

Abstract
β‐Glucocerebrosidase (GBA) is the enzyme that degrades glucosylceramide in lysosomes. Defects in GBA that result in overall loss of enzymatic activity give rise to the lysosomal storage disorder Gaucher disease, which is characterized by the accumulation of glucosylceramide in tissue macrophages. Gaucher disease is currently treated by infusion of mannose receptor‐targeted recombinant GBA. The recombinant GBA is thought to reach the lysosomes of macrophages, based on the impressive clinical response that is observed in Gaucher patients (type 1) receiving this enzyme replacement therapy. In this study, we used cyclophellitol‐derived activity‐based probes (ABPs) with a fluorescent reporter that irreversibly bind to the catalytic pocket of GBA, to visualize the active enzymes in a correlative microscopy approach. The uptake of pre‐labeled recombinant enzyme was monitored by fluorescence and electron microscopy in human fibroblasts that stably expressed the mannose receptor. The endogenous active enzyme was simultaneously visualized by in situ labeling with the ABP containing an orthogonal fluorophore. This method revealed the efficient delivery of recombinant GBA to lysosomal target compartments that contained endogenous active enzyme.
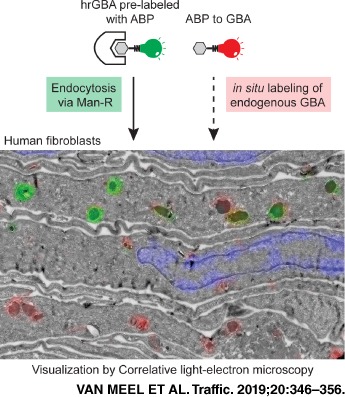
Keywords: activity‐based probes, correlative light and electron microscopy, LIMP II, lysosome, mannose receptor, β‐glucocerebrosidase
1. INTRODUCTION
The lysosomal acid β‐glucosidase, β‐glucocerebrosidase (GBA, EC 3.2.1.45), is essential to the catabolism of glycosphingolipids in lysosomes as it removes the β‐D‐glucopyranose group from glucosylceramide. The enzyme encoded by the GBA gene is synthesized as a 497 amino acid protein in the endoplasmic reticulum. While the majority of the lysosomal enzymes are modified with the mannose 6‐phosphate recognition marker to mediate their transport from the biosynthetic pathway to the lysosomes, GBA does not contain phosphomannosyl residues.1, 2, 3 Instead, GBA is routed via a mannose 6‐phosphate independent targeting pathway, by binding to the lysosomal membrane protein, LIMP II.4 LIMP II interacts with newly synthesized GBA at the site of the endoplasmic reticulum. After passage through the Golgi complex, the LIMP II‐GBA complex is directed to endosomes and lysosomes, likely through a dileucine‐based sorting motif in its C‐terminal cytosolic tail.5, 6 The importance of LIMP II is evident as individuals with LIMP II deficiency develop progressive myoclonic epilepsy with glomerulosclerosis and neurological manifestations, named action myoclonus‐renal failure syndrome (AMRF).7, 8 In AMRF, mutations in the SCARB2 gene encoding LIMP II cause the failure of normally synthesized GBA to interact with LIMP II. As a result, GBA is secreted from the cells, leading to reduced lysosomal levels of the enzyme in various cell types.4, 7, 9, 10
Mutations in GBA result in a prominent loss of GBA in lysosomes and cause the autosomal recessive lysosomal storage disorder Gaucher disease.11 Gaucher disease is characterized by the accumulation of the substrate glucosylceramide in tissue macrophages.12 The clinical manifestations of the disease are remarkably variable, but usually include enlargement of the liver and spleen, infiltration of the bone marrow by storage macrophages, thrombocytopenia, anemia and bone disease, and can include neurological symptoms. Currently approved treatments are substrate reduction therapy aiming to reduce substrate buildup in macrophages and enzyme replacement therapy (ERT), in which human recombinant GBA (hrGBA) is administered intravenously. To mediate uptake by macrophages, hrGBA is modified to expose its mannose moieties, which bind to the mannose receptor (Man‐R) present at the surface of these cells. Endocytosis of the Man‐R delivers hrGBA to the endo‐lysosomal system.
The development of activity‐based probes (ABPs) that covalently and irreversibly tag GBA with high sensitivity and almost complete selectivity has allowed the ultra‐sensitive visualization of active GBA molecules in vitro and in vivo in cells and organisms.13 These cyclophellitol‐derived ABPs react with the catalytic nucleophile Glu340 of GBA to form an enzyme‐substrate complex, linked through an ester bond that is stable under native physiological conditions. When applied to the cell culture medium, the ABPs rapidly enter the cells, a process that appears independent of endocytosis,13 and bind to the intracellular pools of GBA. The presence of a fluorescent reporter allows the detection by light microscopy.13 As fluorescence microscopy does not provide information about the underlying ultrastructure, in this study a correlative light and electron microscopy (CLEM) approach was employed to allow the more detailed localization of active GBA. Moreover, the cellular fate of hrGBA following binding to the Man‐R was investigated. The uptake of ABP‐prelabeled hrGBA was monitored while simultaneously visualizing the endogenous enzyme by using ABPs with different fluorescent reporters. Light microscopy showed colocalization of endocytosed and endogenous GBA, but also distinct punctae for either endocytosed or endogenous enzyme. By CLEM, the endogenous GBA was mainly localized to lysosomes, where it showed substantial overlap with endocytosed hrGBA. Of note, some lysosomes did not appear to be reached by hrGBA during the course of the experiment. In addition, the endocytosed enzyme was detected in earlier compartments of the endo‐lysosomal system that showed no detectable endogenous GBA. This method will be valuable in determining the efficiency of ERT for Gaucher disease and potentially other lysosomal storage diseases.
2. RESULTS
GBA was readily detected in normal human dermal fibroblasts (NHDFs) by fluorescence microscopy upon in situ labeling with the ABP, MDW941 (Figure 1), consistent with previous observations by our lab.13 Under these conditions, approximately 50% of total GBA was labeled, as determined by GBA activity assays. Pre‐incubation with conduritol B epoxide (CBE) for 16 hours prior to the in situ labeling with MDW941, which should block the active site of all available GBA molecules,14 resulted in no detectable signal (Figure S1 in Data S1), neither was any signal detected upon dimethyl sulfoxide (DMSO) or CBE incubation in the absence of probe (Figure S1in Data S1). These data indicate that the fluorescent signal does not represent unbound, endocytosed probe and is specific to active GBA. Importantly, in situ labeled GBA showed clear overlap with total GBA as detected by confocal immunofluorescence microscopy after staining with antibodies to GBA (Figure 1, upper panels, Figure S2A in Data S1). Significant overlap was also detected with the late endosomal/lysosomal marker LAMP‐1, although not all LAMP‐1 containing compartments appeared positive for GBA (Figure 1, lower panels, Figure S2A in Data S1). Loading of the late compartments of the endo‐lysosomal system with the endocytic tracer fluorescein isothiocyanate (FITC)‐dextran (3 hours pulse, 6 hours chase), resulted in substantial colocalization with MDW941, although not all MDW941 positive punctae contained dextran (Figure S3 in Data S1).
