

Abstract
PANTHER Classification System (www.pantherdb.org) is a comprehensive system that combines gene function, ontology, pathways and statistical analysis tools to enable biologists to analyze large-scale genome-wide experimental data. The system is built with 82 complete genomes organized into gene families and subfamilies, and their evolutionary relationships are captured in phylogenetic trees, multiple sequence alignments and statistical models (Hidden-Markov Models, or HMMs). Genes are classified by their function in several different ways: families and subfamilies are annotated with ontology terms (Gene Ontology and PANTHER Protein Class), and sequences are assigned to PANTHER pathways. The PANTHER website includes a suite of tools to allow users to browse and query gene functions, and analyze large-scale experimental data with a number of statistical tests. It is widely used by bench scientists, bioinformaticians, computer scientists and systems biologists. In the 2013 release of PANTHER (v.8.0), besides the update of the data content, the website interface was redesigned to improve the user experience and analytical capability. This protocol will provide a detailed description of how to analyze genome-wide experimental data in the PANTHER Classification System.
INTRODUCTION
The PANTHER Classification System (www.pantherdb.org) is designed to be a comprehensive platform for the analysis of gene function on a genome-wide scale1. Although its initial development aimed to classify gene and protein functions2,3, it has evolved through the years to also serve as an online resource for experimental data analysis4. The easy-to-use user interface and timely user support have made PANTHER one of the most widely used online resources for gene function classification and genome-wide data analysis.
PANTHER was initially released in 2003, and was the first database to combine both phylogenetic and functional data to define protein subfamilies of shared function and sequences3. It was also the first database to associate ontology terms describing function to statistical models (HMMs), which can be used to assign genes -- based on sequence information alone -- to subfamilies and functional classes. The novel concept was to annotate not single genes one at a time, but rather subfamilies of related genes that are likely to share function. We demonstrated the accuracy and comprehensiveness of the classifications on the D. melanogaster genome5. In 2005, we began providing annotations of biochemical pathways, viewable with the new PANTHER Pathway Applet6,7. These pathways were curated from the literature by expert biologists and diagrams were drawn with CellDesigner, a pathway-editing tool using controlled graphical notations to represent pathway knowledge8. In 2006, the first version of gene analysis tools was released4. These were primarily for the analysis of gene expression data. The tools were also designed to handle gene list data from any genome wide experiments. In the 2013 release of PANTHER 8.0, the web interface was redesigned to integrate multiple tools into one user-friendly and flexible interface, so that users can easily access all the tools and choose among them to perform gene list analysis at the genome-wide level1.
Since the initial release in 2003, PANTHER has become one of the most popular online resources for genome wide data analysis. Currently, there are over 7000 registered users, 600–800 daily users (weekdays) and over 14,000 monthly users worldwide. These counts were based on unique Internet Protocol (IP) addresses accessing the site, so the actual number is even greater. PANTHER has been cited in over 3600 publications since 2003 based on Google Scholar, and is growing steadily. Users have successfully analyzed the data from gene expression 9,10, proteomics 11,12 and genome-wide association study (GWAS) experiments13,14 in a diverse area of research, such as cancer research 10,11, neurological disorder studies 15,16, autoimmune diseases 9 and cardiac disease studies14,17.
PANTHER is currently included in a number of large international consortia. PANTHER has been a member of the InterPro Consortium of protein annotation resources since 2005, so PANTHER annotations can be automatically generated from the highly-used InterProScan software18. More recently, PANTHER also became part of the Gene Ontology (GO) Consortium19–21. The phylogenetic-inferred curation paradigm has become part of the GO curation pipeline, and therefore, PANTHER annotation reflects a more up to date GO curation22. Finally, PANTHER Pathway is in the process of being integrated into Pathway Commons23.
The PANTHER System is constructed with three functional modules (Figure 1). The core module is a large protein library that contains all protein-coding genes from 82 organisms organized first into families based on sequence homology, and then into subfamilies based on their shared functions (often a group of orthologous genes). Each family or subfamily is represented by a statistical model (HMM) annotated with ontology terms (GO terms and PANTHER Protein Class terms). The second module is the PANTHER pathway module, which contains 176 expert curated pathways. All pathways are connected, through manual curation, to individual proteins in the protein library through the pathway components, and therefore they are also linked to the phylogenetic information and statistical models. The last module is the website tool suite that contains a collection of bioinformatics tools and software, enabling users to not only query the data and classify genes and proteins, but also visualize, analyze and interpret genome-wide scale experimental data in the context of the enriched data content in the first two modules.
Figure 1. Overview of PANTHER infrastructure.

PANTHER is consisted of three modules. The core module is the PANTHER protein library (yellow shade) that contains a collection of PANTHER families and subfamilies, each of which is represented by a phylogenetic tree, an MSA and an HMM. The second module is the pathway that contains 176 expert-curated pathways (green shade). The pathway components are associated to the protein sequences that are used to build the protein library (the light green shade), and therefore, the pathways are also linked to the subfamilies and HMMs. The third module is the tool suite. In this diagram, the gene list analysis tool is used as an example (blue shade). When the user uploads a gene list to the tool, and if the IDs in the list are from one of 82 organisms in PANTHER, the tool will map the IDs to the IDs in the PANTHER protein library (green arrows). If the uploaded IDs are not from one of the 82 organisms, the user can score the sequences against the PANTHER HMM library and generate the PANTHER Generic Mapping file (see Box 2) (orange arrows). There are three tests in the tool: functional classification, statistical overrepresentation test and statistical enrichment test. Numeric values must be provided in order to use the statistical enrichment test.
PANTHER protein library
The core of the PANTHER system is a collection of phylogenetically-defined protein families and subfamilies generated by computational algorithms, and curated by expert biologist using an extensive software system for associating ontology terms1,3. The current release contains over 640K proteins from 82 genomes, of which 79 are from the Reference Proteome Project (http://www.ebi.ac.uk/reference_proteomes/) (Figure 1). UniProt IDs are used as primary protein identifiers. These proteins are representatives of their respective genes. Therefore, each gene is represented by only one protein. In addition, UniProt IDmapping (http://www.uniprot.org/mapping/) is used to map the primary protein IDs to other IDs from different databases and resources, which expands the capability of PANTHER to support a wider range of ID types (see Supported IDs in Box 1). The proteins are divided into 7729 families, each of which is represented by a phylogenetic tree, an HMM, and a multiple sequence alignment (MSA) (Figure 1). Protein family trees are constructed computationally from sequence data using a phylogenetic tree inference algorithm called GIGA24. Nodes in the tree, corresponding to common ancestors of extant family members, are annotated by expert biologists with their inferred GO terms and PANTHER protein class terms, based on experiments performed on extant proteins. Subfamilies are determined based on the tree structure and often define the orthologous group, especially in organisms in the Deuterostomia superphylum. The functional annotations are propagated from the annotated ancestral nodes to the subfamily nodes, each of which is also represented by an HMM to allow classification of newly discovered protein sequences (Figure 2A). In addition, the HMM library and the scoring pipeline allow users to analyze genome-wide data from any organisms outside of the 82 within the PANTHER system (see below). Phylogenetic tree and functional annotation at the ancestral nodes have great advantage in annotation of previously unclassified genes. The PANTHER phylogenetic trees are currently used by the Gene Ontology Consortium in its curation pipeline 22.
Box 1: Input file.
File format
The input file is a tab-delimited text file (.txt or .tab). Only the data in the columns specified below will be used in the analyses. Data in additional columns are ignored. Microsoft Excel file is not accepted by the tool. Below are three file types that can be used.
ID list – The first column must be the gene or protein identifiers. See below for the supported IDs. A second column of numerical values is required if a user wants to run the Statistical enrichment test.
Previously exported text search results – A text search result can be viewed as a gene list, which can be saved as a text file (see Section A under Anticipated Results). This file contains the gene or protein identifiers in the first column. This file type is not associated with numeric values, so it cannot be used for Statistical enrichment test.
- PANTHER Generic Mapping File – For IDs from organisms other than the 82 organisms in the PANTHER database, a user-generated data containing mappings between those IDs and their corresponding PANTHER IDs can be used (see Box 2 for details about mapping). The file must be tab-delimited and contain the following columns:
-
The first column can contain a list of unique IDs from the user.
-
The second column should be corresponding PANTHER family or subfamily ID (ex: PTHR10078 or PTHR10078:SF1), and is used to look up the association to GO and PANTHER terms (molecular function, biological process, and pathway).
-
If you are uploading data for the “Statistical enrichment test” tool, a third column is required that contains the numeric value of the experiment.
-
Supported IDs
If the “ID list” file type is used, the IDs in the first column of the file must be from one of the following databases that are supported in the PANTHER system.
Ensembl: Ensembl gene identifier. Example: “ENSG00000126243”
Ensembl_PRO: Ensembl protein identifier. Example: “ENSP00000337383”
Ensembl_TRS: Ensembl transcript identifier. Example: “ENST00000391828”
Gene ID: EntrezGene IDs. Examples: “10203” (for Entrez gene GeneID:10203)
Gene symbol: for example, “CALCA”
GI: NCBI GI numbers. Example: “16033597”
HGNC: HUGO Gene Nomenclature ids. Example: “HGNC:16673”
IPI: International Protein Index ids. Example: “IPI00740702”
UniGene: NCBI UniGene ids. Examples: “Hs.654587”, “At.36040”
UniProtKB:UniProt accession. Example: “O80536”
UniProtKB-ID: UniProt ID. Example: “AGAP3_HUMAN”
If you are not certain about the ID type in your uploaded gene list, or when you find that your IDs are not mapped to any PANTHER IDs in the result page, you can simply search your ID at NCBI (http://www.ncbi.nlm.nih.gov/) or a search engine website such as Google. You can find the ID type based on the database source on the result page.
Figure 2. Examples of PANTHER phylogenetic tree and pathway diagram.

(A) A sample phylogenetic tree from PANTHER (PTHR11633, PLATELET-DERIVED GROWTH FACTOR). The family contains three subfamilies (blue arrows). SF1 is annotated as “PDGF/VEGF growth factor related protein 1” based on the annotation in the drosophila and c. elegans sequences (Q9VWP6 and Q9N143, respectively). There is a recent duplication that generates the PDGF A chain (SF3) and the PDGF B chain (SF2). Ontology terms are annotated to the node that represents the common ancestor if the extant family, in this case, AN0 (SF1). The classifications are propagated to all the descent nodes, including the AN4 (SF3) and AN33 (SF2).
(B) An example of PANTHER pathway diagram (P00047, PDGF signaling pathway). The diagram is shown in CellDesigner process diagram, which is similar to the SBGN-PD format, for example (blue circle), a transition of an input (e.g., ERK) to an output (eg., phosphorylated ERK) catalyzed by a modifier (eg., phosphorylated MEK). A pathway component (e.g, PDGF in red circle) is associated with the protein sequences in the protein library (red arrows in 2A) through expert curation. This association is supported by literature evidence. As a result, the pathway component of PDGF can be inferred to other orthologous protein sequences in the subfamilies in the library (SF2 and SF3 in 2A).
PANTHER pathway
The PANTHER pathway dataset uses controlled vocabulary and graphical notation to describe pathway knowledge7. The graphical representation of pathways is generated by CellDesigner, a pathway-editing tool8, and is compliant with the Systems Biology Graphical Notation Process Description standard (SBGN-PD) 25 (Figure 2B). Currently, there are 176 expert-curated pathways in PANTHER. The scope of the pathways is similar to those described in textbooks or review articles, e.g., glycolysis, the PDGF signaling pathway or the p53 pathway. Each pathway contains three key classes of data (Figure 1). First is the pathway component (or molecule class), which represents a specific class of molecules that play the same mechanistic role within a pathway. It can be a protein (e.g., PDGF, Jak), a DNA region, or a simple molecule (e.g., ATP or glucose). If a pathway component is a protein, gene or transcribed RNA, it is associated with the protein sequences in the PANTHER protein library through manual curation (Figure 1 and 2). The individual protein sequences are instances of the pathway component. In these cases, a pathway component is typically a group of homologous/orthologous proteins across various organisms that are involved in the same biochemical reaction within the pathway. The second class is reaction, which represents biochemical relationships among different pathway components. Since all PANTHER pathways are in compliance with SBGN-PD, a typical reaction is a transition of an input (or reactant) to an output (a product) controlled by a modifier (Figure 2B). The last class is the cell/tissue type and cellular component, which provides the location where the reaction occurs.
There are two key features of the PANTHER pathway data model. First, the association between the molecule classes and protein sequences links the pathway to the phylogenetic relationships and statistical models (HMMs) of the protein families. As a result, if a protein is associated with a pathway component via experimental evidence, its orthologs can be inferred to the same component based on the PANTHER phylogenetic tree. Second, PANTHER pathway supports community standards, and all pathways are available in SBML26, BioPAX27 and SBGN25 formats.
PANTHER tools
PANTHER provides a number of useful research tools, including the PANTHER HMM Scoring Tool, cSNP Analysis Tool, and Gene List Analysis Tool. Both the PANTHER HMM Scoring Tool and cSNP Analysis Tools have online and downloadable versions. The online versions allow only one protein sequence to be analyzed at a time, while the downloadable versions allow large batch analyses. A brief description of the downloadable version of PANTHER HMM Scoring Tool can be found in Box 2. The details of how to use these tools can be found in the PANTHER help page (http://www.pantherdb.org/help/PANTHERhelp.jsp) and the user manual. In this protocol, we will focus on the Gene List Analysis Tool. Sample gene list files can be found in the Supplement materials for testing.
Box 2 PANTHER HMM Scoring Tool.
This is the downloadable version of the PANTHER Scoring Tool to allow users to submit a large number of protein sequences in fasta file format, and score against the PANTHER HMM library so that the sequence identifiers can be mapped to PANTHER HMM IDs and used in the gene list analysis tools.
