

Abstract
The costimulatory CD40L–CD40 dyad plays a major role in multiple sclerosis (MS). CD40 is highly expressed on MHCII+ B cells, dendritic cells and macrophages in human MS lesions. Here we investigated the role of the CD40 downstream signaling intermediates TNF receptor‐associated factor 2 (TRAF2) and TRAF6 in MHCII+ cells in experimental autoimmune encephalomyelitis (EAE). Both MHCII–CD40–Traf2 −/− and MHCII–CD40–Traf6 −/− mice showed a reduction in clinical signs of EAE and prevented demyelination. However, only MHCII–CD40–Traf6 −/− mice displayed a decrease in myeloid and lymphoid cell infiltration into the CNS that was accompanied by reduced levels of TNF‐α, IL‐6 and IFN‐γ. As CD40–TRAF6 interactions predominantly occur in macrophages, we subjected CD40 flfl LysM cre mice to EAE. This myeloid‐specific deletion of CD40 resulted in a significant reduction in EAE severity, reduced CNS inflammation and demyelination.
In conclusion, the CD40–TRAF6 signaling pathway in MHCII+ cells plays a key role in neuroinflammation and demyelination during EAE. Concomitant with the fact that CD40–TRAF6 interactions are predominant in macrophages, depletion of myeloid CD40 also reduces neuroinflammation. CD40–TRAF6 interactions thus represent a promising therapeutic target for MS. © 2018 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: CD40, TNF receptor‐associated peptides and proteins, macrophages, experimental autoimmune encephalomyelitis, multiple sclerosis
Introduction
Multiple sclerosis (MS) is a chronic inflammatory disease of the CNS, characterized by a breakdown of the blood–brain barrier, multifocal inflammatory lesions, demyelination, reactive gliosis, axonal degeneration and neurodegeneration 1, 2. MS affects more than two million people worldwide and is one of the most common causes of neurological disability in young adults 2, 3. The exact etiology of MS is unknown, although it is considered to be an autoimmune disease in which lymphocytes programmed to recognize myelin antigen trigger a cascade of events resulting in the formation of acute inflammatory, demyelinating lesions in myelin sheaths in the white matter areas of the CNS 4.
The costimulatory CD40–CD40L dyad has a critical role in driving immune responses and plays a key role in several chronic inflammatory diseases, such as atherosclerosis, obesity, rheumatoid arthritis and MS 5. Both CD40l −/− and CD40 −/− mice are protected against experimental autoimmune encephalomyelitis (EAE) 6, 7. CD40 −/− mice have reduced T cell priming against myelin oligodendrocyte glycoprotein (MOG) and these T cells produce less IFN‐γ 8. Furthermore, CD40 deficiency inhibits leukocyte infiltration into the CNS 8. CD40 is present in MS on different cell types, including B cells, monocytes, macrophages, endothelial cells, T cells and CNS‐resident cells 9, 10. Activation of these cells by CD40L results in the secretion of proinflammatory cytokines and chemokines 11, 12, and T cell expansion 13, which promotes the ongoing inflammation in the CNS. Antibody‐mediated blockade of CD40L blocks CNS inflammation and disease progression in mice with relapsing EAE 14, 15. Accordingly, CD40 antagonists repressed EAE onset and disease severity in marmoset monkeys 16. Fueled by the success of these experimental studies, clinical trials were initiated. The treatment of MS patients with the anti‐CD40L mAb IDEC‐131 was successful in a pilot study, although the subsequent phase II trial was halted due to the occurrence of a case of severe thromboembolism in a parallel Crohn's disease trial that evaluated IDEC‐131 17. Thus, blockade of CD40L is not a feasible therapeutic strategy, as it can result in thromboembolic side‐effects due to the disruption of αIIbβ3–CD40L interactions in arterial thrombi 18, 19. Moreover, complete inhibition of the CD40–CD40L dyad may result in immunosuppression 20. However, modulation of CD40 downstream signaling pathways, preferably in a cell type‐specific manner, may offer a strategy to reduce the risk for these side‐effects 19, 20. Upon binding of CD40L, CD40 recruits TNF receptor‐associated factors (TRAFs) to exert signaling to downstream pathways 20, 21. The intracellular domain of CD40 contains a distal binding domain for TRAF2/3/5 and a proximal domain for TRAF6 20, 21. Which signaling pathway is activated depends on the cell type and the cellular environment. Here we unravel the role of the CD40–TRAF2 and the CD40–TRAF6 signaling pathways in neuroinflammation by subjecting mice with site‐directed mutagenesis for the TRAF6 or TRAF2/3/5 binding site on the CD40 intracellular tail in MHCII+ cells to EAE. We found that both signaling pathways play an important, but distinct, role in EAE disease development, with a prominent role for CD40–TRAF6 signaling in neuroinflammation. As CD40–TRAF6 signaling is especially important in the macrophage 22, 23, we investigated the role of myeloid‐specific CD40 signaling in EAE, using CD40 flfl LysM cre mice and found that macrophage CD40 is indeed an important driver of EAE.
Materials and methods
Mice
CD40 flfl mice were generated by custom design at Ozgene Pty Ltd (Bentley, WA, Australia). A conditional allele was created by inserting loxP sites upstream of exon 2 and downstream of exon 3. Mice carrying the floxed CD40 gene were crossed with LysM cre mice 24 to obtain myeloid‐specific CD40‐deficient mice (CD40 flfl LysM cre mice). Littermates not containing the LysMcre driver were used as controls (CD40 flfl or WT mice). CD40 flfl LysM cre, CD40 flfl, MHCII–CD40–T2/3/5 −/−, MHCII–CD40–T6 −/− and CD40 −/− mice were bred and maintained at the animal facility of the Academic Medical Centre (Amsterdam, the Netherlands). C57Bl6 mice were used as a WT control in the TRAF signaling EAE experiment and purchased from Charles River (Lyon, France). All the experimental procedures were approved by the Ethical Committee for Animal Experiments of the Academic Medical Centre, Amsterdam, the Netherlands (AMC).
