

Abstract
We report a simple protocol for the photochemical Giese addition of C(sp3)‐centered radicals to a variety of electron‐poor olefins. The chemistry does not require external photoredox catalysts. Instead, it harnesses the excited‐state reactivity of 4‐alkyl‐1,4‐dihydropyridines (4‐alkyl‐DHPs) to generate alkyl radicals. Crucial for reactivity is the use of a catalytic amount of Ni(bpy)3 2+ (bpy=2,2′‐bipyridyl), which acts as an electron mediator to facilitate the redox processes involving fleeting and highly reactive intermediates.
Keywords: dihydropyridines, electron mediator, nickel catalysis, photochemistry, synthetic methods
The venerable field of photochemistry is re‐gaining a central role in synthetic endeavors.1 For example, the fast‐moving area of photoredox catalysis is providing modern chemists with the possibility of generating radical species under mild conditions and from bench‐stable reagents.2 A more classical photochemical approach exploits the ability of substrates or reaction intermediates to harvest the energy of the photons and access an electronically excited state (direct photochemistry). The chemical reactivity of excited molecules3 can then be used to unlock reaction manifolds unavailable to conventional ground‐state pathways. In this context, our laboratory recently reported4 how selective excitation with violet light turns 4‐alkyl‐1,4‐dihydropyridines (alkyl‐DHPs, 1) into strong reducing agents, which can activate reagents by single‐electron transfer (SET) manifolds while undergoing a homolytic cleavage to generate C(sp3)‐centered radicals (Figure 1 a).5 This dual photochemical reactivity was used to trigger radical‐based C−C bond‐forming processes.
Figure 1.
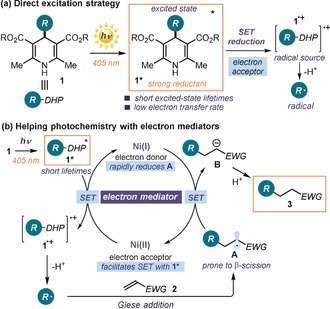
(a) The excited‐state reactivity of 4‐alkyl‐1,4‐dihydropyridines (R‐DHPs, 1): on excitation, they become both photoreductants and precursors of alkyl radicals. (b) Proposed strategy to realize the Giese reaction by combining the photochemistry of 1 and the action of a catalytic nickel complex, which facilitates the redox processes by acting as an electron mediator (EM); NiII rapidly oxidizes the short‐lived, excited R‐DHPs 1* while the ensuing NiI species reduces the highly reactive intermediate A, which has a high tendency to undergo side reactions. The overall sequence affords product 3.
In general, the direct excitation of substrates or intermediates can create new synthetic opportunities for radical chemistry3, 6 while bypassing the need for photoredox catalysts, which are often based on precious transition‐metals. However, there are some challenges intrinsic to this photochemical strategy. The excitation of organic molecules provides strong oxidants and/or reductants. Thermodynamically speaking, this makes it easy to activate reagents by an SET manifold.7 However, these highly reactive excited‐state intermediates may suffer from short lifetimes and low electron‐transfer rates, thus rendering the SET interactions with other reagents kinetically challenging.8
We recently wondered if electron mediators (EMs) could be used to alleviate these limitations, thus expanding the synthetic applicability of the direct photoexcitation approach. Electron mediators have found widespread use in different areas of chemistry, including dye‐sensitized solar cells,9 CO2 reduction,10 and electrochemical synthesis.11 They have also been sporadically applied in photoredox catalysis.12 This is because, by acting as electron shuttles, they can facilitate exergonic redox processes that are kinetically slow. They can also mitigate the occurrence of a back‐electron transfer (BET) and competitive side‐reactivity.
However, to date, there has been little exploration of the potential of EMs to accelerate a thermodynamically feasible but kinetically unfavorable SET event involving the direct excitation of organic intermediates. Herein, we demonstrate the feasibility of this strategy. By combining the excited‐state reactivity of alkyl‐DHPs 1 with the ability of a nickel complex to act as an electron mediator, we have developed a Giese addition process13 without the need for an external photoredox catalyst.14 Figure 1 b details the idea behind our synthetic plan: photoexcitation turns 1 into a strong reductant (1*). The EM facilitates the SET oxidation of this short‐lived electronically excited intermediate 1* to afford the radical cation 1.+, which fragments into a carbon‐centered radical (R.). Interception by the electron‐poor olefin 2 leads to intermediate A, which is a highly reactive radical prone to side‐reactions, including β‐scission and polymerization.15 The reduced form of the EM rapidly engages in a second SET, reducing intermediate A to B while bringing the EM back to the original oxidation state. The SET reduction of A is critical for an effective Giese reaction leading to product 3. Overall, the EM provides a catalytic manifold to secure an efficient SET between 1* and intermediate A. The EM acts as a reservoir for the electron donated by 1*, prolonging its availability for the reduction of A. These redox processes involve two short‐lived intermediates, so they would be problematic without EM.
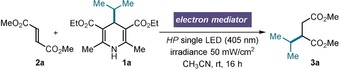
To validate our proposal, we studied the Giese‐type radical conjugate addition to dimethyl fumarate (2 a) using 4‐isopropyl‐DHP 1 a as the radical precursor (Table 1). We conducted our experiments in CH3CN under irradiation by a single high‐power light‐emitting diode (LED, λ max=405 nm, see the Supporting Information for details).
Table 1.
Optimization and control experiments.[a]
| Entry | Conditions | Yield [%][b] |
|---|---|---|
| 1 | No mediator | 32 |
| 2 | p‐Dicyanobenzene (0.2 equiv) | 57 |
| 3 | Methyl viologen dichloride (0.2 equiv) | 41 |
| 4 | Ni(bpy)3(BF4)2 (0.1 equiv) | 85 |
| 5 | Same as entry 4, no light, 50 °C | <5 |
| 6 | Same as entry 4, TEMPO (1 equiv) | 26[c] |
[a] Reactions performed at ambient temperature on a 0.1 mmol scale using 1.5 equiv of 1 a under illumination by a single high‐power (HP) LED (λ max=405 nm) with an irradiance of 50 mW cm−2. [b] Yield determined by 1H NMR analysis of the crude mixture using trichloroethylene as internal standard. [c] The TEMPO‐trapped intermediate (TEMPO‐iPr) was obtained in 53 % NMR yield.
Performing the model reaction in the absence of an electron mediator afforded product 3 a in low yield, while a large amount of reagents 1 a and 2 a remained unreacted (entry 1). We ascribed this low reactivity to the inefficiency of the redox events underlying the overall process.16 We then tested our hypothesis that, by behaving alternately as a donor and an acceptor, an EM could effectively shuttle electrons between the key fleeting intermediates (1 a* and the radical of type A, Figure 1 b). An effective EM must meet several requirements: a) it should easily oxidize 1*; b) its reduced form should selectively reduce intermediate A, but not substrate 2 or the alkyl radical resulting from the fragmentation of 1*; and c) it should not strongly absorb at 405 nm to avoid inner‐filter effects.
In consonance with our design, the presence of organic EMs (20 mol %) substantially improved the efficiency of the Giese process. p‐Dicyanobenzene (E p/2 (EM/EM.−)=−1.64 V vs. SCE)17a increased the yield of 3 a to 57 % (entry 2), while another common redox mediator, methyl viologen dichloride (E 1/2 (EM2+/EM.+)=−0.41 V vs. SCE),17b had only a negligible effect (entry 3). We then turned our attention to transition metal complexes. Based on the aforementioned criteria, Ni(bpy)3(BF4)2 was identified as a promising EM.18 This readily available and bench‐stable NiII complex, which is soluble in acetonitrile, is reduced at E p(NiII/NiI)=−1.35 V versus SCE,19 and absorbs weakly at 405 nm. Gratifyingly, 10 mol % of Ni(bpy)3(BF4)2 increased the yield of product 3 a significantly (up to 85 %, entry 4), and secured full conversion of 1 a and 2 a. Further control experiments showed that the reaction requires light and does not proceed thermally (entry 5). The yield of 3 a was greatly diminished in the presence of 2,2,6,6‐tetramethyl‐1‐piperidinyloxyl (TEMPO, 1 equiv), and we detected the adduct generated upon isopropyl radical trap (53 % yield, entry 6).
