

Abstract
Amperometry with nanotip electrodes has been applied to show cocaine and methylphenidate not only trigger declines in vesicle content and exocytotic catecholamine release in a model cell line but also differentially change the fraction of transmitter released from each individual vesicle. In addition, cocaine accelerates exocytotic release dynamics while they remain unchanged after methylphenidate treatment. The parameters from pre‐spike feet for the two drugs are also in opposition, suggesting this aspect of release is affected differentially. As cocaine and methylphenidate are psychostimulants with similar pharmacologic action but have opposite effects on cognition, these results might provide a missing link between the regulation of exocytosis and vesicles and the effect of this regulation on cognition, learning, and memory. A speculative chemical mechanism of the effect of these drugs on vesicle content and exocytosis is presented.
Keywords: catecholamines, cocaine, exocytosis, methylphenidate, vesicles
Signal transduction and neuronal communication by the conversion of electrical signals into chemical signals occurs through the fundamental process called exocytosis.1 In exocytosis, an action potential triggers vesicles filled with chemical transmitters to fuse with the plasma membrane of a cell and release these molecules to the extracellular environment.2 In the resting stage, neurotransmitter molecules are stored in the essential cell organelle called the synaptic vesicle with nearly uniform size and shape. Owing to its critical involvement in cell communication, the content and the exocytosis process of the synaptic vesicle have drawn a lot of attention to the molecular mechanisms that control the chemical communication between neurons, further influencing cognitive ability.3 This provides us with a pathway to study the chemical‐biological mechanism of cognition‐changing drugs.
The release of a chemical messenger has traditionally been thought to occur through full opening of the vesicle membrane; and, for nearly three decades, the amount of messenger released during the exocytosis process has been routinely measured with amperometry. However, a wealth of recent data, mostly from neuroendocrine cells, strongly suggest that most release occurs through a partial release exocytosis mode, in which only a portion of the transmitter content is expelled.4 This concept of partial release is of significant importance as the amount of exocytotic release in each individual event can be regulated and, therefore, is both a pharmaceutical target and a likely factor in cognition, learning, and disease.
Intracellular vesicle impact electrochemical cytometry (IVIEC), a method recently developed in our group, using conical nanotip electrodes, allows quantification of vesicular content inside the natural environment of the cell.4b, 5 Combined with single‐cell amperometry (SCA), we can measure both the storage content in vesicles and the exocytosis release from them (Scheme S1).6 The high temporal resolution of SCA also allows certain information about the kinetics of the fusion pore and release process to be obtained, and characterization of the spikes allows the quantification of the release amount. By combining these two methods, we can obtain the fraction of transmitter released during exocytosis at the single‐cell level.
We used IVIEC to measure the catecholamine storage of PC12 cell vesicles after treating them with cocaine (COC) or methylphenidate (MPH). Figure 1 A shows traces of release events obtained from control cells or those treated with COC or MPH, in which each current transient corresponds to the total catecholamine content inside a single vesicle. After quantification, a normalized frequency histogram is shown in Figure 1 B. Fitting to a Gaussian distribution, the standard deviation of the Gaussian is 0.278 for COC‐treated, 0.305 for MPH‐treated, and 0.295 for control cells. The similarity of the standard deviation indicates that both COC and MPH equally lowered the catecholamine content of all vesicles in the cells. As shown in Figure 1 C, it is clear that the vesicular catecholamine content decreases significantly after the treatment with either COC or MPH. This is not surprising in the partial release model discussed below. If release is all or none, then remaining vesicles would be expected to have the original content. However, both drugs block catecholamine reuptake into the cells and with partial release, the average vesicle is then not refilled.
Figure 1.
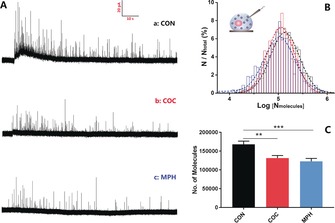
A) Typical traces of vesicle content in cells with a) no drug treatment, b) 10 μm COC, and c) 10 μm MPH. B) Normalized frequency distribution for vesicular content from control (black, n=2568 from 44 cells), COC‐ (red, n=1305 from 39 cells) and MPH‐treated cells (blue, n=1142 from 34 cells). Gaussian fits are shown. C) Average number of catecholamine molecules per vesicle for control and COC‐ and MPH‐treated cells. Error bars=SEM. **: p<0.01; ***: p<0.005.