UNIX and Perl are required on your computer in order to use the tool. The user needs to have the basic knowledge of using UNIX and Perl in order to complete the procedures described in this Box. If you don’t feel you have adequate knowledge in these areas, you may want to get help from your colleague who has the technical expertise and knowledge, such as a bioinformatics support person. You can also send email to feedback@pantherdb.org for help.
Procedures:
-
Download the following scripts and data.
pantherScore script (ftp://ftp.pantherdb.org//hmm_scoring/current_release/ ).
(Timing: 2 minutes)
PANTHER HMM library (ftp://ftp.pantherdb.org/panther_library/current_release/)
(Timing: 15 minutes)
HMMER2 (ftp://selab.janelia.org/pub/software/hmmer/2.3.2/hmmer-2.3.2.tar.gz).
(Timing: 5 minutes)
Note: Please note that this is an archived version of HMMER2. The current release is HGMMER3. The panther scoring script does not support HMMER3.Install PANTHER scoring tool
BLAST (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/2.2.24/ncbi-blast-2.2.24+-x64-linux.tar.gz )
(Timing: 5 minutes)
-
4
Decompress all four downloads. (Timing: 30 minutes)
-
5
Install HMMER2 and BLAST according to the installation instruction coming with the two algorithms. (Timing: 15 minutes)
-
6
Define the location of the HMMER and BLAST binaries in the $PATH variables on your computer. This is usually done quite differently depending on the UNIX shell environment on your computer. Basically, you need to update the $PATH on the UNIX shell files, such as .cshrc (for C shell) or .profile (for Bourne shell). If you are not familiar with $PATH, you need to consult someone with IT knowledge to help you. (Timing: 15 minutes for expert, 1–2 hours for others).
-
7Run the script on UNIX command line. (Timing: each sequence takes 20 seconds to score, therefore, 180 sequences in an hour)
- cd pantherScore
- source panther.cshrc
- ./pantherScore.pl -l <panther_hmm_library> -D B -V -i <fasta file> -o <output file> -n
where
-l The path to the PANTHER HMM library downloaded above.
-D display type for results.
Options: B (best hit), A (all hits)
-i input fasta file to score. A sample fasta file is included in the downloaded called test.fasta
-o the output file
-n to display family and subfamily names in the output file.
Note: If you have a lot of sequences, you can split the fasta file and run the script on multiple computers.
The output file is a tab-delimited file in the following format:
col 1 - sequence ID
col 2 - PANTHER accession if (containts :SF, is a subfamily HMM)
col 3 - PANTHER family or subfamily name
col 4 - HMM evalue score, as reported by HMMER
col 5 - HMM score, as reported by HMMER (not used by PANTHER)
col 6 - alignment range of protein for this particular HMM
This file can be used as PANTHER Generic Mapping File for the gene list analysis tool. If the Statistic enrichment test is used, the numeric values need to be inserted in the 3rd column.
The Gene List Analysis Tool can be accessed directly from the PANTHER home page (www.pantherdb.org) (Figure 3). There are several options for users to input a list of genes (and optionally quantitative data) for analysis. The PANTHER database uses some database identifiers as the primary IDs for each gene, and they are the most common option for input file (e.g., a UniProt identifier, an Entrez Gene identifier, or even a gene symbol). The PANTHER database also maps identifiers from a number of different sources for 82 different organisms using the UniProt IDmapping mechanism, and the identifiers in the user’s list are automatically mapped to the primary IDs in the PANTHER database (Figure 1). A total list of the supported IDs can be found in Box 1. A report is generated that lists not only the mapping of each gene, but also the identifiers that could not be mapped, if any. Since each gene in the PANTHER database belongs to a phylogenetic tree that is annotated with GO and PANTHER ontology terms and pathways, the mapped IDs from the gene list will inherit these annotations. It needs to be pointed out that, in a very rare case, a single gene symbol or its synonym can be mapped to more than one gene from the IDmapping. We are currently working with UniProt to improve such mappings. However, this is so rare that we don’t think it affects significantly to the results from the statistical tests. If the gene list is not from the 82 organisms, a user can still use the PANTHER tools, but they need to map each of their own identifiers to PANTHER identifiers first, using the downloadable PANTHER HMM scoring tool (Figure 1 and Box 2), and creating the Generic PANTHER Mapping File with two columns (the user’s ID, and the ID of the best PANTHER HMM hit) (see Box 1 for more details of the file format). Each gene in the list will inherit the same annotations as those stored in the PANTHER database for the given HMM.
Figure 3.

The PANTHER home page with the Gene List Analysis Tools.
Once the gene list is classified with those functional terms, it can be analyzed in three different ways.
Functional classification – This tool provides the functional classification results of the uploaded list, and displays them in either a gene list page or pie chart.
Statistical overrepresentation test - This tool is based conceptually on the simple binomial test described previously28. It compares a test gene list uploaded by the user to a reference gene list, and determines whether a particular class (e.g., a GO biological process or PANTHER pathway) of genes is over- or under- represented. A more detailed description of the tool can be found in Box 3.
Statistical enrichment test - This tool, based on the work by the PANTHER group29 and Lander’s group30 for two different genomic data analyses at about the same time, uses the Mann-Whitney test31 to determine if any ontology class or pathway has numeric values that are non-randomly distributed with respect to the entire list of values. The numerical data can be normalized raw readouts from the microarray experiments or, more commonly, the fold-change value for each gene in a differential expression experiment, or calculated p-values from GWAS experiment. A more detailed description can be found in Box 4.
Box 3.
Statistic Overrepresentation Test
The input (or test) list is usually a list of genes of your interest. It can be a list of genes that are up-regulated in the gene expression experiment, or have significant p-values from your GWAS experiment. The list is divided into groups based on GO or PANTHER classification (molecular function, biological process, cellular component, PANTHER Protein Class or PANTHER Pathway). As many as four test lists can be uploaded for each analysis. A reference list, which usually contains all the genes/proteins from which the list was drawn, is divided into groups in the same way. PANTHER provides reference proteome data set as default reference lists for all 82 genomes, so uploading a reference list is optional. Then, for each functional category, e.g., protein kinase for molecular function, cell proliferation for biological process, or apoptosis signaling pathway for pathway, the binomial test is applied to determine whether there is a statistical over- or under-representation of genes/proteins in the test list relative to the reference list.
P-value calculation in the overrepresentation test
The expected value is the number of genes you would expect in the test list for a particular PANTHER category, based on the reference list. For example, there are 20,000 genes in the reference list (ex: the human genome). 440 of these genes map to the GO term induction of apoptosis. Based on this, 2.2% (440 divided by 20,000) of the genes in the reference list are involved in induction of apoptosis. Now a test list that contains 500 genes is uploaded. Based on the reference list, it is expected that 11 genes (500 multiplied by 2.2%) in the test list would be involved in induction of apoptosis.
If for this biological process more genes are observed in the test list than expected, you have an over-representation (+) of genes involved in induction of apoptosis. If fewer genes are observed than expected, you have an under-representation (−). A p-value is calculated then to determine whether the over- or under- representation is significant or not. For example, let’s assume that 21 genes are observed in the test list are involved in induction of apoptosis. Although this is almost twice as the expected value, the p-value is large and not significant (the p-value would be 0.722). Alternatively, if 35 genes are observed, this is very different than the expected value, so you would expect a small, significant p-value (the p-value would be 6.21e-7). This small p-value indicates that the result is non-random and potentially interesting, and worth looking at in closer detail. A p-value cutoff of 0.05 is recommended as a start point.
The statistical method used in this test is the binomial test. In the binomial test we assume that under the NULL hypothesis, genes in the test list are sampled from the same general population as genes from the reference set, i.e. the probability p(C) of observing a gene from a particular category C in the test list is the same as in the reference list. We first estimate the probability p(C) from the reference set assuming that it is large and representative:
p(C)=n(C)/N,
where n(C) is the number of genes mapped to category C, and N is the total number of genes in the reference set.
We then use the above estimate to find the p-value: the probability of observing k(C) genes (or a more extreme number) in the uploaded list of size K. Under the NULL hypothesis, the number of genes of mapped to C is distributed binomially with probability parameter p(C) and thus the p-value would be
where the sum runs from k(C) to K in the case of over-representation (i.e. when the number of observed genes k(C) is greater than expected p(C)*K under the NULL hypothesis), and 0 to k(C), in the case of under-representation (i.e. when k(C) is smaller than p(C)*K).
When developing this analysis tool, we tested other statistic methods also. We decided to use the Binomial, since other methods tends to be not as accurate when the population sizes or the expect number is small.
Box 4.
Mann-Whitney Rank-Sum Test (U-Test)
The statistical test is general enough to handle any numerical data, continuous or discontinuous, generated by experiments such as gene expression, proteomics or GWAS. First, a reference distribution is generated using all values from the input data. Then the entire list is divided into groups based on GO or PANTHER classification (molecular function, biological process, cellular component, PANTHER Protein Class or PANTHER Pathway), and the distributions for each group are generated. The probability that the functional category distribution was drawn randomly from the reference distribution is estimated using the Mann-Whitney Rank-Sum Test (U-Test) 29.
To perform the rank sum test, first the values of the genes that map to a given category are combined with the overall list of values that were input. Then, all the values are ranked from smallest to largest, with the smallest value getting a rank of 1. If multiple values are identical, the average of the ranks for these values is used.
Then the rank sum is calculated for this category, by summing up the ranks for all of the genes that map to this category. The average rank, R1 is then calculated by dividing the rank sum by the number of genes, n1, that map to the category. Likewise, the rank sum is calculated for the list of all IDs uploaded, and the average rank, R2, is calculated by dividing the rank sum by the total number of genes uploaded, n2.
Next, the Mann Whitney U statistic is calculated for both populations:
U1 = n1* n2 + (n1 * (n1 + 1)) / 2 - R1
U2 = n2* n2 + (n1 * (n2 + 1)) / 2 - R2
The larger of these two values is the Mann Whitney U-statistic, U, whose distribution for small sample sizes can be found in most statistic books. In our case, our application is for large sample sizes, so we use the normal approximation:
Z-score = (U- (n1* n2)/2)/sqrt(n1*n2*(n1+n2+1)/12).
It follows that the p-value is the integral under the standard normal density.
It is worth pointing out that there are other similar tools to perform overrepresentation and enrichment tests, most notably, GSEA32 and DAVID33. Although all three tools use very similar statistical algorithms in the back end, there are some differences among them. GSEA requires download and installation, so its targeted users are bioinformaticians who are more computer-savvy. Both DAVID and PANTHER are online tools and are more appealing to bench biologists. Compared to DAVID, PANTHER has a few advantages. First, PANTHER is currently part of the GO consortium, and integrates more updated GO curation data with the tools. In fact, DAVID downloads PANTHER data and integrate them in its analysis tools. Second, PANTHER allows users to analyze genome data from 82 organisms using the online tool, and any other organisms using the PANTHER scoring tool. Third, the phylogenetic trees in PANTHER protein library allow users to make more accurate ortholog prediction, and thus greatly enhance the capability of the tools. One weakness of PANTHER is that it cannot analyze data against resources outside of PANTHER, such as KEGG and Reactome.
Workspace
The Workspace is a unique feature in PANTHER that allows users to store their gene lists that they generate for future analysis. Although users do not have to register to use the PANTHER system, registration is required in order to user the workspace. Registration is free. In any PANTHER gene list display page, the user can easily send the list to the Workspace (see below).
MATERIALS
Equipment
Laptop or desktop computer with internet connection. High-speed internet connection is highly recommended.
System Requirements
- Operating System
- Windows XP or Windows 7 for PC users
- MacOS 10.5 or later for Mac users
- Minimum of 2GB RAM recommended
- Browser
- Microsoft Internet Explorer 8 or later (recommended for PC users)
- Safari version 5 (recommended for Mac users)
- Firefox version 19
- Google Chrome version 26
- Java
- Latest version of Java (can be downloaded from http://www.java.com/en/download/)
- JavaScript, Java applets and cookies must be enabled in your browser
- Java applet runtime parameters set to -ms128m -mx512m -Xss16m
PROCEDURE
Note: Sample gene list files can be found in the Supplement materials for you try the tools.
-
1
Access the PANTHER website by entering www.pantherdb.org in your web browser.
-
2Prepare input file(s) according to option A if you are working with one of the 82 genomes in the PANTHER database, or option B if you are working with an organism other than the 82 genomes in the database.
- Prepare input file(s) in simple text file (.txt or .tab) with gene or protein identifiers as the first column, and numeric values as the second column if you want to use the statistical enrichment test. Detailed instruction of file format and supported IDs can be found in Box 1. (Timing: 15–30 minutes)
!! CRITICAL STEP The input file must be in simple text file format (.txt or .tab). It also must use IDs supported in the PANTHER system and the correct tab delimited format as described in Box 1.
-
3Upload the gene list to the PANTHER tool system using option A if you have a small list for functional classification tool, option B if you have a large list and want to analyze using all the gene list analysis tools, or option C if you previously saved the list in the Workspace.
- Paste the ID list prepared in Step 2 into the Enter ID box. Alternatively you can also type IDs, one per line, into the box (blue arrow in Figure 3). Please note that this type of ID upload only supports functional classification tools.
- Upload the list file prepared in Step 2 by clicking the Browse button (red arrow in Figure 3), and follow the online instruction to locate the file.
- If you have previously saved your list into the Workspace, you can use it by clicking the login link (green arrow in Figure 3), and follow the online instruction to locate the file in the Workspace. Please note that numeric values cannot be saved in the Workspace, and therefore this type of upload does not support the Statistical enrichment test.
-
4
Select a corresponding list type in order for the tool to work properly. There are three list types that are supported by the tools: ID List, Previously exported text search results, and PANTHER Generic Mapping File. See Box 1 for details of the list types.
