

ABSTRACT
KCNQ1 and KCNE1 co-assembly generates the IKS K+ current, which is crucial to the cardiac action potential repolarization. Mutations in their corresponding genes cause long QT syndrome (LQT) and atrial fibrillation. The A-kinase anchor protein, yotiao (also known as AKAP9), brings the IKS channel complex together with signaling proteins to achieve regulation upon β1-adrenergic stimulation. Recently, we have shown that KCNQ1 helix C interacts with the KCNE1 distal C-terminus. We postulated that this interface is crucial for IKS channel modulation. Here, we examined the yet unknown molecular mechanisms of LQT mutations located at this intracellular intersubunit interface. All LQT mutations disrupted the internal KCNQ1–KCNE1 intersubunit interaction. LQT mutants in KCNQ1 helix C led to a decreased current density and a depolarizing shift of channel activation, mainly arising from impaired phosphatidylinositol-4,5-bisphosphate (PIP2) modulation. In the KCNE1 distal C-terminus, the LQT mutation P127T suppressed yotiao-dependent cAMP-mediated upregulation of the IKS current, which was caused by reduced KCNQ1 phosphorylation at S27. Thus, KCNQ1 helix C is important for channel modulation by PIP2, whereas the KCNE1 distal C-terminus appears essential for the regulation of IKS by yotiao-mediated PKA phosphorylation.
KEY WORDS: Potassium channel, Long QT, KCNQ, KCNE, Arrhythmia, IKS
INTRODUCTION
The KCNQ (Kv7) subfamily of voltage-gated K+ channels (Kv) comprises five members that play important functions in different tissues including brain and heart (Jentsch et al., 2004). KCNQ1 α-subunits can interact with any one of the five KCNE auxiliary β-subunits, resulting in different channel functional characteristics (Abbott et al., 2001; Nakajo and Kubo, 2011; Sun et al., 2012; Wrobel et al., 2012). Co-assembly of KCNQ1 with KCNE1 generates the IKS K+ current that is vital for the proper repolarization of the cardiac action potential (Barhanin et al., 1996; Nerbonne and Kass, 2005; Sanguinetti et al., 1996). Mutations in either KCNQ1- or KCNE1-encoding genes lead to life-threatening cardiac arrhythmias causing long QT syndrome (LQT) and atrial fibrillation (Peroz et al., 2008). The KCNQ1 subunit possesses a large C-terminus, which has been shown to be important for channel gating, assembly and trafficking (Ghosh et al., 2006; Haitin and Attali, 2008; Shamgar et al., 2006; Wiener et al., 2008). The KCNQ C-terminus comprises proximal α helices A and B that bind calmodulin (CaM), whereas the distal coiled-coil helix C and helix D tetramerize, and crystallographic studies have shown that helix D forms a tetrameric parallel coiled-coil (Howard et al., 2007; Wiener et al., 2008).
The KCNQ C-terminus also appears to be a crucial region for modulation by phosphatidylinositol-4,5-bisphosphate (PIP2), which acts to stabilize the channel open state (Gamper and Shapiro, 2007). Accordingly, the spontaneous rundown of IKS channels observed in excised patches is markedly reduced by cytosolic application of PIP2 (Loussouarn et al., 2003; Park et al., 2005). The site of PIP2 binding on KCNQ2–KCNQ4 channels has been suggested to be located in the linker connecting helix A and B (Hernandez et al., 2008); however, a recent work indicates that this AB helix linker is not required for PIP2 regulation of KCNQ2 (Aivar et al., 2012). The PIP2-binding site also remains elusive for KCNQ1. Recent studies identified clusters of basic residues in KCNQ1 at the segment-2– segment-3 and segment-4–segment-2 intracellular linkers and proximal C-terminus as being involved in PIP2 binding (Thomas et al., 2011; Zaydman et al., 2013). KCNE1 was found to increase PIP2 sensitivity 100-fold over the KCNQ1 α-subunit alone and a juxtamembranous site in the KCNE1 C-terminus is a key structural determinant of PIP2 sensitivity (Li et al., 2011).
The KCNQ1 C-terminus also provides an interface for interacting with signaling proteins (Haitin and Attali, 2008). The scaffolding A-kinase anchor protein (AKAP) called yotiao or AKAP9 brings the IKS channel complex together with protein kinase PKA, protein phosphatase PP1, phosphodiesterase PDE4D3 and adenylate cyclase AC9 (also known as ADCY9) to achieve regulation following β-adrenergic stimulation (Chen and Kass, 2005; Kurokawa et al., 2004; Li et al., 2012; Marx et al., 2002; Terrenoire et al., 2009). The modulation is mediated by the physical interaction of yotiao with the distal helix D, which regulates IKS channel activity following phosphorylation or dephosphorylation of S27 at the N-terminus of KCNQ1 (Kurokawa et al., 2004). S92 at the N-terminus of KCNQ1 is also predicted to be phosphorylated (Lopes et al., 2007; Lundby et al., 2013). We and others have found a direct interaction between the C-termini of KCNQ1 and KCNE (Haitin et al., 2009; Zheng et al., 2010). We have previously shown that the coiled-coil helix C module of KCNQ1 is important for this interaction with KCNE1 distal C-terminus, thereby revealing a new interface between the two subunits (Haitin et al., 2009). We hypothesized that the interface between KCNQ1 helix C and the KCNE1 C-terminus provides a strategic platform for IKS channel modulation. Here, we explored the yet unknown mechanisms of LQT mutations located in this intracellular interface between the IKS α- and β-subunits. LQT1 mutants in KCNQ1 helix C, S546L, K557E, R555H, R555C and R562M showed weaker interaction with KCNE1 C-terminus. They displayed decreased current density and a voltage right-shift of channel activation, which could be accounted for by reduced PIP2 binding. In the distal KCNE1 C-terminus, the LQT mutation P127T showed weaker interaction with KCNQ1 helix C and suppression of cAMP-mediated IKS current upregulation that is caused by a reduced KCNQ1 phosphorylation at S27. Taken together, the data reveal that the intracellular interface between α- and β-subunits is crucial for proper IKS channel function.
RESULTS
IKS currents resulting from LQT5 mutations and truncation mutants at the KCNE1 C-terminus
In our quest to explore the mechanisms underlying LQT mutations located in the intracellular interface between KCNQ1 helix C and the KCNE1 C-terminus, we first examined the LQT5 mutations located at the distal C-terminus of KCNE1 (Fig. 1A). To this end, we probed the effects of co-expressing in CHO cells wild-type (WT) KCNQ1 with either one of the two KCNE1 LQT5 mutations V109I and P127T. The V109I mutant has been described as a heterozygous missense mutation with a borderline QTc interval in a family with a mild phenotype (Ackerman et al., 2003; Schulze-Bahr et al., 2001). The more distal P127T mutation causes a severe Romano-Ward syndrome (Splawski et al., 2000), but it has also been associated in a certain proband with a second mutation in KCNQ1 segment 6, A341V (Westenskow et al., 2004). The KCNE1 V109I LQT5 mutation resulted in currents very similar to those of WT IKS (Fig. 1B,C,G,H; supplementary material Table S1). The KCNE1 P127T LQT5 mutant produced a moderate (≈40%), although significant, reduction of the current density with no substantial change of the voltage dependence of channel activation (Fig. 1B,D,G,H; supplementary material Table S1).
Fig. 1.

Effects of KCNE1 C-terminus deletions and LQT5 mutations on IKS currents. (A) A cartoon of KCNE1, indicating the location of the LQT5 mutations and the deletions in the C-terminus. Representative current traces of WT KCNQ1, co-expressed in CHO cells with WT KCNE1 (B), LQT5 mutant V109I (C), LQT5 mutant P127T (D) or with KCNE1 deletion mutants at the distal C-terminus Δ109–129 (E) or the proximal C-terminus Δ69–77 (F). Cells were held at −90 mV. Membrane voltage was stepped for 3 s from −50 mV to +60 mV (or to +100 mV for Δ69–77) in 10 mV increments and then repolarized for 1.5 s to −60 mV. Current–voltage (G) and conductance–voltage (H) relationships of WT KCNQ1+WT KCNE1 or LQT5 mutants (n = 11–17). Current–voltage (I) and conductance–voltage (J) relationships of WT KCNQ1+WT KCNE1 or deletion mutants (n = 7–10).
Next, we examined, for comparison purposes, the effects of co-expressing WT KCNQ1 with truncation mutants spanning two different regions of the KCNE1 C-terminus. We have previously shown that the KCNQ1 coiled-coil helix C module is essential for the contact between the KCNQ1 C-terminus and the KCNE1 distal C-terminus (Haitin et al., 2009). Deletion of the KCNE1 distal C-terminus (Δ109–129) did not affect the current density (Fig. 1B,E,I). However, it produced a moderate right-shift of the conductance–voltage relationship (+15 mV; Fig. 1J). In contrast, deletion of the KCNE1 proximal C-terminus (Δ69–77) resulted in a drastic reduction (≈70%) of the current density accompanied by a +39.1 mV right-shift of the conductance–voltage relationship (Fig. 1B,F,I,J; supplementary material Table S1) and faster deactivation kinetics (τdeact = 525±55 ms and τdeact = 91±7 ms for WT and Δ69–77, respectively; mean±s.e.m., n = 7–10, P<0.001). This deletion in the KCNE1 proximal C-terminus (Δ69–77) includes a site that was found to be a key determinant of PIP2 sensitivity and is therefore expected to impact on IKS channel gating (Li et al., 2011).
The KCNE1 P127T LQT5 mutation disrupts the interaction between KCNQ1 helix C and the KCNE1 C-terminus but does not affect the channel trafficking to the plasma membrane
Previously, we have shown that deletion of the KCNQ1 helix C is necessary and sufficient to abolish the physical interaction between purified recombinant KCNQ1 and KCNE1 C-termini, suggesting that the KCNE1 C-terminus interacts with the coiled-coil helix C of the KCNQ1 C-terminus (Haitin et al., 2009). Here, we directly show by a GST pulldown assay that purified GST-tagged KCNQ1 helix C specifically interacts with purified MBP–His-tagged KCNE1 C-terminus and not with MBP–His protein alone (n = 4, Fig. 2A). Next, we examined the impact of the LQT5 mutations at the interface of the two subunits. GST pulldown experiments were performed using purified His-tagged WT KCNQ1 C-terminus and purified GST-tagged WT, deletion and LQT5 mutants of the KCNE1 C-terminus (Fig. 2B,C,E,G). We found that the severe KCNE1 LQT5 mutation, P127T, markedly reduced the interaction of KCNE1 with the KCNQ1 C-terminus (as a percentage of WT, P127T = 45%±10, mean±s.e.m., P<0.05; n = 3; Fig. 2E,G), whereas the mild-phenotype LQT5 V109I mutant displayed unaltered binding to the KCNQ1 C-terminus (Fig. 2E,G). In agreement with our previous work (Haitin et al., 2009), deletion of the KCNE1 distal C-terminus (Δ109–129) totally suppressed the interaction with the KCNQ1 C-terminus (Fig. 2B,C). In contrast, deletion of the KCNE1 proximal C-terminus (Δ69–77) significantly enhanced the binding to the KCNQ1 C-terminus (by 2.5±0.2-fold, P<0.001, n = 5; Fig. 2B,C).
Fig. 2.

LQT1 and LQT5 mutations disrupt the interaction between KCNQ1 helix C and KCNE1 C-terminus. (A) Representative immunoblot of GST pulldown (GST PD) (upper row) of the His-MBP-tagged KCNE1 C-terminus (residues 67–129, E1CT in diagram) or control His-MBP peptide, by GST-tagged KCNQ1 helix C (residues 535–572, Q1 Helix C, in diagram), illustrating the direct interaction of KCNE1 C-terminus with KCNQ1 helix C (n = 4). Equal amounts of His-MBP and His-MBP-tagged KCNE1 were used, as seen in the input (lower row). (B) Representative immunoblot of GST pulldown (upper row) of the His-tagged WT KCNQ1 C-terminus (residues 352–622 Δ396–504, Q1CT in diagram) by GST-tagged KCNE1 C-terminus deletion mutants (E1CT in diagram). Inputs are shown in the lower row. (C) Quantification of the pulldowns normalized to input (n = 5). *P<0.05, **P<0.01. (D) Representative immunoblot of GST pulldown (upper row) of the His-tagged WT KCNQ1 C-terminus (residues 352–622 Δ396–504) or LQT1 mutants by GST-tagged KCNE1 C-terminus (77–129). Inputs are shown in the lower row. (E) Representative immunoblot of GST pull-down (upper row) of His-tagged WT KCNQ1 C-terminus (residues 352–622 Δ396–504) by GST-tagged WT KCNE1 C-terminus (residues 67–129) or LQT5 mutants. Inputs are shown in the lower row. (F,G) Quantification of the pulldown shown in D and E, respectively, and normalized to input (n = 3–5). *P<0.05; **P<0.01; ***P<0.001.
We sought to investigate the mechanisms underlying the disrupted function of the severe KCNE1 LQT5 mutation P127T. First, we examined whether defective channel trafficking could account for the decreased current density observed with P127T. For this purpose, we transfected CHO cells with fluorescently tagged WT and mutant subunits (KCNQ1–CFP and KCNE1–YFP) which we imaged using total internal reflection fluorescence (TIRF) microscopy. To validate the TIRF methodology, we used positive and negative controls. The glycosylphosphatidylinositol (GPI)-linked fluorescent protein citrine (GPI-citrine) was used as a positive control for plasma membrane expression. GPI-citrine is known to express very strongly at the plasma membrane of cells (Glebov and Nichols, 2004). As shown in Fig. 3A, the GPI-citrine signal intensity was heavily detected under TIRF microscopy. Co-transfection of KCNQ1–CFP and GPI-citrine revealed that KCNQ1–CFP colocalized only slightly with GPI-citrine, reflecting the known partial processing of WT KCNQ1 to the plasma membrane (Kanki et al., 2004). The substantial cytoplasmic retention of WT KCNQ1 is further illustrated by comparing the somewhat weak TIRF signal relative to the strong epi-fluorescence intensity obtained from a cell expressing KCNQ1–CFP (Fig. 3B, upper panels). The KCNQ1 LQT1 mutation G589D, located in the coiled-coil helix D of the KCNQ1 C-terminus, was used as a negative control, because this mutant has previously been described as a mistrafficked channel that is heavily retained in the endoplasmic reticulum (ER) (Kanki et al., 2004). In agreement with this previous work, we found that the KCNQ1 G589D mutant exhibits a much weaker TIRF signal compared to WT KCNQ1 (13%±4 of WT KCNQ1, mean±s.e.m., n = 16–25, P<0.001) (Fig. 3B,E). The robust ER retention of the G589D mutant is further confirmed by the stronger signal obtained under epi-fluorescence relative to that revealed under TIRF microscopy (Fig. 3B). Importantly, the YFP-tagged KCNE1 LQT5 mutant P127T showed comparable plasma membrane trafficking to that of WT channels (n = 56–60; Fig. 3D,F), suggesting that the decreased current density displayed by the LQT5 mutant does not seem to be accounted for by trafficking defects. However, TIRF does not allow discrimination between the signals from the plasma membrane and those arising from the cortical ER, which is especially relevant for some K+ channels (Fox et al., 2013). Thus, an effect on trafficking cannot be totally excluded.
Fig. 3.

LQT1 and LQT5 mutations do not affect the channel trafficking to the plasma membrane. (A) TIRF fluorescence images of CHO cells co-expressing CFP-tagged WT KCNQ1 and GPI-citrine. (B) Epi-fluorescence (left panels) and TIRF fluorescence (right panels) images of CHO cells expressing CFP-tagged WT KCNQ1+KCNE1 (upper panels) or CFP-tagged G589D LQT1 mutant+KCNE1 (lower panels). (C) TIRF fluorescence images of CHO cells co-expressing CFP-tagged helix C LQT1 mutants with KCNE1. (D) YFP-tagged WT KCNE1 or LQT5 mutant co-expressed with WT KCNQ1. Quantification of the TIRF fluorescent signals of CFP-tagged WT KCNQ1 and LQT1 mutants (E) and of YFP-tagged WT and LQT5 mutant (F) as normalized to membrane signal area (n = 16–60). ***P<0.001. Scale bars: 10 µm.
The P127T LQT5 mutation and Δ109–129 deletion at the KCNE1 distal C-terminus suppress the cAMP-mediated upregulation of the IKS current
As shown above (Fig. 3D,F), the decrease in current density displayed by the KCNE1 P127T LQT5 mutant could not be accounted for by trafficking defects. We hypothesized that the KCNE1 distal C-terminus could be important for cAMP-dependent upregulation of the IKS current, and that mutations in this region might disrupt the modulation. Thus, we tested whether cAMP-stimulation of the P127T KCNE1 mutant was similar to that obtained with the WT IKS channel. CHO cells were transfected with both WT KCNQ1 and WT yotiao, together with WT KCNE1 or its mutants. Transfected CHO cells were stimulated by 200 µM cAMP and 0.2 µM okadaic acid, a PP1 and PP2A phosphatase inhibitor, added in the patch pipette (Marx et al., 2002). WT IKS currents were significantly upregulated by cAMP plus okadaic acid treatment (at +50 mV, 70%±9 stimulation; mean±s.e.m., n = 7, P<0.01; Fig. 4A,B); however, no significant changes in deactivation kinetics and conductance–voltage relations were observed. In striking contrast, the P127T LQT5 mutant completely lost its stimulation upon cAMP plus okadaic acid treatment (Fig. 4C,D). Likewise, the KCNE1 distal C-terminus deletion mutant Δ109–129 was unresponsive to cAMP plus okadaic acid stimulation (Fig. 4E,F). Interestingly, the mild phenotype LQT5 V109I mutant was significantly upregulated by cAMP plus okadaic acid treatment (at +50 mV, 64%±7 stimulation; n = 6, P<0.05; Fig. 4G,H).
Fig. 4.
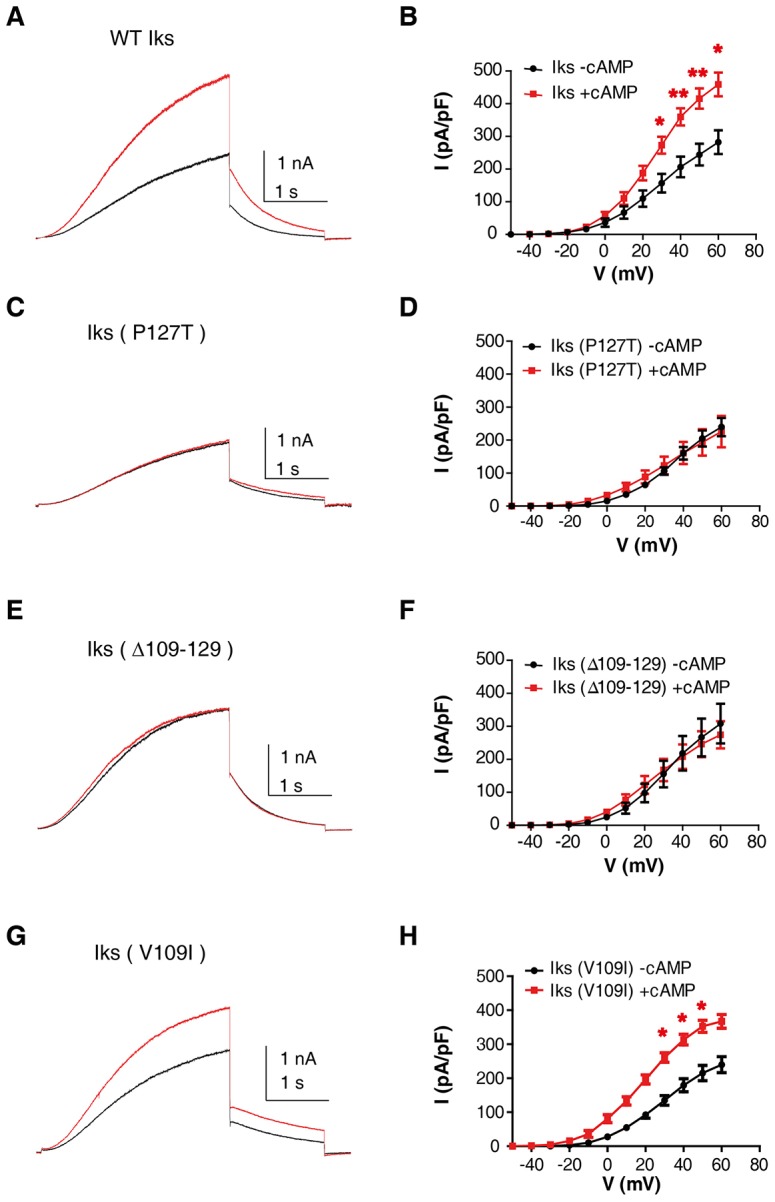
P127T LQT5 mutation and Δ109–129 deletion at the KCNE1 distal C-terminus suppress the cAMP-mediated upregulation of IKS current. Representative current traces recorded from CHO cells co-expressing WT KCNQ1+WT yotiao and WT KCNE1 (A), KCNE1 P127T (C), KCNE1 Δ109–129 (E) and KCNE1 V109I (G). Cells were held at −90 mV and stepped to +30 mV for 3 s and then repolarized to −40 mV for 1.5 s. Cells were recorded in the absence (black traces) or presence (red traces) of 200 µM cAMP+0.2 µM okadaic acid. Current–voltage relationships of WT KCNQ1+WT yotiao and WT KCNE1 (B) or KCNE1 P127T (D) or KCNE1 Δ109–129 (F) or KCNE1 V109I (H). Voltage was stepped for 3 s from −50 mV to +60 mV in 10 mV increments followed by repolarization to −40 mV for 1.5 s. Red and black curves represent recordings with or without cAMP+okadaic acid in the patch pipette, respectively. (n = 6–7). Above +30 mV, the WT IKS and, above +20 mV, the KCNE1 V109I current upregulation induced by cAMP+okadaic acid were significant. *P<0.05, **P<0.01.
P127T LQT5 mutation and deletion mutant Δ109–129 do not disrupt the KCNQ1 interaction with yotiao
To elucidate the possible mechanisms underlying the impaired cAMP-mediated modulation in KCNE1 distal C-terminus mutants, we first examined whether the mutations P127T and Δ109–129 disrupted KCNQ1 interaction with yotiao. To verify proper expression of the proteins in transfected HEK 293 cells, lysates were subjected to western blotting (supplementary material Fig. S1A). Lysates from cells expressing both WT KCNQ1 and WT yotiao, plus WT KCNE1 or its mutants were subjected to immunoprecipitation with anti-KCNQ1 antibodies. Western blots probed with anti-yotiao antibodies, quantitatively tested the interaction of KCNQ1 with yotiao, by normalizing the yotiao signal to the KCNQ1 signal intensity in the precipitate (supplementary material Fig. S1B). The results revealed that when compared to WT KCNE1, there was no significant difference in the ability of KCNQ1 to interact with yotiao when co-expressed with P127T LQT5 mutant or with KCNE1 distal C-terminus deletion mutant Δ109–129 (supplementary material Fig. S1B,C).
The P127T LQT5 mutation and deletion mutant Δ109–129 impair phosphorylation of KCNQ1 at S27 during cAMP-dependent stimulation
Next, we set out to examine whether the KCNE1 distal C-terminus mutations P127T and Δ109–129 disrupted phosphorylation of S27 in KCNQ1, in the presence of yotiao. Transfected HEK 293 cells expressing both WT KCNQ1 and WT yotiao, plus WT KCNE1 or its mutants were treated for 10 min with 250 µM 8-(4-chlorophenylthio) adenosine 3′,5′-cyclic monophosphate (8CPT), a membrane permeable analog of cAMP and with 0.2 µM okadaic acid. Cell lysates were subjected to SDS-PAGE followed by western blotting and blots were probed with anti-KCNQ1, anti-yotiao and anti-KCNE1 antibodies for validating protein expression in the complex and for quantification purposes. Importantly, blots were also probed with a specific antibody against phosphorylated S27 of KCNQ1 to explore the extent of KCNQ1 phosphorylation at S27 under basal and cAMP-stimulated conditions (Kurokawa et al., 2003). Basal S27 phosphorylation was not significantly different when WT KCNQ1 plus WT yotiao were co-expressed with either WT KCNE1 or its mutants. In the WT IKS channel complex co-expressed with yotiao, cAMP-stimulation produced a significant 2.6-fold increase in S27 phosphorylation (n = 5, P<0.0001; Fig. 5A,B). A comparable stimulation was found for the mild-phenotype LQT5 mutation V109I, with a 2-fold increase in S27 phosphorylation upon cAMP treatment (n = 3, P<0.05; Fig. 5A,B). In contrast, for the P127T LQT5 mutation, the increase in S27 phosphorylation following cAMP exposure was lower and not statistically significant (1.65-fold increase, n = 5, P>0.05). Similarly, a weaker and non-significant stimulation of S27 phosphorylation was obtained with the KCNE1 distal C-terminus deletion mutant Δ109–129 (1.7-fold increase, n = 5, P>0.05; Fig. 5A,B). When expressed as the relative cAMP- and okadaic acid-stimulated phosphorylation, the effect of the V109I mutant appears to be larger than that of the deletion mutant Δ109–129, although not statistically different (Fig. 5C). For comparison, we examined whether the LQT1 mutation K557E, residing in KCNQ1 helix C and whose defect involves weaker binding to PIP2 (see below), could alter stimulation of S27 phosphorylation following cAMP treatment. Results show that the KCNQ1 mutant K557E co-expressed with both WT KCNE1 and WT yotiao produced a significant 2.4-fold increase in S27 phosphorylation (n = 4, P<0.01; Fig. 5A,B). We conclude that the LQT5 mutation P127T disrupts yotiao regulation, whereas an unrelated LQT1 mutation (K557E) does not.
Fig. 5.

LQT5 P127T mutant and deletion mutant Δ109–129 impair phosphorylation of KCNQ1 at S27 during cAMP-dependent stimulation. (A) Representative immunoblots (IB) of HEK 293 cells lysates co-expressing yotiao, WT KCNQ1 and WT, mutated (V109I and P127T) or truncated (Δ109–129) KCNE1 from cells treated in the absence and presence of 250 µM 8CPT+0.2 µM okadaic acid and immunoblots of HEK 293 cells lysates co-expressing yotiao WT KCNE1 and WT KCNQ1 or LQT1 mutant K557E from cells treated in the absence and presence of 250 µM 8CPT+0.2 µM okadaic acid. Blots were probed with antibodies against phosphorylated KCNQ1 S27 (first row from top), KCNQ1 (second row from top), yotiao (third row from top) and KCNE1 (fourth row from top). (B) Quantification of phosphorylated KCNQ1 S27 was calculated by dividing phosphorylated signal to KCNQ1 input (anti-KCNQ1 blot) for both unstimulated and cAMP-stimulated cells (250 µM 8CPT+0.2 µM okadaic acid) and normalized to stimulated WT KCNQ1+WT KCNE1+yotiao (n = 3–5). *P<0.05, ** P<0.01, ***P<0.001; ns: not statistically significant. (C) An alternative quantification of phosphorylated KCNQ1 S27 was expressed as the relative cAMP+okadaic acid-stimulated S27 phosphorylation where the extent of induced phosphorylation of WT IKS was equal to 1 (n = 3–5). *P<0.05; ns: not statistically significant.
IKS currents resulting from LQT1 mutations at the KCNQ1 helix C
We then studied the effects of co-expressing WT KCNE1 in CHO cells with five LQT1 mutations, S546L, R555C, R555H, K557E and R562M, located in the KCNQ1 helix C (Fig. 6A; supplementary material Table S1). All mutations markedly reduced the current densities, which were paralleled by potent right-shifts of the voltage dependence of channel activation, slower activation kinetics and faster deactivation (for S546L, R555C, R555H, K557E and R562M, respectively, 75%, 88%, 69%, 99% and 57% inhibition of current density at +60 mV; the ΔV50 was +48 mV, +65.4 mV, +47.1 mV, +92.7 mV and +40.7 mV, respectively, n = 5–10, P<0.001; for WT and the mutants as above, the t1/2 was 0.91±0.07 s, 1.20±0.09 s, 1.36±0.07 s, 1.28±0.07 s, 1.40±0.11 s and 1.41±0.06 s, respectively, P<0.05; for WT and the mutants as above, the τdeact was 675±76 ms, 550±36 ms, 428±26 ms, 460±29 ms and 194±18 ms, respectively, P<0.05; in mutant R562M, τdeact was 720±50 ms, n = 6; all results are mean±s.e.m.) (Fig. 6).
Fig. 6.

Effects of KCNQ1 helix C LQT1 mutations on IKS currents. A cartoon of one KCNQ1 subunit indicating the location of helix C in the C-terminus and its sequence, where LQT1 mutations are situated (A). Representative current traces of WT KCNE1 co-expressed in CHO cells with WT KCNQ1 (B), LQT1 mutants S546L (C), R555C (D), R555H (E), K557E (F) and R562M (G). The voltage-clamp protocol is the same as that described in Fig. 1, except that higher membrane voltages of up to +160 mV were applied for some LQT1 mutations, because of their depolarizing shift. Current–voltage (H) and conductance–voltage relationships of WT and KCNQ1 LQT1 mutants co-expressed with WT KCNE1 (n = 5–9) (I).
The helix C LQT1 mutations disrupt the interaction with the KCNE1 C-terminus but do not affect channel trafficking to the plasma membrane
GST-pulldown experiments revealed that all KCNQ1 helix C LQT1 mutations exhibit significantly weaker interaction with the KCNE1 C-terminus when compared to WT KCNQ1 C-terminus (percentage of WT: S546L = 41%±5, P<0.001; R555C = 65%±8, P<0.05; R555H = 45%±3, P<0.001; K557E = 25%±8, P<0.01; R562M = 52%±8, P<0.01; mean±s.e.m.; n = 3; Fig. 2D,F). Exploring the impact on channel trafficking with TIRF microscopy, we found that the CFP-tagged helix C LQT1 mutants co-expressed with WT KCNE1 exhibited similar trafficking to WT IKS channels to the plasma membrane (n = 8–25; Fig. 3C,E). The normal trafficking to the plasma membrane of the R555C LQT1 mutant is in agreement with a previous report (Kanki et al., 2004). We conclude that the decreased current density displayed by the five LQT1 mutants cannot be accounted for by trafficking defects.
LQT1 mutations in KCNQ1 helix C showed impaired interaction with PIP2
The distal half of KCNQ1 helix C encompasses a cluster of basic residues, which were previously suggested to play a role in channel modulation by PIP2 (Park et al., 2005). As most of the LQT1 mutations in this study neutralize a basic residue and lead to reduced current density and a potent right-shift of the conductance–voltage relations, we hypothesized that the mechanism underlying these helix C mutations might involve an impaired KCNQ1–PIP2 interaction. To test this hypothesis, we first probed the PIP2 sensitivity of WT and mutant channels using the Dr-VSP voltage-sensitive phosphatase (Hossain et al., 2008; Okamura et al., 2009). Fig. 7A shows a standard protocol previously used to monitor WT IKS currents before and following activation of Dr-VSP (Danio rerio VSP, also known as TPTE), where a +10 mV prepulse opens the channel, followed by a +100 mV voltage step that activates the phosphatase and then a return to +10 mV (Kruse et al., 2012). When this protocol was repeatedly applied every 10 s for 2 min, activation of Dr-VSP led to a 64%±4 reduction of WT IKS currents (n = 5; Fig. 7A, right panel, red trace), while in the absence of Dr-VSP a slight current overshoot of 21%±4 was observed (n = 5; Fig. 7A, left panel, red trace). We obtained a relatively lower suppression of WT IKS currents compared to that found by Kruse et al., because our intracellular solution did not contain Mg2+ (see Materials and Methods) as we wanted to prevent a fast run-down of WT IKS current, even in the absence of Dr-VSP. Indeed, Mg2+ has previously been found to reduce the KCNQ currents by electrostatic binding to the negative charges of PIP2, competitively reducing the amount of free PIP2 available for interaction with the channels (Suh and Hille, 2007). However, this +10 mV prepulse protocol could not be applied for the five LQT1 mutants because they exhibit potent right-shifts of the voltage dependence of activation, which prevents them being activated at +10 mV like WT IKS (see Fig. 6). To circumvent this difficulty, we applied repeatedly (every 5 s) a 2-s-long step pulse to +100 mV in order to both open the channel and activate the Dr-VSP. During the successive sweeps, the rate of current decline would be expected to reflect the channel affinity for PIP2. Thus, assuming that the rate of PIP2 re-synthesis is roughly the same, the faster is the rate of current decrease, the lower the channel affinity for PIP2. The kinetics of current decline for WT IKS and mutant channels were analyzed in the absence or presence of Dr-VSP (Fig. 7D,F,H,J,L). The comparison between WT IKS and the five LQT1 mutants (+KCNE1) was quantified at an early time point (10 s) before current drop reached steady state and was expressed as a ratio of currents measured at time 10 s over that recorded at time 0. Representative current traces are also shown at time 0 (Fig. 7B,C,E,G,I,K, black trace) and at time 10 s (Fig. 7B,C,E,G,I,K, red trace). Except for mutant S546L, the results indicate that mutants R555C, R555H, K557E and R562M exhibited a significantly faster current decline compared to WT IKS [at 10 s, ratios of current decline were 0.75±0.05 (n = 5), 0.74±0.02 (n = 7), 0.32±0.01 (n = 3, P<0.01), 0.54±0.06 (n = 4, P<0.05), 0.55±0.04 (n = 7, P<0.05) and 0.48±0.04 (n = 7, P<0.01) for WT, S546L, R555C, R555H, K557E and R562M, respectively (all results are mean±s.e.m.)]. In the absence of Dr-VSP, WT IKS and mutant channels tended to show current overshoot following this protocol (Fig. 7D,F,H,J,L). Next, we assessed whether the higher sensitivity to the Dr-VSP phosphatase displayed by the helix C LQT1 mutants arose from their weaker binding to PIP2. To test this assumption, we performed pulldown experiments of purified His-tagged WT and LQT1 mutant KCNQ1 C-terminus using PIP2-coated agarose beads. We found all mutants to exhibited significantly lower binding to PIP2 as compared to the WT KCNQ1 C-terminus [percentage of WT binding: 43%±12 for S546L, n = 3, P<0.01; 55%±6 for R555C, n = 3, P<0.05; 34%±7 for R555H, n = 3, P<0.001; 17%±6 for K557E, n = 3, P<0.001 and 14%±5 for R562M, n = 3, P<0.001; Fig. 8A,B (all results are mean±s.e.m.)]. These results suggest that LQT1 mutations in KCNQ1 helix C, except S546L, have an impaired interaction with PIP2.
Fig. 7.

LQT1 mutations in KCNQ1 helix C exhibit greater sensitivity to the voltage-sensitive phosphatase Dr-VSP. (A) Representative current traces of WT IKS (KCNQ1+KCNE1) sensitivity to the voltage sensitive phosphatase Dr-VSP recorded by means of a tri-pulse protocol where membrane potential is first stepped to +10 mV (3 s, from −90 mV holding potential) to open the channel, followed by a +100 mV voltage step (2 s) to activate Dr-VSP and then return to +10 mV (3 s). This protocol was repeatedly applied every 10 s for 2 min. Left panel shows WT IKS sensitivity to protocol application in the absence of Dr-VSP at the first (black trace) and last sweeps (red trace). Right panel shows WT IKS sensitivity to protocol application in the presence of Dr-VSP at the first (black trace) and last sweeps (red trace) (n = 5). (B,C,E,G,I,K) Representative current traces of WT IKS, S546L, R555C, R555H, K557E and R562M sensitivity to Dr-VSP recorded by one pulse protocol, where membrane potential is stepped to +100 mV (2 s, from −90 mV holding potential) to both open the channels and activate Dr-VSP. Shown are current traces at time 0 (black trace) and at time 10 s (red trace). Kinetics of current decline for WT IKS and mutant channels are shown in the absence or presence of Dr-VSP. The comparison between WT IKS and the five LQT1 mutants was quantified at an early time point (10 s) before the current drop reached steady state and was expressed as a ratio of currents measured at time 10 s over that recorded at time 0. At 10 s, ratios of current decline were 0.75±0.05 (n = 5), 0.74±0.02 (n = 7), 0.32±0.01 (n = 3, P<0.01), 0.54±0.06 (n = 4, P<0.05), 0.55±0.04 (n = 7, P<0.05) and 0.48±0.04 (n = 7, P<0.01) for WT, S546L, R555C, R555H, K557E and R562M, respectively.
Fig. 8.

LQT1 mutations in KCNQ1 helix C show impaired binding to PIP2. (A) Representative immunoblot of PIP2 pulldown (PIP2 PD) by PIP2-coated agarose beads of His-tagged WT KCNQ1 C-terminus or LQT1 mutants (upper row). Inputs are shown in the lower row. (B) Quantification of the pull-down normalized to input (n = 3). *P<0.05; **P<0.01; ***P<0.001. (C) Cartoon summarizing the two different pathways involved in the mechanisms of LQT mutations located at the interacting KCNE1 distal C-terminus and KCNQ1 helix C module: one involving LQT1 mutants in KCNQ1 helix C that impairs PIP2 interaction and one of the LQT5 mutant P127T at the distal KCNE1 C-terminus, which impedes IKS regulation by yotiao-mediated PKA phosphorylation.
DISCUSSION
In the present work, we provide novel insights into the molecular mechanisms underlying LQT mutations located at a crucial intracellular interface of the subunits forming the cardiac IKS channel and corresponding to the interaction between the KCNE1 distal C-terminus and the KCNQ1 coiled-coil helix C module (Haitin et al., 2009). Here, we show that LQT mutations located on both sides of this interface disrupted IKS channel function. On the one hand, the LQT1 mutants in KCNQ1 helix C (R555C, R555H, K557E and R562M) showed reduced PIP2 binding and a strong right-shift of channel activation. On the other hand, the LQT5 mutation P127T in KCNE1 distal C-terminus, suppressed yotiao-dependent cAMP-mediated upregulation of IKS currents by reducing KCNQ1 phosphorylation at S27 (Fig. 8C).
All members of the KCNQ family require the presence of the membrane phospholipid PIP2 for proper function (Gamper and Shapiro, 2007; Suh and Hille, 2005; Zhang et al., 2003). PIP2 stabilizes the open state of the cardiac IKS KCNQ1–KCNE1 channel, thereby slowing deactivation kinetics and producing a negative shift in the voltage dependence of activation (Loussouarn et al., 2003; Park et al., 2005; Thomas et al., 2011). Different regions endowed with clusters of basic residues have been proposed to interact with PIP2. Hernandez et al., identified a conserved cluster of basic residues within the linker connecting helix A and B in the C-terminus of KCNQ2–KCNQ4 subunits (Hernandez et al., 2008). This basic cluster was suggested to interact with PIP2 through electrostatic and hydrogen-bonding networks (Hernandez et al., 2008). However, a recent study showed that this linker is not required for PIP2 regulation of KCNQ2 (Aivar et al., 2012). Others have identified, in both the KCNQ1 and KCNQ2 proximal C-terminus, a cluster of basic residues just downstream of segment 6 (Telezhkin et al., 2013; Thomas et al., 2011; Zhang et al., 2003). In KCNQ1, residues in the segment-2–segment-3 and segment-4–segment-5 linkers have also been proposed to be important for PIP2 interaction (Matavel et al., 2010; Zaydman et al., 2013). In addition, basic residues have been identified in coiled-coil helix C and have been suggested to be important for PIP2 interaction (Matavel et al., 2010; Park et al., 2005). Notably, in the present study, the LQT1 mutations located in KCNQ1 helix C (R555C, R555H, K557E, R562M), impaired their physical interaction with KCNE1 C-terminus. In addition, these LQT1 mutants co-expressed with WT KCNE1, showed significantly smaller current densities, depolarizing shifts of the conductance–voltage relationship and faster deactivation kinetics. Such features are suggestive of stabilization of the channel closed state.
These properties are in line with those expected for an impaired channel–PIP2 interaction, which led us to hypothesize that this cluster of basic residues in KCNQ1 helix C might be involved in PIP2 modulation. Using two complementary approaches, we found that all LQT1 mutants exhibited significantly lower binding to PIP2 than WT as measured by pulldown experiments and by the use of the voltage-dependent phosphatase Dr-VSP (Okamura et al., 2009). Our results indicate that upon activation of Dr-VSP, mutants R555C, R555H, K557E and R562M exhibited a significant faster current decline compared to WT IKS, which likely reflects their lower affinity for PIP2. Mutant S546L was an exception and displayed a similar sensitivity to Dr-VSP as WT IKS, suggesting that it does not appear to interact functionally with PIP2. However, it is possible that, like mutant R539W, it interacts with cholesterol (Coyan et al., 2014).
Our results suggest that KCNQ1 helix C is crucial for PIP2 modulation (Fig. 8C). They also raise the question of the significance of multiple PIP2-interacting regions in KCNQ1. It is possible that helix C does not bind PIP2 directly but rather contributes allosterically to modulate PIP2 binding to a pocket molded by multiple contact sites (Zaydman et al., 2013), including the conserved membrane regions and regions located downstream segment 6.
The macro-molecular scaffolding complex formed by KCNQ1 and KCNE1, together with yotiao, PKA, PP1, PDE4D3 and AC9, is designed to achieve IKS channel regulation following β1-adrenergic stimulation and can be disrupted in various ways under disease conditions (Chen and Kass, 2005; Kurokawa et al., 2004; Li et al., 2012; Marx et al., 2002; Terrenoire et al., 2009). The LQT1 mutation A341V in KCNQ1 segment 6 has recently been shown to preserve the KCNQ1–yotiao interaction but to reduce cAMP-induced KCNQ1 phosphorylation at S27 and subsequent IKS current upregulation (Heijman et al., 2012). The LQT5 mutation D76N in KCNE1 C-terminus spared the cAMP-mediated KCNQ1 phosphorylation at S27 but failed to upregulate its resulting IKS current. In this study, we found that the LQT5 mutant P127T in KCNE1 distal C-terminus led to a significant decrease in basal IKS currents, which were unable to be stimulated upon cAMP exposure. In addition, the P127T mutation impaired the physical interaction with KCNQ1 helix C. We showed that neither defective channel trafficking nor an impaired KCNQ1–yotiao interaction could account for the compromised P127T mutant channel function. In contrast, we found that mutant P127T significantly decreased KCNQ1 phosphorylation at S27. To our knowledge, the LQT5 mutation P127T is the first described example of a KCNE1 mutant leading to impaired KCNQ1 phosphorylation at S27 following cAMP stimulation (Fig. 8C). To date, only one LQT1 mutation A341V in KCNQ1 (Heijman et al., 2012) has been found to disrupt KCNQ1 phosphorylation at S27, while preserving the KCNQ1–yotiao interaction. Interestingly, the LQT5 mutant P127T has been described both as a single mutation (Splawski et al., 2000) and as being additionally associated with the LQT1 mutation A341V in another proband who presented with syncope and longer QTc interval than his parents (Westenskow et al., 2004). As both mutations impair KCNQ1 phosphorylation at S27 and subsequently prevent current increase upon cAMP stimulation, they might expose the affected individuals to severe clinical phenotypes and increase their susceptibility to ventricular tachyarrhythmias under sympathetic stress conditions. Like S27, S92 has recently been found to be phosphorylated upon β-adrenergic stimulation when the channel is localized at the plasma membrane, and this phosphorylation event resulted in increased IKS current (Lundby et al., 2013). It will be interesting in future work to examine the impact of the LQT5 mutant P127T on the phosphorylation of S92 following cAMP stimulation.
KCNQ1 can be phosphorylated at S27 upon PKA activation, without the need for KCNE1 expression (Kurokawa et al., 2003; Marx et al., 2002). However, in the absence of KCNE1, there is no transduction of PKA-mediated KCNQ1 phosphorylation (Kurokawa et al., 2003). An intact KCNE1 C-terminus has been suggested to be crucial for cAMP-dependent upregulation of IKS currents (Kurokawa et al., 2009), which reflects the crucial role played by KCNE1 in coupling sympathetic β1-adrenergic signaling to post-phosphorylation functional events in the cardiac IKS channel. Here, we mapped the KCNE1 effect to its distal C-terminus by showing that deletion of the KCNE1 C-terminus (Δ109–129) and the LQT5 mutation P127T depress KCNQ1 phosphorylation at S27 and subsequently prevent the cAMP-mediated increase in IKS currents. The molecular mechanism by which this transduction occurs is currently unknown. Previous studies have shown a direct interaction between the KCNQ1 proximal C-terminus with the KCNE1 C-terminus (Zheng et al., 2010). Using FRET and fluorescently labeled subunits, we have previously shown that the interaction between the KCNE1 C-terminus and the KCNQ1 C-terminus in the channel complex shortens the distance between the KCNQ1 N- and C-termini (Haitin et al., 2009). This is probably a crucial step for the yotiao-mediated transfer of PKA from the C-terminus to the N-terminus of KCNQ1 and the subsequent phosphorylation of S27. In a dynamic FRET study, we also found that during IKS channel opening, the KCNQ1 C-terminus and KCNE1 C-terminus draw closer together and that this gated motion is suppressed by the LQT5 mutant D76N (Haitin et al., 2009), a mutation previously shown to cause functional disruption of cAMP-mediated stimulation of IKS current (Kurokawa et al., 2003). Thus, the strategic location of the KCNE1 C-terminus interacting with KCNQ1 helix C and the proximity of KCNQ1 N- and C-termini and their gated motions might be pivotal for transducing the phosphorylation of KCNQ1 S27 into IKS current upregulation.
In summary, this work reveals the molecular mechanisms of LQT mutations located at the interacting KCNE1 distal C-terminus and KCNQ1 helix C module. Our results show that this intracellular interface is a crucial platform for transduction and integration of two modulatory mechanisms, one involving the PKA-mediated increase in IKS currents following β-adrenergic stimulation, and one implying PIP2 modulation (Fig. 8C). Interestingly, these two regulation pathways have recently been suggested to undergo cross talk in such a way that KCNQ1 sensitivity to PIP2 can be modulated by PKA activation (Choveau et al., 2012; Matavel et al., 2010). Consequently, the LQT mutations examined in this study selectively disrupt either one of these two regulatory processes according to their respective location in this important intersubunit interface.
MATERIALS AND METHODS
Molecular biology
Human KCNQ1 and KCNE1 were cloned into pCDNA3 vector to allow eukaryotic expression. The LQT1 mutations in KCNQ1 (S546L, R555C/H, K557E and R562M) and the LQT5 mutations in KCNE1 (V109I and P127T) and KCNE1 C-terminus deletions (69–77 and 109–129) were introduced using the PCR-based Quikchange site-directed mutagenesis (Stratagene). For TIRF microscopy, KCNQ1 and KCNE1 were cloned into pECFP-N1 and pEYFP-N1 vectors, respectively. All constructs were verified by full DNA sequencing.
Production and purification of recombinant proteins
The KCNQ1 C-terminus (residues 352–622), with deletion of the intervening loop between helix A and B (residues 396–504) was cloned into the multiple cloning site I of the pET-Duet vector (Novagen), located downstream of a His8 tag and a tobacco etch virus (TEV) protease site and the calmodulin-encoding gene in multiple cloning site II. LQT1 mutations were introduced to the construct by standard PCR techniques. KCNQ1 C-terminus constructs were co-purified with CaM as previously described (Haitin et al., 2009; Wiener et al., 2008). The KCNE1 C-terminus (residues 67–129 or 77–129) was amplified by PCR with primers containing BamHI and EcoRI sites in the 5′ and 3′ flanking regions, respectively. Digested PCR products were ligated into the pGEX-3X vector (Pharmacia Biotech). The LQT5 mutations were introduced to KCNE1, by standard PCR techniques (Quikchange).
TIRF microscopy
Fluorescence images of CHO cells were taken at room temperature by TIRF microscopy. CHO cells were imaged 48 h after transfection at room temperature (20°C–22°C). Images were taken with a Zeiss inverted microscope (Axio Observer Z1) equipped with an alpha Plan fluar 100× oil immersion objective (NA 1.45). Fluorescent cells were excited with the 458- or 514-nm line of an air-cooled argon-ion laser (LASOS 771) with light intensities of 20%, 5% and 5% for CFP, YFP and GPI-citrine fluorophores, respectively. Emission images were acquired with 485- and 575-nm filter sets (Zeiss, filter sets 51 and 53, respectively) at an exposure time of 100 ms and gain of 400 with an EM CCD camera (QUANT-EM 512SC) controlled by an AxioVision software version 4.8.2, that was also used for quantification.
Pulldown experiments
For GST pulldown, 10 µg of purified WT or mutant GST-tagged KCNE1 C-terminus proteins were incubated with 50 µg of purified WT or mutant His-tagged KCNQ1 C-terminus proteins in PBS (pH 7.4) containing 1% Triton X-100 and 1 mM PMSF for 1.5 h at 4°C under constant rotation. Alternatively, 50 µg of GST-tagged KCNQ1 helix C was incubated with 10 µg of MBP–His-tagged KCNE1 C-terminus in PBS (pH 7.4) containing 0.5% Igepal and 1 mM PMSF. Input samples were taken for quantification purposes. Equal amounts of glutathione–Sepharose (GE Healthcare) were added. After incubation under rotation for 1 h at 4°C and three washes with PBS containing 1% Triton X-100 and 1 mM PMSF, bound complexes were eluted using buffer B (containing in mM, 100 Tris-HCl pH 8, 150 NaCl and 20 glutathione). Eluates were boiled together with Laemmli sample buffer. Detection of western blots was performed using horseradish peroxidase (HRP)-conjugated anti-His antibody and ECL. For PIP2 pulldown, 5 µg of purified WT or LQT1 mutants His-tagged KCNQ1 C-terminus proteins were incubated with equal amounts of PIP2-coated agarose beads (Echelon Biosciences) for 2 h at room temperature in binding buffer containing (in mM): 10 HEPES pH 7.4, 150 NaCl and 0.25% Igepal. After sample centrifugation at 600 g, three washes in binding buffer were performed and followed by boiling with 4× sample buffer at 95°C. Samples were subjected to SDS-PAGE and followed by western blotting. Input samples were taken for quantification purposes. Detection was performed using HRP-conjugated anti-His antibody and ECL.
Electrophysiology
CHO cells were recorded in the whole-cell configuration of the patch-clamp technique as previously described (Meisel et al., 2012). The intracellular solution contained in mM: 130 KCl, 5 Na2ATP, 5 EGTA, 10 HEPES pH 7.3 (adjusted with KOH), and sucrose was added to adjust osmolarity to 290 mOsm. The external solution contained (in mM): 140 NaCl, 4 KCl, 1.2 MgCl2, 1.8 CaCl2, 11 glucose, 5.5 HEPES, pH 7.3 (adjusted with NaOH), and sucrose was added to adjust osmolarity to 320 mOsm. Cells were held at −90 mV. Voltage was stepped for 3 s to +30 mV followed by repolarization to −60 mV for 1.5 s every 30 s until current stabilization. Then, to establish the current–voltage relations, voltage was stepped for 3 s from −50 mV to +60 mV in 10 mV increments followed by repolarization to −60 mV for 1.5 s. To activate PKA, 200 µM cAMP (Sigma) plus 0.2 µM okadaic acid (Sigma) was added to the patch pipette solution. For the treatment with cAMP, the current–voltage relationships were analyzed as for the above, except that the repolarization was at −40 mV.
Data analysis was performed using the Clampfit program (pClamp10, Axon Instruments), Microsoft Excel and Prism 5.0 (GraphPad). Conductance (G) was calculated as G = I/(V−Vrev). G was then normalized to the maximal conductance. Activation curves were fitted to a single Boltzmann distribution according to G/Gmax = 1/{1+exp[(V50−V)/s]}, where V50 is the voltage at which the current is half-activated and s is the slope factor. All data are expressed as mean±s.e.m. Statistically significant differences were assessed by unpaired Student's t-test for two sample assuming unequal variances for comparing WT with mutant channels.
Cell culture and transfections
Chinese hamster ovary (CHO) and human embryonic kidney (HEK 293) cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 2 mM glutamine, 10% fetal calf serum and antibiotics, incubated at 37°C in 5% CO2 as previously described (Meisel et al., 2012). CHO cells were seeded on poly-L-lysine-coated glass coverslips in a 24 multiwell plate and were co-transfected with KCNQ1 and KCNE1 (0.5 and 1 µg, respectively) in the absence or presence of Dr-VSP (2 µg), using Transit LT1-Transfection Reagent (Mirus). In experiments involving cAMP stimulation, 0.5 µg of yotiao was also co-transfected with KCNQ1 and KCNE1. Transfected cells were visualized by co-transfection with the pEYFP-N1 vector (0.15 µg). For TIRF imaging, CHO cells were seeded on poly-L-lysine-coated 1.5-mm glass coverslips in a 12-well multiwall plate and transfected with 1 µg of CFP-tagged WT or mutant KCNQ1 and 2 µg of YFP-tagged WT or mutant KCNE1. For western blotting and immunoprecipitation, HEK 293 cells were seeded on 90-mm dishes and transfected using the calcium phosphate method with 5 µg KCNQ1, 7 µg yotiao and 12 µg KCNE1.
Phosphorylation assays and immunoprecipitations
For immunoprecipitations, HEK 293 cells were washed with ice-cold PBS (with 1.5 mM CaCl2 and 1.5 mM MgCl2) and centrifuged for 8 min at 600 g in 4°C. Pellets were frozen in liquid nitrogen and then lysed under rotation for 1 h at 4°C in 300 µl of immunoprecipitation (IP) buffer containing (in mM): 50 Hepes pH 7.4, 150 NaCl, 10 EDTA, 10% Glycerol, 1% Triton, 1 PMSF and protease inhibitor cocktail (Sigma). Lysates were then centrifuged for 20 min at 10,000 g in 4°C and the supernatant was collected. One mg protein of each cell lysate was incubated with 5 µg of anti-KCNQ1 antibody (Alomone Labs) in 500 µl of IP buffer for overnight at 4°C under rotation. The following day, equal amounts of protein-G–agarose beads (Pierce) were added to the immune complexes and incubated for 2 h at room temperature. Sample centrifugation at 2500 g for 3 min was followed by three washes with 0.5 ml of IP buffer. The final wash was performed with distilled water. Beads were then resuspended in 30 µl of 2× sample buffer, denatured at 95°C for 5 min and samples were subjected to SDS-PAGE and followed by western blotting. For phosphorylation of KCNQ1 channels, HEK293 cells were treated with 250 µM 8CPT plus 0.2 µM okadaic acid in DMEM (without serum and antibiotics) for 10 min in 37°C. Cells were then lysed with IP buffer and lysates were treated with sample buffer, heated for 5 min at 55°C and subjected to SDS-PAGE and western blotting. Primary antibodies were anti-KCNQ1 (1:1000, Alomone), anti-KCNE1 (1:200, Alomone Labs), anti-yotiao (1:1000, Carmen Dessauer, University of Texas Health Science Center, Houston) and anti-KCNQ1 phospho-S27 (1:250, Robert S. Kass, University of Columbia, New York). Secondary antibodies were HRP-conjugated anti-rabbit (1:10,000, Jackson laboratories).
Acknowledgments
We thank Yasushi Okamura (Osaka University, Japan) and Koret Hirschberg (Tel Aviv University, Israel) for their kind gift of the Dr-VSP and GPI-citrine plasmids, respectively.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
M.D., R.S., D.S., I.B.T.C. performed the experiments and analyzed the data. Y.H., C.D., O.P. and R.K. provided invaluable tools and help in this work. J.A.H. and B.A. designed the experiments. B.A. wrote the manuscript.
Funding
This work is supported by the Deutsch-Israelische Projektkooperation DIP fund from the Deutsche Gorschungsgemeinschaft [grant number AT119/1-1 to B.A, O.P. and J.A.H.]: the Israel Science Foundation [grant number ISF 1215/13]; and the Fields Fund for Cardiovascular Research (to B.A.)
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.147033/-/DC1
References
- Abbott G. W., Butler M. H., Bendahhou S., Dalakas M. C., Ptacek L. J., Goldstein S. A. (2001). MiRP2 forms potassium channels in skeletal muscle with Kv3.4 and is associated with periodic paralysis. Cell 104, 217–231. 10.1016/S0092-8674(01)00207-0 [DOI] [PubMed] [Google Scholar]
- Ackerman M. J., Tester D. J., Jones G. S., Will M. L., Burrow C. R., Curran M. E. (2003). Ethnic differences in cardiac potassium channel variants: implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clin. Proc. 78, 1479–1487. 10.4065/78.12.1479 [DOI] [PubMed] [Google Scholar]
- Aivar P., Fernández-Orth J., Gomis-Perez C., Alberdi A., Alaimo A., Rodríguez M. S., Giraldez T., Miranda P., Areso P., Villarroel A. (2012). Surface expression and subunit specific control of steady protein levels by the Kv7.2 helix A-B linker. PLoS ONE 7, e47263 10.1371/journal.pone.0047263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhanin J., Lesage F., Guillemare E., Fink M., Lazdunski M., Romey G. (1996). K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. (see comments). Nature 384, 78–80. 10.1038/384078a0 [DOI] [PubMed] [Google Scholar]
- Chen L., Kass R. S. (2005). A-kinase anchoring proteins: different partners, different dance. Nat. Cell Biol. 7, 1050–1051. 10.1038/ncb1105-1050 [DOI] [PubMed] [Google Scholar]
- Choveau F. S., Abderemane-Ali F., Coyan F. C., Es-Salah-Lamoureux Z., Baró I., Loussouarn G. (2012). Opposite effects of the S4-S5 linker and PIP(2) on voltage-gated channel function: KCNQ1/KCNE1 and other channels. Front Pharmacol 3, 125 10.3389/fphar.2012.00125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyan F. C., Abderemane-Ali F., Amarouch M. Y., Piron J., Mordel J., Nicolas C. S., Steenman M., Mérot J., Marionneau C., Thomas A. et al. (2014). A long QT mutation substitutes cholesterol for phosphatidylinositol-4,5-bisphosphate in KCNQ1 channel regulation. PLoS ONE 9, e93255 10.1371/journal.pone.0093255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox P. D., Haberkorn C. J., Weigel A. V., Higgins J. L., Akin E. J., Kennedy M. J., Krapf D., Tamkun M. M. (2013). Plasma membrane domains enriched in cortical endoplasmic reticulum function as membrane protein trafficking hubs. Mol. Biol. Cell 24, 2703–2713. 10.1091/mbc.E12-12-0895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamper N., Shapiro M. S. (2007). Regulation of ion transport proteins by membrane phosphoinositides. Nat. Rev. Neurosci. 8, 921–934. 10.1038/nrn2257 [DOI] [PubMed] [Google Scholar]
- Ghosh S., Nunziato D. A., Pitt G. S. (2006). KCNQ1 assembly and function is blocked by long-QT syndrome mutations that disrupt interaction with calmodulin. Circ. Res. 98, 1048–1054. 10.1161/01.RES.0000218863.44140.f2 [DOI] [PubMed] [Google Scholar]
- Glebov O. O., Nichols B. J. (2004). Lipid raft proteins have a random distribution during localized activation of the T-cell receptor. Nat. Cell Biol. 6, 238–243. 10.1038/ncb1103 [DOI] [PubMed] [Google Scholar]
- Haitin Y., Attali B. (2008). The C-terminus of Kv7 channels: a multifunctional module. J. Physiol. 586, 1803–1810. 10.1113/jphysiol.2007.149187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haitin Y., Wiener R., Shaham D., Peretz A., Cohen E. B., Shamgar L., Pongs O., Hirsch J. A., Attali B. (2009). Intracellular domains interactions and gated motions of I(KS) potassium channel subunits. EMBO J. 28, 1994–2005. 10.1038/emboj.2009.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijman J., Spätjens R. L., Seyen S. R., Lentink V., Kuijpers H. J., Boulet I. R., de Windt L. J., David M., Volders P. G. (2012). Dominant-negative control of cAMP-dependent IKs upregulation in human long-QT syndrome type 1. Circ. Res. 110, 211–219. 10.1161/CIRCRESAHA.111.249482 [DOI] [PubMed] [Google Scholar]
- Hernandez C. C., Zaika O., Shapiro M. S. (2008). A carboxy-terminal inter-helix linker as the site of phosphatidylinositol 4,5-bisphosphate action on Kv7 (M-type) K+ channels. J. Gen. Physiol. 132, 361–381. 10.1085/jgp.200810007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain M. I., Iwasaki H., Okochi Y., Chahine M., Higashijima S., Nagayama K., Okamura Y. (2008). Enzyme domain affects the movement of the voltage sensor in ascidian and zebrafish voltage-sensing phosphatases. J. Biol. Chem. 283, 18248–18259. 10.1074/jbc.M706184200 [DOI] [PubMed] [Google Scholar]
- Howard R. J., Clark K. A., Holton J. M., Minor D. L., Jr (2007). Structural insight into KCNQ (Kv7) channel assembly and channelopathy. Neuron 53, 663–675. 10.1016/j.neuron.2007.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch T. J., Hübner C. A., Fuhrmann J. C. (2004). Ion channels: function unravelled by dysfunction. Nat. Cell Biol. 6, 1039–1047. 10.1038/ncb1104-1039 [DOI] [PubMed] [Google Scholar]
- Kanki H., Kupershmidt S., Yang T., Wells S., Roden D. M. (2004). A structural requirement for processing the cardiac K+ channel KCNQ1. J. Biol. Chem. 279, 33976–33983. 10.1074/jbc.M404539200 [DOI] [PubMed] [Google Scholar]