EAE induction
To investigate the effects of macrophage CD40 and the CD40–TRAF pathways in EAE, 12‐week‐old female CD40 flfl LysM cre, CD40 flfl, MHCII–CD40–T2/3/5 −/−, MHCII–CD40–T6 −/−, CD40 −/− mice and WT mice had ad libitum access to food and water and were housed under a 12 h light/dark cycle. On day 0, mice were immunized subcutaneously with 200 µg of a MOG peptide (MOG35–55) emulsified in CFA Complete Freund's Adjuvant supplemented with 4 mg/ml Mycobacterium tuberculosis H37Ra (Hooke Laboratories, Lawrence, MA, USA). Mice were injected i.p. on days 0 and 1 with 300 (CD40 flfl LysM cre and CD40 flfl mice, n = 12/group) or 100 ng (MHCII–CD40–T2/3/5 −/−, MHCII–CD40–T6 −/−, CD40 −/− and WT mice, n = 15/group) pertussis toxin (Hooke Laboratories). A control group was included that received only CFA and pertussis toxin (n = 6). Body weight and neurological symptoms were monitored daily. The symptoms were graded using the following scale: 0 = no neurological abnormalities; 0.5 = partial loss of tail tonus; 1 = complete loss of tail tonus; 2 = hind limb paresis; 3 = partial hind limb paralysis; 4 = complete hind limb paralysis; 4.5 = paralysis up to the diaphragm, 5 = death. Scoring of clinical symptoms was performed by an observer who was blinded for the experimental conditions.
Immunohistochemistry
The cerebellum and spinal cord were collected and fixed in 4% paraformaldehyde and embedded in paraffin. Inflammation was graded on 4 µm thick H&E‐stained sections. Per mouse, between four and six sections of the spinal cord were analyzed. Immunohistochemistry was performed for CD45 (30‐F11, 1:5000; BD, Breda, the Netherlands), mac‐3 (M3/84, 1:30; BD), CD3 (CD3‐12, 1:100; AbD Serotec, Puchheim, Germany) and FoxP3 (1:50, FJK‐16s; eBioscience, San Diego, CA, USA). The spinal cord and cerebellum were stained with luxol fast blue (0.1% LFB; Sigma Aldrich, Saint Louis, MO, USA) for observation of myelin loss. All stained sections were analyzed by an observer who was blinded to the experimental conditions.
In vitro bone marrow‐derived macrophage culture
CD40 flfl LysM cre and CD40 flfl mouse bone marrow cells were harvested from the femur and tibia and cultured in RPMI‐1640 with 2 mm l‐glutamine, 10% FCS, penicillin (100 U/ml), streptomycin (100 µg/ml) (Gibco, Waltham, MA, USA) and 15% L929‐conditioned medium. On day 8, cells were harvested, seeded at 500 000 cells/ml and laden with 40 µg/ml (DiI‐labeled) myelin for 1.5 h. Myelin uptake was measured using flow cytometry.
Antibodies and flow cytometry
CD40 flfl LysM cre and CD40 flfl bone marrow‐derived macrophages (BMDMs) were treated with FGK45 (CD40 agonistic antibody, 30 µg/ml), IFN‐γ (5 ng/ml) and LPS (100 ng/ml). After 6 h, cells were incubated with CD16/CD32 (clone 93, 1:100; eBioscience) primary antibody diluted in FACS buffer (PBS containing 0.5% BSA and 2 mm EDTA) to prevent nonspecific binding of antibodies to the Fc receptors. Cells were then incubated with fluorescently labeled antibodies for CD40 (clone 3/23, 1:200; BioLegend, San Diego, CA, USA), CD80 (16‐10A1, 1:200; BD Pharmingen, Breda, the Netherlands), CD86 (GL‐1, 1:100; BioLegend), MHCII (M5/114.15.2, 1:100; eBioscience), CD36 (MF3, 1:100; Bio‐Rad, Hercules, CA, USA) and CD204 (2F8, 1:100; Bio‐Rad). Blood was obtained by cardiac puncture and collected using EDTA‐filled syringes from CD40 flfl LysM cre and CD40 flfl mice, MHCII–CD40–T2/3/5 −/−, MHCII–CD40–T6 −/−, CD40 −/− mice and WT mice. Spleen was collected from MHCII–CD40–T2/3/5 −/−, MHCII–CD40–T6 −/−, CD40 −/− and WT mice. Erythrocytes in blood and spleen were removed by incubation with hypotonic lysis buffer (8.4 g NH4Cl and 0.84 g NaHCO3 per liter of distilled water). Prior to labeling, cell suspensions were incubated with a CD16/32 antibody (clone 93, 1:100; eBioscience). CD45 (30‐F11, 1:200; eBioscience), CD19 (1D3, 1:100; eBioscience), CD3 (145‐2C11, 1:100; BioLegend), CD8 (53–6.7, 1:100; eBioscience), CD4 (GK1.5, 1:100; BD), Ly6G (1A8, 1:200; BD), SiglecF (E50‐2440, 1:100; BD) F4/80 (Cl:A3‐1, 1:100; AbD Serotec) and Ly6C (HK1.4, 1:100; eBioscience) antibodies were incubated with the indicated tissues. Staining was analyzed by flow cytometry (FACSCanto II; BD Biosciences, Breda, the Netherlands) and the FlowJo software version 7.6.5. (Tree Star Inc., Ashland, OR, USA).
Western blot
CD40 flfl LysM cre and CD40 flfl BMDMs were cultured as described above, treated for 10 min or 24 h with FGK45 (30 µg/ml), IFN‐γ (5 ng/ml) and LPS (100 ng/ml) and subsequently lysed in RIPA lysis buffer. Protein concentrations were determined using the Pierce BCA Protein Assay Kit (Thermo Scientific, Waltham, MA, USA). Equal amounts of protein samples were loaded on to a SDS polyacrylamide gel and transferred to a nitrocellulose membrane (Bio‐Rad). After blocking with 5% BSA in PBS containing 0.05% Tween‐20, blots were incubated overnight with anti‐CD40 (Abcam, Cambridge, UK) and anti‐α‐tubulin (Cedarlane, Burlington, ON, Canada). Blots were then washed and incubated with the appropriate HRP‐conjugated secondary antibody (1:800; Thermo Scientific) and visualized using the SuperSignal West Pico Plus Chemiluminescent Substrate kit (Thermo Scientific).
Analysis of Igs
MOG IgG was measured in plasma samples using an anti‐MOG(35–55) IgG Quantitative ELISA Kit (SensoLyte®, Fremont, CA, USA).
Analysis of cytokine profiles
The production of pro‐ and anti‐inflammatory mediators was assessed by ELISA in cell‐free supernatants of 24 h myelin (40 µg/ml), FGK45 (30 µg/ml), IFN‐γ (5 ng/ml) and LPS (100 ng/ml) or IL‐4 (10 ng/ml) treated CD40 flfl LysM cre and CD40 flfl BMDMs using commercial kits for human IL‐12, IL‐6 and the TNF‐α CytoSet ELISA Kit (Biosource, Nivelles, Belgium) according to the manufacturers' protocols. The samples were measured using a Luminex 200 (Bio‐Rad).
RNA isolation and gene expression analysis
Total RNA from the CNS was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA) and RNA from BMDMs was isolated using High Pure RNA Isolation Kits (Roche, Basel, Switzerland). RNA was reverse transcribed using an iScript cDNA synthesis kit (Bio‐Rad) and quantitative (q)PCR was performed using a SYBR Green PCR Kit (Applied Biosystems, Leusden, the Netherlands) on a ViiA7 real‐time PCR system (Applied Biosystems). Expression levels of transcripts obtained using RT‐qPCR were normalized to the mean expression levels of the reference transcripts Gapdh and Rplp0. Primer sequences can be found in supplementary material, Table S1.
Statistical analysis
Results are presented as mean ± SEM. Calculations were performed using GraphPad Prism 7.0 software (GraphPad Software Inc., San Diego, CA, USA). Statistical significance was evaluated using unpaired Student's t‐tests. Clinical parameters were analyzed by nonparametric (Mann–Whitney) test. P values < 0.05 were considered statistically significant. n represents the number of biological replicates.
Results and discussion
Both CD40–TRAF6 and CD40–TRAF2 signaling pathways drive EAE
To define which CD40 signaling pathways are involved in neuroinflammation, we investigated the effects of disruption of either CD40–TRAF6 or CD40–TRAF2 signaling in antigen‐presenting cells in EAE. CD40‐deficient mice expressing a chimeric CD40 transgene with mutations at the TRAF6 or TRAF2/3/5 binding site under the control of the MHCII promotor (MHCII–CD40–T6 −/−, MHCII–CD40–T2/3/5 −/−) 25, CD40‐deficient mice (CD40 −/−) and C57Bl6 WT mice (WT) were subjected to EAE. In accordance with previous studies 8, CD40 −/− mice were completely protected against EAE (Figure 1A and Table 1). Compared with WT mice, disease severity was significantly lower in both the MHCII–CD40–T2/3/5 −/− and MHCII–CD40–T6 −/− mice and was found to be lowest in the MHCII–CD40–T6 −/− mice (maximum disease score 4.2 ± 0.2 in WT versus 2.7 ± 0.4 in MHCII–CD40–T2/3/5 −/− and 1.8 ± 0.3 in MHCII–CD40–T6 −/−, p = 0.0049; Figure 1A and Table 1). Consistently, the cumulative disease score (area under the curve; AUC) of the MHCII–CD40–T2/3/5 −/−, MHCII–CD40–T6 −/− and CD40 −/− mice was significantly lower than the cumulative score of the WT mice (Table 1). Moreover, the average disease onset was >4 days delayed in the MHCII–CD40–T2/3/5 −/− and MHCII–CD40–T6 −/− mice compared with WT mice (onset on day 13.4 ± 0.9 and day 14.6 ± 0.7 versus day 8.9 ± 0.6, p < 0.05) (Figure 1A and Table 1). The incidence of EAE in WT mice was 100.0, 71.4% in both MHCII–CD40–T2/3/5 −/− and MHCII–CD40–T6 −/− mice and 0% in CD40 −/− mice (p < 0.05, Table 1). Survival rates were only 71.4% for WT mice, whereas all MHCII–CD40–T2/3/5 −/−, MHCII–CD40–T6 −/− and CD40 −/− mice survived until the end of the experiment (Table 1). Additionally, in the MHCII–CD40–T6 −/− mice we observed only 5.3 ± 1.9% body weight loss (p < 0.001, Figure 1B and Table 1) at the peak of disease compared with day 0, whereas WT mice suffered an average loss of 16.9 ± 1.4% (Figure 1B and Table 1). Comparable with WT mice, the MHCII–CD40–T2/3/5 −/− suffered an average of 17.6 ± 3.5% body weight loss (Figure 1B and Table 1). CD40 −/− mice only had 1.2 ± 1.1% body weight loss (Figure 1B and Table 1). WT mice displayed extensive CNS demyelination compared with CD40 −/−, MHCII–CD40–T6 −/− and MHCII–CD40–T2/3/5 −/− mice, as shown by LFB staining (see supplementary material, Figure S1A). These data confirm that CD40 plays a pivotal role in EAE development and reveal that both CD40–TRAF2/3/5 the CD40–TRAF6 signaling pathways are involved in its pathogenesis.
Figure 1.
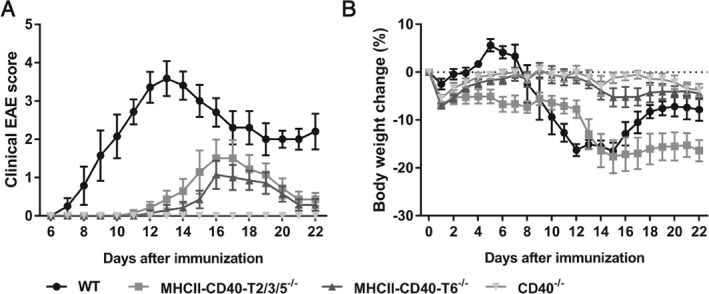
Deficiency of CD40, the CD40–TRAF2/3/5 interaction and the CD40–TRAF6 interaction is protective in EAE. (A) Clinical EAE scores of MHCII–CD40–T2/3/5 −/− (n = 7), MHCII–CD40–T6 −/− (n = 7), CD40 −/− (n = 7) and WT (n = 7) mice 22 days after EAE induction. (B) Percentage body weight change compared with day 0. Mean ± SEM.
Table 1.
EAE disease measurements of MHCII–CD40–T2/3/5 −/−, MHCII–CD40–T6 −/−, CD40 −/− mice and WT mice
| Group | Incidence (%) | Survival (%) | AUC | Mean day of disease onset | Mean maximum disease score | Body weight loss (%) |
|---|---|---|---|---|---|---|
| WT | 100.0 | 71.4 | 30.8 | 8.9 | 4.2 | 16.9 |
| MHCII–CD40–T2/3/5−/− | 71.4 | 100.0 | 8.0** | 13.4** | 2.7* | 17.6 |
| MHCII–CD40–T6−/− | 71.4 | 100.0 | 5.0*** | 14.6** | 1.8** | 5.3*** |
| CD40−/− | 0.0 | 100.0 | 0.0*** | N/A | N/A | 1.2*** |
*p < 0.05, **p < 0.01 as determined by the nonparametric Mann–Whitney U‐test for disease scores.
