

Abstract
Vibrio natriegens has recently emerged as an alternative to Escherichia coli for molecular biology and biotechnology, but low-efficiency genetic tools hamper its development. Here, we uncover how to induce natural competence in V. natriegens and describe methods for multiplex genome editing by natural transformation (MuGENT). MuGENT promotes integration of multiple genome edits at high-efficiency on unprecedented timescales. Also, this method allows for generating highly complex mutant populations, which can be exploited for metabolic engineering efforts. As a proof-of-concept, we attempted to enhance production of the value added chemical poly-β-hydroxybutyrate (PHB) in V. natriegens by targeting the expression of nine genes involved in PHB biosynthesis via MuGENT. Within 1 week, we isolated edited strains that produced ~100 times more PHB than the parent isolate and ~3.3 times more than a rationally designed strain. Thus, the methods described here should extend the utility of this species for diverse academic and industrial applications.
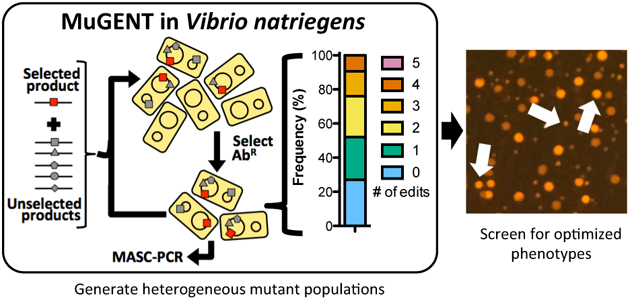
Graphical Abstract
V. natriegens is the fastest growing organism known, with a doubling time of <10 min1, 2. With broad metabolic capabilities, lack of pathogenicity, and its rapid growth rate, it is an attractive alternative to E. coli for diverse molecular biology and biotechnology applications3, 4. Methods for classical genetic techniques have been developed for V. natriegens, but these are relatively laborious, require multiple steps, and must be used sequentially to generate multiple genome edits3, 4. The challenges of these techniques contrast with the ease of genetics in Vibrio species that are naturally transformable. Competent Vibrio species can take up DNA from the environment and integrate it into their genome by homologous recombination; processes known as natural competence and natural transformation, respectively5–8. The inducing cue for natural transformation in competent Vibrio species is growth on the chitinous shells of crustacean zooplankton, which are commonly found in the aquatic environment where these microbes reside5. Chitin induces the expression of the competence regulator TfoX9, 10. In fact, overexpression of TfoX obviates the need for chitin induction, allowing competent Vibrio species to be naturally transformed in rich media5, 9.
As no reports of natural transformation existed for V. natriegens, we first sought to establish whether this was possible. Unlike naturally competent V. cholerae, incubation on chitin did not lead to detectable transformation in V. natriegens (data not shown). However, ectopic expression of TfoX (either the endogenous tfoX gene or one from Vibrio cholerae) on an IPTG-inducible plasmid (pMMB) supported high rates of natural transformation (Fig. 1a). This was tested using a linear PCR product that replaces the gene encoding the DNA endonuclease Dns with an antibiotic resistance (AbR) marker. The dns locus was used as a target for transformation assays throughout this manuscript because loss of this gene does not impact growth or viability in rich medium. Under optimal conditions ~1–10% of the population had integrated the transforming DNA (tDNA), which matches the highest rates of transformation observed among competent species11 (Fig. 1a–c). Natural transformation of V. natriegens required very little transforming DNA (tDNA) (highly efficient with even 1 ng / 108 CFU) and was dependent on the length of homologous sequence surrounding the mutation (Fig. 1b and c). This method could also be used to introduce point mutations into V. natriegens (tested with tDNA containing an rpsL K43R SmR allele); however, this activity was partially suppressed by the mismatch repair system (Fig. 1d).
Fig. 1 – Natural transformation of V. natriegens.

(a-d) Transformation assays of V. natriegens. (a) V. natriegens strains containing a pMMB empty vector or pMMB with the tfoX gene from either V. natriegens (Vn) or V. cholerae (Vc) were transformed with 100 ng of a Δdns::KanR tDNA containing 3 kb flanks of homology on both sides of the mutation (i.e. 3 kb/3 kb). Transformation assay of V. natriegens pMMB-tfoX (Vc) with (b) the indicated concentration of Δdns::KanR (3 kb/3 kb) tDNA or (c) 5 ng of Δdns::KanR tDNA containing the indicated amount of homology on each side of the mutation. (d) Transformation assay in the indicated strain backgrounds with 5 ng of rpsL K43R SmR (3 kb/3 kb) or Δdns::SpecR (3 kb/3 kb) tDNA as indicated. All strains in d harbor Δdns::KanR mutations and pMMB-tfoX (Vc). All data are shown as the mean ± SD and are the result of at least 4 independent biological replicates. ** = p<0.01.
Having demonstrated V. natriegens is naturally competent, we sought to determine if we could use natural transformation to perform scarless multiplex genome editing by natural transformation (MuGENT)12. MuGENT operates under the premise that under competence inducing conditions, only a subpopulation of cells is transformable. Those cells that can be transformed, however, have the capacity to take up and integrate multiple tDNAs12, 13. Thus, during MuGENT, cells are incubated with two types of linear tDNA; (1) a selected product that introduces an antibiotic resistance marker into the genome and (2) unselected products that introduce scarless genome edits of interest at one or more loci.
We first tested the ability of MuGENT to introduce a single unmarked genome edit (also known as cotransformation). To facilitate measurement of cotransformation, we noted this species forms opaque colonies on agar plates (Fig. 2a), which could be due to the production of a capsular polysaccharide. Consistent with this, inactivating a homolog of the essential capsule biosynthesis gene wbfF14 resulted in the formation of transparent colonies on agar plates and loss of expression of a high molecular weight polysaccharide (Fig. 2a and 2b). Thus, to test cotransformation we used an unselected product to replace ~500 bp of the 5ʹ end of the wbfF gene with a premature stop codon and scored cotransformation via colony morphology (opaque vs. transparent) on agar plates (Fig. 3a). We found that cotransformation was remarkably efficient in V. natriegens (up to ~80%), even with low amounts (~25–50 ng / 108 CFU) of the unselected product (Fig. 3b). Also, cotransformation with 1 kb flanks on the unselected product was possible, but at ~6-fold lower frequencies than with 3 kb flanks (Fig. 3c).
Fig. 2 – V. natriegens produces a WbfF-dependent capsular polysaccharide.

(a) Colony morphologies of parent (white arrow) and ΔwbfF (black arrow) strains, which demonstrate the phenotypes screened for in cotransformation assays. (b) Cell lysates of the indicated strains were run on a 4–12% SDS PAGE gel and stained with the carbohydrate stain Alcian blue. The presence of a high molecular weight polysaccharide in the parent is indicated by a red arrow.
Fig. 3 – Cotransformation is highly efficient in V. natriegens.

(a) Cotransformation was tested using a Δdns::KanR (3 kb/3 kb) selected product and an unselected product that deleted ~500 bp of the 5’ end of wbfF gene. Cotransformation assays were performed using 50ng of the Δdns::KanR (3 kb/3 kb) selected product and (b) the indicated amount of the ΔwbfF (3 kb/3 kb) unselected product or (c) 200 ng of ΔwbfF unselected products containing the indicated length of homology on each side of the mutation. Data in b and c are from at least four independent biological replicates and shown as the mean ± SD. (d) Schematic of MuGENT. The selected product is indicated by a red box, while multiple unselected genome edits are depicted by distinct gray shapes. Since there is no selection for genome edits in cis, output mutants can have any number and combination of the unselected genome edits. Circles inside cells represent the two circular chromosomes of V. natriegens. (e and f) MuGENT was performed with 5 unselected genome edits. The selected product was ΔwbfF::KanR, while the unselected products targeted four carbohydrate transporters and dns for inactivation by replacing ~500 bp of the 5’ end of each gene with a premature stop codon. (e) A representative MASC-PCR gel of 24 colonies from the edited population. The targets of each genome edit are indicated on the left and the presence of a band indicates integration of the indicated genome edit. Strains containing 4 genome edits are indicated by the green arrows. (f) Distribution of genome edits in the population determined by MASC-PCR analysis of 48 random mutants.