Figure 1.
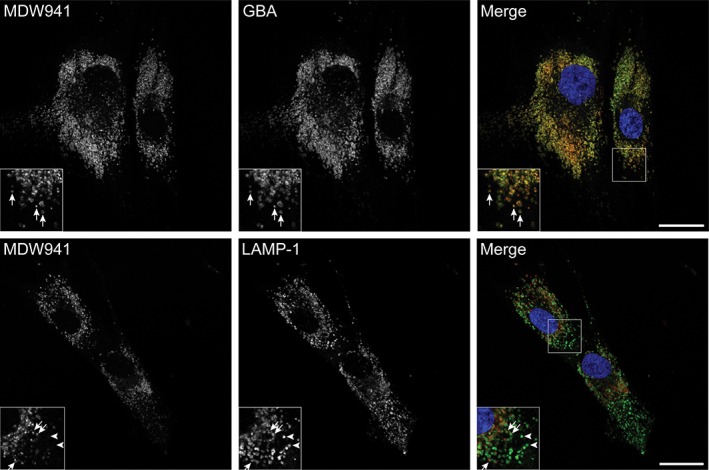
Active GBA accumulates in late endosomes and lysosomes. In situ labeling of active GBA in NHDFs with 5 nM MDW941 for 2 hours shows a punctate staining pattern (red) by confocal fluorescence microscopy. Significant overlap is observed with total GBA (green, see arrows in insets upper panels) and the late endosomal/lysosomal marker LAMP‐1 (green, see arrows in insets lower panels) upon immunostaining with specific antibodies. The arrowheads show LAMP‐1 positive compartments that contain no detectable levels of MDW941. Nuclei were stained with DAPI (blue). Scale bars, 25 μm
To identify the endo‐lysosomal compartments that accumulated active GBA, a correlative microscopy approach was applied. First, GBA was in situ labeled as in the previous experiments, after which the cells were fixed, gelatin embedded and prepared for electron microscopy according to the Tokuyasu technique.15 The ultrathin cryosections on grids were first mounted on glass slides with 50% glycerol to allow their imaging by confocal microscopy. This revealed the presence of numerous fluorescent punctae per cell profile (Figure 2A). Subsequently, the sections were contrasted and embedded with uranyl acetate/methylcellulose and imaged with an electron microscope. Overlay of the fluorescent images and electron micrographs showed that most of the fluorescent signal localized to membrane‐bound compartments (Figure 2B,C), while the CBE‐pre‐treated and DMSO‐treated sections showed no fluorescent signal. The membrane‐bound compartments mainly represented lysosomes, as identified by the presence of the characteristic internal membrane sheets and their electron dense appearance (Figure 2D,E). The earlier compartments of the endo‐lysosomal system, the endosomes, characterized by their more electron lucent appearance and intraluminal vesicles and the absence of internal membrane sheets, occasionally showed low levels of active GBA, but in most cases the signal was below the detection limit (Figure 2D,E). These data indicate that the majority of the active GBA molecules in fibroblasts reside in the lysosomes.
Figure 2.
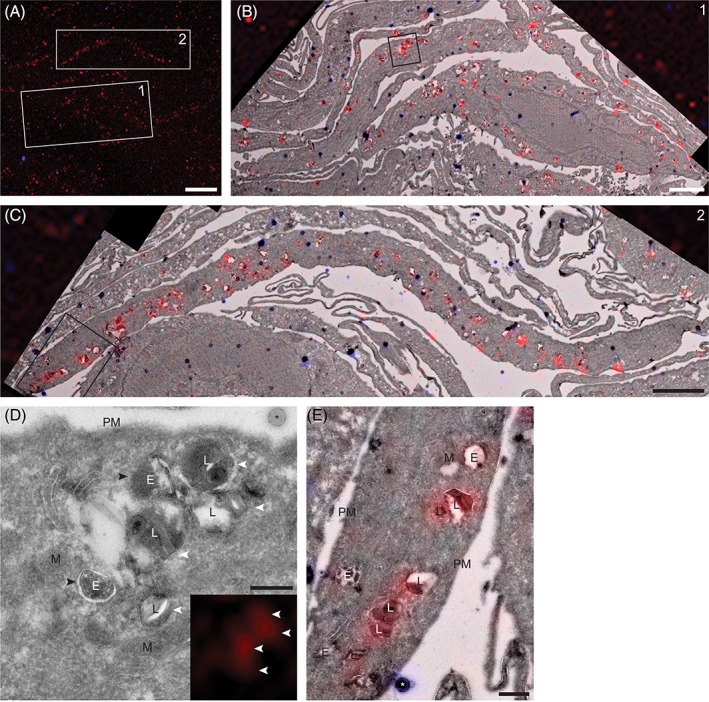
Correlative microscopy shows the accumulation of active GBA in lysosomes of NHDFs. A, Confocal fluorescence image of an ultrathin cryosection of NHDFs in situ labeled with MDW941 for 2 hours at 5 nM. The red punctae represent active GBA (MDW941), while the blue dots represent fluorescent electron dense beads used for correlation of the images. Scale bar, 10 μm. Merged fluorescence and electron microscopy images of boxed areas 1 and 2 are shown in (B) and (C), respectively. Scale bars, 3 μm. D, E, Magnifications of boxed areas in (B) and (C), respectively. MDW941 (red), which labels active GBA, is present in lysosomes. E, endosome; L, lysosome; M, mitochondrion; PM, plasma membrane; *, bead used for correlation. The white arrowheads indicate lysosomes positive for MDW941, recognized by the internal membrane sheets, while the black arrowheads indicate endosomes negative for the ABP. Scale bars, D, 250 nm; E, 500 nm
To allow the efficient internalization of hrGBA by human fibroblasts, cell lines were generated that stably express the mannose receptor (Man‐R) with a C‐terminal V5 tag. In this way, the uptake of hrGBA by the modified fibroblasts mimics the mode of uptake by tissue macrophages, which express Man‐R at their surface. Fibroblasts that were obtained from a patient homozygous for the nonsense mutation p.W178X7 contained no detectable levels of LIMP II by Western blotting (Figure 3A). As a result, the intracellular level of GBA was significantly reduced. In control fibroblasts, three bands were detected for GBA by Western blotting (Figure 3A), that is, the complex glycosylated form, the ER form and the late endosomal/lysosomal form.16 In the LIMP II−/− cells low levels of the ER form were detected (Figure 3A) and the intracellular GBA activity was reduced to approximately 5 ± 0.9% of the level in control fibroblasts (Figure 3B). These findings are consistent with the absence of the lysosomal targeting receptor LIMP II, which results in the increased secretion of GBA by these cells. Lentiviral transduction of both NHDFs and LIMP II−/− fibroblasts with MRC1 resulted in the expression of the Man‐R as determined by anti‐V5 immunoblotting (Figure 3A) and immunofluorescence microscopy (Figure S4 in Data S1), with the LIMP II−/− cells containing slightly higher expression levels of Man‐R than the NHDFs.
Figure 3.
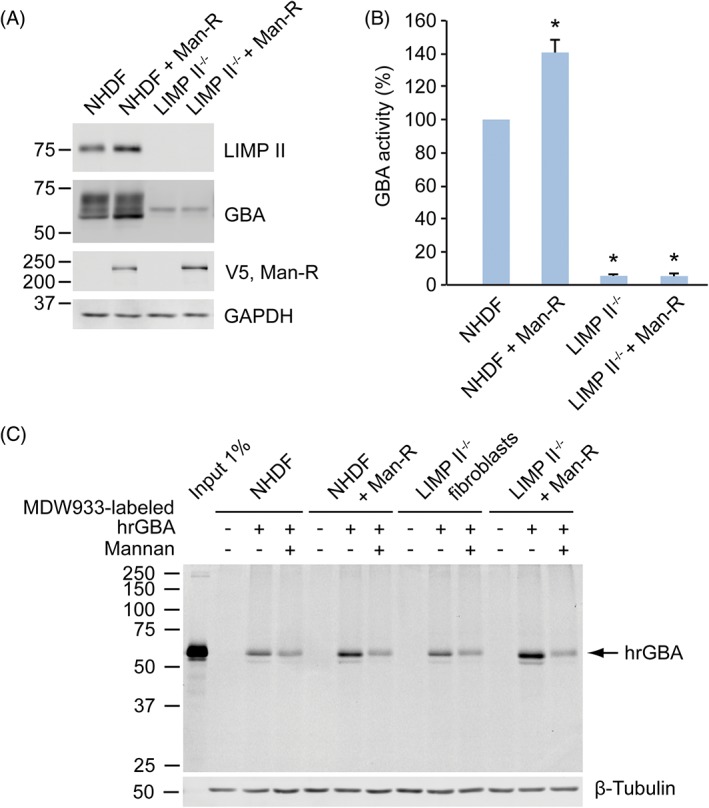
Man‐R expressing fibroblasts endocytose increased amounts of MDW933‐labeled hrGBA. A, NHDFs and LIMP II−/− fibroblast lysates with or without Man‐R with a V5‐tag were subjected to SDS‐PAGE and immunoblotting. No detectable levels of LIMP II are present in the LIMP II−/− cells, while the levels of GBA are significantly reduced (lane 3 and 4). The Man‐R is expressed in the control and LIMP II−/− fibroblast lines (lane 2 and 4), as detected with an antibody to the V5 tag. Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) shows equal protein loading. B, GBA activity in the various cell lines as determined with 4‐methylumbelliferyl‐coupled specific substrate. The values are averages of three independent experiments ± SDs, with the activity in NHDFs set to 100%. The activity in LIMP II−/− fibroblast lines is approximately 5% of NHDFs, while the Man‐R expressing NHDFs show a somewhat increased activity. * P < 0.01. C, Representative wet slab gel of at least three independent experiments shows the increased uptake of hrGBA by Man‐R expressing NHDFs and LIMP II−/− fibroblasts, as compared to the Man‐R negative fibroblasts. The addition of mannan reduces the hrGBA uptake to the same levels in all four cell lines, likely representing the uptake by non‐specific, fluid‐phase endocytosis. 1% of total MDW933‐hrGBA that was added to the culture medium (input) was loaded for comparison. β‐tubulin shows equal protein loading
Subsequently, hrGBA was in vitro labeled with MDW933 (an ABP functionalized with a green fluorescent BODIPY dye13) and, after the removal of unreacted ABP (Figure S5 in Data S1), added to the culture medium to follow its uptake in the various cell lines. A 6 hours‐incubation with MDW933‐hrGBA resulted in the enhanced internalization of hrGBA by fibroblasts that expressed the Man‐R, both NHDFs and LIMP II−/−, as compared to those that did not express the receptor (Figure 3C). Somewhat higher levels of hrGBA were observed in the LIMP II−/− cells as compared to the NHDFs, which is likely because of the higher levels of Man‐R present in the former, resulting in increased uptake. Competition with mannan, to block the Man‐R, reduced the uptake to the same level in all cell lines. This level likely reflects the uptake by fluid‐phase endocytosis. The effect of mannan that was observed in NHDFs and LIMP II−/− fibroblasts that did not overexpress the Man‐R might be caused by the block of another lectin responsible for hrGBA internalization, as these cells are unlikely to express the Man‐R endogenously.
To visualize the delivery of hrGBA to the intracellular target compartments and find out whether it reaches the lysosomes that contain active GBA, MDW933‐labeled hrGBA was applied to the culture medium of the four different cell lines, while endogenous GBA was labeled simultaneously with MDW941. Analysis by SDS‐PAGE showed labeling of the endogenous enzyme with the red probe in NHDFs, identified as two bands representing the complex glycosylated and lysosomal forms of GBA (Figure 4). In LIMP II−/− fibroblasts the GBA levels were in most instances too low to be detected by this method (Figure 4). Importantly, uptake of MDW933‐labeled hrGBA did not result in significant cross‐labeling of the endogenous enzyme because of unreacted MDW933, as only a single band was labeled with MDW933, representing hrGBA. In addition, the signal for MDW941 had a similar intensity in the lanes with or without MDW933‐hrGBA. Likewise, no cross‐labeling of internalized hrGBA with MDW941 appeared to occur.
Figure 4.
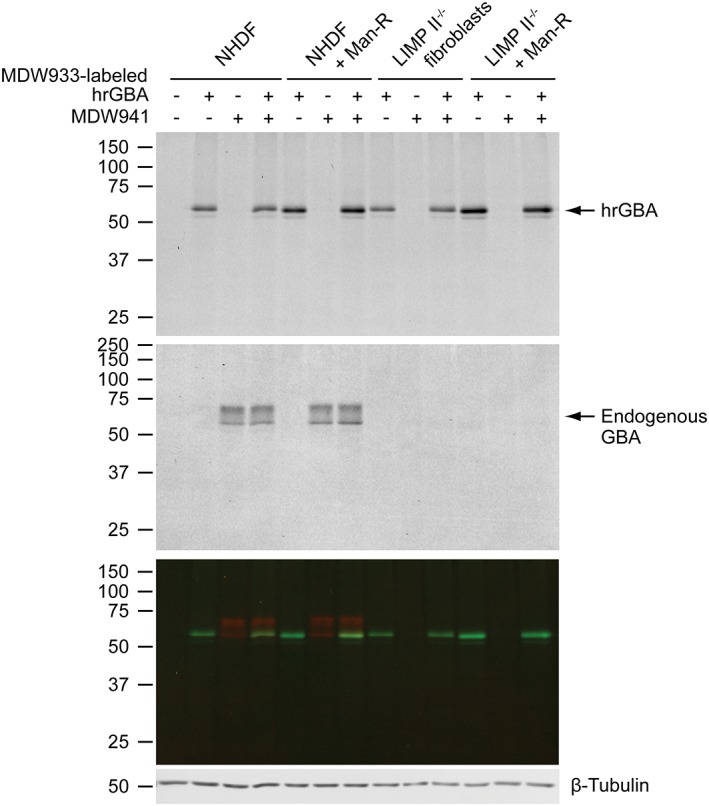
Simultaneous uptake of ABP‐labeled hrGBA and labeling of the active endogenous enzyme. The four different fibroblast cell lines were allowed to take up 2 μg/mL MDW933‐hrGBA for 6 hours, while endogenous GBA was in situ labeled during the final 2 hours with MDW941. A representative wet slab gel shows endocytosed hrGBA (upper panel, scanned green channel MDW933) and labeled endogenous GBA (middle panel, scanned red channel MDW941). The lower panel shows the overlay of the two channels. While endogenous GBA is readily detected in NHDFs, the signal is too low in LIMP II−/− fibroblasts. β‐tubulin shows equal protein loading
Subsequently, the same experimental set‐up was used in confocal fluorescence microscopy. The Man‐R expressing cells represented a heterogeneous population, as they expressed varying levels of receptor. While the signal for MDW933‐labeled hrGBA was below the detection level in all cells without Man‐R, a punctate staining pattern was observed in NHDFs that expressed sufficient levels of Man‐R (Figure 5, upper panels). These punctae significantly overlapped with endogenous GBA as visualized with MDW941, suggesting the delivery of hrGBA to lysosomes. However, some punctae were mainly positive for either hrGBA or endogenous GBA (see insets Figure 5, Figure S2B in Data S1). A similar result was observed in NHDFs that were immunostained for LAMP‐1 after the 6 hours‐uptake with hrGBA (Figure S6 in Data S1). There was partial overlap of hrGBA and LAMP‐1. Importantly, no signal for MDW933‐hrGBA could be visualized after uptake in the presence of mannan, which further suggests that no cross‐labeling of endogenous GBA occurred with MDW933. As expected, endogenous GBA failed to be detected in the LIMP II−/− fibroblasts (Figure 5, middle panels). However, uptake of MDW933‐labeled hrGBA resulted in a punctate pattern that showed partial overlap with LAMP‐1 (Figure 5, lower panels, see insets, Figure S2B in Data S1). These data suggest that hrGBA is delivered to late endosomes/lysosomes in the absence of LIMP II.
Figure 5.
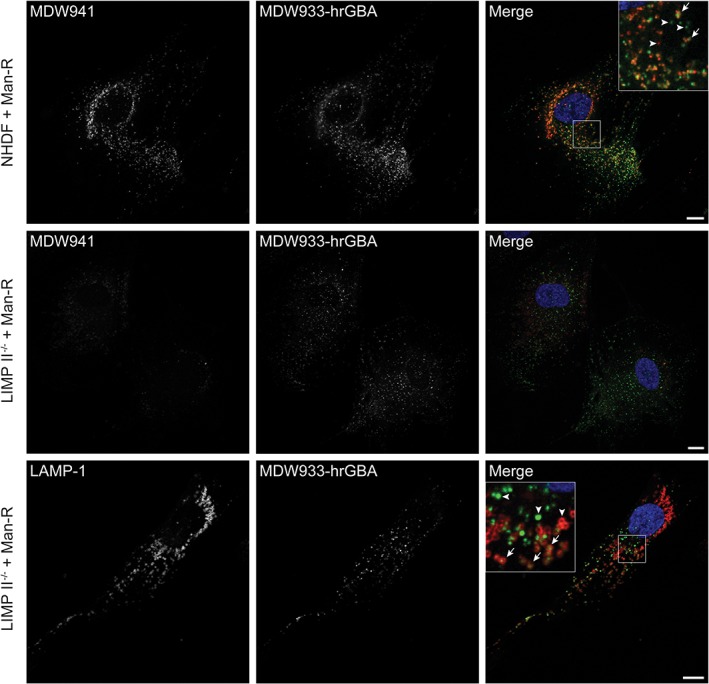
Endocytosed hrGBA partially colocalizes with active GBA and LAMP‐1. Confocal fluorescence microscopy of NHDFs and LIMP II−/− fibroblasts expressing the Man‐R after a 6 hours uptake of 2 μg/mL MDW933‐hrGBA (green) and 2 hours in situ labeling of endogenous GBA (red) with 5 nM MDW941 (upper and middle panels) or immunofluorescent staining of LAMP‐1 (red, lower panels; see also Figure S6 in Data S1 for LAMP‐1 staining in NHDFs). Substantial colocalization was observed in NHDFs between endocytosed hrGBA and endogenous GBA (see arrows in inset upper panels). In LIMP II−/− fibroblasts, the endocytosed enzyme reached a portion of the LAMP‐1 positive compartments (arrows in inset lower panels). The arrowheads indicate compartments positive for either endocytosed hrGBA or endogenous GBA/LAMP‐1. Scale bars, 10 μm
With CLEM the nature of the various fluorescent punctae could be identified. The uptake of labeled hrGBA was continuous for 6 hours, as shorter incubations did not result in sufficient levels of internalized hrGBA that were detectable by our CLEM method. We anticipated that the hrGBA‐containing punctae would represent both early and late compartments of the endo‐lysosomal system. Consistent with this prediction, hrGBA was present in endosomes, as well as in lysosomes (Figure 6A‐E). In addition, significant overlap was observed between endocytosed and endogenous GBA in lysosomes (Figure 6E). Interestingly, some lysosomes appeared to be negative for endocytosed hrGBA (Figure 6E). In agreement with the previous data, no endogenous GBA was detected by CLEM in the LIMP II−/− fibroblasts. However, endocytosed hrGBA was delivered to the endosomes and lysosomes (Figure 7), suggesting that the absence of LIMP II does not have a major effect on the lysosomal delivery of GBA upon endocytosis.
Figure 6.
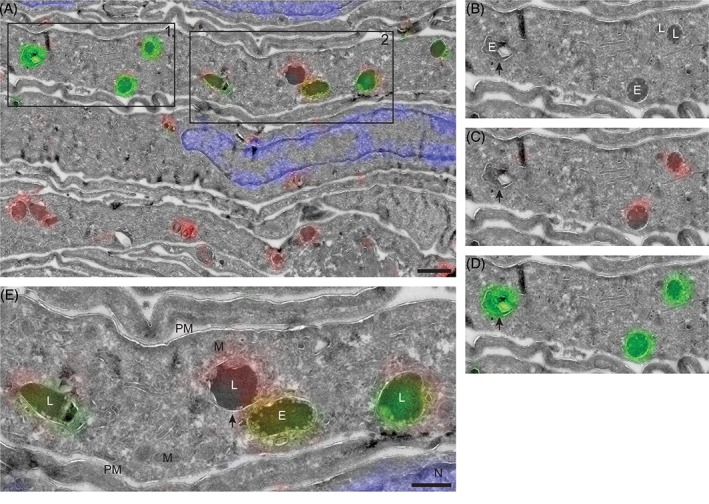
hrGBA is delivered to GBA‐containing lysosomes in NHDFs as determined by CLEM. A‐E, Overlay of electron micrograph and confocal fluorescence image of an ultrathin cryosection of NHDFs expressing the Man‐R. The cells were allowed to take up MDW933‐labeled hrGBA (green) for 6 hours, while endogenous GBA was labeled by adding MDW941 to the culture medium at a concentration of 5 nM for 2 hours (red). Nuclei were stained with DAPI (blue). Scale bar in A, 1 μm. B‐D, Magnification of boxed area 1 from A. E, endosome; L, lysosome. The arrow indicates an endosome that contains endocytosed hrGBA (green). E, Magnification of boxed area 2 from A showing lysosomes with both endocytosed hrGBA (green) and endogenous GBA (red). One lysosome (arrow) contains mainly the endogenous enzyme. M, mitochondrion; PM, plasma membrane. Scale bar, 500 nm
Figure 7.
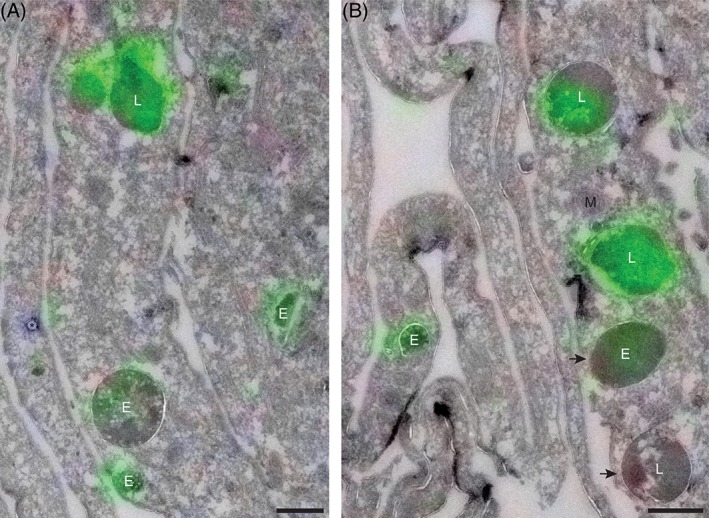
hrGBA is delivered to lysosomes in LIMP II−/− fibroblasts as determined by CLEM. A, B, Overlay of electron micrograph and confocal fluorescent image of an ultrathin cryosection showing LIMP II−/− fibroblasts + Man‐R, after a 6 hours‐uptake of MDW933‐labeled hrGBA (green) and in situ labeling of endogenous GBA (red, not detected). Nuclei were stained with DAPI (blue). Both endosomes (E) and lysosomes (L) are positive for hrGBA, however, not all of them are reached (arrows). M, mitochondrion; *, bead used for correlation. Scale bars, 500 nm
3. DISCUSSION
In this study we have presented a correlative microscopy method that combines the use of ABPs, for the detection of active lysosomal enzyme by fluorescence microscopy, with electron microscopy. This allows the visualization of the positive structures at high resolution. The cyclophellitol‐derived ABPs that irreversibly bind to the active site of the lysosomal β‐glucosidase GBA, readily penetrate through the cellular membranes, which provides the great benefit that they can be applied to living cells.13 In our approach, in situ labeling of human fibroblasts with ABP, equipped with a fluorescent tag, was followed by fixation and sample preparation according to the Tokuyasu technique.15, 17 The ultrathin cryosections were directly imaged for their fluorescent signal by confocal microscopy, without the need of on‐section labeling reactions that could increase background signals or influence the sensitivity of detection. The subsequent imaging by electron microscopy provides a relatively straightforward technique to localize the active enzyme to distinct subcellular compartments.
This correlative approach to localize GBA resulted in the detection of active enzyme mainly in lysosomes. By confocal fluorescence microscopy, the ABP colocalized well with our antibody to GBA and was blocked by pre‐incubation with the inhibitor CBE, showing the specificity of GBA labeling. In addition, the ABP colocalized significantly with the lysosomal membrane protein LAMP‐1, which is present in late endosomes and lysosomes.18, 19 By CLEM we were able to determine that the highest levels of active GBA were present in lysosomes, based on the morphological characteristics of these compartments, while endosomes mostly appeared negative. Thus the LAMP‐1‐containing, ABP‐negative compartments most likely represent late endosomes with GBA levels below the detection limit.
Subsequently, we aimed to follow the uptake of recombinant GBA and relate it to the localization of the endogenous enzyme using two‐color CLEM. To this end, hrGBA was in vitro labeled with ABP containing a green fluorophore, while the endogenous pool of GBA was in situ labeled with a red ABP. By confocal fluorescence microscopy we observed a significant overlap of green and red fluorescent signals, while there were also punctae positive for either fluorophore. Correlative microscopy showed that the overlapping green/red signals mainly localized to lysosomes, while the green dots represented endosomes containing hrGBA and the red dots lysosomes that had not been reached by endocytosed hrGBA. The observation that many, but not all lysosomes were reached by endocytosed hrGBA could be a technicality because of the imaging of sections instead of intact cells, or other factors such as the duration of uptake. Alternatively, an interesting possibility could be that there are different populations of lysosomes, some more and others less accessible by endocytosis.
Altogether, the imaging of GBA with ABPs in this correlative microscopy approach resulted in the localization of the enzyme to specific subcellular compartments, without the need for additional labeling.20, 21 This method could be applied to model organisms as well, as these ABPs can be delivered to various tissues upon intravenous or intracerebroventricular injection in rodents.13, 22 Moreover, correction of lysosomal enzyme deficiency by ERT is presently pursued for several other lysosomal storage disorders, such as Fabry disease and Pompe disease, which are caused by deficiency of lysosomal α‐galactosidase A (GLA) and lysosomal α‐glucosidase (GAA), respectively.23, 24, 25 As ABPs have already been designed for GLA and GAA,26, 27 this method might be applied to analyze the targeting of these expensive drugs. Finally, mutations in GBA have recently been recognized as a risk factor for multiple myeloma and carriers of one mutant GBA allele are more likely to develop Parkinsonism and Lewy body dementia.28, 29 Extending our knowledge of this enzyme could be instrumental to the understanding of these, and possibly other, pathologies.
4. MATERIALS AND METHODS
4.1. Cell lines
Normal human dermal fibroblasts (NHDFs) were obtained from Lonza and LIMP II−/− fibroblasts from a patient homozygous for the mutation c.533G>A, resulting in an early stop codon at W178.7 All cell lines were maintained in Dulbecco's Modified Eagle's Medium with 4.5 g/L glucose, sodium pyruvate and sodium bicarbonate (D6546, Sigma), supplemented with 10% (v/v) fetal bovine serum, 100 000 units/L penicillin, 100 mg/L streptomycin and 2 mM glutamax (Thermo Fisher Scientific Inc.) at 37°C in a 5% CO2 humidified incubator.
NHDFs or LIMP II−/− fibroblasts stably expressing the mannose receptor (Man‐R) were generated by lentiviral transduction. Human spleen mRNA was used to amplify the human MRC1 (MRC1, NM_002438.3) coding sequence by polymerase chain reaction using the following oligonucleotides: sense 5′‐GGG GAC AAG TTT GTA CAA AAA AGC AGG CTT CGG TAC CAC CAT GAG GCT ACC CCT GC ‐3′ and antisense 5′‐GGG GAC CAC TTT GTA CAA GAA AGC TGG GTC GAT GAC CGA GTG TTC ATT CTG ‐3′. The fragment was cloned into pDNOR‐221, and subsequently sub‐cloned into pLenti6.3/TO/V5‐DEST using the Gateway system (Invitrogen), generating a C‐terminal V5 fusion protein. Because of toxicity of the construct in E. coli CopyCutter EPI400 chemically competent E. coli cells were used according to the manufacturer's protocol (Epicentre). The full sequence was verified by DNA sequencing.
To produce lentiviral particles, HEK293T cells in 6‐well plates were transfected at approximately 70% confluency with the envelope and packaging plasmids pMD2.G, pRSV, pMDL‐RRE and pLENTI6.3‐MRC1 at a 1:1 ratio, 3 μg DNA in total, and 9 μg polyethylenimine (PEI 25 K, Polysciences Inc.). The culture medium was replaced 16 hours after transfection with culture medium containing 20 mM HEPES and collected twice after a 24 hours incubation period. The culture medium containing viral particles was passed over a 0.45 μm filter and applied to the control and LIMP II−/− fibroblast lines for 8‐24 hours. Transduced cells were selected for by incubation with 2.5 μg/mL blasticidin (Sigma) containing culture medium for several weeks.
4.2. In situ labeling of GBA and fluorescence microscopy
Cells were grown to approximately 70% confluency on glass coverslips in 12‐well plates. GBA was in situ labeled by adding MDW941 (“red,” BODIPY 573/612) to the culture medium at a concentration of 5 nM. The cells were incubated for 2 hours in a humidified incubator at 37°C with 5% CO2. Pre‐incubation with CBE (Enzo Life Sciences Inc.) was performed at a concentration of 0.3 mM for 16 hours. Unlabeled cells were incubated with similar percentages (v/v) of DMSO. Uptake with FITC‐CM‐dextran (MW 70 000, Sigma) was performed at a concentration of 1 mg/mL for 3 hours (pulse). Subsequently, the cells were washed five times with PBS and incubated in culture medium for 6 hours (chase). In situ labeling with 5 nM MDW941 was performed during the last 2 hours of the chase.
At the end of the incubations, the cells were washed three times with PBS and fixed with 4% (w/v) formaldehyde (Sigma)/PBS for 30 minutes at room temperature. After the fixation, the cells were washed with PBS and distilled H2O, after which they were either stained for immunofluorescence microscopy or mounted directly on a microscope slide with ProLong Diamond antifade reagent containing DAPI (Molecular Probes). Immunofluorescence staining was performed as described.30 Antibodies used were mouse anti‐GBA monoclonal (8E4, generated in the Aerts lab) at dilution 1:500, rabbit anti‐LAMP‐1 (Abcam) at dilution 1:400, mouse anti‐V5 (Invitrogen) at dilution 1:250 and Alexa Fluor conjugated IgGs (H + L) donkey anti‐mouse Alexa 488 and donkey anti‐rabbit Alexa 488 (Invitrogen). The cells were imaged under a Leica TCS SP8 confocal microscope with a 63×/1.40 numerical aperture (NA) HC Plan Apo CS2 oil immersion objective and equipped with a hybrid detector (HyD).
4.3. In vitro labeling of hrGBA with MDW933
hrGBA (Cerezyme) was a kind gift from Genzyme. Five microliter hrGBA at a concentration of 1 μg/μL was incubated for 16 hours at 37°C with 40 μL of 150 mM McIlvaine buffer pH = 5.2, 0.2% (w/v) sodium taurocholate (Calbiochem), 0.1% (v/v) Triton X‐100 and 2 μM MDW933 (“green,” BODIPY 493/503), while protected from the light. After the incubation, an aliquot was kept to confirm that the residual hrGBA activity was reduced >99% (see below). The remainder was passed over a protein desalting spin column, MWCO 7 K (Thermo Scientific) according to the manufacturer's protocol, to remove any unreacted probe (see also Figure S3 in Data S1).
4.4. GBA activity assay
The enzymatic activity of hrGBA preparations or GBA in cell lysates was measured by incubating 10‐20 μL sample with 100 μL substrate mix, which consisted of 3.75 mM 4‐methylumbelliferyl β‐D‐glucopyranoside (Glycosynth Limited), 0.2% (w/v) sodium taurocholate (Calbiochem), 0.1% (w/v) BSA, 0.1% (v/v) Triton X‐100 in 150 mM McIlvaine buffer pH = 5.2, in a 96‐well plate at 37°C. The reactions were stopped with 200 μL 1 M glycine‐NaOH pH = 10.3 and read in an LS55 fluorescence spectrometer (PerkinElmer) at λex 366 nm, λem 445 nm.
4.5. Uptake of MDW933‐labeled hrGBA and in situ labeling of endogenous GBA
All uptake experiments were performed with 2 μg/mL MDW933‐labeled hrGBA for 6 hours. When indicated, MDW941 was added during the last 2 hours of the incubation at a concentration of 5 nM. To block uptake of hrGBA by the Man‐R, mannan from Saccharomyces cerevisiae (Sigma) was added simultaneously at 3 mg/mL. At the end of the incubations, the cells were washed three times with PBS and either lysed (for the GBA activity assay/SDS‐PAGE) or fixed for microscopy.
4.6. SDS‐PAGE and Western blotting
Cells were lysed by the addition of ice‐cold lysis buffer, that is, 25 mM potassium phosphate buffer pH = 6.5, 0.1% Triton X‐100 and a protease inhibitor cocktail (cOmplete, Roche Diagnostics GmbH). Total protein concentration in the lysates was determined by the bicinchoninic acid (BCA) protein assay (Pierce BCA protein assay kit; Thermo Fisher Scientific Inc.) in a 96‐well plate format, according to the manufacturer's protocol. The samples were read in an EMax Plus microplate reader (Molecular Devices). Equal protein amounts (5 μg) were resolved on 10% polyacrylamide gels and transferred onto 0.2 μm nitrocellulose membranes (Amersham, GE Healthcare). Antibodies used were rabbit anti‐LIMP II (Novus Biologicals), mouse anti‐GBA (8E4, generated in the Aerts lab), rabbit anti‐GAPDH (Cell Signaling), mouse anti‐V5 (Invitrogen) and rabbit anti‐β‐tubulin (Cell Signaling) and secondary antibodies used were donkey anti‐rabbit Alexa Fluor 647 IgG (H + L) (Invitrogen), goat anti‐mouse IgG (H + L)‐HRP conjugate and goat anti‐rabbit IgG (H + L)‐HRP conjugate (BioRad). Horseradish peroxidase (HRP)‐conjugated antibodies were visualized by chemiluminescence (Pierce ECL Plus Western blotting substrate, Thermo Fisher Scientific Inc.). The wet slab gels and immunoblots were scanned on a Typhoon FLA 9500 scanner (GE Healthcare).
4.7. Correlative microscopy
NHDFs were grown in 100 mm culture dishes to approximately 70% confluency. GBA was in situ labeled with 5 nM MDW941 for 2 hours at 37°C, either with or without pre‐incubation with 0.3 mM CBE for 16 hours as a control for the specificity of the signal. To check for autofluorescence, one dish was incubated for 2 hours with a similar percentage (v/v) of DMSO. The uptake experiments were performed with NHDFs + Man‐R and LIMP II−/− fibroblasts + Man‐R, with 2 μg/mL MDW933‐labeled hrGBA for 6 hours. During the last 2 hours endogenous GBA was in situ labeled with 5 nM MDW941. As a control for the Man‐R‐specific uptake of MDW933‐hrGBA, the uptake in NHDFs + Man‐R, followed by the in situ labeling was performed in the presence of 3 mg/mL mannan.
At the end of the incubations, the culture medium was removed and the cells were washed three times quickly with culture medium at 37°C. Subsequently, the cells were fixed with 2% (w/v) formaldehyde +0.2% glutaraldehyde (Electron Microscopy Sciences) in 0.1 M PHEM buffer for 2 hours at room temperature, while protected from the light. Embedding of the samples and cryosectioning was performed as described.15 For correlation purposes, fluospheres carboxylate‐modified microspheres blue (350/440) with a diameter of 100 nm (Thermo Fisher Scientific Inc.) were added to the grids, which were coated with a formvar film, either before or after collection of the sections by incubating the grids for 2 minutes on the beads (1:2000 dilution of 2% solids). Subsequently, the sections were incubated on 2% gelatin for 30 minutes at 37°C and on 15 mM glycine/PBS for 10 minutes at room temperature. The nuclei were stained with 50 ng/mL DAPI (Sigma)/PBS for 5 minutes. After a quick wash with distilled H2O, the ultrathin cryosections on titanium grids (Agar Scientific) were mounted on microscope glass slides in 50% glycerol/H2O and imaged under a Leica TCS SP8 confocal microscope with a 63×/1.40 NA HC Plan Apo CS2 oil immersion objective. Immediately after imaging, the glycerol was removed from the grids by washing in distilled H2O. The sections were contrasted with 1.8% methylcellulose/0.6% uranyl acetate/H2O at pH = 4.8 and analyzed in a FEI Tecnai 12 BioTwin electron microscope at 120 kV. Correlation of the images was performed using Adobe Photoshop CC 2015.5.
4.8. Labeling specificity in correlative microscopy
The specificity of the green and red fluorescent signals for hrGBA and endogenous GBA, respectively, was determined in the following way. First of all, the in vitro labeling reaction was optimized to label over 99% of hrGBA, which was confirmed by an enzymatic activity assay to result in near absence of hrGBA activity. In this way, cross‐labeling of hrGBA with red ABP was prevented during the in situ labeling. Consistent herewith, no red ABP signal was detected in fibroblasts that lack GBA, because of a mutation in the receptor LIMP II, upon endocytosis of labeled hrGBA. Secondly, any unreacted green ABP was removed from the preparation before addition to the culture medium. SDS‐PAGE analysis showed substantial reduction of unreacted ABP. Any remaining unreacted probe was not sufficient to result in detectable cross‐labeling of endogenous GBA as determined by SDS‐PAGE of the cell lysates. Finally, the uptake of ABP‐labeled hrGBA in the presence of mannan did not result in a detectable green fluorescent signal in the cells by microscopy, indicating that the endogenous enzyme was not labeled with green probe.
AUTHOR CONTRIBUTIONS
E.v.M., H.O., S.v.K., A.K. and J.A. conceptualized and designed the project. E.v.M., E.B. and M.v.d.L. performed the experiments and analyzed the data. E.v.M. prepared the manuscript. E.v.M., E.B., M.v.d.L., H.O., S.v.K., A.K. and J.A. finalized the manuscript. All authors read and approved the final manuscript.
Supporting information
Data S1.
ACKNOWLEDGMENTS
We thank our colleagues for helpful discussions and Dr. Rolf Boot for providing the MRC1 construct. This work was supported by ERC advanced grant 290836 CHEMBIOSPHING awarded to Herman Overkleeft and Johannes Aerts.
van Meel E, Bos E, van der Lienden MJC, et al. Localization of active endogenous and exogenous β‐glucocerebrosidase by correlative light‐electron microscopy in human fibroblasts. Traffic. 2019;20:346–356. 10.1111/tra.12641
Funding information European Research Council, Grant/Award Number: ERC advanced grant / 290836 CHEMBIOSPHING
Peer Review The peer review history for this article is available at https://publons.com/publon/10.1111/tra.12641
REFERENCES
- 1. Aerts JM, Schram AW, Strijland A, et al. Glucocerebrosidase, a lysosomal enzyme that does not undergo oligosaccharide phosphorylation. Biochim Biophys Acta. 1988;964:303‐308. [DOI] [PubMed] [Google Scholar]
- 2. Rijnboutt S, Aerts HM, Geuze HJ, Tager JM, Strous GJ. Mannose 6‐phosphate‐independent membrane association of cathepsin D, glucocerebrosidase, and sphingolipid‐activating protein in HepG2 cells. J Biol Chem. 1991;266:4862‐4868. [PubMed] [Google Scholar]
- 3. Rijnboutt S, Kal AJ, Geuze HJ, Aerts H, Strous GJ. Mannose 6‐phosphate‐independent targeting of cathepsin D to lysosomes in HepG2 cells. J Biol Chem. 1991;266:23586‐23592. [PubMed] [Google Scholar]
- 4. Reczek D, Schwake M, Schroder J, et al. LIMP‐2 is a receptor for lysosomal mannose‐6‐phosphate‐independent targeting of beta‐glucocerebrosidase. Cell. 2007;131:770‐783. [DOI] [PubMed] [Google Scholar]
- 5. Ogata S, Fukuda M. Lysosomal targeting of Limp II membrane glycoprotein requires a novel Leu‐Ile motif at a particular position in its cytoplasmic tail. J Biol Chem. 1994;269:5210‐5217. [PubMed] [Google Scholar]
- 6. Sandoval IV, Arredondo JJ, Alcalde J, et al. The residues Leu(Ile)475‐Ile(Leu, Val, Ala)476, contained in the extended carboxyl cytoplasmic tail, are critical for targeting of the resident lysosomal membrane protein LIMP II to lysosomes. J Biol Chem. 1994;269:6622‐6631. [PubMed] [Google Scholar]
- 7. Balreira A, Gaspar P, Caiola D, et al. A nonsense mutation in the LIMP‐2 gene associated with progressive myoclonic epilepsy and nephrotic syndrome. Hum Mol Genet. 2008;17:2238‐2243. [DOI] [PubMed] [Google Scholar]
- 8. Berkovic SF, Dibbens LM, Oshlack A, et al. Array‐based gene discovery with three unrelated subjects shows SCARB2/LIMP‐2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am J Hum Genet. 2008;82:673‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gaspar P, Kallemeijn WW, Strijland A, et al. Action myoclonus‐renal failure syndrome: diagnostic applications of activity‐based probes and lipid analysis. J Lipid Res. 2014;55:138‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Malini E, Zampieri S, Deganuto M, et al. Role of LIMP‐2 in the intracellular trafficking of beta‐glucosidase in different human cellular models. FASEB J. 2015;29:3839‐3852. [DOI] [PubMed] [Google Scholar]
- 11. Brady RO, Kanfer JN, Bradley RM, Shapiro D. Demonstration of a deficiency of glucocerebroside‐cleaving enzyme in Gaucher's disease. J Clin Invest. 1966;45:1112‐1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grabowski GA, Petsko GA, Kolodny EH. Gaucher disease In: Valle D, Beaudet AL, Vogelstein B, et al., eds. The Online Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw‐Hill; 2014. [Google Scholar]
- 13. Witte MD, Kallemeijn WW, Aten J, et al. Ultrasensitive in situ visualization of active glucocerebrosidase molecules. Nat Chem Biol. 2010;6:907‐913. [DOI] [PubMed] [Google Scholar]
- 14. Daniels LB, Glew RH, Radin NS, Vunnam RR. A revised fluorometric assay for Gaucher's disease using conduritol‐beta‐epoxide with liver as the source of beta‐glucosidase. Clin Chim Acta. 1980;106:155‐163. [DOI] [PubMed] [Google Scholar]
- 15. Slot JW, Geuze HJ. Cryosectioning and immunolabeling. Nat Protoc. 2007;2:2480‐2491. [DOI] [PubMed] [Google Scholar]
- 16. Van Weely S, Aerts JM, Van Leeuwen MB, et al. Function of oligosaccharide modification in glucocerebrosidase, a membrane‐associated lysosomal hydrolase. Eur J Biochem. 1990;191:669‐677. [DOI] [PubMed] [Google Scholar]
- 17. Tokuyasu KT. A technique for ultracryotomy of cell suspensions and tissues. J Cell Biol. 1973;57:551‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barriocanal JG, Bonifacino JS, Yuan L, Sandoval IV. Biosynthesis, glycosylation, movement through the Golgi system, and transport to lysosomes by an N‐linked carbohydrate‐independent mechanism of three lysosomal integral membrane proteins. J Biol Chem. 1986;261:16755‐16763. [PubMed] [Google Scholar]
- 19. Marsh M, Schmid S, Kern H, et al. Rapid analytical and preparative isolation of functional endosomes by free flow electrophoresis. J Cell Biol. 1987;104:875‐886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ngo JT, Adams SR, Deerinck TJ, et al. Click‐EM for imaging metabolically tagged nonprotein biomolecules. Nat Chem Biol. 2016;12:459‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Elsland DM, Bos E, de Boer W, Overkleeft HS, Koster AJ, van Kasteren SI. Detection of bioorthogonal groups by correlative light and electron microscopy allows imaging of degraded bacteria in phagocytes. Chem Sci. 2016;7:752‐758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Herrera Moro Chao D, Kallemeijn WW, Marques AR, et al. Visualization of active glucocerebrosidase in rodent brain with high spatial resolution following in situ labeling with fluorescent activity based probes. PLoS One. 2015;10:e0138107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha‐galactosidase A replacement therapy in Fabry's disease. N Engl J Med. 2001;345:9‐16. [DOI] [PubMed] [Google Scholar]
- 24. Schiffmann R, Kopp JB, Austin HA 3rd, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285:2743‐2749. [DOI] [PubMed] [Google Scholar]
- 25. Van den Hout JM, Reuser AJ, de Klerk JB, Arts WF, Smeitink JA, Van der Ploeg AT. Enzyme therapy for pompe disease with recombinant human alpha‐glucosidase from rabbit milk. J Inherit Metab Dis. 2001;24:266‐274. [DOI] [PubMed] [Google Scholar]
- 26. Jiang J, Kuo CL, Wu L, et al. Detection of active mammalian GH31 alpha‐glucosidases in health and disease using in‐class, broad‐spectrum activity‐based probes. ACS Cent Sci. 2016;2:351‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Willems LI, Beenakker TJ, Murray B, et al. Potent and selective activity‐based probes for GH27 human retaining alpha‐galactosidases. J Am Chem Soc. 2014;136:11622‐11625. [DOI] [PubMed] [Google Scholar]
- 28. Aflaki E, Westbroek W, Sidransky E. The complicated relationship between Gaucher disease and parkinsonism: insights from a rare disease. Neuron. 2017;93:737‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nair S, Branagan AR, Liu J, Boddupalli CS, Mistry PK, Dhodapkar MV. Clonal immunoglobulin against Lysolipids in the origin of myeloma. N Engl J Med. 2016;374:555‐561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Qian Y, Flanagan‐Steet H, van Meel E, Steet R, Kornfeld SA. The DMAP interaction domain of UDP‐GlcNAc:lysosomal enzyme N‐acetylglucosamine‐1‐phosphotransferase is a substrate recognition module. Proc Natl Acad Sci U S A. 2013;110:10246‐10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.