the latent phenotype approach was 99% compared to 94% for the next best method (biomarkers with MI). Results for classification accuracy were similar in the high and low missingness scenarios. Notably, the latent phenotype approach was the only classification method that was able to achieve both good sensitivity and good overall classification accuracy. Results for simulations conducted under MNAR missingness were very similar (Figure 2). The only notable difference was that both the latent phenotype and biomarkers with MI strategies displayed increased variability under MNAR missingness. Results of sensitivity analyses investigating the effect of higher prevalence, dichotomization of the latent phenotyping approach using the estimated prevalence, and higher biomarker variance among cases were extremely similar to results of the primary analysis (Web Figures 1 to 3).
Figure 1.
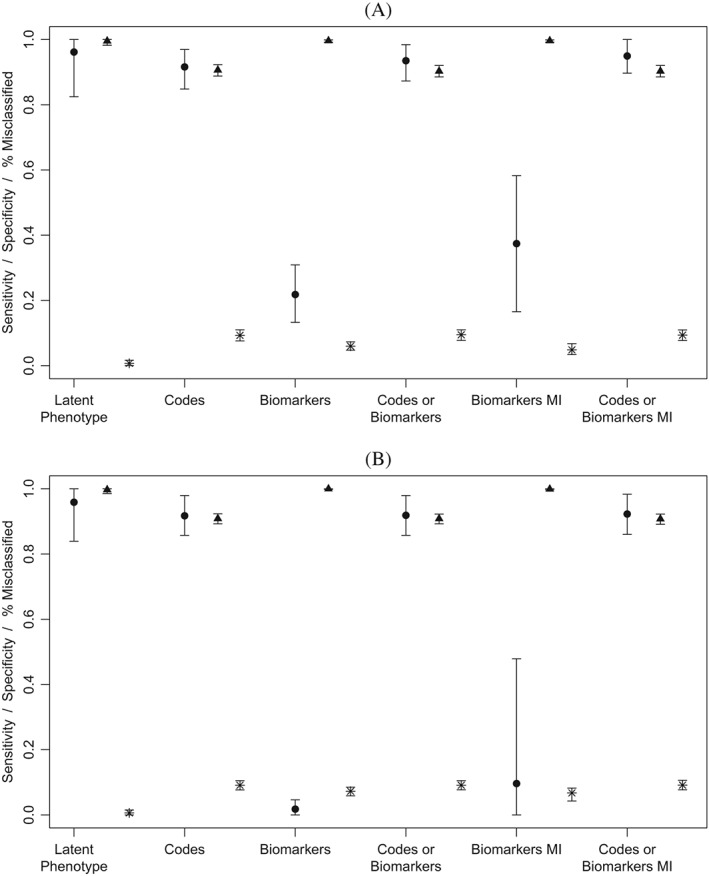
Sensitivity and specificity of methods for identifying patients with type 2 diabetes based on 100 simulations per scenario. Biomarkers were simulated under MAR missingness with average missingness in biomarkers of 48% for HbA1c and 25% for glucose in Scenario 1 (Panel A) and 95% for HbA1c and 81% for glucose in Scenario 2 (Panel B). Circle = sensitivity, Triangle = specificity, Star = percent misclassified. MI, multiple imputation
Figure 2.