We next tested the full multiplex genome editing capacity of MuGENT to simultaneously cotransform multiple scarless genome edits into the genome in a single step12, 15. Since there is no selection for integration of the unselected genome edits in cis during MuGENT, output populations are highly heterogeneous and individual mutants contain any number and combination of the multiplexed genome edits. Also, this process can be carried out in multiple iterative cycles to further increase the complexity of genome edits in the population (Fig. 3d)12.
As an initial test of multiplex genome editing, we targeted 5 genes whose mutagenesis was considered unlikely to affect viability or growth in LB. These targets included four carbohydrate transporters (specific for mannitol, fructose, sucrose, and trehalose – all of which are absent in LB) and the dns gene. All genes were targeted for inactivation by replacing ~500 bp of the 5’ end of each gene with a premature stop codon. Integration of genome edits was determined by multiplex allele-specific colony PCR (MASC-PCR)16 (Fig. 3e). Following one cycle of MuGENT, we found that ~70% of the population contained at least 1 genome edit, with ~25% of the population containing 3–4 genome edits (Fig. 3f). A quadruple mutant from this experiment was isolated and whole genome sequencing of this strain did not reveal any off-target mutations. Thus, MuGENT rapidly generated V. natriegens strains with multiple large (0.5 kb) scarless genome edits at high-efficiency without off-target effects, and can be used to make highly complex mutant populations.
As a second demonstration of multiplex genome editing, we demonstrated its utility in metabolic engineering by attempting to rapidly enhance production of a value-added chemical in V. natriegens. This species naturally accumulates low levels of the bioplastic precursor poly-β-hydroxybutyrate (PHB) as a storage polymer17. PHB is derived from the condensation and subsequent NADPH-dependent reduction of acetyl-CoA precursors18. Thus, for our targets, we tuned the expression (swap out Pnative for IPTG-inducible Ptac) or inactivated genes that we hypothesized would affect NADPH and/or acetyl-CoA availability. The targets for promoter swaps were the PHB synthesis operon (phaBAC), NAD kinase (nadK), and two transhydrogenases (pntAB and udhA), while targets for inactivation were phosphoglucose isomerase (pgi), citrate synthase (gltA), phosphotransacetylase (pta), isocitrate lyase (aceA), and lactate dehydrogenase (ldhA) (Fig. 4a). Thus, there were 512 possible combinations for these 9 genome edits. We performed multiple cycles of MuGENT to introduce these genome edits into a competent population of V. natriegens. At each cycle, the selected product was designed to swap the AbR marker at the dns locus to maintain coselection at each step. Following four cycles of MuGENT, which took just 5 days to perform, ~50% of the population had 3 or more genome edits and ~10% contained 5+ genome edits (Fig. 4b). To select mutants with increased PHB production, we then plated this output population onto media containing Nile red, which stains PHB granules19. Nile red fluorescence on these plates was highly heterogeneous, suggesting that some genotypes produced more PHB than the parent isolate (Fig. 4c). A number of highly fluorescent colonies were picked and the genotypes determined by MASC-PCR. Also, PHB in these select strains was directly measured by HPLC. Cumulatively, these analyses rapidly revealed genotypes that produced ~100-fold more PHB than the parent and ~3.3-fold more than a strain with just the Ptac-phaBAC mutation (Fig. 4d). Overexpression of the phaBAC locus is a commonly used rational approach for enhancing PHB production18, 20. Thus, this result demonstrates that MuGENT can allow for rapid isolation of genotypes associated with enhanced phenotypes (e.g. enhanced PHB production) compared to rationally engineered strains (e.g. a Ptac-phaBAC mutant) without prior knowledge of effective combinations of individual mutations.
Fig. 4 – MuGENT rapidly enhances PHB production in V. natriegens.

(a) The indicated targets were subjected to either a promoter swap (top) or inactivation by replacing ~500bp of the 5’ end of each gene with a short sequence to introduce a premature stop codon (bottom). (b) Distribution of the 9 genome edits in a population of cells following four cycles of MuGENT. (c) Representative image of the mutant pool generated in b plated on Nile red containing plates, which stain PHB granules. White arrows indicate colonies with increased fluorescence intensity compared to the parent. (d) PHB content of select MuGENT optimized strains is shown as the % of dry cell weight (DCW). The genotype of each mutant is shown below each bar where a filled box indicates the presence of the genome edit indicated on the left. Data are shown as the mean ± SD and are from at least 2 independent biological replicates. * = p<0.05.
While many methods for multiplex genome editing in bacterial systems have been described21, many of these are limited to small changes such as SNPs. MuGENT, on the other hand, can efficiently swap, insert, or remove whole promoters or coding sequences as demonstrated above. Furthermore, one of the major limitations to other multiplex genome editing methods is that mutagenesis must be performed in strains lacking DNA repair pathways to allow for high-efficiency integration of genome edits, which results in a large number of off-target mutations16, 21. MuGENT in V. natriegens is performed in DNA repair sufficient backgrounds, thus, little to no off target mutations are introduced during the procedure as indicated above. Also, unlike other multiplex editing approaches, MuGENT requires no specialized equipment and, thus, has the potential to make multiplex genome editing commonplace.
In conclusion, this study demonstrates that MuGENT is a rapid, efficient, and simple tool for engineering the V. natriegens genome. This microbe is already being developed as an alternative to E. coli, and we believe that the ease and speed of MuGENT will extend the use of V. natriegens as a novel chassis for diverse molecular biology and biotechnology applications.
METHODS
Bacterial strains and culture conditions
The parent V. natriegens strain used throughout this study was a spontaneous rifampicin-resistant derivative of ATCC 140482. For a list of all strains used / generated in this study, see Table S1. Strains were routinely grown in LB+v2 salts (LBv2)3, which is LB Miller broth (BD) supplemented with 200 mM NaCl, 23.14 mM MgCl2, and 4.2 mM KCl. LBv2 was supplemented with 100 μM IPTG, 50 μg/mL kanamycin (Kan), 200 μg/mL spectinomycin (Spec), 100 μg/mL rifampicin (Rif), 100 μg/mL streptomycin (Sm), or 100 μg/mL carbenicillin (Carb) as appropriate.
Generation of mutant strains and constructs
Mutant constructs were generated by splicing-by-overlap extension (SOE) PCR exactly as previously described22. Briefly, for three-piece mutant constructs (i.e. for constructs where a gene of interest is replaced with an AbR cassette or where the native promoter is swapped for a Ptac promoter) segments were designated UP, MIDDLE, and DOWN and correspond to: (1) UP = the upstream region of homology amplified with F1 and R1 primers, (2) DOWN = the downstream region of homology amplified with F2 and R2 primers, and (3) MIDDLE = the AbR marker or promoter swap fragment. For two-piece mutant constructs (i.e. for constructs where ~501 bp of the 5’ end of a gene is replaced with a stop codon), the mutation of interest is incorporated into the R1 and F2 primers used to amplify the upstream and downstream regions of homology, respectively. Gel purified segments were then mixed in equal ratios and used as template for a SOE PCR reaction with the F1 and R2 primers. All mutant constructs were made using Phusion polymerase. These were introduced into the V. natriegens genome via natural transformation as described below. All primers used to generate mutant constructs are listed in Table S2.
Natural transformation / MuGENT assays
Strains harboring pMMB-tfoX (Vn tfoX or Vc tfoX) were induced to competence by growing overnight (12–18 hours) in LBv2+100 μg/mL carbenicillin+100 μM IPTG in a rollerdrum at 30°C. Then, ~108 CFUs of this overnight culture (~3.5 μL) were diluted directly into 350 μL of instant ocean medium (28 g/L; Aquarium Systems Inc.) supplemented with 100 μM IPTG. Transforming DNA (tDNA) was then added as indicated, and reactions were incubated statically at 30°C for 5 hours. Next, 1 mL of LBv2 was added and reactions were outgrown at 30°C with shaking (250 rpm) for ~1–2 hrs. Then, reactions were plated for quantitative culture onto media to select for integration of tDNA (i.e. LB+drug = transformants) and onto nonselective media (i.e. plain LB = total viable counts). Transformation efficiency is shown as: transformants / total viable counts.
For MuGENT, transformation assays were conducted exactly as described above. Unless otherwise specified, ~50 ng of the selected product was incubated with cells along with ~200 ng of each unselected product. After outgrowth, 1/10th of the reaction was removed and plated for MASC-PCR analysis (described below). If multiple cycles of MuGENT were performed, the rest of the reaction was grown overnight in LBv2 supplemented with 100 μM IPTG, 100μg/mL carbenicillin (to maintain pMMB-tfoX), and the antibiotic to select for integration of the selected product. The following day, the population was then subjected to another round of MuGENT as described above using a selected product containing a different AbR marker to maintain coselection at each cycle.
Integration of genome edits was detected via MASC-PCR exactly as previously described12, 16. Briefly, colonies were boiled in 50 μL of sterile water, vortexed, and then 2 μL were used as template in a 25 μL PCR reaction. PCR was conducted with Taq polymerase (SydLabs) using a modified 5X Taq buffer: 200 mM Tris pH 8.8, 100 mM KCl, 100 mM (NH4)2SO4, 30 mM MgSO4, and 1% Triton X-100. The total primer used in each MASC-PCR reaction (regardless of the number of multiplexed products being detected) was 1200 nM (i.e. for detection of 4 multiplexed genome edits, 300 nM of each genome edit-specific primer pair was used). The cycling conditions used were: 95°C 3 min; 26 × [95°C 40s, 58°C 30s, 72°C 3 min]; 72°C 3 min; 12°C hold. Reactions were then run on 2% agarose gels and imaged with GelGreen dye according to manufacturer’s instructions (Biotium). For a list of all primers used for MASC-PCR see Table S2.
Alcian blue stained gels
To prepare cell lysates, ~109 cells of the indicated V. natriegens strains were pelleted and then resuspended in 180 μL of Buffer ATL (Qiagen). Then, 20 μL of a 20 mg/mL proteinase K stock solution was added to each reaction and incubated at 56°C for 20 mins. Samples were then boiled in 2X SDS PAGE sample buffer and separated on 4–12% SDS PAGE gels. Gels were then stained with 0.1% Alcian Blue 8GX in 40% ethanol/3% acetic acid as previously described23. The gel was then destained in a 40% ethanol/3% acetic acid and imaged on a Biorad ChemiDoc MP Imaging system.
Whole genome sequencing
Genomic DNA was prepped from strains and sequencing libraries were prepped via homopolymer-tail mediated ligation exactly as previously described24. Single-end 50 bp reads were collected on the Illumina platform. Then, data was analyzed for small indels and single nucleotide variants using CLC Genomics Workbench exactly as previously described15, 25.
Qualitative and quantitative assessment of PHB production
PHB was qualitatively assessed in MuGENT edited populations of V. natriegens by plating onto Nile red containing medium with excess glucose as a carbon source and 100 μM IPTG to induce Ptac-containing genome edits = recipe per L: 28 g instant ocean, 2.5 g tryptone, 1 g yeast extract, 20 g glucose, 15 g agar, and 1 mg Nile red. Fluorescence of colonies was detected using a PrepOne Sapphire LED blue light base (475 nm ± 30 nm) and amber filter (530 nm long pass) (Embi Tec).
For quantitative assessment of PHB levels, the indicated strains were grown overnight in M9 minimal medium (BD) supplemented with 2 mM MgSO4, 100 μM CaCl2, 200 mM NaCl, 30 μM FeSO4, 100 μM IPTG, 1% tryptone, and 2% glucose. Approximately 8 × 109 cells were then pelleted, resuspended with 50 μL water and transferred to pre-weighed glass screw-cap tubes. Cell suspensions were dried for 5 h at 80°C and then the tubes were weighed again to determine dry cell weights. PHB was then hydrolyzed and extracted as crotonic acid by boiling the dried cells in 1 ml of pure sulfuric acid. Extracts were chilled on ice and diluted with 4 ml ice-cold water. Aliquots were further diluted 10-fold with water, centrifuged, filtered, and then crotonic acid was quantified by HPLC as described26.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Tufts TUCF Genomics and the Indiana University CGB for assistance with whole genome sequencing of strains. This work was supported by US National Institutes of Health Grant AI118863 to ABD. JBM and ABD were also supported by the Indiana University College of Arts and Sciences.
Footnotes
COMPETING FINANCIAL INTERESTS
MuGENT is the subject of a pending patent application.
SUPPORTING INFORMATION
Supplementary protocol: Natural transformation / MuGENT in V. natriegens.
Supplementary Tables S1–S2: strains and primers used in this study.
REFERENCES
- [1].Eagon RG (1962) Pseudomonas natriegens, a marine bacterium with a generation time of less than 10 minutes, J Bacteriol 83, 736–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Payne WJ, Eagon RG, and Williams AK (1961) Some observations on the physiology of Pseudomonas natriegens nov. spec, Antonie Van Leeuwenhoek 27, 121–128. [DOI] [PubMed] [Google Scholar]
- [3].Weinstock MT, Hesek ED, Wilson CM, and Gibson DG (2016) Vibrio natriegens as a fast-growing host for molecular biology, Nat Methods 13, 849–851. [DOI] [PubMed] [Google Scholar]
- [4].Lee HH, Ostrov N, Wong BG, Gold MA, Khalil A, and Church GM (2016) Vibrio natriegens, a new genomic powerhouse, bioRxiv, doi: 10.1101/058487. [DOI] [Google Scholar]
- [5].Meibom KL, Blokesch M, Dolganov NA, Wu CY, and Schoolnik GK (2005) Chitin induces natural competence in Vibrio cholerae, Science 310, 1824–1827. [DOI] [PubMed] [Google Scholar]
- [6].Chen Y, Dai J, Morris JG Jr., and Johnson JA (2010) Genetic analysis of the capsule polysaccharide (K antigen) and exopolysaccharide genes in pandemic Vibrio parahaemolyticus O3:K6, BMC Microbiol 10, 274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gulig PA, Tucker MS, Thiaville PC, Joseph JL, and Brown RN (2009) USER friendly cloning coupled with chitin-based natural transformation enables rapid mutagenesis of Vibrio vulnificus, Appl Environ Microbiol 75, 4936–4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pollack-Berti A, Wollenberg MS, and Ruby EG (2010) Natural transformation of Vibrio fischeri requires tfoX and tfoY, Environ Microbiol 12, 2302–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dalia AB, Lazinski DW, and Camilli A (2014) Identification of a membrane-bound transcriptional regulator that links chitin and natural competence in Vibrio cholerae, MBio 5, e01028–01013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yamamoto S, Mitobe J, Ishikawa T, Wai SN, Ohnishi M, Watanabe H, and Izumiya H (2014) Regulation of natural competence by the orphan two-component system sensor kinase ChiS involves a non-canonical transmembrane regulator in Vibrio cholerae, Mol Microbiol 91, 326–347. [DOI] [PubMed] [Google Scholar]