Percentage body weight loss was calculated from day 15 compared with day 0 and significance was determined by Student's t‐test; ***p < 0.001 versus WT.
CD40‐, CD40–TRAF6‐ and CD40–TRAF2/3/5‐deficient mice have impaired anti‐MOG IgG production
As CD40 signaling plays an important role in isotype switching 26 we analyzed MOG‐specific IgG levels in the plasma of mice immunized for EAE. Twenty‐eight days after EAE induction, MOG‐specific IgG titers were significantly lower in mice lacking CD40, CD40–T2/3/5 or CD40–T6 signaling compared with WT mice (Figure 2), confirming the impaired Ig isotype switching in the absence of CD40, CD40–TRAF2/3/5 or CD40–TRAF6 signaling.
Figure 2.
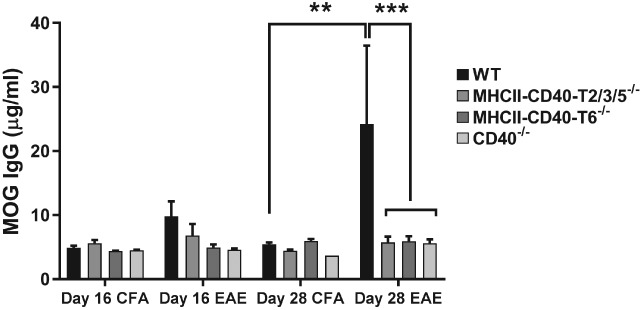
CD40‐, CD40–TRAF6‐ and CD40–TRAF2/3/5‐deficient mice have impaired anti‐MOG IgG production. MOG‐specific IgG concentrations in serum of MHCII–CD40–T2/3/5 −/−, MHCII–CD40–T6 −/−, CD40 −/− and WT mice 16 days after induction (n = 8/group) and 28 days after induction (n = 7/group). Mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 versus WT.
CD40‐ and CD40–TRAF6‐deficient mice are protected against neuroinflammation
Analysis of the CNS (cerebellum and spinal cord) shows that the number of total leukocytes (CD45+ cells), macrophages (Mac3+ cells) and T cells (CD3+ cells) that infiltrated the CNS was significantly reduced in MHCII–CD40–T6 −/− and CD40 −/− mice compared with WT mice subjected to EAE (Figure 3A,B). Although MHCII–CD40–T2/3/5 −/− mice exhibited significantly milder EAE disease symptoms than WT mice, the amount of immune cell infiltration in the CNS did not differ (Figure 3A,B). Moreover, although not significant, the number of CD45+ cells in the spinal cord of WT versus MHCII–CD40–TRAF2/3/5 −/− mice even seemed to be increased (Figure 3A,B), whereas clinical symptoms were ameliorated. One of the possible reasons why this increase in CD45+ cells did not result in an aggravated EAE response is that the number of immune‐regulatory FoxP3+ T cells was also increased (see supplementary material, Figure S1B), resulting in a ‘balanced’ immune response 27. In a previous study, in which MHCII–CD40–T2/3/5 −/− mice were subjected to atherosclerosis, we also observed an increase in CD3+ T cells during disease and found that the increase was due to an increase in regulatory T cells 22. The balancing of the immune response is also reflected by the fact that gene expression analysis also showed no differences in the expression of inflammatory genes in the CNS of these mice (Figure 3C). The reduction in EAE severity in the MHCII–CD40–T2/3/5 −/− mice might be explained by the reduced MOG IgG production by the B cells (Figure 2).
Figure 3.
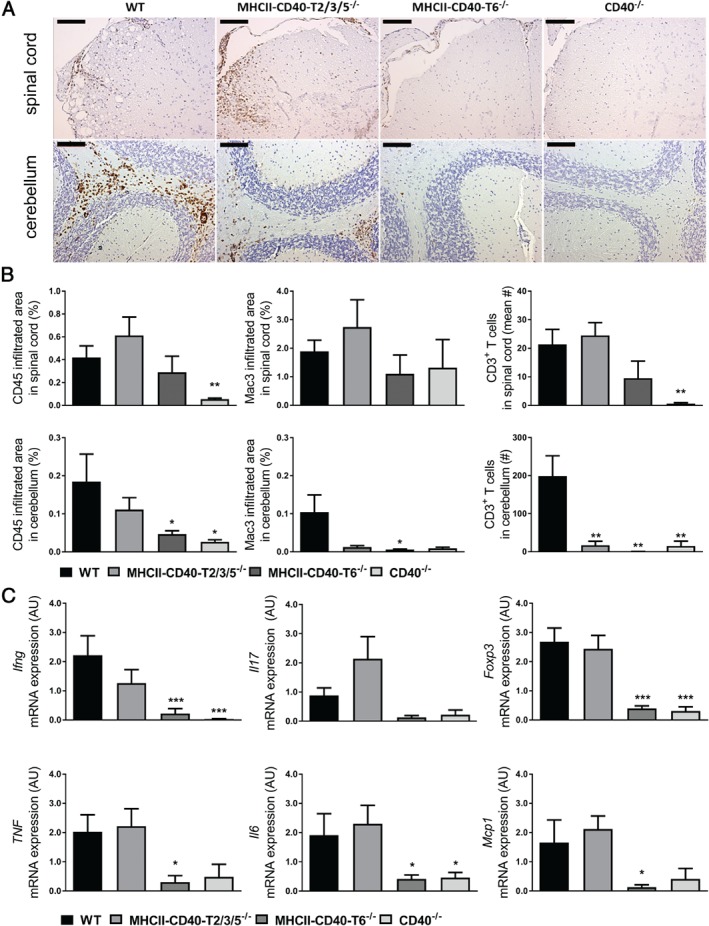
CD40‐ and CD40–TRAF6‐deficient mice exhibit reduced neuroinflammation. (A) Representative images of CD45+ staining of spinal cord and cerebellum sections, bars 100 µm. (B) Quantification of CD45+, mac‐3+ and CD3+ immune cell infiltrates in the spinal cord and cerebellum from mice sacrificed at day 16 (n = 8/group). (C) mRNA expression levels of inflammatory genes in cerebellum obtained 16 days after induction of EAE, presented as relative expression compared with Gapdh and Rplp0. CD40 −/− and MHCII–CD40–T6 −/− mice, but not MHCII–CD40–T2/3/5 −/− mice, had significantly reduced inflammatory cytokine expression in the CNS during EAE compared with WT mice (n = 8/group). Mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 versus WT.
Both CD40 −/− and MHCII–CD40–T6 −/− mice had significantly reduced Ifng, Foxp3, Tnf, Il6 and Mcp1 expression in the CNS compared with WT or MHCII–CD40–T2/3/5 −/− mice during the peak of disease (Figure 3C). MHCII–CD40–T2/3/5 −/− mice did not show any changes in CNS cytokine levels (Figure 3C). Analysis of the peripheral immune response in blood and spleen showed only minor changes (see supplementary material, Figure S2). In MHCII–CD40–T6 −/− mice, we observed an increase in the percentage of CD19+ (B) cells in blood and spleen (see supplementary material, Figure S2A,B) and a decreased percentage of Ly6G+ cells (neutrophils) in blood compared with WT mice during EAE. This decrease in Ly6G+ cells (neutrophils) was also observed in MHCII–CD40–T2/3/5 −/− mice.
Taken together, our data reveal that both CD40–TRAF6 and CD40–TRAF2/3/5 signaling pathways in MHCII+ cells are involved in the pathogenesis of EAE. Although both MHCII–CD40–T2/3/5 −/− and MHCII–CD40–T6 −/− mice exhibited a lower disease score, only MHCII–CD40–T6 −/− mice were protected against EAE‐associated body weight loss and CNS inflammation, indicating that CD40–TRAF6 signaling is more relevant in EAE than CD40–TRAF2/3/5 signaling.
The CD40–TRAF6 pathway is especially known to be important for CD40 signaling in the myeloid lineage. CD40–TRAF6 interactions induce classical monocyte activation, monocyte recruitment and macrophage activation via canonical NF‐κB signaling 23, 27, 28, 29. Although the role of CD40–CD40L signaling in MS is mostly attributed to T cells 30 and B cells 31, the main cell types expressing CD40 in human MS lesions are monocytes, macrophages and activated microglia 7. Infiltrating macrophages and activated microglia present in active human MS lesions contribute to CNS injury and have mainly M1 characteristics, including high CD40 expression 32, 33, 34. Although macrophage CD40 is abundantly present in MS lesions, its role in the pathogenesis of MS is currently unknown.
Myeloid CD40‐deficient mice develop less severe EAE and exhibit decreased CNS inflammation
To study the role of myeloid CD40 on neuroinflammation in vivo, CD40 flfl LysM cre mice were generated, which exhibited an 84–89% reduction in CD40 expression in macrophages (Figure 4A,B). We immunized CD40 flfl LysM cre and CD40 flfl (WT) mice for induction of EAE. Deficiency of myeloid CD40 resulted in significant lower disease severity (maximum disease score 3.0 ± 0.2) and cumulative disease score (AUC 14.4 ± 2.0) compared with WT controls (maximum score 4.0 ± 0.3, p = 0.010, AUC 24.7 ± 1.9, p = 0.0013; Figure 4C and Table 2). Moreover, survival of CD40 flfl LysM cre mice was increased compared with WT mice (100% versus 75%, Table 2). In WT mice, the maximum body weight loss was 13.9 ± 2.0% and resulted in a 4.3 ± 3.4% weight loss at the end of the experiment. CD40 flf LysM cre mice had significantly less severe weight loss, with a maximum of only 7.9 ± 1.7% loss and complete recovery of body weight at the end of the experiment (p < 0.05, Figure 4D and Table 2). As expected from a macrophage‐specific CD40 knockout, IgM, total IgG and anti‐MOG IgG levels did not differ between CD40 flfl LysM cre and WT mice (see supplementary material, Figure S3). The CNS of CD40 flfl LysM cre mice exhibited a significant reduction in CD45+ leukocyte infiltration, which was due to a reduction in Mac3+ macrophages/active microglia and CD3+ T cells (Figure 4E,F), revealing a reduction in CNS inflammation in the absence of macrophage CD40. No differences in peripheral immune responses were observed (see supplementary material, Figure S4C). In addition to the previously described role of CNS CD40 on microglial cell activation during EAE 35, we here show that loss of peripheral macrophage CD40 signaling ameliorates EAE and prevents inflammation, revealing a key role for macrophage CD40 signaling in neuroinflammation.
Figure 4.
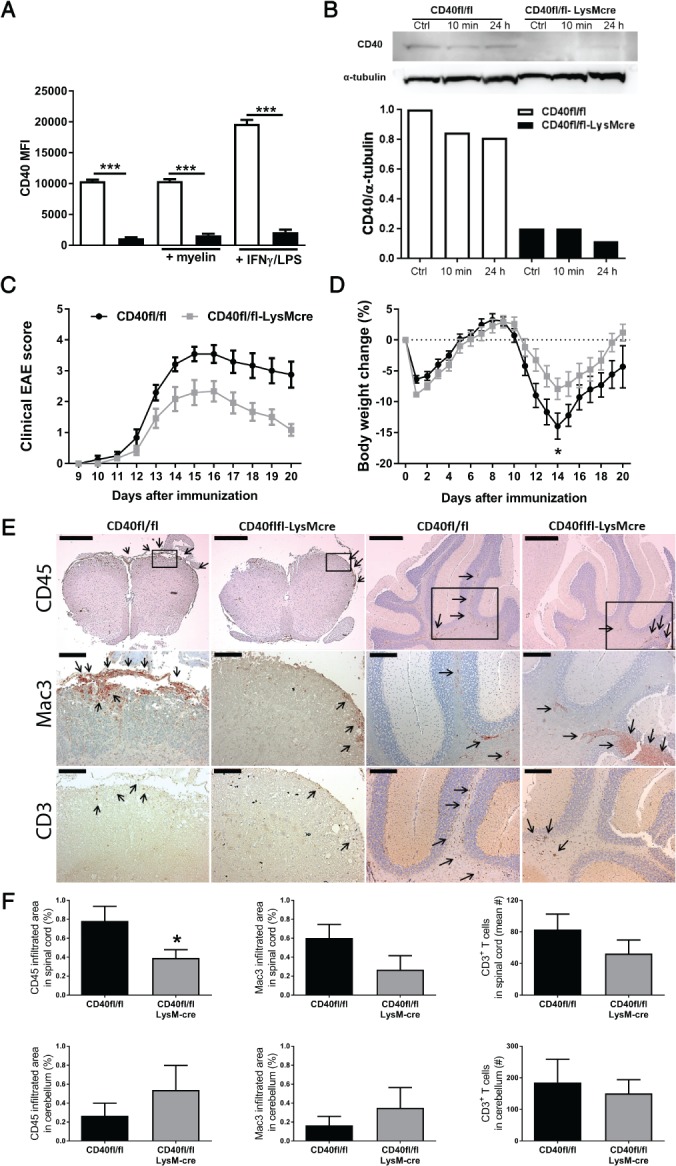
Myeloid CD40 deficiency ameliorates EAE. BMDMs from CD40 flfl LysM cre mice and CD40 flfl (WT) mice were stimulated for 6 h with FGK45 (30 µg/ml) and myelin (40 µg/ml) or IFN‐γ (5 ng/ml) and LPS (100 ng/ml). (A) MFI of CD40 measured by flow cytometry. (B) Western blot of CD40 (40 kDa band size) and α‐tubulin (50 kDa band size) and quantification of CD40 protein expression in CD40 flfl LysM cre mice and CD40 flfl (WT) BMDMs stimulated with FGK45 for 10 min or 24 h. (C) Clinical EAE scores of CD40 flfl LysM cre mice and CD40 flfl (WT) mice during the 20 days after induction (n = 12/group). (D) Percentage body weight change compared with the start weight at day 0. (E) Representative images of CD45, MAC3 and CD3 immunostaining of spinal cord and cerebellum sections. CD45: bars, spinal cord and cerebellum, 500 µm. MAC3 and CD3: bars, spinal cord, 100 µm; cerebellum, 200 µm. (F) Counts of CD45 (leukocytes), mac‐3 (macrophages) and CD3 (T cells) immune cell infiltrates in the spinal cord and cerebellum. Means ± SEM. The in vitro results in (A) and (B) are means ± SEM from experimental triplicates and are representative of at least three independent experiments. *p < 0.05 versus WT.
Table 2.
EAE disease measurements of CD40 flfl LysM cre mice and WT mice
| Group | Incidence (%) | Survival (%) | AUC | Mean day of disease onset | Mean maximum disease score | Body weight loss (%) |
|---|---|---|---|---|---|---|
| WT | 85.7 | 75.0 | 24.7 | 12.2 | 4.0 | 13.9 |
| CD40flflLysMcre | 80.0 | 100.0 | 14.4** | 12.8 | 3.0* | 7.9* |
Percentage body weight loss was calculated from day 14 compared with day 0.
*p < 0.05, **p < 0.01 versus WT as determined by the nonparametric Mann–Whitney U‐test for disease scores or Student's t‐test for body weight loss.
In EAE, myeloid cells cause CNS damage by presenting antigen, secretion of cytokines and phagocytosis of cell debris, resulting in the production of free radicals 36. In EAE lesions, macrophages phagocytose myelin and allow the removal of cell debris, critical for effective repair 33. CNS LFB staining displayed that CD40 flfl LysM cre mice were protected against EAE‐induced myelin loss (Figure 5A). Flow cytometry analysis of myelin‐laden BMDMs from CD40 flfl LysM cre and WT mice show that CD40 flfl LysM cre BMDMs ingested significantly less myelin than WT BMDMs (54.8 ± 8.6% reduction, n = 4) (Figure 5B). CD40 flfl LysM cre BMDMs also expressed less CD36, suggesting that macrophage CD40 mediates clearance of myelin debris (Figure 5C). Earlier research using bone marrow chimeric mice showed that CD40 expression on hematopoietic cells is essential for the priming of encephalitogenic T cells 8. FACS analysis revealed that CD40 flfl LysM cre BMDMs exhibited a reduced expression of the classically activated macrophage markers CD80 and CD86, an unchanged expression of MHCII and a decrease in the scavenger receptor and, alternatively, the activated macrophage marker CD204 (Figure 5D–G). The reduced expression of costimulatory molecules supports the results on the reduced EAE development in these mice as costimulation is an essential signal for T cell activation 37, 38. Gene expression analysis of BMDMs incubated with myelin showed no significant differences (see supplementary material, Figure S4A,B). mRNA levels of Il12b were decreased in CD40 flfl LysM cre macrophages treated with LPS/IFN‐γ, whereas Il6, Tnf and Nos2 mRNA levels were increased (see supplementary material, Figure S4A). However, protein levels of these cytokines were not affected (see supplementary material, Figure S4C). CD40 flfl LysM cre macrophages treated with IL‐4 showed increased Cdh1 gene expression compared with WT BMDMs, but no differences were observed in mRNA levels of other immunomodulatory genes, including Cd204, Il10 and Tgfb (see supplementary material, Figure S4B). In MS lesions, human foamy macrophages and microglia express proinflammatory (CD40 and iNOS) markers as well as anti‐inflammatory markers (MR) 33. Macrophages are diverse and plastic, their phenotype is dependent on cues from the microenvironment, including inflammatory signals 39. This might explain why we do not observe a reduced inflammatory phenotype in vitro, whereas the CD40 flfl LysM cre mice display reduced neuroinflammation and are protected against clinical symptoms of EAE in vivo.
Figure 5.
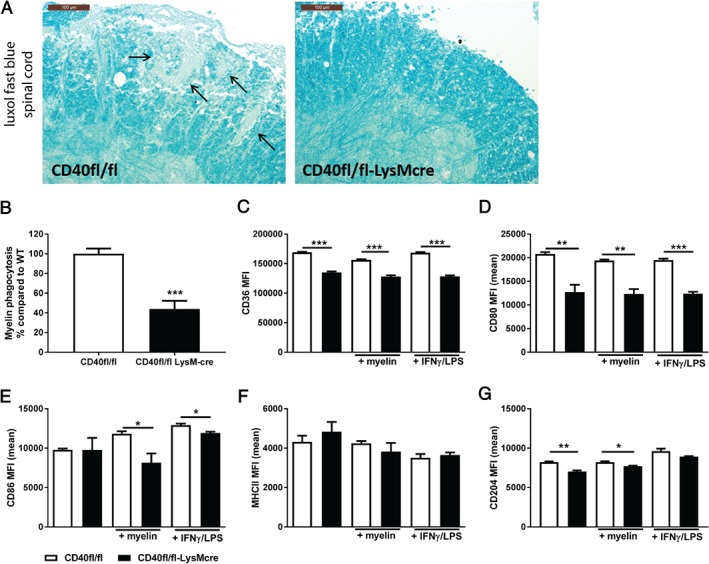
CD40‐deficient macrophages have decreased myelin phagocytosis capacity and display reduced costimulatory capacity. (A) Representative LFB staining of spinal cord; arrows indicate areas with loss of myelin; bars 100 µm. (B) Myelin phagocytosis capacity of CD40 flfl LysM cre mice and CD40 flfl (WT) BMDMs. BMDMs from CD40 flfl LysM cre mice and WT mice were stimulated for 6 h with FGK45 (30 µg/ml) and myelin (40 µg/ml) or IFN‐γ (5 ng/ml) and LPS (100 ng/ml). Flow cytometry analysis of (C) CD36, (D) CD80, (E) CD86, (F) MHCII, and (G) CD204 expression of CD40 flfl LysM cre mice and WT BMDMs. In vitro data in (B)–(F) are means ± SEM from experimental triplicates and are representative of at least two independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 versus WT.
Conclusions
We have shown that macrophage CD40 plays a key role during neuroinflammation, probably via CD40–TRAF6 signaling. However, the reduction in neuroinflammation was stronger in mice lacking CD40–TRAF6 signaling in all MHCII+ cells than in mice lacking CD40 signaling in macrophages, suggesting that both CD40–TRAF6 signaling in macrophages and other antigen‐presenting cells including B cells and dendritic cells drive neuroinflammation. Recently, we have developed small molecule inhibitors of the CD40–TRAF6 interaction, TRAF‐STOPs, and we were able to show that these small molecule inhibitors can reduce atherosclerosis, peritonitis, sepsis and metabolic complications of diet‐induced obesity in mice 23, 28, 29. Interestingly, TRAF‐STOP treatment was also able to reduce CNS leukocyte infiltration during EAE in both mice and rats but unfortunately failed to ameliorate disease severity 40. Further optimization of TRAF‐STOPs for more optimal delivery in the CNS may be required to achieve better therapeutic results.
Our data reveal that blocking CD40 in a cell type‐specific manner, or inhibition of parts of the CD40 signaling, has great therapeutic potential for the treatment of MS, as it will cause limited immune‐suppressive side‐effects compared with untargeted inhibition of the entire CD40 pathway.
Author contributions statement
The study presented here was carried out in collaboration among all authors. All authors read and approved the final manuscript. SA and TS contributed to the study concept and design, performing experiments, acquisition of data, analysis and interpretation of data, drafting of the manuscript and statistical analysis. PK, CT and MR performed experiments and acquisition of data. MT, LB and CR carried out the acquisition and immunohistochemistry of the CNS. MW, GK and EL contributed to the study concept and design, analysis and interpretation of data, drafting of the manuscript and critical revision of the manuscript for important intellectual content, obtained funding and offered study supervision.
SUPPLEMENTARY MATERIAL ONLINE.
Supplementary figure legends
Figure S1. CNS inflammation of MHCII–CD40–T2/3/5−/−, MHCII–CD40–T6−/−, CD40−/− and WT mice
Figure S2. Peripheral immune responses in MHCII–CD40–T2/3/5 −/−, MHCII–CD40–T6 −/−, CD40 −/−, CD40 flfl LysM cre and WT mice
Figure S3. Normal Ig isotype switching in CD40 flfl LysM cre mice
Figure S4. CD40‐deficient macrophages have a diverse inflammatory pattern
Table S1. Primer sequences
Supporting information
Supplementary figure legends
Figure S1. CNS inflammation of MHCII–CD40–T2/3/5 −/−, MHCII–CD40–T6 −/−, CD40 −/− and WT mice
Figure S2. Peripheral immune responses in MHCII–CD40–T2/3/5 −/−, MHCII–CD40–T6 −/−, CD40 −/−, CD40 flfl LysM cre and WT mice
Figure S3. Normal Ig isotype switching in CD40 flfl LysM cre mice
Figure S4. CD40‐deficient macrophages have a diverse inflammatory pattern
Table S1. Primer sequences
Acknowledgements
We acknowledge the support from the Netherlands CardioVascular Research Initiative, the Dutch Heart Foundation, Dutch Federation of University Medical Centers, the Netherlands Organization for Health Research and Development and the Royal Netherlands Academy of Sciences for the GENIUS project ‘Generating the best evidence‐based pharmaceutical targets for atherosclerosis’ (CVON2011‐19). This work was supported by the Dutch MS Research Foundation [13‐809MS], the Deutsche Forschungs Gemeinschaft (SFB 1123 to EL) and the European Research Council [ERC Consolidator grant to EL].
No conflicts of interest were declared.
References
- 1. Compston A, Coles A. Multiple sclerosis. Lancet 2002; 359: 1221–1231. [DOI] [PubMed] [Google Scholar]
- 2. Peterson JW, Trapp BD. Neuropathobiology of multiple sclerosis. Neurol Clin 2005; 23: vi–vii. [DOI] [PubMed] [Google Scholar]
- 3. Stys PK, Zamponi GW, van Minnen J, et al Will the real multiple sclerosis please stand up? Nat Rev Neurosci 2012; 13: 507–514. [DOI] [PubMed] [Google Scholar]
- 4. Frohman EM, Racke MK, Raine CS. Multiple sclerosis – the plaque and its pathogenesis. N Engl J Med 2006; 354: 942–955. [DOI] [PubMed] [Google Scholar]
- 5. Peters AL, Stunz LL, Bishop GA. CD40 and autoimmunity: the dark side of a great activator. Semin Immunol 2009; 21: 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chitnis T, Khoury SJ. Role of costimulatory pathways in the pathogenesis of multiple sclerosis and experimental autoimmune encephalomyelitis. J Allergy Clin Immunol 2003; 112: 837–849. [DOI] [PubMed] [Google Scholar]
- 7. Gerritse K, Laman JD, Noelle RJ, et al CD40‐CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci U S A 1996; 93: 2499–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Becher B, Durell BG, Miga AV, et al The clinical course of experimental autoimmune encephalomyelitis and inflammation is controlled by the expression of CD40 within the central nervous system. J Exp Med 2001; 193: 967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aarts SABM, Seijkens TTP, van Dorst KJF, et al The CD40–CD40L dyad in experimental autoimmune encephalomyelitis and multiple sclerosis. Front Immunol 2017; 8: 1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vaitaitis GM, Yussman MG, Waid DM, et al Th40 cells (CD4+CD40+ Tcells) drive a more severe form of experimental autoimmune encephalomyelitis than conventional CD4 T cells. PLoS One 2017; 12: e0172037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. D'Aversa TG, Weidenheim KM, Berman JW. CD40‐CD40L interactions induce chemokine expression by human microglia: implications for human immunodeficiency virus encephalitis and multiple sclerosis. Am J Pathol 2002; 160: 559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Goer de Herve MG, Delfraissy JF, Taoufik Y. Following direct CD40 activation, human primary microglial cells produce IL‐12 p40 but not bioactive IL‐12 p70. Cytokine 2001; 14: 88–96. [DOI] [PubMed] [Google Scholar]
- 13. Girvin A, Dal Canto M, Miller S. CD40/CD40L interaction is essential for the induction of EAE in the absence of CD28‐mediated co‐stimulation. J Autoimmun 2002; 18: 83–94. [DOI] [PubMed] [Google Scholar]
- 14. Howard LM, Dal Canto MC, Miller SD. Transient anti‐CD154‐mediated immunotherapy of ongoing relapsing experimental autoimmune encephalomyelitis induces long‐term inhibition of disease relapses. J Neuroimmunol 2002; 129: 58–65. [DOI] [PubMed] [Google Scholar]
- 15. Howard LM, Miga AJ, Vanderlugt CL, et al Mechanisms of immunotherapeutic intervention by anti‐CD40L (CD154) antibody in an animal model of multiple sclerosis. J Clin Invest 1999; 103: 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Laman J, 't Hart B, Brok H, et al Protection of marmoset monkeys against EAE by treatment with a murine antibody blocking CD40 (mu5D12). Eur J Immunol 2002; 32: 2218–2128. [DOI] [PubMed] [Google Scholar]
- 17. Couzin J. Drug discovery. Magnificent obsession. Science 2005; 307: 1712–1715. [DOI] [PubMed] [Google Scholar]
- 18. Andre P, Prasad KS, Denis CV, et al CD40L stabilizes arterial thrombi by a beta3 integrin‐dependent mechanism. Nat Med 2002; 8: 247–252. [DOI] [PubMed] [Google Scholar]
- 19. Kawai T, Andrews D, Colvin RB, et al Thromboembolic complications after treatment with monoclonal antibody against CD40 ligand. Nat Med 2000; 6: 114. [DOI] [PubMed] [Google Scholar]
- 20. Engel D, Seijkens T, Poggi M, et al The immunobiology of CD154‐CD40‐TRAF interactions in atherosclerosis. Semin Immunol 2009; 21: 308–312. [DOI] [PubMed] [Google Scholar]
- 21. Kusters PJH, Lutgens E, Seijkens TTP. Exploring immune checkpoints as potential therapeutic targets in atherosclerosis. Cardiovasc Res 2018; 114: 368–377. [DOI] [PubMed] [Google Scholar]
- 22. Lutgens E, Lievens D, Beckers L, et al Deficient CD40‐TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J Exp Med 2010; 207: 391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seijkens TTP, van Tiel CM, Kusters PJH, et al Targeting CD40‐induced TRAF6 signaling in macrophages reduces atherosclerosis. J Am Coll Cardiol 2018; 71: 527–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Clausen BE, Burkhardt C, Reith W, et al Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 1999; 8: 265–277. [DOI] [PubMed] [Google Scholar]
- 25. Ahonen C, Manning E, Erickson LD, et al The CD40‐TRAF6 axis controls affinity maturation and the generation of long‐lived plasma cells. Nat Immunol 2002; 3: 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Elgueta R, Benson MJ, de Vries VC, et al Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev 2009; 229: 152–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mukundan L, Bishop GA, Head KZ, et al TNF receptor‐associated factor 6 is an essential mediator of CD40‐activated proinflammatory pathways in monocytes and macrophages. J Immunol 2005; 174: 1081–1090. [DOI] [PubMed] [Google Scholar]
- 28. Chatzigeorgiou A, Seijkens T, Zarzycka B, et al Blocking CD40‐TRAF6 signaling is a therapeutic target in obesity‐associated insulin resistance. Proc Natl Acad Sci U S A 2014; 111: 2686–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zarzycka B, Seijkens T, Nabuurs SB, et al Discovery of small molecule CD40‐TRAF6 inhibitors. J Chem Inf Model 2015; 55: 294–307. [DOI] [PubMed] [Google Scholar]
- 30. Jensen J, Krakauer M, Sellebjerg F. Increased T cell expression of CD154 (CD40‐ligand) in multiple sclerosis. Eur J Neurol 2001; 8: 321–328. [DOI] [PubMed] [Google Scholar]
- 31. Chen D, Ireland SJ, Remington G, et al CD40‐mediated NF‐kappaB activation in B cells is increased in multiple sclerosis and modulated by therapeutics. J Immunol 2016; 197: 4257–4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Peferoen LA, Vogel DY, Ummenthum K, et al Activation status of human microglia is dependent on lesion formation stage and remyelination in multiple sclerosis. J Neuropathol Exp Neurol 2015; 74: 48–63. [DOI] [PubMed] [Google Scholar]
- 33. Vogel DY, Vereyken EJ, Glim JE, et al Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J Neuroinflammation 2013; 10: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kigerl KA, Gensel JC, Ankeny DP, et al Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci 2009; 29: 13435–13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ponomarev ED, Shriver LP, Dittel BN. CD40 expression by microglial cells is required for their completion of a two‐step activation process during central nervous system autoimmune inflammation. J Immunol 2006; 176: 1402–1410. [DOI] [PubMed] [Google Scholar]
- 36. Atkins G, Amor S, Fletcher J, et al The Biology of Multiple Sclerosis. Cambridge University Press: Cambridge, 2012. [Google Scholar]
- 37. Grewal IS, Flavell RA. CD40 and CD154 in cell‐mediated immunity. Annu Rev Immunol 1998; 16: 111–135. [DOI] [PubMed] [Google Scholar]
- 38. Kambayashi T, Laufer TM. Atypical MHC class II‐expressing antigen‐presenting cells: can anything replace a dendritic cell? Nat Rev Immunol 2014; 14: 719–730. [DOI] [PubMed] [Google Scholar]
- 39. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 2008; 8: 958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aarts SABM, Seijkens TTP, Kusters PJH, et al Inhibition of CD40‐TRAF6 interactions by the small molecule inhibitor 6877002 reduces neuroinflammation. J Neuroinflammation 2017; 14: 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure legends
Figure S1. CNS inflammation of MHCII–CD40–T2/3/5 −/−, MHCII–CD40–T6 −/−, CD40 −/− and WT mice
Figure S2. Peripheral immune responses in MHCII–CD40–T2/3/5 −/−, MHCII–CD40–T6 −/−, CD40 −/−, CD40 flfl LysM cre and WT mice
Figure S3. Normal Ig isotype switching in CD40 flfl LysM cre mice
Figure S4. CD40‐deficient macrophages have a diverse inflammatory pattern
Table S1. Primer sequences