-
5
Select an organism from the drop-down menu, which lists the 12 model organisms first, followed by the rest 70 organisms ordered alphabetically.
-
6
Note: There are two purposes to select an organism at this point. First, some identifiers, such as gene symbols, are not organism specific. By selecting an organism here, it ensures that the IDs are mapped to those in the organism you are interested in. Second, if the statistical overrepresentation test is selected, the default reference gene list is based on the selected organism.
-
7In the Select Analysis box, select one of the following 4 analyses by clicking the radial button, and then click the submit button.
- Select Functional classification viewed in gene list if you want to find out the functional classification of the genes in your list (Figure 4). See the next section for more details about how to interpret the results.
- Select Functional classification viewed in pie chart to get the functional classification of the genes in your list displayed as a pie chart (Figure 5).
- Select Statistical overrepresentation test.
- On the next page (Figure 6), you can select additional gene lists for the analysis. A total of 4 test gene lists can be uploaded for this tool. To do so,
- Click Browse button
- Select the gene list from your computer
- Select the organism. The default is the organism selected when the first gene list is uploaded.
- Select the List type. The default is the one selected when the first gene list is uploaded.
- Click Upload list.
- After all lists are uploaded, click the Finish selecting lists button. The tool will take you to the next page, which allows you to make the following selections.
- Select a reference list. The default is always the entire proteome of the organism selected above. If you choose to change it, click the Select reference list button.
- You can then select a reference list from another organism, or upload your own using an interface similar to one to upload test gene list.
- Select an ontology to analyze. There are five options.
- PANTHER Pathway
- GO molecular function
- GO biological process
- GO cellular location
- PANTHER protein class
- Multiple test correction (Bonferroni correction) is selected by default.
- Click the Launch analysis button.
- On the results page (Figure 7A), you can visualize the results with the following options:
- You can export the result table in a tab-delimited file by clicking the Export results button.
- You can also view the results in graphs by using the View drop-down menu.
- If your analysis is done in pathway as shown here, you can click the pathway name and display the pathway diagram. The pathway components that have genes in your test list will be highlighted. The color of the highlighted component can be defined at the top of the page (Figure 7A, red circle). A total of 4 test lists can be analyzed and viewed at the same time.
- Select Statistical enrichment test
- After clicking the submit button, on the next page, select an ontology to analyze.
- Click the Launch analysis button.
- On the results page (Figure 8A), you can visualize the results with the following options:
- You can export the result table in a tab-delimited file clicking the Export results button.
- You can compare the distribution curve in graph view. To do so, you can check the box in front of the category or pathway of your interest, and then click the Graph selected categories button (Figure 8B).
- If your analysis is done in pathway as shown here, you can click the pathway name and display the pathway diagram. The pathway components are colored in “heat-map” based on the input numeric values (Figure 8C). Click the Specify color ranges of pathway diagrams button on the result page to view or specify the color ranges.
Figure 4.
User interface of the statistical overrepresentation test to allow user to select additional test gene lists.
Figure 5.

Results of functional classification displayed as a gene list page. The results are based on the sampleTestList_NP_500 file in the Supplemental Materials.
Figure 6.

PANTHER pie chart results from the sampleTestList_NP_500 file in the Supplemental Materials. You can use the Select ontology drop-down menu to switch to the pie chart of different ontologies. Click on the pie chart section to display the child categories. Click on the legends on the right side to retrieve the list of the genes for that category.
Figure 7. Result from the statistical overrepresentation test. The results are based on the sampleTestList_NP_500 file in the Supplemental Materials.

(A) The summary of the results is displayed in a table. You can export the table in a tab-delimited file by clicking the Export results button. You can also view the results in other views by using the View drop-down menu. If your analysis is done in pathway as shown here, you can click the pathway name and display the pathway diagram. The pathway components that have genes in your test list will be highlighted. The color of the highlighted component can be defined at the top of the page (red circle). A total of 4 test lists can be analyzed and viewed at the same time.
(B) The results viewed in the PANTHER pathway Heterotrimeric G-protein signaling pathway – Gi alpha and Gs alpha mediated pathway (P00026). The components that contain the genes in the test gene list are highlighted in red.
Figure 8.
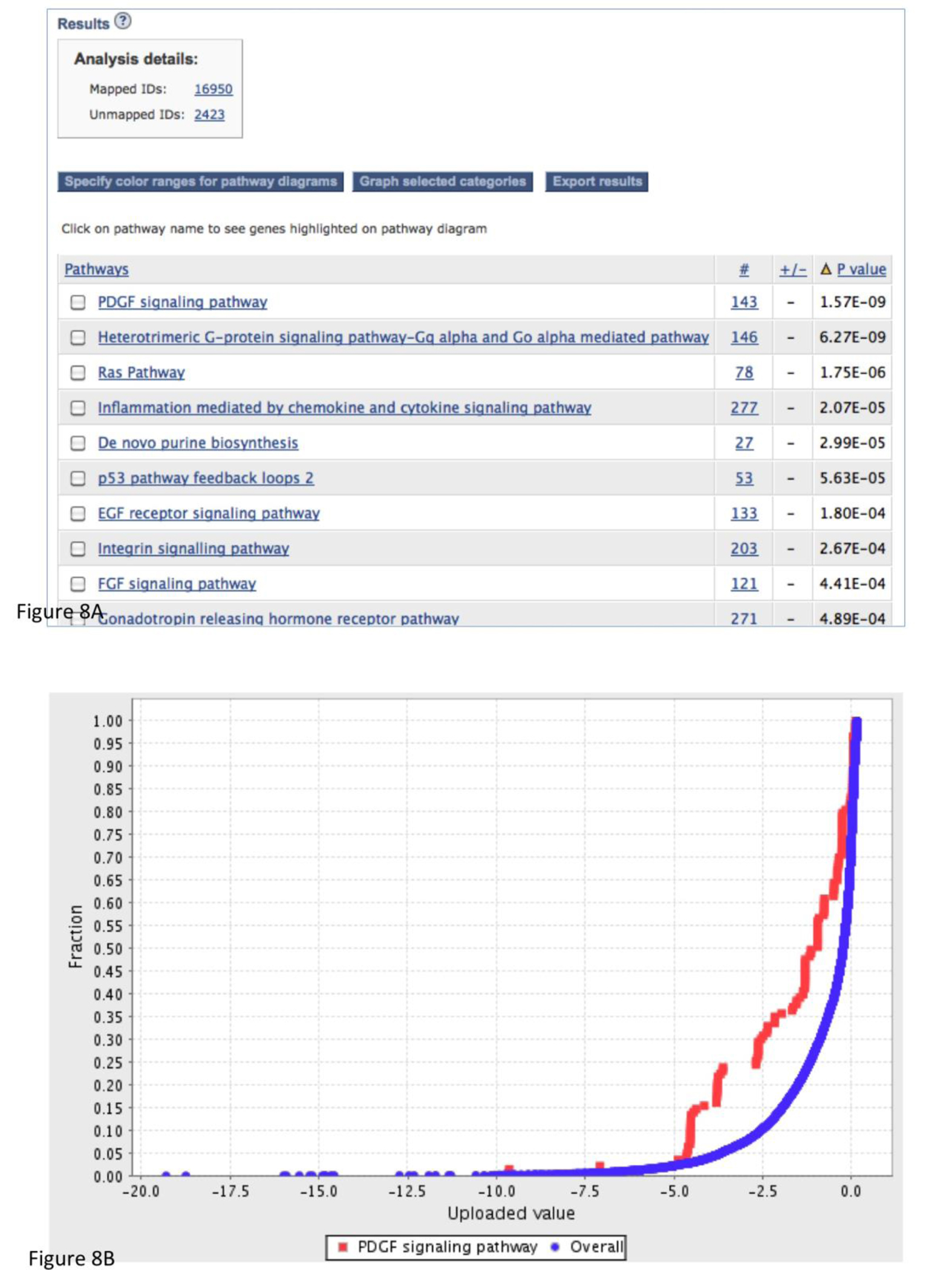

The results from the statistical enrichment test. The results are based on the sampleTestList_NP file in the Supplemental Materials. (A) The output of the tool with a list of P-values for each comparison between a functional category distribution and the reference distribution. (B) Comparison of the distributions from PDGF signaling pathway (red) and reference (blue) in graph view. (C) A pathway diagram of PDGF signaling pathway that is visualized using an interactive pathway Java applet that colors the pathway using a “heat map” derived from the input values.
Note: In order to use this tool, make sure that the uploaded gene list contains a second column with numerical values.
Timing
Step 1, launch website: instant.
Step 2A, prepare input file: 15–30 minutes
Step 2B, prepare input file by scoring the PANTHER HMM library: varies based on the number of sequences. On average, 180 sequences per hour, plus 1.5–3.5 hour tool installation.
Step 3–6, using online tools: 5 minutes.
TROUBLESHOOTING
Table 1 lists some of the common problems and the possible solutions.
Table 1.
Troubleshooting
| Step | Problem | Possible reasons and solutions |
|---|---|---|
| 3 | Fail to upload the file | This is usually because the input file is in the wrong file format. Possible solutions: 1. Make sure that your file is in simple text format (.txt or .tab). 2 If you are uploading a file with numeric values for the enrichment test, make sure that the second column contains only numeric numbers. Any rows with no values should be removed instead of leaving it blank or mark it as “n/a”, etc. 3. Make sure that there is no blank rows in the first column. |
| 6A, 6Cvii, 6Dii. | IDs in the uploaded file don’t have a mapped ID in PANTHER | The current PANTHER data is based on the April 2012 release of Reference Proteome Project and its ID mapping. It is possible that a small fraction of the IDs may not map due to the outdated data either in the PANTHER database or your uploaded file. There is no solution to this. If you believe that you are using the current IDs, please do the followings: 1. Make sure that the IDs in the uploaded file are supported by PANTHER. Refer to Box 1 for Supported IDs. 2. IDs from certain database may contain a version number at the end of them, e.g., NP_000242.1. Do not include the version number “.1” in the ID, and just use NP_000242. 3. Send a feedback to feedback@pantherdb.org. |
| 6Cviiic, 6Diiic | Pathway Applet can not be launched to view the pathway diagrams | This is most likely that your Java plug-in is outdated. Read the system requirement and make sure that your computer has the most updated Java version. |
ANTICIPATED RESULTS
A. Functional classification tool viewed in gene list page
This tool returns the results as a gene list webpage (Figure 5). The page displays all the IDs from the uploaded gene list and their mapped PANTHER sequence IDs as well as ontology and pathway terms. The page contains the following information (Figure 5).
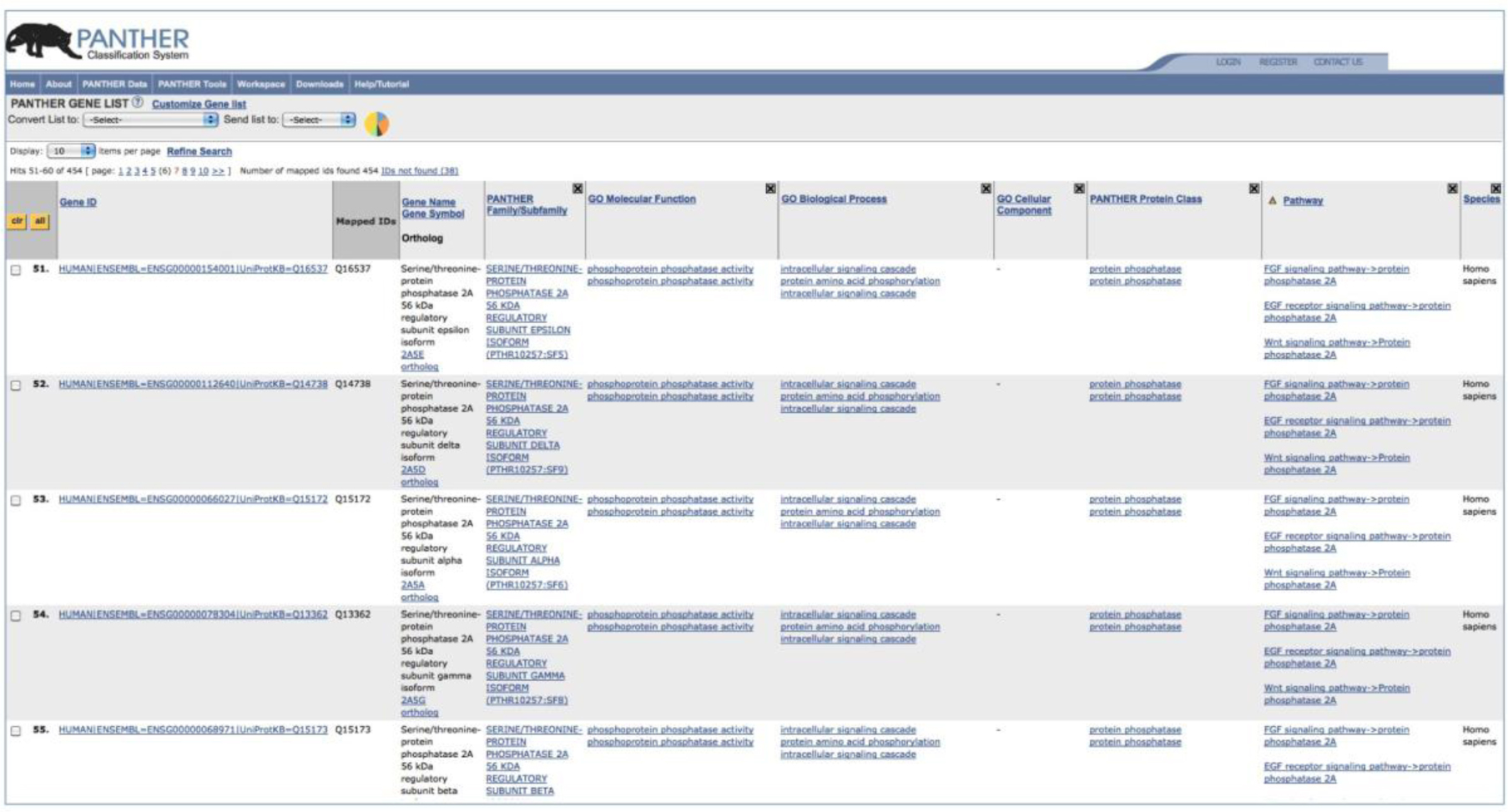
Gene ID – This is the identifier for genes in the PANTHER library. The format is as follows: organism|gene database source=gene id|protein database source=protein id. For example, HUMAN|ENSEMBL=ENSG00000111262|UniProtKB=Q09470 is a human se- quence, the gene sequence is from ENSEMBL with id ENSG00000111262, and the protein sequence is from UniProt with id Q09470.
Mapped IDs – IDs from the uploaded gene list that are mapped to the gene ids in the first column.
Gene Name/Gene Symbol – The Entrez gene definition and gene symbol.
PANTHER Family/Subfamily – The name and identifier of the PANTHER family or subfamily where the gene in the first column is in.
GO Molecular Function, Biological Process, Cellular Component: These are Gene Ontology terms from PANTHER GO Slim describing the function of the gene product.
PANTHER Protein Class – This is a PANTHER Index terms describing protein classes.
Pathway – Pathway and pathway component that are linked to the PANTHER subfamily in column 4. The subfamily is linked to the pathway component when at least one of its member genes is associated to the component directly by manual curation.
Species – The organism of the gene in Column 1.
Once you reach the gene list page, you can make following changes to view the results.
Sort the list – You can always sort the list by clicking on any of the underlined column names. A yellow triangle appears in front of the column name that you choose to sort. The orientation of the triangle indicates the sort is ascending or descending.
Customize columns – You can click on the “x” button next to the column names to collapse the column.
Converting a list to another list type – Select the genes you want to convert by clicking the checkboxes. The default is for all genes in the list.
- Saving the list. Select the genes you want to save by clicking the check- boxes. The default is for all genes in the list. You can select one of the followings from the pull-down menu as the destination:
- Workspace – You need to register to save data to the workspace. The registration is free. When you make this selection, a pop-up window will ask you to name the list and add any comments. The name and comments can be edited at any time in the future from the Workspace page. Once the gene list is saved in your workspace, it can be returned to at any time. Only the IDs are stored, and they are mapped to the internal PANTHER gene ids, so when you access a list in the future, all information will be updated and current.
- Exporting a list to a file – The list will be exported as a tab-delimited file. You can now import the file into Excel or perform any post-processing you wish.
- View the list as text on the website.
Use the pie chart view by clicking the colorful pie chart icon (see Glossary for the meaning of the abbreviations). See the next section below for details about how to interpret the pie chart.
B. Functional classification tool viewed in pie chart
This tool returns the results in a pie chart, which displays an overview of all ontology terms at the first (or most general) level within the same ontology (Figure 6). When a slice of the pie chart, which represents an ontology term, is clicked, a new pie chart will appear that contains its child ontology terms. Since one gene can be classified to more than one term, the pie chart is calculated based on the number of “hits” to the terms over the total number of class hits. Class hit means independent ontology terms. For example, if a gene is classified to 2 ontology terms that are not parent or child to each other, it counts as 2 class hits.
When you place the computer mouse pointer over a slice, the category name and a series of counts are displayed. In our example in Figure 6, the name is a GO term apoptosis (GO:0006915) followed by
the number of genes (70) from the uploaded list that are classified to the term apoptosis;
the percent (14%) of genes classified to apoptosis (70) over the total number of genes (500);
the percent (4.7%) of genes classified to this apoptosis (70) over total number of class hits (1494).
C. Statistical overrepresentation test
The results of this analysis tool are displayed in a table (Figure 7A). If one test gene list is uploaded, the table contains six essential columns of data:
The first column contains the name of the PANTHER classification category. If you are doing this analysis in pathways, you can click on the pathway name to view the corresponding pathway diagram.
The second column contains the number of genes in the reference list that map to this particular PANTHER classification category.
The third column contains the number of genes in your uploaded list that map to this PANTHER classification category.
The fourth column contains the expected value (see Box 3), which is the number of genes you would expect in your list for this PANTHER category, based on the reference list.
The fifth column has either a + or −. A plus sign indicates over-representation of this category in the test list: you observed more genes than expected based on the reference list (for this category, the number of genes in your list is greater than the expected value). Conversely, a negative sign indicates under-representation, i.e. fewer genes than expected.
The sixth column is the p-value as determined by the binomial statistic (see Box 3). This is the probability that the number of genes you observed in this category occurred by chance (randomly), as determined by your reference list. A small p-value indicates that the number you observed is significant and potentially interesting. A cutoff of 0.05 is recommended as a starting point.
If more than one test list is uploaded, columns 3 to 6 are repeated for each list.
From this result page, various statistics can be exported by using the drop-down menu next to the “Export results” button. The list of genes/proteins in any functional group can be viewed by clicking on the listed counts. When pathways are chosen as the functional categories, clicking on the pathway name brings up pathway diagrams colored according to preferences specified by the user (Figure 7B). The resulting pathway diagram can be exported as an image file (.png) by choosing “File -> Export image” from the Applet menu.
D. Statistical enrichment test
The returned results are displayed in a table with four essential columns of data (Figure 8A):
The first column contains the name of the PANTHER classification category. If you are doing this analysis in terms of pathways, you can click on the pathway name to view the pathway diagram. The genes in the pathway diagram are colored according to the numeric value provided in the uploaded gene list file, and the rules for this can be specified by clicking on the ‘Specify color ranges’ button.
The second column contains the number of genes that map to this particular PANTHER classification category.
The third column has either a + or −. A plus sign indicates that for this category, the distribution of values for your uploaded list is shifted towards greater values than the overall distribution of all genes that were uploaded. A negative sign indicates that the uploaded list is shifted towards smaller values than the overall list.
The fourth column contains the p-value as calculated from the Mann-Whitney U Test (Wilcoxon Rank-Sum test) (Box 4). A large p-value indicates that the genes for this category have a distribution that is similar to randomly choosing genes from the overall distribution. In other words, the values of the uploaded genes for this category have a similar distribution to the overall list of values that were input. A small, significant p-value indicates that the distribution for this category is non-random and different than the overall distribution. A cutoff of 0.05 is recommended as a starting point.
To have a visual representation of these distributions, select the checkboxes of the categories of interest, and click on the ‘Graph selected categories’ button. The graph will be displayed in a new window (Figure 8B). The x-axis is your uploaded value. The y-axis is the cumulative fraction. The blue curve is the overall distribution for all genes. The red curve is the selected functional category. In this case, it is PDGF signaling pathway. If you look at the data point x = −2.5, y is 0.3 for the red curve and 0.1 for the blue curve. This means that 30% of your uploaded genes have a value of −0.25 or smaller, but only 10% of the overall genes have a value of −0.25 or smaller. In other words, it shows that the distribution of the category tends to be smaller than the overall distribution. We find that this is critical for interpreting any deviation between the functional category distribution and the overall distribution.