- Kruse M., Hammond G. R., Hille B. (2012). Regulation of voltage-gated potassium channels by PI(4,5)P2. J. Gen. Physiol. 140, 189–205. 10.1085/jgp.201210806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa J., Chen L., Kass R. S. (2003). Requirement of subunit expression for cAMP-mediated regulation of a heart potassium channel. Proc. Natl. Acad. Sci. USA 100, 2122–2127. 10.1073/pnas.0434935100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa J., Motoike H. K., Rao J., Kass R. S. (2004). Regulatory actions of the A-kinase anchoring protein Yotiao on a heart potassium channel downstream of PKA phosphorylation. Proc. Natl. Acad. Sci. USA 101, 16374–16378. 10.1073/pnas.0405583101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa J., Bankston J. R., Kaihara A., Chen L., Furukawa T., Kass R. S. (2009). KCNE variants reveal a critical role of the beta subunit carboxyl terminus in PKA-dependent regulation of the IKs potassium channel. Channels (Austin) 3, 16–24. 10.4161/chan.3.1.7387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Zaydman M. A., Wu D., Shi J., Guan M., Virgin-Downey B., Cui J. (2011). KCNE1 enhances phosphatidylinositol 4,5-bisphosphate (PIP2) sensitivity of IKs to modulate channel activity. Proc. Natl. Acad. Sci. USA 108, 9095–9100. 10.1073/pnas.1100872108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Chen L., Kass R. S., Dessauer C. W. (2012). The A-kinase anchoring protein Yotiao facilitates complex formation between adenylyl cyclase type 9 and the IKs potassium channel in heart. J. Biol. Chem. 287, 29815–29824. 10.1074/jbc.M112.380568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes C. M., Remon J. I., Matavel A., Sui J. L., Keselman I., Medei E., Shen Y., Rosenhouse-Dantsker A., Rohacs T., Logothetis D. E. (2007). Protein kinase A modulates PLC-dependent regulation and PIP2-sensitivity of K+ channels. Channels (Austin) 1, 124–134. 10.4161/chan.4322 [DOI] [PubMed] [Google Scholar]
- Loussouarn G., Park K. H., Bellocq C., Baró I., Charpentier F., Escande D. (2003). Phosphatidylinositol-4,5-bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage-gated potassium channels: a functional homology between voltage-gated and inward rectifier K+ channels. EMBO J. 22, 5412–5421. 10.1093/emboj/cdg526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundby A., Andersen M. N., Steffensen A. B., Horn H., Kelstrup C. D., Francavilla C., Jensen L. J., Schmitt N., Thomsen M. B., Olsen J. V. (2013). In vivo phosphoproteomics analysis reveals the cardiac targets of β-adrenergic receptor signaling. Sci. Signal. 6, rs11 10.1126/scisignal.2003506 [DOI] [PubMed] [Google Scholar]
- Marx S. O., Kurokawa J., Reiken S., Motoike H., D'Armiento J., Marks A. R., Kass R. S. (2002). Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 295, 496–499. 10.1126/science.1066843 [DOI] [PubMed] [Google Scholar]
- Matavel A., Medei E., Lopes C. M. (2010). PKA and PKC partially rescue long QT type 1 phenotype by restoring channel-PIP2 interactions. Channels (Austin) 4, 3–11. 10.4161/chan.4.1.10227 [DOI] [PubMed] [Google Scholar]
- Meisel E., Dvir M., Haitin Y., Giladi M., Peretz A., Attali B. (2012). KCNQ1 channels do not undergo concerted but sequential gating transitions in both the absence and the presence of KCNE1 protein. J. Biol. Chem. 287, 34212–34224. 10.1074/jbc.M112.364901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajo K., Kubo Y. (2011). Nano-environmental changes by KCNE proteins modify KCNQ channel function. Channels (Austin) 5, 397–401. 10.4161/chan.5.5.16468 [DOI] [PubMed] [Google Scholar]
- Nerbonne J. M., Kass R. S. (2005). Molecular physiology of cardiac repolarization. Physiol. Rev. 85, 1205–1253. 10.1152/physrev.00002.2005 [DOI] [PubMed] [Google Scholar]
- Okamura Y., Murata Y., Iwasaki H. (2009). Voltage-sensing phosphatase: actions and potentials. J. Physiol. 587, 513–520. 10.1113/jphysiol.2008.163097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K. H., Piron J., Dahimene S., Mérot J., Baró I., Escande D., Loussouarn G. (2005). Impaired KCNQ1-KCNE1 and phosphatidylinositol-4,5-bisphosphate interaction underlies the long QT syndrome. Circ. Res. 96, 730–739. 10.1161/01.RES.0000161451.04649.a8 [DOI] [PubMed] [Google Scholar]
- Peroz D., Rodriguez N., Choveau F., Baró I., Mérot J., Loussouarn G. (2008). Kv7.1 (KCNQ1) properties and channelopathies. J. Physiol. 586, 1785–1789. 10.1113/jphysiol.2007.148254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti M. C., Curran M. E., Zou A., Shen J., Spector P. S., Atkinson D. L., Keating M. T. (1996). Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384, 80–83. 10.1038/384080a0 [DOI] [PubMed] [Google Scholar]
- Schulze-Bahr E., Schwarz M., Hauenschild S., Wedekind H., Funke H., Haverkamp W., Breithardt G., Pongs O., Isbrandt D. (2001). A novel long-QT 5 gene mutation in the C-terminus (V109I) is associated with a mild phenotype. J. Mol. Med. 79, 504–509. 10.1007/s001090100249 [DOI] [PubMed] [Google Scholar]
- Shamgar L., Ma L., Schmitt N., Haitin Y., Peretz A., Wiener R., Hirsch J., Pongs O., Attali B. (2006). Calmodulin is essential for cardiac IKS channel gating and assembly: impaired function in long-QT mutations. Circ. Res. 98, 1055–1063. 10.1161/01.RES.0000218979.40770.69 [DOI] [PubMed] [Google Scholar]
- Splawski I., Shen J., Timothy K. W., Lehmann M. H., Priori S., Robinson J. L., Moss A. J., Schwartz P. J., Towbin J. A., Vincent G. M. et al. (2000). Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 102, 1178–1185. 10.1161/01.CIR.102.10.1178 [DOI] [PubMed] [Google Scholar]
- Suh B. C., Hille B. (2005). Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr. Opin. Neurobiol. 15, 370–378. 10.1016/j.conb.2005.05.005 [DOI] [PubMed] [Google Scholar]
- Suh B. C., Hille B. (2007). Electrostatic interaction of internal Mg2+ with membrane PIP2 Seen with KCNQ K+ channels. J. Gen. Physiol. 130, 241–256. 10.1085/jgp.200709821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X., Zaydman M. A., Cui J. (2012). Regulation of voltage-activated K(+) channel Gating by transmembrane β subunits. Front Pharmacol 3, 63 10.3389/fphar.2012.00063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telezhkin V., Thomas A. M., Harmer S. C., Tinker A., Brown D. A. (2013). A basic residue in the proximal C-terminus is necessary for efficient activation of the M-channel subunit Kv7.2 by PI(4,5)P(2). Pflugers Arch [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrenoire C., Houslay M. D., Baillie G. S., Kass R. S. (2009). The cardiac IKs potassium channel macromolecular complex includes the phosphodiesterase PDE4D3. J. Biol. Chem. 284, 9140–9146. 10.1074/jbc.M805366200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A. M., Harmer S. C., Khambra T., Tinker A. (2011). Characterization of a binding site for anionic phospholipids on KCNQ1. J. Biol. Chem. 286, 2088–2100. 10.1074/jbc.M110.153551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenskow P., Splawski I., Timothy K. W., Keating M. T., Sanguinetti M. C. (2004). Compound mutations: a common cause of severe long-QT syndrome. Circulation 109, 1834–1841. 10.1161/01.CIR.0000125524.34234.13 [DOI] [PubMed] [Google Scholar]
- Wiener R., Haitin Y., Shamgar L., Fernández-Alonso M. C., Martos A., Chomsky-Hecht O., Rivas G., Attali B., Hirsch J. A. (2008). The KCNQ1 (Kv7.1) COOH terminus, a multitiered scaffold for subunit assembly and protein interaction. J. Biol. Chem. 283, 5815–5830. 10.1074/jbc.M707541200 [DOI] [PubMed] [Google Scholar]
- Wrobel E., Tapken D., Seebohm G. (2012). The KCNE tango – how KCNE1 interacts with Kv7.1. Front Pharmacol 3, 142 10.3389/fphar.2012.00142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaydman M. A., Silva J. R., Delaloye K., Li Y., Liang H., Larsson H. P., Shi J., Cui J. (2013). Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proc. Natl. Acad. Sci. USA 110, 13180–13185. 10.1073/pnas.1305167110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Craciun L. C., Mirshahi T., Rohács T., Lopes C. M., Jin T., Logothetis D. E. (2003). PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37, 963–975. 10.1016/S0896-6273(03)00125-9 [DOI] [PubMed] [Google Scholar]
- Zheng R., Thompson K., Obeng-Gyimah E., Alessi D., Chen J., Cheng H., McDonald T. V. (2010). Analysis of the interactions between the C-terminal cytoplasmic domains of KCNQ1 and KCNE1 channel subunits. Biochem. J. 428, 75–84. 10.1042/BJ20090977 [DOI] [PMC free article] [PubMed] [Google Scholar]