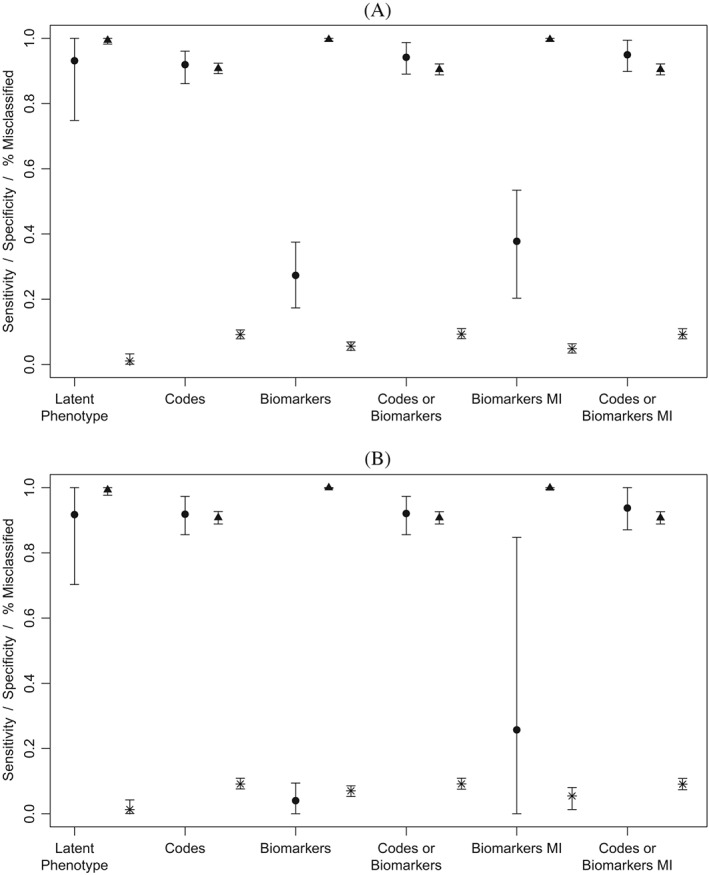
Sensitivity and specificity of methods for identifying patients with type 2 diabetes based on 1000 simulations per scenario. Biomarkers were simulated under missing not at random missingness with average missingness in biomarkers of 48% for HbA1c and 25% for glucose in Scenario 3 (Panel A) and 95% for HbA1c and 81% for glucose in Scenario 4 (Panel B). Circle = sensitivity, Triangle = specificity, Star = percent misclassified. MI, multiple imputation
Figure 3.
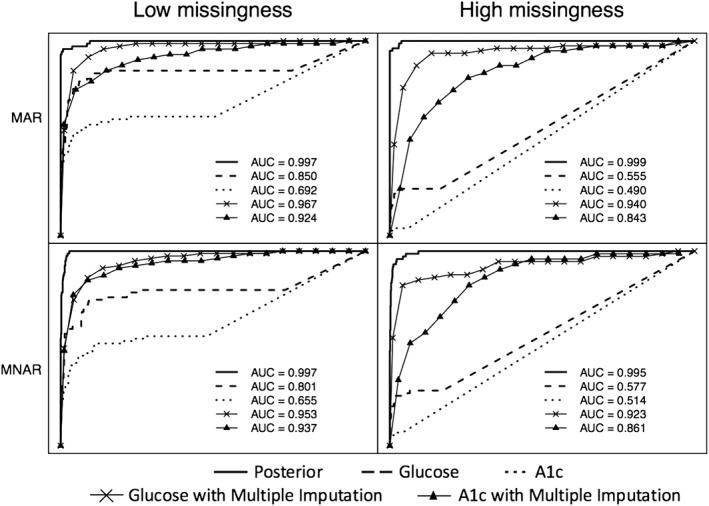
Receiver operating characteristic curves for identifying patients with type 2 diabetes from four example simulated data sets. Biomarkers were simulated under missing at random (MAR) (first row) and missing not at random (MNAR) (second row) missingness with average missingness in biomarkers of 48% for HbA1c and 25% for glucose in Scenario 3 (left column) and 95% for HbA1c and 81% for glucose in Scenario 4 (right column). AUC, area under the curve.
We also compared the classification accuracy of continuous measures (posterior means from Bayesian latent phenotype approach, biomarkers, and biomarkers with missing values imputed via MI) using AUC. Figure 3 provides example ROC curves for one simulated data set from each of the four simulation scenarios. Classification accuracy based on the latent phenotype posterior probabilities is very good in all four scenarios. In the low missingness scenario, glucose with imputed missing values also performs very well. Because of the higher proportion of missing values for HbA1c compared to glucose, this biomarker performs relatively more poorly both with and without imputation. Table 3 summarizes AUC across simulations for each approach in each of the four simulation scenarios.