The genes/proteins in each category can also be viewed from the output page by clicking on the listed counts. In addition, for pathways, clicking on the pathway name will bring up an interactive Java applet that colors the pathway using a “heat map” derived from the input values (Figure 8C). The resulting pathway diagram can be exported as an image file (.png) by choosing “File -> Export image” from the Applet menu.
Supplementary Material
Acknowledgements
The authors would like to thank the Reference Proteome team, especially Craig McAnulla and Maria Martin, for their support to provide up-to-date Reference Proteome dataset, and Yukiko Matsuoka and Katoh Manami from the SBI Japan for their support on CellDesigner and pathway file update. This work is supported by NIH/NIGMS GM081084 to P.D.T.]. Funding for open access charge: University of Southern California.
Glossary
- BioPAX
Biological Pathway Exchange format
- BP
biological process or GO biological process
- CC
cellular component or GO cellular component
- cSNP
coding SNP
- HMM
Hidden Markov Model
- GO
gene ontology
- GWAS
genome-wide association study
- ID
identifier
- IP
internet protocol
- MF
molecular function or GO molecular function
- MSA
multiple sequence alignment
- NGS
next generation sequencing
- PANTHER
Protein Annotation through Evolutionary Relationship
- PC
protein class or PANTHER protein class
- PDGF
platelet-derived growth factor
- Png
portable network graphics
- SBGN
Systems Biology Graphical Notation
- SBGN-PD
Systems Biology Graphical Notation Process Description
- SBML
Systems Biology Markup Language
- SNP
single nucleoside polymorphism
- VEGF
vascular endothelial growth factor
Footnotes
Competing financial interests
The authors declare that they have no competing financial interests.
REFERENCES
- 1.Mi H, Muruganujan A & Thomas PD PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Research 41, D377–D386 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Venter JC, Adams MD, Myers EW, Li PW, et al. The sequence of the human genome. Science 291, 1304–1351 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Thomas PD, Campbell MJ, Kejariwal A, Mi H, et al. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13, 2129–2141 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas PD, Kejariwal A, Guo N, Mi H, et al. Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res 34, W645–W650 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mi H, Vandergriff J, Campbell M, Narechania A, et al. Assessment of genome-wide protein function classification for Drosophila melanogaster. Genome Res 13, 2118–2128 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mi H, Lazareva-Ulitsky B, Loo R, Kejariwal A, et al. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res 33, D284–D288 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mi H & Thomas P PANTHER pathway: an ontology-based pathway database coupled with data analysis tools. Methods Mol Biol 563, 123–140 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Funahashi A, Matsuoka Y, Jouraku A, Morohashi M, et al. CellDesigner 3.5: A Versatile Modeling Tool for Biochemical Networks. Proceedings of the IEEE 96, 1254–1265 (2008). [Google Scholar]
- 9.van Baarsen LGM, Bos WH, Rustenburg F, van der Pouw Kraan TCTM, et al. Gene expression profiling in autoantibody-positive patients with arthralgia predicts development of arthritis. Arthritis & Rheumatism 62, 694–704 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Verma G, Bhatia H & Datta M Gene expression profiling and pathway analysis identify the integrin signaling pathway to be altered by IL-1β in human pancreatic cancer cells: role of JNK. Cancer Lett 320, 86–95 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Boyer AP, Collier TS, Vidavsky I & Bose R Quantitative proteomics with siRNA screening identifies novel mechanisms of trastuzumab resistance in HER2 amplified breast cancers. Mol Cell Proteomics 12, 180–193 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stützer I, Selevsek N, Esterhazy D, Schmidt A, et al. Systematic proteomic analysis identifies beta-site amyloid precursor protein cleaving enzyme 2 and 1 (BACE2 and BACE1) substrates in pancreatic beta-cells. Journal of Biological Chemistry (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi Y, Zhao H, Shi Y, Cao Y, et al. Genome-wide association study identifies eight new risk loci for polycystic ovary syndrome. Nature Genetics 44, 1020–1025 (2012). [DOI] [PubMed] [Google Scholar]
- 14.den Hoed M, Eijgelsheim M, Esko T, Brundel BJJM, et al. Identification of heart rate–associated loci and their effects on cardiac conduction and rhythm disorders. Nature Genetics (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng J, Zhou Y, Campbell SL, Le T, et al. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci 13, 423–430 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hek K, Demirkan A, Lahti J, Terracciano A, et al. A Genome-Wide Association Study of Depressive Symptoms. Biological Psychiatry 73, 667–678 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neely GG, Kuba K, Cammarato A, Isobe K, et al. A Global In Vivo Drosophila RNAi Screen Identifies NOT3 as a Conserved Regulator of Heart Function. Cell 141, 142–153 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McDowall J & Hunter S InterPro protein classification. Methods Mol Biol 694, 37–47 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Gene Ontology Consortium The Gene Ontology: enhancements for 2011. Nucleic Acids Res 40, D559–D564 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mi H, Dong Q, Muruganujan A, Gaudet P, et al. PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res 38, D204–D210 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reference Genome Group of the Gene Ontology Consortium The Gene Ontology’s Reference Genome Project: a unified framework for functional annotation across species. PLoS Comput Biol 5, e1000431 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaudet P, Livstone MS, Lewis SE & Thomas PD Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Brief Bioinform 12, 449–462 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cerami EG, Gross BE, Demir E, Rodchenkov I, et al. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Research 39, D685–D690 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas PD GIGA: a simple, efficient algorithm for gene tree inference in the genomic age. BMC Bioinformatics 11, 312 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le Novere N BioModels Database - A Database of Annotated Published Models Available on the internet at: http://www.ebi.ac.uk/biomodels-main/static-pages.do?page=home (2011).
- 26.Hucka M, Finney A, Sauro HM, Bolouri H, et al. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics 19, 524–531 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Demir E, Cary MP, Paley S, Fukuda K, et al. The BioPAX community standard for pathway data sharing. Nat Biotech 28, 935–942 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cho RJ & Campbell MJ Transcription, genomes, function. Trends in Genetics 16, 409–415 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Clark AG, Glanowski S, Nielsen R, Thomas PD, et al. Inferring nonneutral evolution from human-chimp-mouse orthologous gene trios. Science 302, 1960–1963 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Mootha VK, Lepage P, Miller K, Bunkenborg J, et al. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci U S A 100, 605–610 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mann HB & Whitney DR On a Test of Whether one of Two Random Variables is Stochastically Larger than the Other. The Annals of Mathematical Statistics 18, 50–60 (1947). [Google Scholar]
- 32.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sherman B, Huang D, Tan Q, Guo Y, et al. DAVID Knowledgebase: a gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinformatics 8, 426 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.