To measure the catecholamine release, we used single‐cell amperometry. After stimulation with a high‐concentration K+ solution, the vesicle membrane fuses with the cell membrane and releases part of the vesicle content, which is recorded as a trace of current transients, each of which represents a single exocytotic release event. Typical traces obtained from the control (curve a), COC (curve b) and MPH (curve c) treated cells are shown in Figure 2 A, in which each spike represents a single exocytotic release event. There were fewer events and, consistent with the lower vesicle content observed, lower transient currents for COC‐ or MPH‐treated cells compared to the control. Figure 2 B is the normalized frequency histogram of number of molecules released per event, which provides a near‐Gaussian distribution with similar standard deviations but different mean values of the distribution. Furthermore, in order to minimize the impact of cell‐to‐cell variation, the mean values of the average number of molecules from treated and untreated (control) single cells were also compared and shown in Figure 2 C. Fewer molecules were released from cells after COC or MPH treatment.
Figure 2.
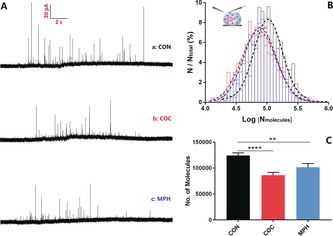
A) Typical traces of exocytotic release from cells treated with a) no drug, b) 10 μm COC, and c) 10 μm MPH. B) Normalized frequency histograms for molecules released from control cells (black, n=636 from 22 cells) and COC‐ (red, n=458 from 23 cells) and MPH‐treated cells (blue, n=398 from 17 cells). Gaussian fits are shown. C) Average number of catecholamine molecules per exocytotic event from control and COC‐ and MPH‐treated cells. Error bars=SEM. **: p<0.01; ****: p<0.001.
Exocytosis has traditionally been thought to occur through full distention of the vesicle membrane with the plasma membrane. This assumes that it is an all or none event; however, the vast majority of exocytosis events in these cells have recently been shown to involve only partial release of the transmitter content of a vesicle.4 This means that the release amount can be regulated. In this case, the fraction released could be important because a higher fraction released might lead to more molecules per exocytosis event and therefore fewer events are needed to elicit a minimum post‐synaptic response. We studied the effect of COC and MPH on the release fraction by combination of IVIEC and SCA. The data in Table 1 show that for treatment with 10 μm COC or MPH, both the vesicle content and the exocytotic release decrease; however, the change in the release fraction is in opposite direction, with COC treatment decreasing the fraction released and MPH treatment increasing it. Additionally, the fraction released during exocytosis upon treatment with different concentrations of COC or MPH was studied (Supporting Information, Figure S1). The changes of the fraction after COC or MPH treatment were concentration‐dependent and clearly trend exponentially in opposite direction, further indicating the differential effects of COC and MPH on the fraction released.
Table 1.
The effects of COC and MPH on exocytotic release fraction.
| Control | Cocaine | Methylphenidate | |
|---|---|---|---|
| n_intra [103 mole] | 168±8 | 132±7 (−21.43 %**) | 123±8 (−26.79 %****) |
| n_ex [103 mole] | 124±5 | 86±6 (−30.65 %***) | 102±7 (−17.74 %**) |
| Fraction released [%] | 74±5 | 65±5 | 83±8 |
[a] The data of n_intra and n_ex are presented as mean of the average number of molecules released±SEM. The SEM of the release fraction was obtained through Monte Carlo simulations. The pairs of data sets were compared using a two‐tailed Wilcoxon–Mann–Whitney rank‐sum test. **, p<0.01; ***, p<0.005; ****, p<0.001.