Table 3.
Mean and standard deviation (SD) for area under the curve (AUC) based on posterior probability of type 2 diabetes mellitus from latent phenotype analysis, glucose, hemoglobin A1c (HbA1c), glucose with missing imputed via multiple imputation (MI), and HbA1c with missing imputed via MI based on simulated data. Means and standard deviations were computed across 100 simulated data sets for each scenario
| AUC (SD× 10 3) | ||||
|---|---|---|---|---|
| Scenario 1 | Scenario 2 | Scenario 3 | Scenario 4 | |
| Low MAR Missingness | High MAR Missingness | Low MNAR Missingness | High MNAR Missingness | |
| Latent phenotype | 0.999 (5.98) | 1.000 (2.60) | 0.998 (4.66) | 0.997 (7.40) |
| Glucose | 0.761 (40.11) | 0.513 (28.64) | 0.851 (36.45) | 0.592 (31.62) |
| HbA1c | 0.571 (102.88) | 0.499 (12.14) | 0.735 (42.67) | 0.530 (19.36) |
| Glucose MI | 0.954 (14.01) | 0.920 (28.66) | 0.959 (13.20) | 0.931 (21.97) |
| HbA1c MI | 0.933 (18.12) | 0.811 (107.93) | 0.944 (15.78) | 0.883 (69.61) |
Abbreviations: MAR, missing at random; MNAR, missing not at random.
3.2. Application to pediatric T2DM
Data from the PEDSnet sample included 68 265 children. Characteristics of patients included in the study sample are provided in Table 4. This sample included 5043 patients (7.4%) who had biomarkers or codes suggestive of possible T2DM (ie, no T1DM codes and at least one of abnormal glucose, abnormal HbA1c, T2DM codes, or a metformin prescription). Children with codes or biomarkers suggestive of possible T2DM were more likely to be female and to have made visits to an endocrinologist. Overall, only 16.6% of patients had glucose measurements and 8.8% had HbA1c measurements.
Table 4.
Characteristics of study population of pediatric patients at risk for type 2 diabetes mellitus (T2DM) stratified according to absence of codes for type 1 diabetes mellitus and presence of codes for T2DM, metformin prescription, or elevated hemoglobin A1c or glucose
| Total | Codes or Biomarkers Suggesting T2DM | ||
|---|---|---|---|
| Yes | No | ||
| N = 68 265 | N = 5043 | N = 63 222 | |
| N (%) | N (%) | N (%) | |
| Male | 36 836 (53.96) | 2026 (40.17) | 34 810 (55.06) |
| White | 35 740 (52.35) | 2886 (57.23) | 32 854 (51.97) |
| Endocrinologist | 5338 (7.82) | 510 (63.43) | 4828 (7.16) |
| Metformin | 764 (1.12) | 675 (83.96) | 89 (0.13) |
| Insulin | 727 (1.06) | 154 (19.15) | 573 (0.85) |
| T1D codes | 632 (0.93) | 0 (0) | 632 (0.94) |
| T2D codes | 275 (0.4) | 221 (27.49) | 54 (0.08) |
| Any glucose measurement | 11 325 (16.59) | 355 (44.15) | 10 970 (16.26) |
| Any HbA1c measurement | 6031 (8.83) | 397 (49.38) | 5634 (8.35) |
| Mean (SD) | Mean (SD) | Mean (SD) | |
| Age | 11.90(2.50) | 13.79 (2.58) | 11.87 (2.49) |
| BMI | 2.02 (0.30) | 2.27 (0.36) | 2.01 (0.30) |
| Glucose | 94.31 (32.51) | 141.39 (104.47) | 92.79 (27.44) |
| Hemoglobin A1c | 5.79 (1.25) | 6.93 (1.94) | 5.71 (1.15) |
Abbreviations: BMI, body mass index; SD, standard deviation; T1D, type 1 diabetes; T2D, type 2 diabetes.
We applied the Bayesian latent phenotyping approach to the PEDSnet data to obtain posterior probabilities of T2DM for this sample. The model indicated large shifts in the means of glucose (90.6, 95% credible interval [CI] [90.2‐91.0]) and HbA1c (3.2, 95% CI [3.1‐3.2]) associated with assignment to the latent T2DM class (Table 5). Making a visit to an endocrinologist had high sensitivity and fair specificity, while presence of codes for T2DM, metformin prescriptions, and insulin prescriptions had lower sensitivity and nearly perfect specificity relative to the latent T2DM status. The very high specificity for T2DM codes and prescription medications is likely due to their low prevalence in the data set (∼1%). The latent T2DM class was also negatively associated with missingness in biomarkers suggesting an MNAR missingness mechanism. Glucose was less likely to be missing for patients assigned to the latent T2DM class (OR = 0.52). This negative association between latent T2DM status and missingness was even stronger for HbA1c (OR = 0.03). The mean posterior probability of T2DM was 4.5% among patients with codes or biomarkers suggestive of T2DM and 3.4% among patients with no codes or abnormal biomarkers.
Table 5.
Posterior means and 95% credible intervals (CI) for model parameters for analysis of pediatric T2DM in the PEDSnet sample
| Posterior | 95% CI | |
|---|---|---|
| Mean | ||
| Mean shift in glucose ( ) | 90.62 | (90.25, 91.00) |
| Mean shift in HbA1c ( ) | 3.15 | (3.06, 3.24) |
| T2DM code sensitivity ( ) | 0.17 | (0.15, 0.20) |
| T2DM code specificity (1‐ ) | 1.00 | (1.00, 1.00) |
| Endocrinologist visit code sensitivity ( ) | 0.94 | (0.92, 0.95) |
| Endocrinologist visit code specificity (1‐ ) | 0.93 | (0.93, 0.94) |
| Metformin code sensitivity ( | 0.31 | (0.28, 0.35) |
| Metformin code specificity (1‐ ) | 0.99 | (0.99, 0.99) |
| Insulin code sensitivity ( ) | 0.66 | (0.61, 0.70) |
| Insulin code specificity (1‐ ) | 1.00 | (1.00, 1.00) |
| OR missing glucose ( ) | 0.52 | (0.44, 0.61) |
| OR missing HbA1c ( ) | 0.03 | (0.02, 0.04) |
Abbreviations: HbA1c, hemoglobin A1c; T2DM, type 2 diabetes mellitus; OR, odds ratio.
We applied the six approaches described in Section 2.3 to estimate prevalence of T2DM in the PEDSnet data set (Table 6). Relying on biomarkers alone, with or without MI, resulted in very low prevalence estimates (<1%). Similarly, in this sample, few patients had T2DM codes or metformin prescriptions leading to low prevalence estimates based on these codes (1.1%). The Bayesian latent phenotyping approach estimated the prevalence of T2DM in this cohort to be 3.5%.
Table 6.
Prevalence of pediatric type 2 diabetes mellitus in the PEDSnet sample according to six phenotyping approaches
| N | Prevalence (%) | |
|---|---|---|
| Latent phenotype | 2362 | 3.5 |
| Codes | 722 | 1.1 |
| Biomarkers | 209 | 0.3 |
| Codes or biomarkers | 804 | 1.2 |
| Biomarkers MI | 424 | 0.6 |
| Codes or biomarkers MI | 995 | 1.5 |
Abbreviation: MI, multiple imputation.
4. DISCUSSION
In this paper, we investigated alternative strategies for constructing EHR‐derived phenotypes when no gold‐standard validation data are available and in the presence of high levels of possibly MNAR missingness in important predictors. Overall, we found that the latent phenotyping approach incorporating explicit models for availability of missing predictors in relation to the underlying condition of interest achieved better discrimination than any of the comparator approaches. In simulation studies, this approach was able to correctly classify the majority of cases while still retaining high specificity and overall classification accuracy. This approach combines information from high quality biomarkers, which are available for only a limited subset of patients, with information from weaker predictors that is broadly available. In comparison to a strict dichotomization on the basis of diagnostic or procedure codes, the Bayesian approach allows for the possibility that these codes may be related to the condition of interest without requiring a deterministic classification of the condition of interest. This greater flexibility improved on the specificity of clinical decision rules without sacrificing sensitivity.
In the case of adult T2DM, many prior studies have explored phenotypes based on claims codes or EHR data (see, eg, other works7, 19, 20, 21, 22, 23). In general, prior approaches have treated missing data as indicative of no evidence of T2DM. A review of data elements commonly included in T2DM phenotypes identified clinical codes for diabetes, HbA1c, fasting plasma glucose, random plasma glucose, oral glucose tolerance test results, and use of diabetes‐associated medications.19 Diabetes codes can be further subdivided into codes that are specific to T2DM and those that are either nonspecific or specific to T1DM. Alternative rule‐based uses of these codes result in substantial disagreement between diabetes phenotypes.19 The latent phenotyping approach proposed here has the advantage of potentially learning from the data in settings such as pediatric T2DM where the information content of some potential predictors may be uncertain. For instance, in our analysis of the PEDSnet data, we incorporated information on visits to an endocrinologist, which is likely to be related to T2DM diagnosis but may be nonspecific since patients with T1DM and other conditions would also be treated by an endocrinologist.
Latent class approaches are subject to the label switching problem. We have addressed this problem by constraining the prevalence of the phenotype of interest to be <50% by using a prior distribution with mass on a finite range. While this proved effective in simulation studies for phenotypes with prevalence of approximately 5% and 20%, it may be less effective for phenotypes with prevalence close to 50%. Several alternative approaches to the label switching problem in Bayesian mixture models have been proposed.18 In cases with prevalence close to 50%, a relabeling approach may be preferable.24
A strength of the proposed approach is that it provides a probability of case status rather than a dichotomous classification. While these probabilities can be dichotomized, there are several advantages to preserving the posterior probability in continuous form. This probability encapsulates information about the relative certainty of disease classification. If incorporated as a predictor or outcome in subsequent analyses, this uncertainty can be propagated through the analysis to achieve valid inference. Several prior studies have proposed approaches to incorporating predicted probabilities derived from EHR data into subsequent research studies in order to decrease bias and improve efficiency.25, 26, 27 If a dichotomous classification is required, for instance, to identify patients for inclusion in a survey or patient outreach effort, a cut point can be chosen by the investigator in order to identify a target number or proportion of patients or to achieve a desired balance between sensitivity and specificity. In simulation studies, we found that dichotomizing based on either a known population prevalence or estimated population prevalence based on the mean of the posterior means resulted in good classification accuracy. In practice, the choice of cut point can be varied depending on the objective of a given project and the relative costs of false‐negative and false‐positive classifications.
This study has several limitations. Our Bayesian latent class model assumed conditional independence of data elements related to an underlying T2DM phenotype. Prior work has noted that latent class–based estimates of sensitivity and specificity of diagnostic tests are sensitive to violation of the conditional independence assumption.28 In the case of an EHR‐based study, because the number of available data elements related to the underlying phenotype is potentially very large, it should be possible to distinguish between alternative dependence structures. Patient‐specific random effects could be introduced into the model formulation to induce dependence if needed. Additionally, data from PEDSnet do not include gold‐standard information on T2DM diagnosis. Thus, while we were able to estimate posterior probabilities and compare these to data elements suggestive of possible T2DM, we cannot compute operating characteristics of the posterior probabilities relative to true T2DM status. However, simulation studies indicated that classification accuracy of this approach is good and superior to rule‐based approaches. Additionally, the proposed Bayesian model is relatively complex, requiring MCMC estimation methods that are computationally intensive and may be infeasible in extremely large data sets. Our analysis of the PEDSnet data that includes approximately 70 000 patients took 29 hours to run on a desktop computer with a 3.5 GHz Intel Core i7 processor and 32 GB memory. In larger data sets including millions of patients, simpler models making use of conjugate priors to obtain closed form posterior distributions or variational Bayes approximations are recommended to avoid the computational challenges of MCMC.
There are a variety of future directions for EHR‐based phenotyping that can be explored. Notably, we have investigated the case of a phenotype that was assumed to be time invariant. In longitudinal studies of disease onset or progression, it is necessary to make both a disease status classification and to identify a time of onset. Additionally, there are a variety of ways that EHR‐derived phenotypes can be incorporated into subsequent research studies including as outcomes, exposures, or inclusion/exclusion criteria. Work is needed to characterize bias and efficiency of alternative approaches to using EHR‐derived phenotypes in each of these settings.
In conclusion, the proposed latent phenotyping approach provides a means of combining data elements with variable availability across patients and can perform well even when some data elements are missing for almost all patients and when data are MNAR. Given the complexity of the processes that give rise to data in the EHR, a flexible approach to incorporating variably available information that takes into account data provenance, the process by which data are generated and captured, may improve our ability to identify patients with phenotypes of interest relative to existing rule‐based approaches.
Supporting information
SIM_7953‐Supp‐0001‐pepper_aim1_online_supplement.pdf
ACKNOWLEDGEMENTS
This research was supported by Patient‐Centered Outcomes Research Institute (PCORI) Awards (ME‐1511‐32666, CDRN‐306‐01556). The statements presented in this article are solely the responsibility of the authors and do not necessarily represent the views of the Patient‐Centered Outcomes Research Institute (PCORI), its Board of Governors, or Methodology Committee.
Hubbard RA, Huang J, Harton J, et al. A Bayesian latent class approach for EHR‐based phenotyping. Statistics in Medicine. 2019;38:74–87. 10.1002/sim.7953
REFERENCES
- 1. Duan R, Cao M, Wu Y, et al. An empirical study for impacts of measurement errors on EHR based association studies. Paper presented at: AMIA Annual Symposium Proceedings, Vol. 2016; 2016; Chicago, IL. [PMC free article] [PubMed] [Google Scholar]
- 2. Huang J, Duan R, Hubbard R, et al. PIE: a prior knowledge guided integrated likelihood estimation method for bias reduction in association studies using electronic health records data. J Am Med Inform Assoc. 2018;25(3):345‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weiskopf NG, Hripcsak G, Swaminathan S, Weng C. Defining and measuring completeness of electronic health records for secondary use. J Biomed Inform. 2013;46(5):830‐836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Weiskopf NG, Weng C. Methods and dimensions of electronic health record data quality assessment: enabling reuse for clinical research. J Am Med Inform Assoc. 2013;20(1):144‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Haneuse S, Daniels M. A general framework for considering selection bias in EHR‐based studies: what data are observed and why. eGEMs. 2016;4(1):1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goldstein BA, Bhavsar NA, Phelan M, Pencina MJ. Controlling for informed presence bias due to the number of health encounters in an electronic health record. Am J Epidemiol. 2016;184(11):847‐855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shivade C, Raghavan P, Fosler‐Lussier E, et al. A review of approaches to identifying patient phenotype cohorts using electronic health records. J Am Med Inform Assoc. 2014;21(2):221‐230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Buuren S. Multiple imputation of discrete and continuous data by fully conditional specification. Stat Methods Med Res. 2007;16(3):219‐242. [DOI] [PubMed] [Google Scholar]
- 9. Wells BJ, Chagin KM, Nowacki AS, Kattan MW. Strategies for handling missing data in electronic health record derived data. eGEMs. 2013;1(3):1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hripcsak G, Albers DJ. Next‐generation phenotyping of electronic health records. J Am Med Inform Assoc. 2012;20(1):117‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. He Y, Landrum MB, Zaslavsky AM. Combining information from two data sources with misreporting and incompleteness to assess hospice‐use among cancer patients: a multiple imputation approach. Statist Med. 2014;33(21):3710‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen B, Zhou XH. A latent‐variable marginal method for multi‐level incomplete binary data. Statist Med. 2012;31(26):3211‐3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Coley RY, Fisher AJ, Mamawala M, Carter HB, Pienta KJ, Zeger SL. A Bayesian hierarchical model for prediction of latent health states from multiple data sources with application to active surveillance of prostate cancer. Biometrics. 2016;73(2):625‐634. [DOI] [PubMed] [Google Scholar]
- 14. Dabelea D, Mayer‐Davis EJ, Saydah S, et al. Prevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. J Am Med Assoc. 2014;311(17):1778‐1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hanson RL, Nelson RG, McCance DR, et al. Comparison of screening tests for non‐insulin‐dependent diabetes mellitus. Arch Intern Med. 1993;153(18):2133‐2140. [PubMed] [Google Scholar]
- 16. Faraggi D, Reiser B. Estimation of the area under the ROC curve. Statist Med. 2002;21(20):3093‐3106. [DOI] [PubMed] [Google Scholar]
- 17. Gustafson P, Gelfand AE, Sahu SK, et al. On model expansion, model contraction, identifiability and prior information: two illustrative scenarios involving mismeasured variables [with comments and rejoinder]. Stat Sci. 2005:111‐140. [Google Scholar]
- 18. Jasra A, Holmes CC, Stephens DA. Markov chain Monte Carlo methods and the label switching problem in Bayesian mixture modeling. Stat Sci. 2005:50‐67. [Google Scholar]
- 19. Richesson RL, Rusincovitch SA, Wixted D, et al. A comparison of phenotype definitions for diabetes mellitus. J Am Med Inform Assoc. 2013;20(e2):e319‐e326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kho AN, Hayes MG, Rasmussen‐Torvik L, et al. Use of diverse electronic medical record systems to identify genetic risk for type 2 diabetes within a genome‐wide association study. J Am Med Inform Assoc. 2012;19(2):212‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hebert PL, Geiss LS, Tierney EF, Engelgau MM, Yawn BP, McBean AM. Identifying persons with diabetes using medicare claims data. Am J Med Qual. 1999;14(6):270‐277. [DOI] [PubMed] [Google Scholar]
- 22. Newton KM, LaCroix AZ, Heckbert SR, Abraham L, McCulloch D, Barlow W. Estrogen therapy and risk of cardiovascular events among women with type 2 diabetes. Diabetes Care. 2003;26(10):2810‐2816. [DOI] [PubMed] [Google Scholar]
- 23. Flory JH, Roy J, Gagne JJ, et al. Missing laboratory results data in electronic health databases: implications for monitoring diabetes risk. J Comp Eff Res. 2017;6(1):25‐32. [DOI] [PubMed] [Google Scholar]
- 24. Stephens M. Dealing with label switching in mixture models. J R Stat Soc Series B Stat Methodol. 2000;62(4):795‐809. [Google Scholar]
- 25. McCaffrey DF, Elliott MN. Power of tests for a dichotomous independent variable measured with error. Health Serv Res. 2008;43(3):1085‐1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sinnott JA, Dai W, Liao KP, et al. Improving the power of genetic association tests with imperfect phenotype derived from electronic medical records. Hum Genet. 2014;133(11):1369‐1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hubbard RA, Johnson E, Chubak J, et al. Accounting for misclassification in electronic health records‐derived exposures using generalized linear finite mixture models. Health Serv Outcomes Res Methodol. 2017;17(2):101‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Albert PS, Dodd LE. A cautionary note on the robustness of latent class models for estimating diagnostic error without a gold standard. Biometrics. 2004;60(2):427‐435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SIM_7953‐Supp‐0001‐pepper_aim1_online_supplement.pdf