

Abstract
Cardiogenic shock (CS) is a complex and highly morbid entity conceptualised as a vicious cycle of injury, cardiac and systemic decompensation, and further injury and decompensation. The pathophysiology of CS is incompletely understood but limited clinical trial experience suggests that early and robust support of the failing heart to allow for restoration of systemic homoeostasis appears critical for survival. We review the pathophysiology, clinical features and trial data to construct a contemporary model of CS as a systemic process characterised with maladaptive compensatory mechanisms requiring prompt and appropriately tailored medical and mechanical support for optimal outcomes. We conclude with an algorithmic approach to acute CS incorporating clinical and haemodynamic data to match the patient’s cardiac and systemic needs as a template for contemporary management.
Keywords: cardiogenic shock, myocardial infarction, heart failure, mechanical circulatory support, temporary circulatory support
Introduction
Though there have been significant advances in reperfusion therapy and percutaneous mechanical circulatory support (MCS) devices, mortality among patients presenting with acute cardiogenic shock (CS) remain obstinately high, ranging from 25% to 50%.1–3 In a recent large cohort of 21 210 patients in London with ST segment elevation myocardial infarction (STEMI), CS was observed in 8.9% of patients with the incidence increasing over time and high mortality of 45%–70%.4 Significant effort has been dedicated to the development of management guidelines and treatment algorithms to improve survival from this highly lethal condition. Despite increasing awareness, a paucity of high-quality clinical trial data and wide practice variation exists. Though the exact pathophysiology of this multifactorial, haemodynamically diverse population remains poorly understood, early recognition and intervention to interrupt the devastating ‘cardiogenic shock spiral’ are critical to survival.5
The generally accepted definition of CS is a state in which ineffective cardiac output (CO) due to a primary cardiac dysfunction results in inadequate end-organ perfusion. Current CS-defining criteria used in clinical trials and guidelines are varied, and recommendations are largely based on data from patients with CS due to acute coronary syndrome (ACS).1 2 6 Considerably less is understood regarding the identification and outcomes in the non-ACS population, particularly acute decompensation in patients with underlying congestive heart failure–the so-called, ‘acute-on-chronic’ CS. Improved understanding of the pathophysiological process and identification of specific criteria for classification in this widely heterogeneous population is critical for early identification and appropriate management of patients with CS.
Pathophysiology
The pathophysiology of CS is poorly understood owing to a paucity of high-quality clinical data. Even for the most common cause, ACS, significant heterogeneity (eg, STEMI vs type I and type II non-STEMI with more specific variables such as postcardiac arrest or renal failure) exists that likely informs management and influences outcomes. Ideally, management of CS would integrate general supportive measures such as pharmacological circulatory support and MCS titrated to interventions directed at treating specific mechanisms in increasingly granular detail such as interrupting the cellular, metabolic and inflammatory pathways. In the absence of robust multicentre cohort studies providing detailed patient-level phenotyping, current management of CS necessarily focuses on the quantifiable and modifiable parameters of CS as gleaned from invasive haemodynamic assessment. In general, CS is characterised by an initial insult resulting in impaired CO followed by progressive injury culminating in inadequate and ultimately maladaptive compensatory mechanisms and rapid deterioration to end-organ hypoperfusion and complete cardiovascular collapse.7–9 This is conceptualised as a vicious cycle of cardiac injury, systemic deterioration and further cardiac impairment (figure 1). Perhaps more than any single feature, this self-perpetuating feedback loop, encompassing the heart and the whole patient, is what underlies CS. Interrupting this ‘shock spiral’ and restoring cardiovascular homoeostasis is central to the overarching treatment paradigm of CS. Any cause of acute, severe impairment of CO can trigger this cascade and precipitate CS.
Figure 1.

Conceptual algorithm for the management of cardiogenic shock (CS). The pathophysiology of CS is characterised by impaired cardiac output, SIRS, end-organ hypoperfusion and maladaptive compensatory mechanisms. Prevention of progressive cardiac and systemic compromise requires early recognition typically requiring right and left catheterisation and interruption of the vicious cycle by addressing underlying insults and initiation of mechanical circulatory support matched to the degree of clinical decompensation. Clinical indices such as CPI for LV function, PAPi for right ventricular function, and presence of malignant clinical features such as arrhythmia and hypoxaemia may help guide the decision for the most appropriate MSC modality. Bi-V, biventricular; CPI, Cardiac Power Index; LV, left ventricular; MCS, mechanical circulatory support; MI, myocardial infarction; PAPi, Pulmonary Artery Pulsatility Index; SIRS, systemic inflammatory response syndrome.
Acute myocardial infarction (AMI) with left ventricular (LV) failure represents 60%–80% of CS6 10 (figure 2). In a classic study of 22 patients who succumbed to CS following AMI, clinical, physiological and pathological examination was methodically performed.11 Compared with control hearts from patients who died suddenly following AMI without interval shock, hearts with CS showed massive loss of functional myocardium involving roughly 50% of the LV. At the tissue level, progressive expansion of the initial infarction into the peri-infarct ischaemic myocardium occurs in two complementary directions. First, at the peripheral edges of injury in the subendocardium, infarction extends outwards to become transmural. Second, the lateral edges of the transmural infarct extend circumferentially outwards. Extension of infarct is mediated by decreased CO and tissue perfusion, including to the heart itself due to (A) Infarction. (B) Increased oxygen demand and consumption of the remaining viable myocardium. (C) Further ischaemia as the heart continues to fail.12 This process frequently occurs silently without evidence of ongoing ischaemia or infarction. The ECG is suggestive of ongoing injury in only half of patients in CS.13 Thus in the context of ongoing shock, persistent myocardial injury should be assumed and occurs without overt clinical signs. The myriad factors governing myocardial metabolic demand and supply is comprised of local actors within the central cardiovascular system and the peripheral vasculature as well as paracrine and hormonal signalling from the neurovascular and innate immune systems.
Figure 2.

Causes of cardiogenic shock (adapted from Harjola et al [10]). ACS, acute coronary syndrome; MI, myocardial infarction.
Although AMI is the most common aetiology, consideration of other cardiac (eg, right heart failure,14 myopericarditis, arrhythmia, takotsubo cardiomyopathy, hyperthrophic cardiomyopathy, valvular heart disease, postpartum cardiomyopathy) and extracardiac (eg, aortic dissection with acute aortic stenosis or myocardial infarction, traumatic chordal rupture, massive pulmonary embolism) causes must also be rapidly diagnosed in parallel and if present, managed uniquely.15 Consideration of these non-ischaemic aetiologies serve as a reminder that while CS is typically accompanied by acutely reduced ejection fraction, CS is a physiological condition of depressed CO ill-defined in anatomical terms and may occur with only modestly impaired ejection fraction.16
Maladaptive compensatory mechanisms accelerate the self-perpetuation of injury. Local and systemic release of catecholamines transiently increase myocardial chronotropy and ionotropy at the cost of increased rates of arrhythmia and peripheral vasoconstriction, exacerbating afterload and myocardial perfusion mismatch. The systemic inflammatory response system, in contrast, causes inappropriate vasodilation, capillary leak, microvascular dysfunction and hypoperfusion of end organs, most gravely in the intestinal tract, which predisposes to gut translocation and sepsis.17 Later evolution of systemic inflammatory response syndrome related effectors, including tumour necrosis factor-α and interlukin 6, are further proinflammatory and cardiodepressive. Neurohormonal activation of the renin-angiotensin system results in increased salt and water retention adding to the burden of preload and decompensated heart failure. The ability to interrupt these processes and prevent or reverse the extension of myocardial injury before unsalvageable damage is central to the hypothesis that CS is treatable. The goal of therapies is thus to rescue, support and optimise the remaining viable myocardium.
Evolving management
Given the difficulty in performing randomised trials in CS, current management recommendations are largely empirical or based on results of the few trials adequately powered to detect differences in clinical outcomes. Moreover, the only intervention with proven mortality benefit is early revascularisation either with percutaneous coronary intervention (PCI) or coronary artery bypass grafting: the SHould we emergently revascularise Occluded Coronaries for cardiogenic shocK (SHOCK) Trial failed to demonstrate mortality benefit of early revascularisation over initial medical stabilisation at 30 days; however, significant mortality benefit with early revascularisation was seen at 6 months, 1 year and 6 years2.18 Rates of revascularisation in CS have steadily increased but remain suboptimal at approximately 50% (with only 60% of patients receiving cardiac catheterisation at all).19 Access to emergent revascularisation thus remains a significant obstacle.20 Though benefit of culprit vessel revascularisation is accepted as standard practice, revascularisation of multivessel coronary artery disease (CAD) in patients with CS remains of unclear benefit (multivessel revascularisation, Class IIa B). Data suggest that patients with multivessel CAD have a higher mortality21 and in a meta-analysis of patients with STEMI with multivessel disease without CS, emergent culprit vessel revascularisation with staged multivessel complete revascularisation within the index hospitalisation showed lower mortality.22 For STEMI with CS, registry experience from France23 and more recently Korea24 support multivessel revascularisation. However, the Culprit Lesion only PCI vs multivessel PCI in Cardiogenic Shock (CULPRIT-SHOCK) Trial showed higher risk of all-cause mortality and need for renal replacement therapy in the multivessel PCI group.25 Additionally, in the Evaluating Xience and Left Ventricular Function in Percutaneous Coronary Intervention on Occlusions after ST-Elevation Myocardial Infarction (EXPLORE) trial, non-culprit chronic total occlusion (CTO) revascularisation within a week of STEMI revascularisation did not improve LV size or function. However, the study is limited by unclear completeness of revascularisation, randomisation to non-culprit CTO PCI before culprit revascularisation, and slow enrolment. Subgroup analysis showed a trend towards benefit with the left anterior descending (LAD) artery CTO revascularisation cohort where the most functional benefit would be expected. The role for complete multivessel revascularisation in CS remains uncertain.
Role of MCS
Knowledge of fundamental haemodynamic principles in CS is critical to appreciate the utility of MCS devices and aids in management decisions. Details of CS haemodynamics have been summarised previously26 and are briefly reviewed here. The LV pressure-volume loop and changes that occur with CS provide a foundation for understanding the haemodynamic perturbations of CS and the mechanism of support devices. In CS, end diastolic and systolic volumes increase, stroke volume decreases, and end systolic pressure decreases reflecting the overall reduction in LV contractility and output.27 MCS devices alter haemodynamics in an attempt to restore CO and normalise perfusion pressures.28
For many decades, LV unloading with counterpulsation via the intra-aortic balloon pump (IABP) was the only percutaneous device short of full MCS with extracorporeal membrane oxygenation (ECMO). In one of the largest randomised trials of patients with CS, the intra-aortic balloon counterpulsation in AMI complicated by the CS (IABP-SHOCK II) Trial, 600 patients were randomised to either IABP or medical therapy. No significant difference was seen in the primary end point of 30-day all-cause mortality.1 Follow-up analysis also showed no difference in mortality at 12 months.29 Results of these clinical trials are reflected in the current European Society of Cardiology guidelines (early culprit revascularisation, Class 1B; IABP use in CS, Class III).6 30
MCS devices for management of CS have advanced significantly over the past six decades. While devices have shown improvement in haemodynamics, randomised trials have failed to show a mortality benefit with use of these devices.1 31–34 Though haemodynamic factors represent a critical component of CS pathophysiology, the fact that MCS devices can improve haemodynamics without impacting mortality suggests that CS pathophysiology is incompletely understood and therapies directed at supporting only the LV, while leaving the right ventricle (RV) unaddressed, provide only partial support, may be inadequate and have a limited window for correction before irreversible injury occurs. Using strict haemodynamic definitions may limit our ability to recognise and intervene early in the pathophysiological course before end-organ hypoperfusion occurs. Additionally, haemodynamic profiles in subpopulations of CS without ACS are poorly understood, and extrapolation of ACS CS profiles to these patients may not be relevant.
Clinical trials evaluating MCS devices also suffer from limited enrolment and largely include only patients presenting with CS due to AMI (table 1). One of the more recent trials assessing contemporary MCS, the Impella CP vs IABP in AMI complicated by cardiogenic shock (IMPRESS) Trial, showed no difference in the primary end point of 30-day mortality with use of the Impella CP device compared with the IABP.34 Patients included in this trial were considered to have severe shock, with all patients mechanically ventilated prior to randomisation and 92% suffering out-of-hospital cardiac arrest, raising concerns about futility in the trial population and leaving questions about specificity of target populations unanswered. Clinical trials currently enrolling aim at addressing the benefit of earlier intervention,35 upfront full support with venoarterial (VA)-ECMO,36 and combination therapy with VA-ECMO and Impella CP. For clinical practice, an expanded view of CS incorporating the full gamut of cardiac and non-cardiac aetiologies as well as optimal utilisation of pharmacological and mechanical support is needed.
Table 1.
Summary of mechanical circulatory support (MCS) device trials
| Year | n | Study information | Design | Primary end point | Results | |
| IABP-SHOCK II1 | 2012 | 600 | IABP vs MT AMI with CS and revascularisation |
Multicentre RCT |
30-day mortality | No difference in mortality (39.7% IABP vs 41.3% MT) |
| Thiele H et al31 | 2005 | 41 | TH vs IABP AMI with CS |
Single centre RCT |
Cardiac Power Index and haemodynamics | TH improved haemodynamics 2◦ end point 30-day mortality: no difference (TH 43% vs IABP 45%) |
| TandemHeart Investigators Group32 | 2006 | 42 | TH vs IABP Within 24 hours of CS 70% AMI, 30% HF |
Multicenter RCT | Haemodynamics CI, MAP, PCWP |
TH improved haemodynamics 2◦ end point 30 day mortality: no difference (TH 47% vs IABP 36%) |
| ISAR-Shock33 | 2008 | 26 | Impella 2.5 vs IABP AMI with CS |
Multicentre RCT |
Change in Cardiac Index from baseline to 30 min | Impella 2.5 improved haemodynamics 2◦ end point 30-day mortality: no difference (46% both groups) |
| IMPRESS in severe shock34 | 2016 | 48 | Impella CP vs IABP AMI with STEMI and CS |
Multicentre RCT | 30-day mortality | No difference in 30-day mortality (50% CP vs 46% IABP) |
| National Cardiogenic Shock Initiative (NCSI)35 (NCT03677180) |
Recruiting | 500 | Early initiation of MCS | Multicentre cohort | Survival to hospital discharge | Ongoing study |
| ECMO-CS36 (NCT02301819) | Recruiting | 120 | VA-ECMO CS |
Multicentre RCT |
30-day mortality, cardiac arrest and additional MCS | Ongoing study |
| Danish CS Trial (DanShock) (NCT01633502) | Ongoing | 360 | Impella CP vs IABP AMI with STEMI and CS |
Multicentre RCT |
6-month mortality | Ongoing study |
| REVERSE Trial (NCT03431467) | Recruiting | 96 | Impella CP with VA-ECMO CS |
Single-centre RCT |
30-day recovery | Ongoing study |
AMI, acute myocardial infarction;CS, cardiogenic shock;HF, heart failure; IABP, intraoartic balloon pump;MAP, mean arterial pressure;MT, medical therapy; PCWP, pulmonary capillary wedge pressure; RCT, randomised control trial;SHOCK, SHould we emergently revascularise Occluded Coronaries for cardiogenic shocK; TH, Tandem Heart;VA-ECMO, venoarterial extracorporeal membrane oxygenation.
Additional haemodynamic parameters
Though several scoring systems have been proposed to predict clinical outcomes in CS,25 37 a uniform set of objective parameters for identification of this diverse patient population has not been established. Cardiac power output (CPO) (calculated as mean arterial pressure (MAP) × CO/451) and Cardiac Power Index (CPI) (calculated as MAP × Cardiac Index/451) where MAP = ((systolic blood pressure – diastolic blood pressure) / 3) + diastolic blood pressure. The CPO and CPI are representations of mean hydraulic energy based on the physical rule of fluids that power = flow × pressure. Namely, the pumping ability of the heart can be quantified as the simultaneous product of CO (flow) and MAP. Under resting conditions for a well-compensated, average size adult man, the CPO and CPI are approximately 1 W38 and 0.5–0.7 W/m2, respectively. Under stress or exercise, the CPO augments considerably up to 6 W. This capacity to increase output is severely diminished in chronic heart failure and has been shown to predict long-term outcomes.39 The resting CPO in chronic heart failure is preserved and allows for haemostasis and haemodynamic stability. The resting CPO in normal and chronic heart failure, therefore, may be indistinguishable. In acute heart failure, however, any capacity to increase output is recruited to maintain vital organ perfusion and the resting CPO is often compromised, reflecting the severity of the decompensated state. Univariate and multivariate analysis of the SHOCK Trial registry data identified CPO <0.53 and CPI<0.33 as the strongest independent haemodynamic correlates of in-hospital mortality in CS.40 While Cardiac Index and MAP are included in accepted definitions of CS (table 2), this relatively contemporary concept of cardiac power is not. The interdependence of Cardiac Index and MAP with respect to CPI is shown in figure 3 and demonstrates the importance of integrating both flow and pressure when assessing overall cardiac function. Indeed, CS clinically represents a continuum from pure pump failure (eg, low CO but high MAP following acute infarction) to pure vasoplegia (eg, relatively preserved CO and low MAP following reperfusion or end-stage heart failure) that is unified by underlying cardiac injury.
Table 2.
Current clinical and trial definitions of cardiogenic shock
| Clinical definition | SHOCK Trial2 | IABP-SHOCK II1 | ESC heart failure guidelines6 |
| Cardiac disorder that results in both clinical and biochemical evidence of tissue hypoperfusion | Clinical criteria: MI complicated by left ventricular dysfunction SBP <90 mm Hg for >30 min or support to maintain SBP >90 mm Hg and end-organ hypoperfusion (urine output <30 mL/hour or cool extremities) Haemodynamic criteria: Cardiac Index <2.2 L/min/m2 and PCWP >15 mm Hg |
Clinical criteria: Acute MI SBP <90 mm Hg or >30 min or catecholamines to maintain SBP >90 mm Hg and clinical pulmonary congestion and impaired end-organ perfusion (altered mental status, cold/clammy skin and extremities, urine output <30 mL/hour, or lactate >2.0 mmol/L) |
Clinical criteria: SBP <90 mm Hg with adequate volume and clinical or laboratory signs of hypoperfusion Clinical hypoperfusion: Cold extremities, oliguria, mental confusion, dizziness, narrow pulse pressure Laboratory hypoperfusion: Metabolic acidosis, elevated serum lactate, elevated serum creatine |
ESC, European Society of Cardiology; IABP, intra-aortic balloon pump; MI, myocardial infarction; PCWP, pulmonary capillary wedge pressure; SBP, systolic blood pressure; SHOCK, SHould we emergently revascularise Occluded Coronaries for cardiogenic shocK.
Figure 3.
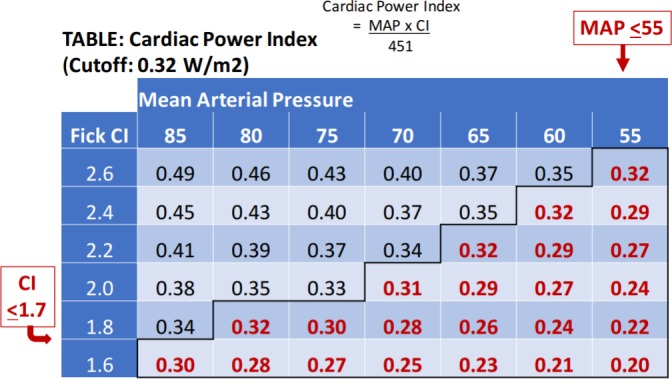
Cardiac index (CI) and mean arterial pressure (MAP) correlation to Cardiac Power Index (CPI).
Future directions
The central paradigm in the treatment of CS is interrupting the self-perpetuating feedback loop of myocardial insult, worsening cardiac power, and further coronary and systemic perfusion mismatch in the context of increasingly maladaptive compensatory mechanisms (figure 1). Interventions are thus directed at limiting myocardial damage after index insult, supporting the failing heart, and interrupting the processes responsible for progressive dysfunction before accumulating an unsurvivable burden of injury. Although the diagnosis can often be made rapidly on a clinical basis alone, invasive haemodynamic monitoring to assess CPI and pulmonary capillary wedge pressure are critical to tailoring a multidisciplinary approach involving procedural, mechanical and pharmacological interventions. Early consideration and initiation of therapies are targeted to correct the initial insult, limit further myocardial damage and adequately support the failing heart and end organs as necessary to allow for recovery. Concurrent with considerations of emergent revascularisation, supporting the heart and end organs is critical to preventing progressive cardiovascular collapse and forestall irreversible end-organ injury.
Ionovasodilator and vasopressor therapies have demonstrated limited benefit in CS and are often counterproductive.41 42 MCS to improve haemodynamics and perfusion is increasingly available following proliferation of rapidly initiated percutaneous left ventricular assist devices (pLVAD). While initially implemented as adjunctive therapy after revascularisation, maximal pharmacological therapy, medical optimisation and placement of an IABP, the first-line use of pVAD devices may have superior outcomes. The Detroit Cardiogenic Shock Initiative was a regional feasibility protocol of early initial MCS with the Impella CP microaxial pLVAD Impella (Abiomed, Danvers, Massachusetts, USA) prior to emergent revascularisation that suggested benefit.35 The results of this pilot study requires validation in the ongoing National Cardiogenic Shock Initiative (NCT03677180) and within a broader context, but the potential benefit of early MCS reinforces the concept of CS as a systemic derangement of the cardiovascular system with a limited window for global haemodynamic stabilisation, possibly before addressing the specific insults responsible such as ongoing ischaemia.
The University of Washington (UW) has developed a systematic and consistent approach to acute CS applying this contemporary pathophysiological model and incorporates validated indices of LV and RV dysfunction. The UW CS algorithm features a multidisciplinary shock team comprised of members from advanced heart failure cardiology, interventional cardiology, cardiothoracic surgery and critical care services to risk-stratify and develop an integrated approach using patient-specific factors from clinical data and invasive haemodynamics. The major determination is the degree of LV instability as assessed by CPI or presence of high-risk features such as haemodynamically significant atrial fibrillation or malignant ventricular tachyarrhythmias. Severe LV instability is further stratified by the degree of RV involvement using the surrogate index Pulmonary Artery Pulsatility Index (ratio of right atrial pressure and pulmonary artery systolic pressure).43 The recommended intervention is then tailored to the degree of LV and RV failure ranging from partial LV unloading (eg, Impella CP) for moderate LV instability without RV involvement, full LV unloading (eg, Impella 5.0) for more severe LV instability, and finally cannulation for VA-ECMO for severe biventricular failure or hypoxaemic respiratory failure. It should be stated that this paradigm is not applied to patients with acute-on-chronic decompensated heart failure leading to CS. While they are also undoubtedly at risk, specific haemodynamic profiles and thresholds for intervention as well as anticipated outcomes are likely far different.
Our algorithm emphasises rapid identification of the patient’s haemodynamic and critical care needs and deployment of appropriately tailored interventions. Implementation of such an approach is resource-intensive, drawing expertise from multiple specialties to simultaneously consider multiple MCS modalities and the offer the most appropriate support. This recognises the importance of rapidly matching the patient’s haemodynamic needs to optimise outcomes irrespective of resource availability. Regionalisation for the treatment of CS is thus required and allows for the comprehensive and individualised care of the patient. Certainly, our proposed approach requires rigorous investigation to assess feasibility and validate efficacy. To that end, trials specific to CS are required given the unique characteristics of the patients and pathophysiology at play.
Conclusions
The pathophysiology of CS is fundamentally a systemic derangement of maladaptive compensatory mechanisms resulting in perpetuation of coronary and systemic perfusion mismatch. Although the optimal treatment approach is incompletely defined, especially in non-ACS presentations, clinical trials are difficult to conduct in this patient population. While percutaneous MCS device trials to date have failed to show survival benefit in small randomised trials, haemodynamics improve with use of these devices. Current understanding of the pathophysiological process of CS suggests that interrupting the ‘shock spiral’ and restoration of cardiovascular homoeostasis early before end-organ hypoperfusion occurs may be ultimately critical to survival. To ensure appropriate use of these devices and uniformity in CS management, a systematic and consistent approach using contemporary haemodynamic parameters to guide management should be considered. In addition, risk/safety as well as cost must be factored into the institutional approach to CS before tailoring to the specific needs of the patient.
Footnotes
TLJ and KN contributed equally.
Contributors: TLJ, KN and JMM conceived the presented concepts. TLJ and KN performed the literature review and drafted the manuscript and figures. KN and JMM revised the final manuscript. KN and JMM are responsible for the overall content as guarantor.
Funding: Publication fees were supported by the Bruce-Laughlin Fellow Award at the University of Washington, Division of Cardiology.
Competing interests: None declared.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: All data relevant to the study are included in the article or uploaded as supplementary information.
References
- 1.Thiele H, Zeymer U, Neumann F-J, et al. Intraaortic balloon support for myocardial infarction with cardiogenic shock. N Engl J Med 2012;367:1287–96. 10.1056/NEJMoa1208410 [DOI] [PubMed] [Google Scholar]
- 2.Hochman JS, Sleeper LA, Webb JG, et al. Early revascularization in acute myocardial infarction complicated by cardiogenic shock. SHOCK Investigators. Should We Emergently Revascularize Occluded Coronaries for cardiogenic shock. N Engl J Med 1999;341:625–34. 10.1056/NEJM199908263410901 [DOI] [PubMed] [Google Scholar]
- 3.Stretch R, Sauer CM, Yuh DD, et al. National trends in the utilization of short-term mechanical circulatory support: incidence, outcomes, and cost analysis. J Am Coll Cardiol 2014;64:1407–15. 10.1016/j.jacc.2014.07.958 [DOI] [PubMed] [Google Scholar]
- 4.Rathod KS, Koganti S, Iqbal MB, et al. Contemporary trends in cardiogenic shock: incidence, intra-aortic balloon pump utilisation and outcomes from the London heart attack group. Eur Heart J Acute Cardiovasc Care 2018;7:16–27. 10.1177/2048872617741735 [DOI] [PubMed] [Google Scholar]
- 5.van Diepen S, Katz JN, Albert NM, et al. Contemporary management of cardiogenic shock: a scientific statement from the American Heart Association. Circulation 2017;136:e232–68. 10.1161/CIR.0000000000000525 [DOI] [PubMed] [Google Scholar]
- 6.Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2016;37:2129–22. 10.1093/eurheartj/ehw128 [DOI] [PubMed] [Google Scholar]
- 7.Reynolds HR, Hochman JS. Cardiogenic shock: current concepts and improving outcomes. Circulation 2008;117:686–97. 10.1161/CIRCULATIONAHA.106.613596 [DOI] [PubMed] [Google Scholar]
- 8.Califf RM, Bengtson JR. Cardiogenic shock. N Engl J Med 1994;330:1724–30. 10.1056/NEJM199406163302406 [DOI] [PubMed] [Google Scholar]
- 9.Swan HJ, Forrester JS, Diamond G, et al. Hemodynamic spectrum of myocardial infarction and cardiogenic shock. A conceptual model. Circulation 1972;45:1097–110. 10.1161/01.CIR.45.5.1097 [DOI] [PubMed] [Google Scholar]
- 10.Harjola V-P, Lassus J, Sionis A, et al. Clinical picture and risk prediction of short-term mortality in cardiogenic shock. Eur J Heart Fail 2015;17:501–9. 10.1002/ejhf.260 [DOI] [PubMed] [Google Scholar]
- 11.Alonso DR, Scheidt S, Post M, et al. Pathophysiology of cardiogenic shock. Quantification of myocardial necrosis, clinical, pathologic and electrocardiographic correlations. Circulation 1973;48:588–96. [DOI] [PubMed] [Google Scholar]
- 12.Page DL, Caulfield JB, Kastor JA, et al. Myocardial changes associated with cardiogenic shock. N Engl J Med 1971;285:133–7. 10.1056/NEJM197107152850301 [DOI] [PubMed] [Google Scholar]
- 13.Levine HD, Phillips E. An appraisal of the newer electrocardiography: correlations in one hundred and fifty consecutive autopsied cases. N Engl J Med 1951;245:833–42. 10.1056/NEJM195111292452201 [DOI] [PubMed] [Google Scholar]
- 14.Jacobs AK, et al. Cardiogenic shock caused by right ventricular infarction: a report from the shock registry. J. Am. Coll. Cardiol 2003;41:1273–9. [DOI] [PubMed] [Google Scholar]
- 15.Hochman JS, et al. Cardiogenic shocK complicating acute myocardial infarction-etiologies, management and outcome: a report from the shocK trial registry. Should We Emergently Revascularize Occluded Coronaries for cardiogenic shocK? J. Am. Coll. Cardiol 2000;36:1063–70. [DOI] [PubMed] [Google Scholar]
- 16.Chatterjee K, McGlothlin D, Michaels A. Analytic reviews: cardiogenic shock with preserved systolic function: a reminder. J Intensive Care Med 2008;23:355–66. 10.1177/0885066608324250 [DOI] [PubMed] [Google Scholar]
- 17.Kohsaka S, et al. Systemic inflammatory response syndrome after acute myocardial infarction complicated by cardiogenic shock. Arch Intern Med 2005;165:1643–50. 10.1001/archinte.165.14.1643 [DOI] [PubMed] [Google Scholar]
- 18.Hochman JS, Sleeper LA, Webb JG, et al. Early revascularization and long-term survival in cardiogenic shock complicating acute myocardial infarction. JAMA 2006;295:2511–5. 10.1001/jama.295.21.2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goldberg RJ, Makam RCP, Yarzebski J, et al. Decade-long trends (2001–2011) in the incidence and hospital death rates associated with the in-hospital development of cardiogenic shock after acute myocardial infarction. Circ Cardiovasc Qual Outcomes 2016;9:117–25. 10.1161/CIRCOUTCOMES.115.002359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rab T, Ratanapo S, Kern KB, et al. Cardiac shock care centers: JACC review topic of the week. J Am Coll Cardiol 2018;72:1972–80. 10.1016/j.jacc.2018.07.074 [DOI] [PubMed] [Google Scholar]
- 21.Sanborn TA, Sleeper LA, Webb JG, et al. Correlates of one-year survival inpatients with cardiogenic shock complicating acute myocardial infarction: angiographic findings from the shock trial. J Am Coll Cardiol 2003;42:1373–9. [DOI] [PubMed] [Google Scholar]
- 22.Iqbal MB, Nadra IJ, Ding L, et al. Culprit vessel versus multivessel versus in-hospital staged intervention for patients with ST-Segment Elevation myocardial infarction and multivessel disease: stratified analyses in high-risk patient groups and anatomic subsets of nonculprit disease. JACC Cardiovasc Interv 2017;10:11–23. 10.1016/j.jcin.2016.10.024 [DOI] [PubMed] [Google Scholar]
- 23.Mylotte D, et al. Primary percutaneous coronary intervention in patients with acute myocardial infarction, resuscitated cardiac arrest, and cardiogenic shock: the role of primary multivessel revascularization. JACC Cardiovasc. Interv 2013;6:115–25. [DOI] [PubMed] [Google Scholar]
- 24.Lee JM, Rhee T-M, Hahn J-Y, et al. Multivessel percutaneous coronary intervention in patients with ST-segment elevation myocardial infarction with cardiogenic shock. Journal of the American College of Cardiology 2018;71:844–56. 10.1016/j.jacc.2017.12.028 [DOI] [PubMed] [Google Scholar]
- 25.Thiele H, Akin I, Sandri M, et al. PCI strategies in patients with acute myocardial infarction and cardiogenic shock. N Engl J Med Overseas Ed 2017;377:2419–32. 10.1056/NEJMoa1710261 [DOI] [PubMed] [Google Scholar]
- 26.Burkhoff D. Hemodynamic Support: Science and Evaluation of the Assisted Circulation with Percutaneous Assist Devices. Interv. Cardiol. Clin 2013;2:407–16. [DOI] [PubMed] [Google Scholar]
- 27.Burkhoff D, Sayer G, Doshi D, et al. Hemodynamics of mechanical circulatory support. J Am Coll Cardiol 2015;66:2663–74. 10.1016/j.jacc.2015.10.017 [DOI] [PubMed] [Google Scholar]
- 28.Burkhoff D. Where next in cardiogenic shock owing to myocardial infarction? Nat Rev Cardiol 2015;12:383–5. 10.1038/nrcardio.2015.81 [DOI] [PubMed] [Google Scholar]
- 29.Thiele H, Zeymer U, Neumann F-J, et al. Intra-aortic balloon counterpulsation in acute myocardial infarction complicated by cardiogenic shock (IABP-SHOCK II): final 12 month results of a randomised, open-label trial. The Lancet 2013;382:1638–45. 10.1016/S0140-6736(13)61783-3 [DOI] [PubMed] [Google Scholar]
- 30.Ibanez B, James S, Agewall S, et al. 2017 ESC guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: the task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of cardiology (ESC). Eur Heart J 2018;39:119–77. 10.1093/eurheartj/ehx393 [DOI] [PubMed] [Google Scholar]
- 31.Thiele H, Sick P, Boudriot E, et al. Randomized comparison of intra-aortic balloon support with a percutaneous left ventricular assist device in patients with revascularized acute myocardial infarction complicated by cardiogenic shock. Eur Heart J 2005;26:1276–83. 10.1093/eurheartj/ehi161 [DOI] [PubMed] [Google Scholar]
- 32.Burkhoff D, Cohen H, Brunckhorst C, et al. A randomized multicenter clinical study to evaluate the safety and efficacy of the TandemHeart percutaneous ventricular assist device versus conventional therapy with intraaortic balloon pumping for treatment of cardiogenic shock. Am Heart J 2006;152:469.e1–469.e8. 10.1016/j.ahj.2006.05.031 [DOI] [PubMed] [Google Scholar]
- 33.Seyfarth M, Sibbing D, Bauer I, et al. A randomized clinical trial to evaluate the safety and efficacy of a percutaneous left ventricular assist device versus intra-aortic balloon pumping for treatment of cardiogenic shock caused by myocardial infarction. Journal of the American College of Cardiology 2008;52:1584–8. 10.1016/j.jacc.2008.05.065 [DOI] [PubMed] [Google Scholar]
- 34.Ouweneel DM, Eriksen E, Sjauw KD, et al. Percutaneous mechanical circulatory support versus intra-aortic balloon pump in cardiogenic shock after acute myocardial infarction. J Am Coll Cardiol 2017;69:278–87. 10.1016/j.jacc.2016.10.022 [DOI] [PubMed] [Google Scholar]
- 35.Basir MB, Schreiber T, Dixon S, et al. Feasibility of early mechanical circulatory support in acute myocardial infarction complicated by cardiogenic shock: the Detroit cardiogenic shock initiative. Catheterization and Cardiovascular Interventions 2018;91:454–61. 10.1002/ccd.27427 [DOI] [PubMed] [Google Scholar]
- 36.Ostadal P, Rokyta R, Kruger A, et al. Extra corporeal membrane oxygenation in the therapy of cardiogenic shock (ECMO-CS): rationale and design of the multicenter randomized trial. Eur J Heart Fail 2017;19 Suppl 2:124–7. 10.1002/ejhf.857 [DOI] [PubMed] [Google Scholar]
- 37.Pöss J, Köster J, Fuernau G, et al. Risk Stratification for Patients in Cardiogenic Shock After Acute Myocardial Infarction. Journal of the American College of Cardiology 2017;69:1913–20. 10.1016/j.jacc.2017.02.027 [DOI] [PubMed] [Google Scholar]
- 38.Tan LB, Littler WA. Measurement of cardiac reserve in cardiogenic shock: implications for prognosis and management. Heart 1990;64:121–8. 10.1136/hrt.64.2.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams S, et al. Peak exercise cardiac power output; a direct indicator of cardiac function strongly predictive of prognosis in chronic heart failure. European Heart Journal 2001;22:1496–503. 10.1053/euhj.2000.2547 [DOI] [PubMed] [Google Scholar]
- 40.Fincke R, Hochman JS, Lowe AM, et al. Cardiac power is the strongest hemodynamic correlate of mortality in cardiogenic shock: a report from the shock trial registry. Journal of the American College of Cardiology 2004;44:340–8. 10.1016/j.jacc.2004.03.060 [DOI] [PubMed] [Google Scholar]
- 41.De Backer D, Biston P, Devriendt J, et al. Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010;362:779–89. 10.1056/NEJMoa0907118 [DOI] [PubMed] [Google Scholar]
- 42.Levy B, Clere-Jehl R, Legras A, et al. Epinephrine Versus Norepinephrine for Cardiogenic Shock After Acute Myocardial Infarction. Journal of the American College of Cardiology 2018;72:173–82. 10.1016/j.jacc.2018.04.051 [DOI] [PubMed] [Google Scholar]
- 43.Korabathina R, Heffernan KS, Paruchuri V, et al. The pulmonary artery Pulsatility Index identifies severe right ventricular dysfunction in acute inferior myocardial infarction. Catheterization and Cardiovascular Interventions 2012;80:593–600. 10.1002/ccd.23309 [DOI] [PubMed] [Google Scholar]