We used the high temporal resolution of SCA to obtain kinetic information about exocytotic release. Figure 3 A shows the average peak shape obtained from exocytosis for control cells (curve a), compared to those treated with COC (curve b), leading to lower amplitude and narrower exocytotic events, whereas MPH (curve c) causes very little change in the transients. Figure 3 B shows the peak parameters evaluated and those for the main release event are summarized in Figure 3 C–F. A decrease in the value of I max is observed after COC or MPH incubation (Figure 3 C), which is in agreement with the depletion of single vesicle content caused by both drugs. The value of t half (Figure 3 D) significantly decreases after COC treatment but is not significantly changed after MPH treatment, which means that the rate of exocytosis release becomes faster with perhaps a less stable fusion pore being formed after COC treatment and MPH appears not to affect the dynamics of exocytosis. The values of t rise and t fall are characteristics of fusion pore opening and closing, respectively. There is a slight but not significant decrease in t rise (Figure 3 E) following COC incubation suggesting that the opening process of the fusion pore might be slightly affected, but not greatly. However, an obvious decrease in the value of t fall (Figure 3 F) is observed after COC treatment, which implies that the closing of the fusion pore has been accelerated and the pore stays open for a shorter time compared to control or MPH‐treated cells. The values of t rise and t fall remained the same following MPH treatment, suggesting that the dynamics of pore opening and closing are not influenced, although the ratio of released amount to vesicle content peak increases (by comparison of Figures 1 and 2, see above). This might result from a pore that is opened more after MPH and less after COC. The pre‐spike feet from single‐cell amperometry (small current prior to the main current transient, see Figure 3 B) were also analysed to gain more insight into the opening phase of the exocytosis event when affected by increased COC or MPH, as discussed in more detail in the Supporting Information (Figure S2).
Figure 3.
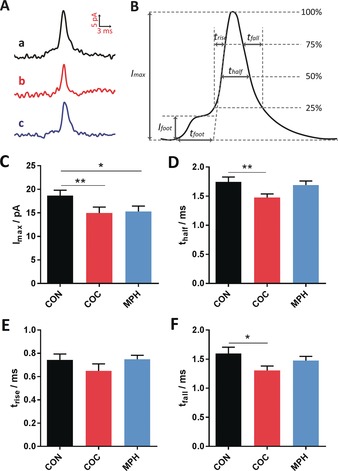
A) Average peaks obtained from single‐cell amperometry: a) control, b) COC, and c) MPH. B) Scheme showing the different parameters used for the peak analysis for exocytosis. Comparisons of C) peak current, I max; D) half peak width, t half; E) 25–75 % rise time, t rise; and F) fall time, t fall, from single‐cell amperometry; control (22 cells), COC (23 cells), MPH (17 cells). Error=SEM. *: p<0.05; **: p<0.01.
It is fascinating that these two drugs have similar effects on dopamine uptake and therefore transmission but opposite effects on cognition. A speculative chemical mechanism for how these might work is shown in Figure 4. First, the dopamine transporter (DAT) is a membrane‐spanning protein that pumps the released neurotransmitter dopamine back into cells. Previous studies with animal models have shown that COC and MPH are both psychostimulants that inhibit DAT, which means that they block the inward transport (re‐uptake) of dopamine.7 However, direct measurements regarding the effect of COC or MPH on catecholamine levels in single cells and especially in single vesicles have not been reported. Our results show that the vesicular catecholamine content is decreased in COC‐treated or MPH‐treated PC12 cells compared to control cells consistent with the inhibition of uptake of released dopamine.
Figure 4.
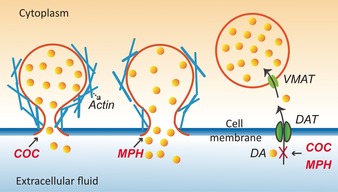
Proposed scheme for the different effects of COC and MPH on vesicle content and exocytosis. COC appears decrease the pore opening with a lower fraction released. MPH increases the pore opening with a higher fraction released.
Second, dynamin and actin have been reported to have an important role in exocytosis and have been found to be involved in opening and closing of the pore, respectively.1b, 6a, 8 A decrease in values of t half and t fall was observed after COC treatment, suggesting that COC might speed up the closing process of the pore and the observed effect might be due to a cocaine–actin interaction. The level of filamentous‐actin (F‐actin) has been shown to increase upon COC administration.9 This could result in an accelerated constriction of fusion pore and change the vesicular fraction of neurotransmitter release. Protein kinase C (PKC) was found to regulate the morphology of the F‐actin cytoskeleton and thereby influence the formation of F‐actin microfilaments.10 In previous work, added zinc was shown to affect exocytosis, and it was speculated that zinc enhances the activity of cytosolic PKC.4a PKC was also reported as a regulator of exocytosis with cisplatin treatment.1b This leads to speculation that the effect of COC on exocytotic dynamics is related to the action of PKC. PKC could phosphorylate adducin, an actin capping protein found at spectrin–actin junctions, decreasing the binding affinity of adducin for the barbed end of actin,11 thereby allowing actin polymerization. Adducin also binds calmodulin and is an in vivo substrate for PKC. We hypothesize a mechanism for the effect of COC on exocytosis in which COC enhances the levels of F‐actin induced by PKC, possibly by affecting adducin. In contrast, it has been reported that MPH does not change actin immunoreactivity,12 suggesting that this protein does not change after MPH. This is consistent with our data showing that the kinetics of release remains the same after MPH treatment.
The opposite effects on the release fraction for COC and MPH suggest a third molecular mechanism for their action in exocytosis in addition to DAT inhibition and actin modulation. Our group has recently shown that MPH and COC have opposite effects on the lipid structure of the fly brain by using mass spectrometry imaging.13 MPH appears to increase the lipids like phosphatidylethanolamine (PE) and phosphatidylinositol (PI) that have unequal head‐to‐tail‐group size and fit better in membrane regions of high curvature and to decrease the lipids associated with flat membrane regions, like phosphatidylcholine (PC), in the fly brain. However, the effects of COC on the lipids of the central area of the fly brain are strikingly opposite and statistically different. Interestingly, the amount of neurotransmitter released per event and dynamics can be changed by influencing the lipid composition of the plasma membrane, providing direct evidence regarding the regulation of the release process at the level of an individual event.14 It is thought that COC and MPH can alter the PC and PE abundance and influence the asymmetry of the bilayer leaflets, in an opposite way. This would govern the biophysical properties of the cell membrane, including bilayer curvature, strength, and plasticity, which further affect the fusion pore formed during exocytosis. This leads us to speculate that a less stable and smaller release pore is formed after COC incubation, while MPH treatment triggers a more stable and larger release pore during exocytosis. These different pore sizes might lead to the observed opposite effects on the fraction released. This is also consistent with a larger relative current for MPH versus COC in the foot events (Figure S2) despite the lower vesicle content for MPH. Thereby, the altered lipid composition of the plasma membrane influences the release pore formed during exocytosis, and the pore can potentially govern the amount of neurotransmitter that is released from the vesicle, leading to the control of release fraction. As COC and MPH are considered to affect cognition, it is possible that the change in release fraction in PC12 cells following COC or MPH treatment might be an important factor in cognition, learning, and memory.
In summary, single‐cell amperometry and intracellular vesicle impact electrochemical cytometry were applied with nanotip electrodes to investigate the effects of the cognition‐changing drugs (COC and MPH) on exocytotic release and vesicle content in PC12 cells. Our data underlines that, although both COC and MPH decrease the vesicle content and the amount of catecholamine released in each event, they show opposite effects on the release fraction during exocytosis. Additionally, COC changes the rate of release to induce faster events, whereas MPH does not. With the similar effects on the neurotransmitter uptake exhibited by COC and MPH but opposite cognitive effects, the release fraction during exocytosis and the kinetics of the release event are likely to be important factors in cognition, learning, and memory. A more stable and larger fusion pore following MPH incubation, possibly caused by the alteration of lipid composition, might cause the increased release fraction. It is enticing to speculate that an increased fraction of release leads to fewer exocytosis events needed to build plasticity and therefore enhances cognition. Thus, these fundamental data might be helpful for understanding the relationship between regulation of vesicles, exocytosis, and cognition at the single‐cell level.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We acknowledge support from the European Research Council (ERC Advanced Grant), the Knut and Alice Wallenberg Foundation in Sweden, the Swedish Research Council (VR), and the U.S. National Institutes of Health (NIH).
W. Zhu, C. Gu, J. Dunevall, L. Ren, X. Zhou, A. G. Ewing, Angew. Chem. Int. Ed. 2019, 58, 4238.
Contributor Information
Prof. Xuemin Zhou, Email: xueminzhou001_001@hotmail.com
Prof. Andrew G. Ewing, Email: andrew.ewing@chem.gu.se.
References
- 1.
- 1a. Ren L., Mellander L. J., Keighron J., Cans A., Kurczy M. E., Svir I., Oleinick A., Amatore C., Ewing A. G., Q. Rev. Biophys. 2016, 49, 1; [DOI] [PubMed] [Google Scholar]
- 1b. Li X., Dunevall J., Ewing A. G., Angew. Chem. Int. Ed. 2016, 55, 9041; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9187. [Google Scholar]
- 2.
- 2a. Phan N. T. N., Li X., Ewing A. G., Nat. Rev. Chem. 2017, 1, 48; [Google Scholar]
- 2b. Dunevall J., Fathali H., Najafinobar N., Lovric J., Wigstrom J., Cans A. S., Ewing A. G., J. Am. Chem. Soc. 2015, 137, 4344. [DOI] [PubMed] [Google Scholar]
- 3. Lovinger D. M., Neuropharmacology 2010, 58, 951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Ren L., Pour M. D., Majdi S., Li X., Malmberg P., Ewing A. G., Angew. Chem. Int. Ed. 2017, 56, 4970; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5052; [Google Scholar]
- 4b. Amatore C., Oleinick A. I., Svir I., ChemPhysChem 2010, 11, 159; [DOI] [PubMed] [Google Scholar]
- 4c. Omiatek D. M., Dong Y., Heien M. L., Ewing A. G., ACS Chem. Neurosci. 2010, 1, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dunevall J., Majdi S., Larsson A., Ewing A., Curr. Opin. Electrochem. 2017, 5, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Li X., Majdi S., Dunevall J., Fathali H., Ewing A. G., Angew. Chem. Int. Ed. 2015, 54, 11978; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12146; [Google Scholar]
- 6b. Majdi S., Najafinobar N., Dunevall J., Lovric J., Ewing A. G., ChemBioChem 2017, 18, 1898. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Farnsworth S. J., Volz T. J., Hanson G. R., Fleckenstein A. E., J. Pharmacol. Exp. Ther. 2009, 328, 807; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Volz T. J., Farnsworth S. J., Hanson G. R., Fleckenstein A. E., Ann. N. Y. Acad. Sci. 2008, 1139, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Shin W., Ge L., Arpino G., Villarreal S. A., Hamid E., Liu H., Zhao W., Wen P. J., Chiang H., Wu L., Cell 2018, 173, 934; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Wen P. J., Grenklo S., Arpino G., Tan X., Liao H., Heureaux J., Peng S., Chiang H., Hamid E., Zhao W., Shin W., Näreoja T., Evergren E., Jin Y., Karlsson R., Ebert S. N., Jin A., Liu A. P., Shupliakov O., Wu L., Nat. Commun. 2016, 7, 12604; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Trouillon R., Ewing A. G., ACS Chem. Biol. 2014, 9, 812; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8d. Berberian K., Torres A. J., Fang Q., Kisler K., Lindau M., J. Neurosci. 2009, 29, 863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Toda S., Shen H. W., Peters J., Cagle S., Kalivas P. W., J. Neurosci. 2006, 26, 1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Larsson C., Cell. Signalling 2006, 18, 276. [DOI] [PubMed] [Google Scholar]
- 11. Matsuoka Y., Li X., Bennett V., Cell. Mol. Life Sci. 2000, 57, 884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Volz T. J., Farnsworth S. J., King J. L., Riddle E. L., Hanson G. R., Fleckenstein A. E., J. Pharmacol. Exp. Ther. 2007, 323, 738. [DOI] [PubMed] [Google Scholar]
- 13. Philipsen M. H., Phan N. T. N., Fletcher J. S., Malmberg P., Ewing A. G., ACS Chem. Neurosci. 2018, 9, 1462. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Mellander L. J., Trouillon R., Svensson M. I., Ewing A. G., Sci. Rep. 2012, 2, 907; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b. Uchiyama Y., Maxson M. M., Sawada T., Nakano A., Ewing A. G., Brain Res. 2007, 1151, 46; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14c. Amatore C., Arbault S., Bouret Y., Guille M., Lemaître F., Verchier Y., ChemBioChem 2006, 7, 1998. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary