

Summary
Objective
The Epilepsy Genetics Initiative (EGI) was formed in 2014 to create a centrally managed database of clinically generated exome sequence data. EGI performs systematic research‐based reanalysis to identify new molecular diagnoses that were not possible at the time of initial sequencing and to aid in novel gene discovery. Herein we report on the efficacy of this approach 3 years after inception.
Methods
One hundred sixty‐six individuals with epilepsy who underwent diagnostic whole exome sequencing (WES) were enrolled, including 139 who had not received a genetic diagnosis. Sequence data were transferred to the EGI and periodically reevaluated on a research basis.
Results
Eight new diagnoses were made as a result of updated annotations or the discovery of novel epilepsy genes after the initial diagnostic analysis was performed. In five additional cases, we provided new evidence to support or contradict the likelihood of variant pathogenicity reported by the laboratory. One novel epilepsy gene was discovered through dual interrogation of research and clinically generated WES.
Significance
EGI's diagnosis rate of 5.8% represents a considerable increase in diagnostic yield and demonstrates the value of periodic reinterrogation of whole exome data. The initiative's contributions to gene discovery underscore the importance of data sharing and the value of collaborative enterprises.
Keywords: data sharing, seizures, whole exome sequencing
Key Points.
The Epilepsy Genetics Initiative (EGI) is a centrally managed database of clinically generated exome sequence data
EGI performs systematic research‐based reanalysis to identify new molecular diagnoses that were not possible at the time of initial sequencing and to aid in novel gene discovery
EGI has facilitated 8 new diagnoses from 139 cases that were unresolved following diagnostic whole exome sequencing (WES) and has contributed to novel gene discovery through dual interrogation of research and clinically generated WES
1. INTRODUCTION
The Epilepsy Genetics Initiative (EGI) was established to facilitate two main goals: (a) to provide a mechanism for periodic reanalysis of whole exome sequencing (WES) data of individuals with epilepsy for whom diagnostic genetic testing was initially inconclusive, and (b) to amass this sequencing data in a standardized repository to allow for aggregate analysis and novel gene discovery in epilepsy. EGI is a signature program of Citizens United for Research in Epilepsy (CURE) (http://www.cureepilepsy.org/egi/index.html).
A broad range in the diagnostic rate of WES in epilepsy has been described in the scientific and medical literature, a result of the variable definition of each cohort depending on factors such as type of epilepsy, phenotypic features, disease severity, prior genetic screening, and so on. In focal epilepsy, one group reported genetic diagnoses in 12.5% of cases1; in another study, a clinical diagnostic laboratory in the United States reported a genetic diagnosis in 43% of cases with epileptic encephalopathy (EE) and in 33% of their epilepsy cohort overall.2 Clinical diagnostic sequencing laboratories will often perform a one‐time reanalysis at no cost at the ordering physician's request; however, there are often limitations on when this request can be initiated. Although ongoing reanalysis is not standard for clinical diagnostic exome sequencing companies, reinterrogation of sequence data in unsolved exome cases can increase diagnostic yield.3, 4, 5, 6, 7
Reanalysis can increase diagnostic yield in broad cohorts; however, it has not been investigated specifically in epilepsy. Reanalysis and diagnosis are particularly important in epilepsy due to the rapid rate of gene discovery and potential for treatment implications.8 EGI was established as a mechanism to provide dynamic reanalysis of unsolved cases through iterative and contemporary reinterrogation of existing WES data. Herein we describe our experience reanalyzing exome data from 139 unsolved epilepsy cases, including 96 trios and 43 nontrios. We also demonstrate how merging of research and clinical data can lead to novel gene discovery.
2. METHODS
Participants were enrolled at one of a number of established EGI enrollment sites: Columbia University Irving Medical Center (CUIMC), University of California San Francisco, Children's Hospital of Philadelphia, Boston Children's Hospital, New York University Langone Medical Center, Ann & Robert H. Lurie Children's Hospital of Chicago, University of Melbourne, University of Iowa, Children's Hospital Colorado, and Duke University. Several patients enrolled at CUIMC were referred by one of EGI's established referral partners, including Stanford University and the University of Virginia. Study participants were referred by their physician or identified EGI on their own and requested their physician's participation in the enrollment process. This study was approved by the respective ethics review board at each institution. All participants consented to take part in the work of EGI. The consent included the use of the participant's sequence and phenotypic data for research purposes and the return of clinically relevant results to the participant through their physician.
The EGI analyzed exome sequence data from 166 families: 117 trios (proband and both parents) and 49 nontrios (proband only). Two of the families reviewed in the trio analysis were enrolled as quads (proband, both parents, and an affected sibling) and full diagnostic review was performed for both the proband and affected siblings. Having a genetic diagnosis made at the time of clinical WES was not an exclusion criterion of the study, since in some cases a genetic diagnosis may be incorrect, and as EGI grows, this cohort could be used to evaluate genetic modifiers. The majority of individuals had sequencing performed at a diagnostic clinical laboratory; we also enrolled one trio and one proband‐only nontrio that had sequencing performed as part of a research study. Sequence data obtained and used as part of this study were generated at GeneDx, Ambry Genetics, Baylor Genetics, Laboratory of Personalized Genomic Medicine at Columbia University, Claritas Genomics, Children's Hospital of Philadelphia, University of California Los Angeles, Iowa Institute of Human Genetics, University of Chicago, Emory Genetics Laboratory, Broad Institute, Yale University DNA Diagnostics Laboratory, MedGenome, Centogene, the Epilepsy Research Centre at the University of Melbourne, and the Center for Advanced Studies, Research and Development in Sardinia (CRS4). Exome data files were transferred to the EGI repository at the Institute for Genomic Medicine (IGM) at CUIMC from the respective diagnostic companies in FASTQ or BAM file format.
Data were analyzed at the IGM utilizing an updated version of our established pipeline, where variants are filtered based on quality and allele frequency using the Genome Analysis Toolkit (GATK) best‐practices protocol and prioritized based on bioinformatic signatures as described previously.9 Prioritized variants were interrogated by the genetic counseling and bioinformatics teams via thorough review of most recent OMIM, ClinVar, HGMD, and PubMed databases and the IGM's internal database to determine gene‐disease association, inheritance pattern, disease, and mechanism. We also evaluated whether the variant was reported in the subject's clinical WES report. Candidate variants were discussed at a multidisciplinary team meeting comprising genetic counselors, bioinformaticians, geneticists, epileptologists, and clinicians. Although we did not rely strictly on the ACMG (American College of Medical Genetics) guidelines for variant classification,10 we attempted to follow these standards in our interpretations and had an experienced clinical molecular genetics laboratory director among our team. Variants of interest deemed likely or definitely causative by group consensus were reported to referring providers via a research analysis summary letter from the EGI team at CUIMC; these included new variants not described by the clinical laboratory that were considered likely to be contributing to disease, as well as variants identified originally for which additional evidence became available to either support pathogenicity of a variant in question or weaken the support for pathogenicity. The referring physician was asked to provide their clinical input/interpretation, request an amended report by the clinical laboratory if in agreement with our research assessment, and ultimately to return the result to the family if warranted (ie, confirmed/supported by the clinical laboratory) (see Figure 1).
Figure 1.
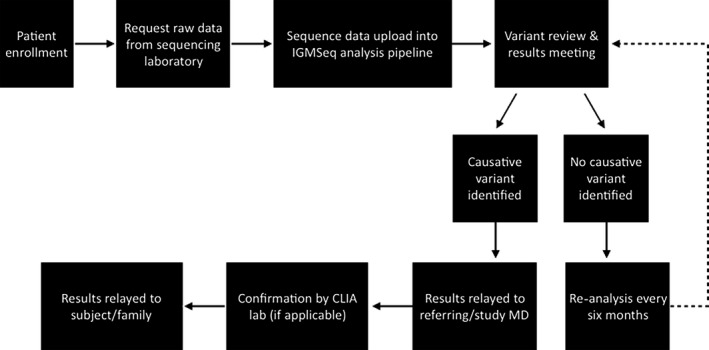
Epilepsy Genetics Initiative (EGI) work flow
All 166 EGI cases, including those enrolled with a clinical genetic diagnosis, were analyzed and reviewed by the EGI team. By removing cases enrolled in EGI with a secure genetic diagnosis (n = 27) from the overall cohort (n = 166), we arrived at a total of 139 unsolved cases that were reinterrogated by EGI. We defined a new “EGI diagnosis” as a variant deemed to be causative by the EGI team that was: (a) not reported on the original diagnostic WES report, (b) reported by the clinical laboratory as a variant of uncertain significance (VUS), or (c) reported by the clinical laboratory as a variant in a gene whose association with disease or the patient's phenotype was unclear.
3. RESULTS
Of the 166 probands, >70% identified as Caucasian, 4% identified as Asian, and 3% identified as African American, with the remainder from other ethnic backgrounds. The ages ranged from <1 year up to 40 years of age, with >70% under age 10 at enrollment. Of participants with age at seizure onset reported, 65% (101/155) had their first unprovoked seizure before age 1 year, with a mean age of seizure onset of 1.96 years. The cohort was almost evenly split between male (n = 86) and female (n = 80) participants. More than 30% of our cohort had a family history of epilepsy as reported by the referring provider. Ninety‐five percent of our participants had more than one seizure type. The majority (~90%) of participants had some degree of delay or cognitive impairment including gross motor, fine motor, language, personal‐social and global delays, intellectual disability, or learning difficulty. Broad electroclinical classification of the cases showed that 114 had an epileptic encephalopathy, 17 had focal epilepsy, 12 had generalized epilepsy, and 23 were unclassified.
3.1. New genetic diagnoses
Of the 139 probands evaluated who did not have a secure diagnosis at the time of enrollment, 8 had a putative genetic diagnosis made by EGI (Table 1). Two of these diagnoses were possible due to the identification of a variant in the alternative exon 5A of SCN8A, as reported previously by EGI.11 In two cases, we reported on a variant in PPP3CA, an epilepsy gene newly described through a collaborative effort that involved EGI.16 Additional diagnoses include one case each with a variant identified in the FGF12, HNRNPU, SATB2, or STAG2 genes. See Table 1 for additional details.
Table 1.
New EGI diagnoses
| Case | Gene | Variant details | Variant type | Inheritance | Patient phenotype | Month/year of clinical WES | Month/year of reanalysis | Clinical classification | Clinical laboratory notes | Additional evidence considered by EGI team |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | FGF12 |
3‐192053223‐C‐T ENST00000454309.2:c.341G>A ENSP00000413496.2:p.Arg114His |
Missense | De novo | Early onset epileptic encephalopathy (seizure onset 1 month; drug resistant), GDD | 2014 (month unknown) | 8/2017 | Not reported clinically | New OMIM entry, new literature reports: Now associated with disease in OMIM (#617166 Epileptic encephalopathy, early infantile 47, created in 2016). This variant has been reported in multiple individuals in the literature as a recurrent gain‐of‐function de novo variant.13, 14, 15 | |
| 2 | HNRNPU |
1‐245027356‐T‐TC ENST00000283179.9:c.253dupG ENSP00000283179.9:p.Glu85GlyfsTer |
Frameshift | Inheritance unknown (proband only) | Mixed epilepsy with both generalized and focal seizures, primarily occurring in the context of fever (onset 11 months; drug resistant), GDD | 10/2016 | 8/2017 | Not reported clinically | Laboratory interrogated pediatric neurology regions of interest only, this gene was not included | New OMIM entry: Now associated with disease in OMIM (#617391 Epileptic encephalopathy, early infantile, 54, created in 2017), which may explain why gene was not considered a gene of interest by clinical laboratory. |
| 3 | PPP3CA |
4‐101953430‐G‐A ENST00000394854.3:c.1333C>T ENSP00000378323.3:p.Gln445Ter |
Stop gain | De novo | West syndrome/infantile spasms (onset 6 weeks; drug resistant), GDD | 8/2014 | 2/2017 | VUS | Gene not yet associated with human disease | New observation based on EGI and collaborator data: This case was included in a cohort of 6 cases with de novo PPP3CA variants, securely implicating gene as a cause of early‐onset refractory seizures.12 |
| 4 | PPP3CA |
4‐101950354‐T‐C ENST00000394854.3:c.1340‐2A>G |
Splice site acceptor | De novo | West syndrome/infantile spasms (onset 5 months; drug resistant), GDD | 6/2014 | 8/2017 | VUS in a candidate gene | Gene not yet associated with human disease | New literature report: This additional case of a de novo PPP3CA variant was identified after submission of manuscript, which secured gene as a cause of epileptic encephalopathy.12 |
| 5 | SATB2 |
2‐200213837‐G‐A ENST00000417098.1:c.760C>T ENSP00000401112.1:p.His254Tyr |
Missense | De novo | Unclassified epilepsy with infantile spasms and complex partial seizures (onset 5 months; drug resistant), GDD | 1/2015 | 2/2017 | Variant, likely mutation, in a gene possibly associated with reported phenotype | New literature reports: New literature reports show that de novo missense variants in this gene cause disease. Additionally, these reports further describe the associated phenotype, which overlaps with that of our participant.21,22,26 | |
| 6 | SCN8A |
12‐52082841‐A‐G ENST00000551216.1:c.61A>G E.1:p.Arg21Gly |
Missense | De novo | West syndrome (onset 6 months; drug resistant), GDD | 6/2014 | 2/2017 | Not reported clinically | New observation based on EGI data: Observation of 2 de novo missense variants in highly expressed alternative exon 5 A of SCN8A out of 54 EGI trios.11 | |
| 7 | SCN8A |
12‐52082806‐T‐C ENST00000551216.1:c.26T>C ENSP00000447567.1:p.Val9Ala |
Missense | De novo | Epileptic encephalopathy (onset 3 months; drug resistant), GDD | 9/2014 | 2/2017 | VUS | Reported as an intronic variant | New observation based on EGI data: See above explanation (Case 6). |
| 8 | STAG2 |
X‐123196750‐AG‐A ENST00000218089.9:c.1639‐1delG |
Splice site acceptor | De novo | Focal seizures with secondary generalization, gelastic seizures (age of onset 6 years; drug resistant), GDD, other features (MRI abnormalities, dysmorphic features, sensorineural hearing loss) | 10/2015 | 8/2017 | VUS in a candidate gene | Gene not yet associated with human disease | New literature report: 3 females reported with de novo STAG2 variants, reported phenotypes overlap with participant's phenotype.27 |
GDD, global developmental delay; VUS, variant of unknown significance.
For each of the new diagnoses in SCN8A, both de novo variants were located in an alternative version of exon 5. This novel exon was not included in the consensus coding sequence (CCDS) and was not routinely interrogated by clinical laboratories.11 The PPP3CA diagnoses were possible due to new observations based on EGI and collaborator data, and EGI Case 3 was included in a paper to implicate variants in the gene as causative for epilepsy.12 Many of the additional diagnoses were not possible at the time of clinical sequencing, as the respective genes were not implicated with disease until after the initial diagnostic WES. For example, in EGI Case 1 (see Table 1), the subject's clinical WES in 2014 yielded several variants of uncertain significance but no definitive genetic diagnosis. Upon reanalysis by EGI, we identified a de novo heterozygous missense variant (NM_004113.5:c.155G>A, p.R52H) in FGF12 that was not reported by the clinical laboratory. The same variant was described in 2016 in seven individuals with epileptic encephalopathy and was functionally characterized to disrupt the gene via a gain‐of‐function mechanism.13, 14, 15
Following receipt of our EGI analysis summary, the referring provider for Case 2 (Table 1) submitted a sample for targeted testing of the HNRNPU frameshift variant; this finding was confirmed by the clinical laboratory and classified as a likely pathogenic variant. The referring provider for Case 6 (Table 1) requested a clinical reanalysis of the patient's exome data and received an amended report that confirmed the SCN8A variant identified by EGI and classified the finding as a pathogenic variant. The referring provider for Case 4 (Table 1) requested a clinical reanalysis and reinterpretation from the diagnostic laboratory, which resulted in the PPP3CA variant being reclassified from a “variant of uncertain significance in a candidate gene with a potential relationship to phenotype” to a “likely pathogenic variant in a disease gene associated with reported phenotype.” The referring provider for Case 8 (Table 1) also requested a clinical reanalysis and reinterpretation for the STAG2 variant; however, the laboratory maintained their classification of the variant as a VUS. Regardless, the provider agreed with EGI's interpretation of the variant based on the new evidence provided in the summary letter and relayed to the family that this finding is likely the cause of the patient's epilepsy.
3.2. Additional findings
In addition to making new genetic diagnoses, the EGI team also highlighted new gene‐disease associations for three subjects. This included two cases in which a frameshift variant in SMC1A was identified by the clinical laboratories and called “likely positive” and “likely mutation,” respectively; at the time of clinical testing, the described gene‐disease association for SMC1A was with Cornelia de Lange syndrome, which is typically caused by missense variants in the gene. A recent publication demonstrated an association between heterozygous truncating SMC1A variants and epileptic encephalopathy,16 which is a better fit for these individuals based on both disease mechanism and phenotype. For a third EGI case, the clinical laboratory reported a homozygous missense variant in the PROSC gene and classified this finding as a VUS in a gene with an unknown association to disease. Upon reanalysis, EGI learned that PROSC is now known to be associated with early onset pyridoxine‐dependent epilepsy and represents a high clinical fit with our subject.
There were also instances in which the EGI team identified new evidence that weakened the support of a particular diagnosis. One subject was enrolled with a CHD4 variant classified as likely pathogenic by the clinical laboratory; however, the patient's geneticist questioned whether the variant represented a secure diagnosis. This subject was initially reported to have a history of seizures, speech delay with regression, celiac disease, possible hearing loss, and a diagnosis of Landau‐Kleffner syndrome but was later reported to have normal hearing. CHD4 is associated with an autosomal dominant intellectual disability syndrome, with variable features and affected systems including cardiac, skeletal, and urogenital; additional findings can include hearing loss, macrocephaly, short stature, and nonspecific dysmorphic features.17 During reanalysis, EGI identified the same variant in three individuals in gnomAD. An experienced clinical molecular geneticist on our team determined that the variant meets criteria for a VUS per ACMG guidelines for variant interpretation.10 Given the frequency of the variant in control databases and the low clinical fit, the EGI team communicated to the referring provider our interpretation that the variant is unlikely to be causative for the subject's phenotype.
In another case, a variant in VHL reported as likely pathogenic on the clinical report (reported as a secondary finding based on ACMG guidelines) was later described as a VUS by the same laboratory in ClinVar. Although this variant would be predicted to cause loss of function of the protein in the primary VHL isoform, the variant is upstream of the start codon of the shorter, clinically relevant isoform and would therefore not be expected to affect protein function.18 Our interpretation that there is insufficient evidence to support pathogenicity of this variant was relayed back to the referring provider, who contacted the clinical sequencing laboratory and received an amended report indicating that the variant is classified as a VUS and is no longer a medically actionable finding.
4. DISCUSSION
The two broad goals of EGI are to make new genetic diagnoses and facilitate gene discovery. After analysis of our first 166 probands, both goals were achieved.
Of the 139 unresolved cases for which the EGI team performed systematic reanalysis of diagnostic sequencing data, EGI was able to return 8 new diagnoses. This represents a diagnosis rate of 5.8% (8/139) and a marked increase in diagnostic yield overall.
Two crucial elements allowed us to make these new diagnoses: prioritization of variants according to the bioinformatic signatures described by Petrovski et al (2012) and an updated literature review since the initial diagnostic sequencing. Of our eight new diagnoses, five of the variants exhibited a strong bioinformatic signature; the SATB2 and SCN8A findings were each de novo missense “hot zone” variants, defined by having a PolyPhen2 score of greater than 0.995 and a Residual Variance Intolerance Score (RVIS) in the 25th percentile.19 The PPP3CA variants also had a compelling bioinformatic signature, as both were de novo loss of function (LoF) variants in a gene depleted of LoF variation in the general population.20 For several cases, literature reports published after the initial diagnostic sequencing allowed for a secure diagnosis upon reevaluation by EGI. The FGF12 variant was reported in the literature as a recurrent de novo missense mutation with an association with disease in 2016,13, 14, 15 after the subject's diagnostic testing in 2014. Similarly, a disease association for the STAG2 and HNRNPU genes had not been securely established at the time of clinical testing. Regarding SATB2, new literature expanding the phenotype and reporting at least six other de novo missense variants in the gene,21, 22 coupled with the strong bioinformatic signature, allowed the EGI team to interpret this finding as causative for disease. These cases demonstrate the utility of bioinformatics and the value of new and updated literature in making diagnoses for previously unsolved exomes.
By identifying two cases of de novo variants in the alternatively spliced exon 5A of SCN8A (EGI Cases 6 and 7), we were able to demonstrate that variants in this exon can be causative for epileptic encephalopathy.11 This allowed us to not only return these findings to the respective providers but also to provide evidence that this exon should be interrogated in all diagnostic exomes. Our experience implicating the alternative exon 5A demonstrates the value of collaboration between researchers, providers, and clinical laboratories to combine expertise and resources toward the mutual goals of diagnosis and novel insight into disease.
Toward the goal of gene discovery, the additional value of EGI is that this cohort ultimately becomes part of a larger set of epilepsy genetics studies, including Epi4K, Epi25, and EPIGEN.23 When reanalyzing and reinterpreting our results in the context of other studies, we increase the power of our ability to make new discoveries. PPP3CA was one of the first genes in which a candidate variant was identified in the EGI cohort. This resulted in a collaborative publication describing the association of PPP3CA with severe neurodevelopmental disease with seizures12 and allowed for a new diagnosis for two EGI families (Cases 3 and 4). Combining exome sequence data from many different studies or databases has historically been extremely valuable in facilitating the discovery of novel genes, like it was for PPP3CA, and will likely continue to be in the short term. However, we note that inevitably there will be diminishing returns as larger consortium level efforts, like Epi25, that seek to analyze thousands of samples simultaneously, approach discovering all the genes where the signal is coming at least in part from protein‐coding variants detectable with short‐read next‐generation sequencing. Once we near this asymptote, improving diagnosis rate of exome negative cases will require the generation of new genomic datasets, including whole genome and long‐read next‐generation sequencing data that will capture noncoding variation and protein coding variation that is overlooked by short‐read sequencing technology.
In several cases, we identified a variant with a strong bioinformatic signature in a gene not yet securely associated with disease. Although there was not enough evidence in the literature to reclassify these variants as disease‐causing, we added several of these findings to our internal “watch list,” which consists of a new submission to GeneMatcher24 and an alert for new publications on PubMed. Genes added to this watch list thus far include EGR3, PNPLA8, HTT, BCOR, SSBP3, and ADAMTS3. The EGI process will allow us to reevaluate these strong candidate variants as new disease associations are secured. Proof of this concept has already been demonstrated by the two diagnoses in PPP3CA; at the time of our initial reanalysis, these findings were classified as variants with a strong bioinformatic signature but were lacking a known disease association.
Beyond the established goals of EGI, we have learned that the process of reanalysis can be impactful in other meaningful ways. By informing the referring physician of the newly described disease association between PROSC and early onset vitamin pyridoxine‐dependent epilepsy in a subject with a homozygous PROSC variant (described as a VUS in a gene with an unknown association to disease at the time of clinical testing), we have provided crucial information for this physician to consider when making a clinical interpretation of this patient's genetic test results. This is of particular significance in this case, as affected individuals have an immediate response to pyridoxine, with several patients described as having an improvement in seizure control following a change in treatment from pyridoxine to pyridoxal‐5‐phosphate.25 This case underscores the utility of reanalysis in the potential for clinical management implications for patients with epilepsy. We anticipate the long‐term implications of such work to lead to increasing opportunities for targeted treatment and precision medicine.
Although, unquestionably, our data further support that periodic reinterrogation of unresolved exomes is critical to improving the diagnostic rate3, 4, 5, 6, 7 it is important to note that 10 of the 13 new and revised diagnoses that arose from this analysis came from reassessment of a variant that was initially provided to the clinicians in the original clinical report (SATB2, STAG, PROSC, VHL, CHD4, both SMC1A diagnoses, one of two of the SCN8A diagnoses, and both PPP3CA diagnoses, assuming that PPP3CA would have been discovered eventually without the involvement of EGI). In these 13 instances, a clinician with experience interpreting genetic diagnoses would have the potential to perform a similar review. For the additional three new or revised diagnoses, a deeper review of the sequence data, including reassessment of annotated variants (FGFR2 and HNRNPU) and reannotation in one case (SCN8A), would be required from the diagnostic sequencing company. This suggests that data centralization may not be needed outside of the novel gene discovery endeavors; however, it warrants thoughtful consideration of to whom the responsibility of initiation of reanalysis should fall. There are a number of possible models that include the patient/family, physician, clinical laboratory, centralized repository, or other entity, and this framework is likely to evolve over time.
A generalizable long‐term strategy for how to provide patients and physicians with a practical means for reevaluating existing sequence data has not yet evolved. The development and implementation of EGI highlights one model allowing for a mutually beneficial, synergistic partnership between families, clinicians, researchers, and clinical laboratories. This partnership has led to new discoveries in the area of epilepsy genetics and has provided clinicians and families genetic diagnoses through iterative reanalysis. Our results demonstrate the power of centralization of research and clinical exome sequencing data in epilepsy and also suggest that this approach may be equally powerful in a range of diseases where exome sequencing is frequently being performed clinically, including intellectual disability, autism, amyotrophic lateral sclerosis (ALS), movement disorders, and beyond.
DISCLOSURE OF CONFLICTS OF INTEREST
D.B.G. is a founder of and holds equity in Pairnomix and Praxis; has research supported by Janssen, Gilead, Biogen, AstraZeneca, and UCB; and has an advisor role on Apostle. S.P. is an employee of AstraZeneca and serves on the advisory board and is an equity holder of Pairnomix. O.D. serves on the advisory board of Pairnomix. J.S. conducted a webinar for Invitae on genetic testing in epilepsy and received monetary compensation. The remaining authors have no conflicts of interest. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
AUTHOR CONTRIBUTIONS
The Steering Committee conceived, designed, and implemented the activities of EGI. The Enrollment Group consists of delegates at each site responsible for the enrollment of patients, phenotyping, and returning significant results to patients. Patients who participated specifically in this study were enrolled by Columbia University Irving Medical Center, University of California San Francisco, Children's Hospital of Philadelphia, Boston Children's Hospital, New York University Langone Medical Center, University of Melbourne, Ann & Robert H. Lurie Children's Hospital of Chicago, University of Iowa, Children's Hospital Colorado, and Duke University. Sequence and phenotypic data transfer was overseen by L.B., M.E.E., and T.M.A. Data analysis and interpretation were performed by L.B., M.E.E., N.C.L., M.S.M., A.R.P., P.H., N.S., D.B.G., E.L.H., V.A., D.H.L., and T.S. M.E. drafted the manuscript with support from N.C.L. and M.S.M. All contributors discussed the results and participated in the development of the manuscript.
ACKNOWLEDGMENTS
The Epilepsy Genetics Initiative (EGI) would like to thank our patients and families for their participation in this research and their role in advancing our understanding of the genetic causes of epilepsy. We would also like to thank the following external providers and clinicians for connecting their patients to EGI and facilitating their enrollment in our study via the provision of medical history and relevant follow‐up care: Cigdem Akman, Arthur Mandel, Melodie Winawer, James Riviello, Tiffani McDonough, Alejandro Iglesias, Kwame Yeboa, Aliza Alter, Wendy Chung, Michelle Primiano, Priyanka Ahimaz, Ashley Wilson, Natalia Gomez, Paul Mark, Mark Nunes, Matthew Wong, Uta Lichter‐Konecki, Luis Rohena, Patrick Mabray, Ilana Kahn, Michael Johnston, Nicola Specchio, Mark Tarnopolsky, Lauren Brady, Katherine Dawson, Tracy McGregor, SakkuBai Naidu, Karina Osipova, Marcus Wheeler, and Peter Karachunski. We want to recognize Citizens United for Research in Epilepsy (CURE; 339143), the National Institute for Neurological Disease and Stroke (NINDS; U01‐NS077303‐04S1), and The John and Barbara Vogelstein Foundation for providing the funding to make this research possible.
APPENDIX 1.
Contributors
EGI Steering Committee: Samuel F. Berkovic,1 David B. Goldstein,2 Erin L. Heinzen,2 Brandon L. Laughlin,3 Daniel H. Lowenstein,4 Laura Lubbers,3 Randall Stewart,5 Vicky Whittemore.5
EGI Enrollment Group: Kaitlin Angione,6 Carl W. Bazil,7 Louise Bier,2 Judith Bluvstein,8 Elise Brimble,9 Colleen Campbell,10 Gianpiero Cavalleri,11,12 Chelsea Chambers,13 Hyunmi Choi,7 Maria Roberta Cilio,4,14 Michael Ciliberto,15 Susannah Cornes,4 Norman Delanty,11,12 Scott Demarest,6 Orrin Devinsky,8 Dennis Dlugos,16 Holly Dubbs,16 Patricia Dugan,8 Michelle E. Ernst,2 Melissa Gibbons,6 Howard P. Goodkin,13 Ingo Helbig,16 Laura Jansen,13 Kaleas Johnson,4 Charuta Joshi,6 Natalie C. Lippa,2 Eric Marsh,16,17 Alejandro Martinez,16 John Millichap,18,19 Maureen S. Mulhern,2 Adam Numis,4,14 Kristen Park,6 Tommaso Pippucci,20 Annapurna Poduri,21 Brenda Porter,9 Brigid Regan,1 Tristan T. Sands,2,22 Ingrid E. Scheffer,1,23,24 John M. Schreiber,25 Beth Sheidley,21 Nilika Singhal,4,14 Lacey Smith,21 Joseph Sullivan,4,14 Alan Taylor,21 Patricia Tolete8
Other Contributors: Tahseen M. Afgani,2 Vimla Aggarwal,2,26 Rosemary Burgess,1 Tracy Dixon‐Salazar,3 Parisa Hemati,2 Julie Milder,3 Slavé Petrovski,27,28 Anya Revah‐Politi,2 Nicholas Stong2
1Department of Medicine, Epilepsy Research Center, The University of Melbourne and Austin Health, Heidelberg, Victoria, Australia
2Institute for Genomic Medicine, Columbia University Irving Medical Center, New York, New York
3Citizens United for Research in Epilepsy (CURE), Chicago, Illinois
4Department of Neurology, University of California San Francisco, San Francisco, California
5National Institute for Neurological Disorders and Stroke, Bethesda, Maryland
6Department of Pediatrics, Section of Neurology, University of Colorado School of Medicine, Aurora, Colorado
7Division of Epilepsy, Department of Neurology, Columbia University Irving Medical Center, New York, New York
8New York University Langone Medical Center, New York, New York
9Department of Neurology and Neurological Sciences, Stanford Medicine, Stanford, California
10Iowa Institute of Human Genetics, Carver College of Medicine, University of Iowa, Iowa City, Iowa
11Department of Molecular and Cellular Therapeutics, Royal College of Surgeons in Ireland, Dublin, Ireland
12Department of Neurology, Beaumont Hospital, Dublin, Ireland
13Division of Pediatric Neurology, Department of Neurology, University of Virginia, Charlottesville, Virginia
14Department of Pediatrics, Benioff Children's Hospital, University of California San Francisco, San Francisco, California
15Department of Pediatrics, University of Iowa Hospitals and Clinics, Iowa City, Iowa
16Division of Neurology, The Children's Hospital of Philadelphia, Philadelphia, Pennsylvania
17Departments of Neurology and Pediatrics, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, Pennsylvania
18Division of Neurology, Ann & Robert H. Lurie Children's Hospital of Chicago, Chicago, Illinois
19Departments of Pediatrics and Neurology, Northwestern University Feinberg School of Medicine, Chicago, Illinois
20Medical Genetics Unit, S. Orsola‐Malpighi Hospital, University of Bologna, Bologna, Italy
21Epilepsy Genetics Program, Boston Children's Hospital, Boston, Massachusetts
22Department of Neurology, The Neurological Institute of New York, Columbia University Irving Medical Center, New York, New York
23The Florey Institute of Neuroscience and Mental Health, Melbourne, Victoria, Australia
24Departments of Paediatrics and Neurology, The Royal Children's Hospital, The University of Melbourne, Melbourne, Victoria, Australia
25Center for Neuroscience, Children's National Health System, George Washington University, Washington, District of Columbia
26Department of Pathology and Cell Biology, Columbia University Irving Medical Center, New York, New York
27IMED Biotech Unit, Centre for Genomics Research, Precision Medicine and Genomics, AstraZeneca, Cambridge, UK
28Department of Medicine, Royal Melbourne Hospital, The University of Melbourne, Victoria, Australia
Epilepsy Genetics Initiative . The Epilepsy Genetics Initiative: Systematic reanalysis of diagnostic exomes increases yield. Epilepsia. 2019;60:797–806. 10.1111/epi.14698
Contributor Information
Epilepsy Genetics Initiative, Email: Igm-contact@cumc.columbia.edu.
Epilepsy Genetics Initiative:
Samuel F. Berkovic, David B. Goldstein, Erin L. Heinzen, Brandon L. Laughlin, Daniel H. Lowenstein, Laura Lubbers, Randall Stewart, Vicky Whittemore, Kaitlin Angione, Carl W. Bazil, Louise Bier, Judith Bluvstein, Elise Brimble, Colleen Campbell, Gianpiero Cavalleri, Chelsea Chambers, Hyunmi Choi, Maria Roberta Cilio, Michael Ciliberto, Susannah Cornes, Norman Delanty, Scott Demarest, Orrin Devinsky, Dennis Dlugos, Holly Dubbs, Patricia Dugan, Michelle E. Ernst, Melissa Gibbons, Howard P. Goodkin, Ingo Helbig, Laura Jansen, Kaleas Johnson, Charuta Joshi, Natalie C. Lippa, Eric Marsh, Alejandro Martinez, John Millichap, Maureen S. Mulhern, Adam Numis, Kristen Park, Tommaso Pippucci, Annapurna Poduri, Brenda Porter, Brigid Regan, Tristan T. Sands, Ingrid E. Scheffer, John M. Schreiber, Beth Sheidley, Nilika Singhal, Lacey Smith, Joseph Sullivan, Alan Taylor, Patricia Tolete, Tahseen M. Afgani, Vimla Aggarwal, Rosemary Burgess, Tracy Dixon‐Salazar, Parisa Hemati, Julie Milder, Slavé Petrovski, Anya Revah‐Politi, and Nicholas Stong
REFERENCES
- 1. Perucca P, Scheffer IE, Harvey AS, James PA, Lunke S, Thorne N, et al. Real‐world utility of whole exome sequencing with targeted gene analysis for focal epilepsy. Epilepsy Res. 2017;131:797–8. [DOI] [PubMed] [Google Scholar]
- 2. Helbig KL, Farwell Hagman KD, Shinde DN, Mroske C, Powis Z, Li S, et al. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med. 2016;18:898–905. [DOI] [PubMed] [Google Scholar]
- 3. Eldomery MK, Coban‐Akdemir Z, Harel T, Rosenfeld JA, Gambin T, Stray‐Pedersen A, et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017;9:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wenger AM, Guturu H, Bernstein JA, Bejerano G. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med. 2017;19:209–14. [DOI] [PubMed] [Google Scholar]
- 5. Wright CF, McRae JF, Clayton S, Gallone G, Aitken S, FitzGerald TW, et al. Making new genetic diagnoses with old data: iterative reanalysis and reporting from genome‐wide data in 1,133 families with developmental disorders. Genet Med. 2018;20(10):1216–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Need AC, Shashi V, Schoch K, Petrovski S, Goldstein DB. The importance of dynamic re‐analysis in diagnostic whole exome sequencing. J Med Genet. 2017;54:155–6. [DOI] [PubMed] [Google Scholar]
- 7. Gibson KM, Nesbitt A, Cao K, Yu Z, Denenberg E, DeChene E, et al. Novel findings with reassessment of exome data: implications for validation testing and interpretation of genomic data. Genet Med. 2018;20(10):1298. [DOI] [PubMed] [Google Scholar]
- 8. Helbig I, Heinzen EL, Mefford HC, ILAE Genetics Commission . Primer Part 1‐The building blocks of epilepsy genetics. Epilepsia. 2016;57:861–8. [DOI] [PubMed] [Google Scholar]
- 9. Zhu X, Petrovski S, Xie P, Ruzzo EK, Lu YF, McSweeney KM, et al. Whole‐exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med. 2015;17:774–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Epilepsy Genetics Initiative . De novo variants in the alternative exon 5 of SCN8A cause epileptic encephalopathy. Genet Med. 2017;20:275–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Myers CT, Stong N, Mountier EI, Helbig KL, Freytag S, Sullivan JE, et al. De novo mutations in PPP3CA cause severe neurodevelopmental disease with seizures. Am J Hum Genet. 2017;101:516–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Al‐Mehmadi S, Splitt M, Ramesh V, For DDD Study group , DeBrosse S, Dessoffy K, et al. FHF1 (FGF12) epileptic encephalopathy. Neurol Genet. 2016;2:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guella I, Huh L, McKenzie MB, Toyota EB, Bebin EM, Thompson ML, et al. De novo FGF12 mutation in 2 patients with neonatal‐onset epilepsy. Neurol Genet. 2016;2:e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Siekierska A, Isrie M, Liu Y, Scheldeman C, Vanthillo N, Lagae L, et al. Gain‐of‐function FHF1 mutation causes early‐onset epileptic encephalopathy with cerebellar atrophy. Neurology. 2016;86:2162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Symonds JD, Joss S, Metcalfe KA, Somarathi S, Cruden J, Devlin AM, et al. Heterozygous truncation mutations of the SMC1A gene cause a severe early onset epilepsy with cluster seizures in females: detailed phenotyping of 10 new cases. Epilepsia. 2017;58:565–75. [DOI] [PubMed] [Google Scholar]
- 17. Weiss K, Terhal PA, Cohen L, Bruccoleri M, Irving M, Martinez AF, et al. De novo mutations in CHD4, an ATP‐dependent chromatin remodeler gene, cause an intellectual disability syndrome with distinctive dysmorphisms. Am J Hum Genet. 2016;99:934–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9:e1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Petrovski S, Gussow AB, Wang Q, Halvorsen M, Han Y, Weir WH, et al. The intolerance of regulatory sequence to genetic variation predicts gene dosage sensitivity. PLoS Genet. 2015;11:e1005492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zarate YA, Fish JL. SATB2‐associated syndrome: mechanisms, phenotype, and practical recommendations. Am J Med Genet A. 2017;173:327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zarate YA, Kalsner L, Basinger A, Jones JR, Li C, Szybowska M, et al. Genotype and phenotype in 12 additional individuals with SATB2‐associated syndrome. Clin Genet. 2017;92:423–9. [DOI] [PubMed] [Google Scholar]
- 23. Epi4K Consortium . Epi4K: gene discovery in 4,000 genomes. Epilepsia. 2012;53:1457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36:928–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Darin N, Reid E, Prunetti L, Samuelsson L, Husain RA, Wilson M, et al. Mutations in PROSC disrupt cellular pyridoxal phosphate homeostasis and cause vitamin‐B6‐dependent epilepsy. Am J Hum Genet. 2016;99:1325–37. [DOI] [PMC free article] [PubMed] [Google Scholar]