- [11].Lorenz MG, and Wackernagel W (1994) Bacterial gene transfer by natural genetic transformation in the environment, Microbiol Rev 58, 563–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dalia AB, McDonough E, and Camilli A (2014) Multiplex genome editing by natural transformation, Proc Natl Acad Sci U S A 111, 8937–8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Erickson RJ, and Copeland JC (1973) Congression of unlinked markers and genetic mapping in the transformation of Bacillus subtilis 168, Genetics 73, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Bik EM, Bunschoten AE, Willems RJ, Chang AC, and Mooi FR (1996) Genetic organization and functional analysis of the otn DNA essential for cell-wall polysaccharide synthesis in Vibrio cholerae O139, Mol Microbiol 20, 799–811. [DOI] [PubMed] [Google Scholar]
- [15].Hayes CA, Dalia TN, and Dalia AB (2017) Systematic genetic dissection of PTS in Vibrio cholerae uncovers a novel glucose transporter and a limited role for PTS during infection of a mammalian host, Mol Microbiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, and Church GM (2009) Programming cells by multiplex genome engineering and accelerated evolution, Nature 460, 894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chien CC, Chen CC, Choi MH, Kung SS, and Wei YH (2007) Production of poly-beta-hydroxybutyrate (PHB) by Vibrio spp. isolated from marine environment, J Biotechnol 132, 259–263. [DOI] [PubMed] [Google Scholar]
- [18].Centeno-Leija S, Huerta-Beristain G, Giles-Gomez M, Bolivar F, Gosset G, and Martinez A (2014) Improving poly-3-hydroxybutyrate production in Escherichia coli by combining the increase in the NADPH pool and acetyl-CoA availability, Antonie Van Leeuwenhoek 105, 687–696. [DOI] [PubMed] [Google Scholar]
- [19].Spiekermann P, Rehm BH, Kalscheuer R, Baumeister D, and Steinbuchel A (1999) A sensitive, viable-colony staining method using Nile red for direct screening of bacteria that accumulate polyhydroxyalkanoic acids and other lipid storage compounds, Arch Microbiol 171, 73–80. [DOI] [PubMed] [Google Scholar]
- [20].Kidwell J, Valentin HE, and Dennis D (1995) Regulated expression of the Alcaligenes eutrophus pha biosynthesis genes in Escherichia coli, Appl Environ Microbiol 61, 1391–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Csorgo B, Nyerges A, Posfai G, and Feher T (2016) System-level genome editing in microbes, Curr Opin Microbiol 33, 113–122. [DOI] [PubMed] [Google Scholar]
- [22].Dalia AB, Lazinski DW, and Camilli A (2013) Characterization of undermethylated sites in Vibrio cholerae, J Bacteriol 195, 2389–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mercaldi MP, Dams-Kozlowska H, Panilaitis B, Joyce AP, and Kaplan DL (2008) Discovery of the dual polysaccharide composition of emulsan and the isolation of the emulsion stabilizing component, Biomacromolecules 9, 1988–1996. [DOI] [PubMed] [Google Scholar]
- [24].Lazinski DW, and Camilli A (2013) Homopolymer tail-mediated ligation PCR: a streamlined and highly efficient method for DNA cloning and library construction, Biotechniques 54, 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Seed KD, Yen M, Shapiro BJ, Hilaire IJ, Charles RC, Teng JE, Ivers LC, Boncy J, Harris JB, and Camilli A (2014) Evolutionary consequences of intra-patient phage predation on microbial populations, eLife 3, e03497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Karr DB, Waters JK, and Emerich DW (1983) Analysis of Poly-beta-Hydroxybutyrate in Rhizobium japonicum Bacteroids by Ion-Exclusion High-Pressure Liquid Chromatography and UV Detection, Appl Environ Microbiol 46, 1339–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.