Having identified Ni(bpy)3(BF4)2 as a suitable EM, we evaluated the synthetic potential of the photochemical Giese reaction (Figure 2). We first demonstrated that a high efficiency was maintained when running the reaction on a 2 mmol scale. This experiment required the use of commercially available LEDs (see section B5 in the Supporting Information) and it afforded product 3 a in 78 % yield (294 mg). Concerning the reaction scope, a variety of α,β‐unsaturated nitriles and esters can be successfully functionalized using 4‐isopropyl‐DHP 1 a as the radical precursor (products 3 a–3 f). We then explored the possibility of using substrates 1 bearing alkyl fragments other than isopropyl at the C4‐position. Secondary alkyl (3 g–h), benzyl (3 i,j), and α‐heterosubstituted primary radicals are tolerated well in this transformation. Noteworthy examples include the addition of α‐oxygen (3 k–n) and α‐nitrogen radicals (3 o–t), which bring about the facile introduction of synthetically interesting motifs such as benzotriazoles (3 s,t), phthalimides (3 o,p), and dioxolanes (3 k,l). Appealing features of this protocol include the need for a slight excess (1.5 equiv) of the radical precursor 1. In contrast to other Giese addition protocols, there is no need for stoichiometric amounts of additional reagents, such as bases, reductants or hydrogen‐atom donors, or for photoredox catalysts.14
Figure 2.
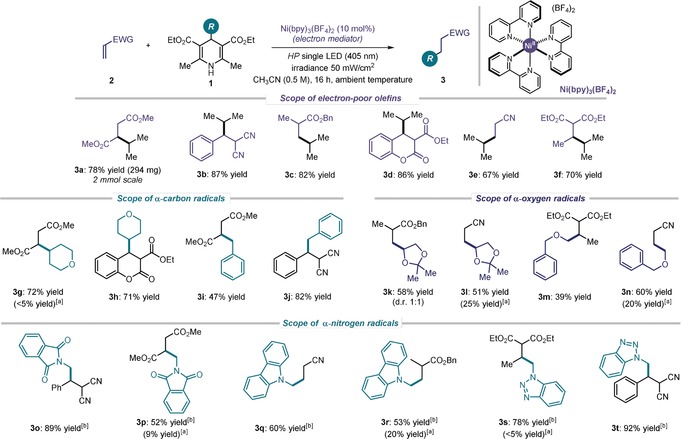
Survey of the electron‐poor olefins 2 and the dihydropyridines 1 that can participate in the photochemical Giese addition. Reactions performed at 0.1 mmol scale using 1.5 equiv of 1; yields refer to isolated products 3 after purification (average of two runs per substrate). [a] Yields of products obtained in the absence of Ni(bpy)3(BF4)2. [b] Conducted in a 1:1 CH3CN:CH2Cl2 (0.25 m) solvent mixture due to the low solubility of the corresponding radical precursor 1.
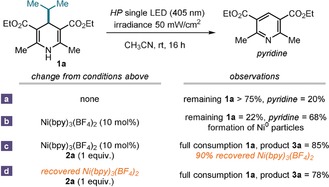
We next conducted extensive mechanistic investigations to clarify the role played by the nickel catalyst and to establish its ability to act as an EM. The first crucial step of the mechanism proposed in Figure 1 b requires Ni(bpy)3 2+ to oxidize the excited state of DHP (1 a*). Based on UV/Vis and electrochemical data, the redox potential of 1 a* (E (1 a.+/1 a*)) was estimated to be −1.9 V versus SCE according to the Rehm–Weller approximation.20 Therefore, the reduction of NiII (E p(NiII/NiI)=−1.35 V vs. SCE)19 by 1 a* is thermodynamically feasible. We further investigated this photoreduction using the following experiment: direct illumination of a CH3CN solution of 4‐isopropyl‐DHP 1 a at 405 nm in the absence of the nickel(II) complex led to very low conversion of 1 a into the corresponding pyridine (Scheme 1 a). The relative photostability of 1 a implies that the redox processes underlying the generation of the isopropyl radical from 1 a* are inefficient in the absence of an EM. The addition of Ni(bpy)3(BF4)2 (10 mol %) greatly increased the degradation of 1 a (Scheme 1 b). In this experiment, because of the lack of an oxidation mechanism to regenerate the NiII complex, we observed the formation of Ni0 particles, which otherwise were never observed during the progress of the catalytic reaction. When repeating the experiment (Scheme 1 c) in the presence of dimethyl fumarate 2 a, which provides a mechanism for regenerating the EM (see Figure 1 b), we recovered Ni(bpy)3(BF4)2 in 90 % yield.
Scheme 1.
Mechanistic experiments; NMR yields are reported.
The identity of the nickel(II) complex, which crashed out from the reaction mixture upon dilution with Et2O, was confirmed by UV/Vis and IR spectroscopy. Furthermore, the recovered Ni(bpy)3(BF4)2 was successfully reused to promote another Giese reaction, affording product 3 a in high yield (Scheme 1 d).
Overall, the experiments detailed in Scheme 1 suggest that Ni(bpy)3(BF4)2 can effectively mediate the SET oxidation of 1 a* and that its regeneration is only possible in the presence of olefin 2 (Schemes 1 c,d). The latter observation is very relevant to the overall mechanism (Figure 1 b): to act as an effective EM, it is essential that the NiI complex reduces the highly reactive intermediate A, emerging from the radical addition to 2. The feasibility of this crucial redox step, which turns back the nickel‐based EM in the original oxidation state, is supported by the redox potentials of the key intermediates involved (Scheme 2). The data in Scheme 2 indicate that the excited 1 a* can reduce Ni(bpy)3 2+. The resulting NiI complex is not able to reduce typical radicals generated upon fragmentation of 1 a* (for example E 1/2 (Bn./Bn−)=−1.45 V21 and E calc (iPr./iPr−)=−2.42 V vs. SCE).22 However, it can easily reduce radicals α to electron withdrawing groups, such as intermediate A. This mechanistic possibility is supported by the reduction potential of the radical resulting from the addition of iPr. to 2 a, estimated by DFT calculations (E calc (A ./A −)=−0.54 V vs. SCE).22 An alternative scenario, based on a self‐propagating radical manifold, could be envisaged in an SET reduction of A from the ground state of 1 a, but this pathway can be excluded based on the redox potential of 1 a (E p/2 (1 a+ ./1 a)=+1.04 V vs. SCE). The quantum yield measured for the model reaction (Φ=0.014) is also incongruent with a radical chain process.
Scheme 2.
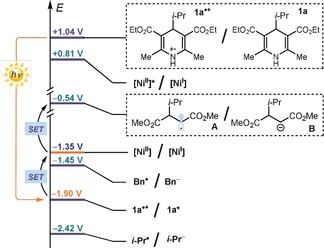
Redox potentials of the species involved in the proposed mechanism.
We also obtained evidence to support the intermediacy of the anionic intermediate B, arising from the SET reduction of A (Scheme 2). We performed deuteration experiments by adding CD3OD to the reaction medium. A high deuterium incorporation (>80 %) was observed at the α‐carbon of product 3 a (Scheme 3). No deuterium incorporation took place at the β‐carbon, indicating that deuteration occurs solely via the in situ‐generated anion.
Scheme 3.
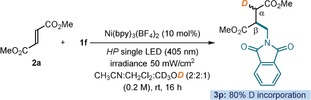
Deuteration experiments.
We deem it very unlikely that the reaction proceeds via the intermediacy of organonickel species. The trapping of photogenerated alkyl radicals with Ni complexes has been the subject of recent studies.23 While the majority of these procedures employ a metal‐to‐ligand ratio of 1:1 or 1:1.5, the present chemistry works when Ni is coordinatively saturated. The addition of extra bpy (up to 10 equiv with respect to Ni(bpy)3 2+) did not result in a significant drop of yield. Additional observations are incongruent with the involvement of organometallic intermediates: a) Ni(bpy)3(BF4)2 can be recovered almost quantitatively at the end of the reaction and reused; b) in situ monitoring of the reaction by the Evans NMR method (see Section C5 in the Supporting Information for details) reveals no significant change in the magnetic susceptibility of the reaction mixture, suggesting that the resting state of the catalyst is Ni(bpy)3 2+; c) organic electron mediators (Table 1, entries 2–3) promote the model reaction in the absence of nickel.
We finally considered the possibility that Ni(bpy)3 2+ could act as photoredox catalyst. The irradiation of the model reaction at 530 nm, where only the Ni complex absorbs, did not result in the conversion of 1 a or the formation of 3 a. This result is not surprising, given that the electronic structure and photophysical properties of Ni(bpy)3 2+ (a d8, high spin complex, paramagnetic) are very different from those of typical photoredox catalysts, including Ru(bpy)3 2+ (d6 low spin, diamagnetic).2a This is why the sporadic applications of nickel‐based photoredox catalysts have required meticulous engineering of the ligands to ensure a low‐spin NiII centers in a square planar coordination environment.24 We also observed that, in consonance with the notion that the reaction is triggered by direct excitation of alkyl‐DHP 1 and not of Ni(bpy)3 2+,25 the optimal irradiation wavelength changes depending on the nature of the substrate. For example, dihydropyridine 4 absorbs at significantly shorter wavelength (λ max=323 nm) than esters 1 (λ max=335 nm for 1 a). Irradiation at 405 nm does not promote the formation of the Giese addition product 5 at all, while a high yield is obtained using a 365 nm LED (Scheme 4).
Scheme 4.
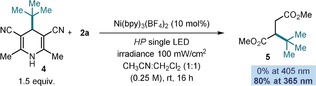
Reactivity/wavelength correlation for substrate 4.
In conclusion, we have reported a photochemical strategy to perform the Giese addition of C(sp3) radicals to a variety of electron‐poor olefins. Mechanistic investigations suggest that readily available Ni(bpy)3 2+ acts as an electron mediator to facilitate the redox processes between fleeting and highly reactive intermediates. These findings could be relevant in the design of other photochemical strategies based on the direct excitation of organic molecules or intermediates. In addition, they may have mechanistic implications in the context of the combination of nickel and photoredox catalysis,23 since nickel complexes could be involved in kinetically enhancing electron‐transfer processes.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support was provided by MICIU (CTQ2016‐75520‐P), the AGAUR (Grant 2017 SGR 981), and the European Research Council (ERC‐2015‐CoG 681840—CATA‐LUX).
T. van Leeuwen, L. Buzzetti, L. A. Perego, P. Melchiorre, Angew. Chem. Int. Ed. 2019, 58, 4953.
Contributor Information
Dr. Thomas van Leeuwen, http://www.iciq.org/research/research_group/prof‐paolo‐melchiorre/
Prof. Dr. Paolo Melchiorre, Email: pmelchiorre@iciq.es.
References
- 1.
- 1a. Handbook of Synthetic Photochemistry (Eds.: A. Albini, M. Fagnoni), Wiley-VCH, Weinheim, 2010; [Google Scholar]
- 1b. Schultz D. M., Yoon T. P., Science 2014, 343, 1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For selected reviews:
- 2a. Shaw M. H., Twilton J., MacMillan D. W. C., J. Org. Chem. 2016, 81, 6898–6926; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Matsui J. K., Lang S. B., Heitz D. R., Molander G. A., ACS Catal. 2017, 7, 2563–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Modern molecular photochemistry of organic molecules (Eds.: N. J. Turro, V. Ramamurthy, J. C. Scaiano), University Science Books, Sausalito, 2010. [Google Scholar]
- 4. Buzzetti L., Prieto A., Roy S. R., Melchiorre P., Angew. Chem. Int. Ed. 2017, 56, 15039–15043; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15235–15239. [Google Scholar]
- 5.For examples of 4-alkyl-1,4-dihydropyridines serving as radical precursors under photochemical conditions, see:
- 5a. Nakajima K., Nojima S., Sakata K., Nishibayashi Y., ChemCatChem 2016, 8, 1028–1032; [Google Scholar]
- 5b. Chen W., Liu Z., Tian J., Ma J., Cheng X., Li G., J. Am. Chem. Soc. 2016, 138, 12312–12315; [DOI] [PubMed] [Google Scholar]
- 5c. Badir S. O., Dumoulin A., Matsui J. K., Molander G. A., Angew. Chem. Int. Ed. 2018, 57, 6610–6613; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 6720–6723; [Google Scholar]
- 5d. de Assis F. F., Huang X., Akiyama M., Pilli R. A., Meggers E., J. Org. Chem. 2018, 83, 10922–10932; [DOI] [PubMed] [Google Scholar]
- 5e. Wu Q.-Y., Min Q.-Q., Ao G.-Z., Liu F., Org. Biomol. Chem. 2018, 16, 6391–6394; [DOI] [PubMed] [Google Scholar]
- 5f. McDonald B. R., Scheidt K. A., Org. Lett. 2018, 20, 6877–6881; [DOI] [PubMed] [Google Scholar]
- 5g. Verrier C., Alandini N., Pezzetta C., Moliterno M., Buzzetti L., Hepburn H. B., Vega-Peñaloza A., Silvi M., Melchiorre P., ACS Catal. 2018, 8, 1062–1066; For a review, see: [Google Scholar]
- 5h. Huang W., Cheng X., Synlett 2017, 28, 148–158. [Google Scholar]
- 6. Silvi M., Melchiorre P., Nature 2018, 554, 41–49. [DOI] [PubMed] [Google Scholar]
- 7.For selected examples, see:
- 7a. Emmanuel M. A., Greenberg N. R., Oblinsky D. G., Hyster T. K., Nature 2016, 540, 414–417; [DOI] [PubMed] [Google Scholar]
- 7b. Jung J., Kim J., Park G., You Y., Cho E. J., Adv. Synth. Catal. 2016, 358, 74–80; [Google Scholar]
- 7c. Silvi M., Verrier C., Rey Y. P., Buzzetti L., Melchiorre P., Nat. Chem. 2017, 9, 868–873. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Electron Transfer in Chemistry (Ed: V. Balzani), Wiley-VCH, Weinheim, 2001; [Google Scholar]
- 8b. Kavarnos G. J. in Fundamentals of Photoinduced Electron Transfer, VCH-Publishers, Weinheim, 1993. [Google Scholar]
- 9. Grätzel M., J. Photochem. Photobiol. C 2003, 4, 145–153. [Google Scholar]
- 10.
- 10a. Kumar B., Llorente M., Froehlich J., Dang T., Sathrum A., Kubiak C. P., Annu. Rev. Phys. Chem. 2012, 63, 541–569; [DOI] [PubMed] [Google Scholar]
- 10b. Wu J., Huang Y., Ye W., Li Y., Adv. Sci. 2017, 4, 1700194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yan M., Kawamata Y., Baran P. S., Chem. Rev. 2017, 117, 13230–13319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Skubi K. L., Blum T. R., Yoon T. P., Chem. Rev. 2016, 116, 10035–10074, and references therein; For selected examples, see: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Riener M., Nicewicz D. A., Chem. Sci. 2013, 4, 2625–2629; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c. Tyson E. L., Niemeyer Z. L., Yoon T. P., J. Org. Chem. 2014, 79, 1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Giese B., Angew. Chem. Int. Ed. Engl. 1983, 22, 753–764; [Google Scholar]; Angew. Chem. 1983, 95, 771–782. [Google Scholar]
- 14.For examples of previous methods for Giese-type additions required the use of an external photoredox catalyst, see:
- 14a. Lackner G. L., Quasdorf K. W., Overman L. E., J. Am. Chem. Soc. 2013, 135, 15342–15345; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b. Chu L., Ohta C., Zuo Z., MacMillan D. W. C., J. Am. Chem. Soc. 2014, 136, 10886–10889; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14c. Millet A., Lefebvre Q., Rueping M., Chem. Eur. J. 2016, 22, 13464–13468; Other non-photochemical methodologies have also been reported, which require stoichiometric amounts of reductants, see:27321136 [Google Scholar]
- 14d. Qin T., Malins L. R., Edwards J. T., Merchant R. R., Novak A. J. E., Zhong J. Z., Mills R. B., Yan M., Yuan C., Eastgate M. D., Baran P. S., Angew. Chem. Int. Ed. 2017, 56, 260–265; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 266–271. [Google Scholar]
- 15. Chiefari J., Jeffery J., Mayadunne R. T. A., Moad G., Rizzardo E., Thang S. H., Macromolecules 1999, 32, 7700–7702. [Google Scholar]
- 16.The reduction of 2 a (E p/2 (2 a/2 a .−)=−1.39 V vs. SCE) by the excited 1 a* is exergonic but apparently slow, given that only a fraction of 1 a and 2 a reacted after 16 h of irradiation. The reduction potential of 1 a* was estimated to be −1.9 V versus SCE based on UV/Vis and electrochemical data and according to the Rehm–Weller approximation, see Section C3 of the Supporting Information.
- 17.
- 17a. Roth H., Romero N., Nicewicz D., Synlett 2015, 27, 714–723; [Google Scholar]
- 17b. Bockman T. M., Kochi J. K., J. Org. Chem. 1990, 55, 4127–4135. [Google Scholar]
- 18.For the use of Ni(bpy)3(BF4)2 as a mediator in electrosynthesis, see:
- 18a. Franco D., Duñach E., Tetrahedron Lett. 2000, 41, 7333–7336; [Google Scholar]
- 18b. Stepanov A. A., Grinberg V. A., Russ. J. Electrochem. 2003, 39, 1347–1350. [Google Scholar]
- 19. Bartlett P. N., Eastwick-Field V., Electrochim. Acta 1993, 38, 2515–2523. [Google Scholar]
- 20. Rehm D., Weller A., Isr. J. Chem. 1970, 8, 259–271. [Google Scholar]
- 21. Wayner D. D. M., McPhee D. J., Griller D., J. Am. Chem. Soc. 1988, 110, 132–137. [Google Scholar]
- 22.Estimated by DFT computations at the B3LYP/6–311++G(2d,2p)//B3LYP/6-31+G(d) level, with PCM solvation. See the Supporting Information for computational details (Section D1) and validation of the theoretical approach (Section D2).
- 23. Milligan J. A., Phelan J. P., Badir S. O., Molander G., Angew. Chem. Int. Ed. 2019, 10.1002/anie.201809431; [DOI] [Google Scholar]; Angew. Chem. 2019, 10.1002/ange.201809431. [DOI] [Google Scholar]
- 24.
- 24a. Cameron L. A., Ziller J. W., Heyduk A. F., Chem. Sci. 2016, 7, 1807–1814; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24b. Grübel M., Bosque I., Altmann P. J., Bach T., Hess C. R., Chem. Sci. 2018, 9, 3313–3317; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24c. Shields B. J., Kudisch B., Scholes G. D., Doyle A. G., J. Am. Chem. Soc. 2018, 140, 3035–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.For additional mechanistic discussion, including triplet quenching experiments, see the Supporting Information, Section C6.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary