

Abstract
Enzymes are complex biological catalysts and are critical to life. Most oxidations of chemicals are catalyzed by cytochrome P450 (P450, CYP) enzymes, which generally utilize mixed-function oxidase stoichiometry, utilizing pyridine nucleotides as electron donors: NAD(P)H + O2 + R → NAD(P)+ + RO + H2O (where R is a carbon substrate and RO is an oxidized product). The catalysis of oxidations is largely understood in the context of the heme iron-oxygen complex generally referred to as Compound I, formally FeO3+, whose basis was in peroxidase chemistry. Many X-ray crystal structures of P450s are now available (≥ 822 structures from ≥146 different P450s) and have helped in understanding catalytic specificity. In addition to hydroxylations, P450s catalyze more complex oxidations, including C-C bond formation and cleavage. Enzymes derived from P450s by directed evolution can even catalyze more unusual reactions, e.g. cyclopropanation. Current P450 questions under investigation include the potential role of the intermediate Compound 0 (formally FeIII-O2−) in catalysis of some reactions, the roles of high- and low-spin forms of Compound I, the mechanism of desaturation, the roles of open and closed structures of P450s in catalysis, the extent of processivity in multi-step oxidations, and the role of the accessory protein cytochrome b5. More global questions include exactly how structure drives function, prediction of catalysis, and roles of multiple protein conformations.
Keywords: Cytochrome P450, enzymology, cytochrome b5, Compound I, directed evolution, processivity, kinetics, oxidation
Graphical Abstract
Background and Introduction
Catalysts come in many forms, and enzymes are the catalysts in most biological reactions. Like all catalysts, they are not consumed in reactions. Enzymes can control regioselectivity and stereospecificity of reactions of complex organic molecules and, because they are chiral catalysts (i.e., built of L-amino acids), they can induce stereoselectivity into seemingly non-chiral molecules (e.g., distinguish between pro-R and pro-S hydrogens on a simple methylene group such as that in ethanol1,2). Unlike most other catalysts, enzymes are synthesized in biological systems, coded for by genes.
The functions of enzymes vary, and several types of enzymes catalyze oxidations and reductions.2 Of these, ≥ 95% of those reported in the literature are done by cytochrome P450 (P450, CYP) enzymes.3 Most of the P450 reactions are oxidations and involve the use of molecular oxygen, i.e. O2. P450s almost always act as monooxygenases, or mixed-function oxidases, using the stoichiometry shown in Equation 1 and utilizing the pyridine nucleotide NADH or NADPH as a cofactor (used to deliver electrons via a flavoprotein, sometimes via an iron-sulfur protein)
| (1) |
(where R is an organic substrate and RO is the reaction product), as opposed to the stoichiometry used in dioxygenases (Equation 2)
| (2) |
or (Equation 3)
| (3) |
where both oxygen atoms are incorporated into organic substrates.2,4–6
An important concept to remember in catalysis involving oxygen is that O2 is in the triplet state and its direct reaction with singlet organic molecules is spin-forbidden.5–7 Metalloproteins such as P450s overcome this barrier by complexing the oxygen to a metal (iron), so that the metal-oxygen complex can react with carbon substrates, as well as some heteroatoms and even metals.8
The first reports of P450 were published in 1962 and 1964.9–11 Before that time, the concepts of dioxygenases and mixed-function oxidases had been established by Hayaishi, Mason, and others.4,12,13 The concept of generating oxygenated compounds by activated oxygen species was in contrast to earlier views by Wieland and others that these alcohols and other oxygenated chemicals were generated from the activation of carbon and subsequent reaction with water.6,12–14 The connection between P450 and mixed-function oxidation (of a steroid) was made later by Cooper et al.15 using photochemical action spectra; i.e. light reversal of inhibition of a hydroxylation reaction by carbon monoxide.
Subsequent work with bacterial and mammalian systems led to the purification of many P450s. Early proposals about the generation of mobile oxygen species and their roles16,17 have been abandoned in favor of high-valent iron oxygen complexes that do the oxidation chemistry, overcoming the problem of spin-forbidden reaction of singlet (carbon) substrates with triplet oxygen and restricting the regioselectivity of the reaction to a specific site(s) on a substrate.2,5,6 The oxygenated forms of P450 enzymes have considerable similarity to those used by other iron-based monooxygenases and dioxygenases.6,18
Current Understanding
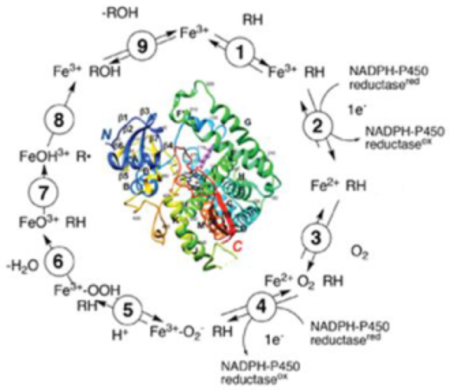
The catalytic cycle shown in Scheme 1 is generally accepted for most P450- catalyzed oxidations. The intermediates between steps 4 and 8 are unstable and have been difficult to characterize. There is some ambiguity in the exact location of the electronic charge in these entities, and variations are seen in the literature, i.e. in the distribution of charge in the iron atom, the Fe-O bond, and the porphyrin ring.
Scheme 1.

General catalytic cycle for P450 reactions.6,19 In some cases cytochrome b5 (b5) can donate the electron in step 4. b5 can also donate the electron in step 2 although not as efficiently because of the thermodynamic barrier. In some P450s (some bacterial and mitochondrial P450s) steps 2 and 4 involve electron donation from ferredoxin proteins. The exact electronic distribution in the Fe-O entities between steps 4–8 is not well established in most cases.
Most P450 oxidations are considered to occur via Compound I, the formal FeO3+ entity shown in Scheme 1 (i.e., steps 7 and 8), named because of its history in peroxidase chemistry.20 Much of our understanding of the catalysis came from work on biomimetic models; i.e. model metalloporphyrin oxide complexes.21,22 Compound I has been characterized in some bacterial P450s by Green and associates23–25 and by Makris and associates.26 Compound I abstracts a hydrogen atom from an alkyl group (e.g., methyl, methylene) yielding a carbon radical (step 7 in Scheme 1). The formal Fe-OH complex reacts rapidly with this radical to generate the product, often an alcohol, in a so-called “oxygen rebound” step (step 8 in Scheme 1). In cryo-annealing/γ-ray reduction studies by Hoffman and Sligar, it has been possible to characterize dioxygen intermediates of P450 101A1 (P450cam) and P450 11A1 by ENDDR spectroscopy and to document catalytic competence as judged by product formation.27–29
At this point it should be mentioned that many X-ray crystal structures are now available (≥ 822 structures for ≥ 146 different P450s currently in the Protein Data Bank, with 112 uploaded but not yet released as of the date of revision of this manuscript). These have generally similar folds but the size and shape of the active site vary considerably, yielding catalytic specificity among P450s.30 Most of the structures are of ferric enzymes, many with substrates or inhibitors present. Some structures of ferrous and ferrous-oxygen complexes of bacterial P450s 101A1 (P450cam) and 107A1 (P450eryF) are available (e.g., PDB 1DZ8, 1T88, 2A1M, 2A1N, 2A1O, 1Z8O, 1Z8P, 1Z8Q)30–33
Not all P450 reactions are carbon hydroxylations, but Compound I is involved in these as well. Compound I has a high oxidation-reduction potential and, when in the proper position, can abstract electrons from amines34–36 and other relatively low potential reductants, e.g. certain strained alkanes and substituted aromatic ring systems.37,38 The abstraction of an electron from an amine is the initial step in many N-dealkylation reactions.19,34,39,40 With some of these amines and also carbon substrates, the carbon radical generated in step 7 (Scheme 1) can rearrange before step 8 occurs,41 even though the rebound reaction is generally fast. This competition between oxygen rebound and rearrangement can be “clocked” with strained alkanes having known rates of rearrangement in solution.42,43 Other competing reactions with Compound I are reduction to water44 and abstraction of a phenolic hydrogen atom from a (P450) tyrosyl group, which confounded early efforts to characterize Compound I.25,45–48
For more extensive lists of rearrangements and more unusual oxidation reactions catalyzed by P450s, see references 6, 8, 19, 49, 50. Most of these can be rationalized by mechanisms involving Compound I, with Compound 0 (Scheme 1) possibly involved in others.
Current Questions
Compound I vs. Compound 0.
Compound 0 (formally FeIII-O2− or its protonated form) has been invoked in some P450 reactions that have been difficult to explain with Compound I.8,19 The difficulty in discriminating between Compound 0 and Compound I derives from instability of these species and the transformation of Compound 0 to Compound I in the catalytic cycle (Scheme 1). Any effort to discern these by site-directed mutagenesis of a P450 has the caveat that other aspects (not only proton transfer) have been modified. Compound I is an inherently electrophilic species and (unprotonated) Compound 0 is nucleophilic. The use of “oxygen surrogates” can be useful, with appropriate caveats, e.g. iodosylbenzene (and alkyl hydroperoxides) can generate Compound I (or a closely-related structure51,52) but cannot produce the dioxygenated complex Compound 0 (Scheme 1).
One of the historically dominant pieces of evidence supporting a role for Compound 0 was 18O2 labeling work with P450 19A1, in which one atom of 18O was reported to be incorporated in the product formic acid in the third and final step of the steroid aromatase reaction.53–55 Conclusions about the mechanism were extrapolated from this result to other P450 reactions.56 However, when the original experiments with P450 19A1 were repeated under more technically sophisticated conditions, no 18O incorporation was observed and the results are only consistent with a role for Compound I and not Compound 0.6,57
Another P450 C-C cleavage reaction, the periodate-like cleavage of 20,22- dihydroxycholesterol to two carbonyl products, is done by P450 11A1 Compound I, as demonstrated by a relatively high yield (~ 50%) of product formed by radiolytically-generated Compound I.29
The case of the P450 17A1-catalyzed cleavage of 17α-hydroxy steroids (“lyase” reaction) is more problematic, in that the documented incorporation of one atom of 18O58 has been repeated52 but this result is not unambiguous regarding the mechanism. Raman spectral evidence for a Compound 0 species has been generated in the presence of substrate but this entity has not been shown to be catalytically competent (i.e., converted to product).59–61 lodosylbenzene and 17α-hydroperoxy steroids (oxygen surrogates) both yield the 19-carbon lyase reaction products,52,62 but the involvement of Compound 0 in the normal reaction cannot be ruled out.6
The purported roles of Compound 0 in other reactions remain to be investigated, e.g. lanosterol 14 α-demethylation (P450 51 species)63 and the cleavage of nabumetone.64,65
Multiple States of Compound I.
Theoretical calculations, primarily by Shaik and his associates, have largely discounted all potential oxygenated intermediates other than Compound I in the catalytic cycle (Scheme 1) as unable to be involved in P450 oxidations, including Compound 0 and Compound II (formally FeOH3+, following step 7 in Scheme 1).66 Theory has also led to the concept of “two-state reactivity,” i.e. the view that different forms of Compound I are involved in different reactions, even with a single substrate. This subject (which is too complex to fully consider here) is often approached in the context of “high-spin” and “low-spin” states of Compound I and their reactivity, but the possibilities for electronic distribution and multiple forms of Compound I are more complex. Shaik and associates use the term “chameleon nature” of Compound I, meaning that Compound I can change its character depending upon its environment. That is, the juxtaposition of a substrate in the enzyme can shift the balance of states of Compound I and affect the mechanism.66
The concept has attraction in explaining multiple reaction courses. For instance, epoxidation of aryl compounds is observed along with formation of phenols and ketones, but the epoxide does not necessarily form the products nor does a Meissenheimer complex necessariily form epoxides. Epoxidation of olefins is often accompanied by 1,2- shifts indicative of cationic intermediates (plus heme destruction).67–69 These sets of reactions have often been attributed to a single P450 intermediate that collapses in different ways (Scheme 2A)8,70 but the chameleon theory would suggest different electronic distributions of Compound I participating in formation of different products (Scheme 2B).
Scheme 2.

An example of the differences between explaining multiple reaction products by (A) a common intermediate, with variations due to the protein,8,67 and (B)”two-state” (actually multistate) theory.66.
This concept sounds very reasonable but unfortunately it is not very possible to prove experimentally (unless one considers more theoretical work experimental). The theoretical considerations are based largely on rationalization of prior experimental data, and development of new predictions that could distinguish among alternate mechanisms is difficult. In principle, the barrier to one reaction could be raised and the result observed, in terms of product redistribution (Scheme 2). However, the result could be explained by either (i) shifting the positioning of the substrate in the active site to favor one reaction (due to substrate motion) or (ii) to the ability of alternate forms of Compound I to do the reaction (at the same site in the protein). Although bond strength is one issue in influencing product reactivity (e.g., the preference of allylic sites for hydroxylation71), it is not the only one and examples of deficiencies in prediction are considered later (vide infra,).72
In the author’s opinion, these issues may ultimately be addressed experimentally in studies with artificially-generated Compound I (vide supra), in which detailed spectral analysis and product measurements are done.23–26,29
Open vs. Closed Structures.
More crystal structures (≥ 119) of bacterial P450 101A1 (P450cam) are available than for any other P450. These structures, with the substrate camphor and other ligands (e.g., PDB 2CPP, 7CPP, 1PHA, 1PHB, 1PHC, 5IK1), can be grouped into three conformational states: open, closed, and intermediate.30,73–76 The states involve differential movements of the F and G helices that cover the substrate binding channel. The studies support a view that this protein exists in a small set of distinct conformations, rather than a more continuous distribution of malleable, induced-fit states.76 How this paradigm applies to mammalian and other eukaryotic P450s is unknown, although there is considerable evidence that those enzymes also have at least open and closed conformations.30
One issue is the effect of the auxiliary protein putidaredoxin on the structure of P450cam. Putidaredoxin has long been known to be the immediate electron donor to P450cam,77 but it has also been known for many years to have an additional “effector” role in catalysis, in which it enhances product formation in a role separate from electron delivery.78 Although this role has been known since 1972,78 the exact mechanism has remained unclear. The general consensus is that oxidized putidaredoxin remains bound to the reduced P450 Fe2+O2 (or Fe3+-O2−) complex long enough to alter its conformation and facilitate steps 5–8 of the catalytic cycle (Scheme 1). Several structural studies have provided contrasting answers to how this happens. A crystallographic study with a crosslinked P450cam-putidaredoxin complex indicated that P450cam was in the open form (with product bound),79 as did an independent double-electron-electron resonance (DEER) study with spin labels attached to P450cam.80 However, an NMR complex yielded an opposite result, i.e. a closed complex with P450cam.81
These studies leave questions open about the catalytic mechanism, and the exact relevance to most eukaryotic P450s is unknown. Further DEER studies on P450cam with different spin labels also show that oxidized putidaredoxin induces reduced P450cam to change to the open form.76 Carbon monoxide binding somehow prevents this shift. Further DEER measurements have indicated that putidaredoxin binds to the same site of P450cam in both the open and closed forms.82 Apparently this binding triggers the conformational change through rather subtle structural interactions.82 Exactly how this information is relevant to the human P450 redox partner interactions is still unclear (i.e., NADPH-P450 reductase and cytochrome b5 (b5) in the microsomal P450s and adrenodoxin in the mitochondrial P450s).
Desaturation.
Numerous examples of P450-catalyzed desaturations are known, and the process does not involve dehydration of an alcohol product.8 In many cases, the desaturated product is minor relative to the alcohol, consistent with the view that a common intermediate (step 7, Scheme 1) leads to both.83–85 In several cases,86–90 including fish and human P450s,91,92 the major or only product is the desaturated product.
Presumably the reaction involves the abstraction of a second hydrogen atom in the FeOH3+ radical intermediate (Schemes 1, 3). Whether this is actually a hydrogen atom abstraction or proton-coupled electron transfer is not clear. Site-directed mutagenesis work with (non-heme) iron-based fatty acid desaturases and the iron/α- ketoglutarate dioxygenase AsqJ indicates that the balance between desaturation and hydroxylation is rather sensitive to minor changes,86,93–95 and P450s are probably similar in this regard (Scheme 3).
Scheme 3.

Potential desaturation mechanisms for oxygenases. (A) P450s; (B) non-heme diiron enzymes; (C) iron/α-ketoglutarate (α-KG) dioxygenases.93
Processivity.
Many P450 reactions involve multiple steps, i.e. a product of one reaction is a substrate for a subsequent reaction by the same enzyme. An issue is the degree to which the two reactions are “processive,” i.e. the extent to which the first product is released by the enzyme and must re-bind to generate the final product. If a reaction is not processive, it is “dissociative” (Scheme 4). The balance is not simply a matter of kinetic curiosity, but the coupling of reactions (i.e., degree of processivity) is an indication of whether a biological process is deterministic or stochastic. A more deterministic sequence of reactions may be important, in terms of biological function. The nature of the processivity can also be an issue when considering development of enzyme inhibitors.96,97
Scheme 4.

Processivity in multistep reactions. The enzyme (E) catalyzes the conversion of A → B → C, e.g, P450 17A1.97 The degree of processivity is dominated by the ratio k4 k-3, although other rate constants can contribute.104
This issue has been investigated in several cases involving steroid oxidations.97–102 The design of such experiments is critical, and pre-steady-state kinetic approaches are necessary.103 Our own work led to the conclusion that the three steps in the P450 19A1 steroid aromatase reaction are distributive.102 Studies on animal and human P450 17A1 reactions (steroid 17α-hydroxylation and subsequent C17 α,20 cleavage (lyase reaction)) indicate partial processivity,97–101 although only in the case of the human P450 17A1 studies was the highly stimulatory protein b5 included.97
Pulse-chase experiments indicated that the P450 2E1-catalyzed oxidations of ethanol and N,N -diethylnitrosamine to acetic acid are both processive, with the latter being less processive than the oxidation of N,N-dimethylnitrosamine to formic acid (Scheme 4).105,106 P450 2A6 also showed processivity in the same oxidation of the above two nitrosamines to 1- and 2-carbon carboxylic acids.107 However, in these reactions the results were not fit to quantitative models with rate constants for both product dissociation and the second oxidation step. These situations have a dilemma in that there was no evidence for high-affinity binding of acetaldehyde or formaldehyde.
Other P450s known to catalyze sequential oxidations with endogenous chemicals include 11A1, 11B1, 11B2, 24A1, 27A1, 46A1, and 51A1. Human P450s catalyzing sequential reactions with xenobiotics include 1A1, 1A2, 1B1, 2A6, 2A13, 2C9, 2E1, and 3A4.
Role of b5.
b5 is a microsomal heme protein that has an electron transfer role in fatty acid desaturation and some other enzymatic reactions.2 In 1971, Estabrook and Hildebrandt108 postulated that b5 could be involved in P450 reactions in rat liver microsomes, on the basis of results of stimulation with the cofactor NADH, which only interacts poorly with NADPH-P450 reductase to reduce P450s but reduces NADH- b5 reductase and then b5. With the purification and reconstitution of P450s and b5, a number of results have been reported. Depending upon the system, investigators have reported either no effect, stimulation, or inhibition with the addition of b5.109–112 Even with a given P450, the results can vary depending upon the reaction under consideration.110,113
The original postulate was that b5 is transferring the second electron (to the Fe2+O2 (or Fe3+−O2−) complex, step 4 in Scheme 1).108 However, with several P450s either apo-b5 (devoid of heme) or Mn2+-substituted b5 can stimulate as well as holo-b5 (with the normal heme).114–117 A proposal that heme is transferred from the P450s to the apo-b5 during the course of enzyme reactions118 was shown to be untenable.119
At least four mechanisms for b5 stimulation have been proposed: (i) electron transfer (to the Fe2+O2 (or Fe3+−O2−) complex, step 4, Scheme 1); (ii) an allosteric effect that favors a more active conformation of the P450s; (iii) an effect of decreasing the abortive use of electrons to generate H2O2, avoiding uncoupling; and (iv) facilitating the protonation of the hydroperoxo intermediate (step 5 in Scheme 1). Waskell and colleagues have provided EPR evidence for the latter proposal in the case of rabbit P450 2B4-catalyzed d-benzphetamine N-demethylation,120 although this reaction is not strongly influenced by the presence of b5 under all conditions.110 Enhanced protonation of the peroxo complex would actually be inhibitory to the strong stimulation of the lyase activity of the P450 17A1 if the proposed nucleophilic Compound 0 mechanism58–61 is viable, in that decomposition to Compound I would be facilitated. Apo-b5 can stimulate reactions as well as b5 with several P450s114,117,119 but is ineffective with some P450 reactions,117,121–124 thus implicating electron transfer in those.
There are several experimental deficiencies in the b5 field. Clearly b5 must bind to P450s, as shown by NMR perturbation125 and crosslinking126 studies, but estimates of binding parameters have only been made in two cases, and then by surface plasmon resonance,127,128 which can be notoriously inaccurate due to surface artifacts.104 No crystal structures of b5:P450 complexes are known (other than computational). Also, kinetic investigations with b5 and high-valent P450 intermediates are limited because b5 must be preincubated with P450 in vesicles (i.e., unlike a ferredoxin it cannot be added quickly in a stopped-flow experiment). NMR investigations by Estrada and Scott125 also indicate that b5 and NADPH-P450 reductase compete for binding at the same site of P450 17A1, precluding some experimental kinetic designs (see also ref. 129).
To add to the complexity are in vivo transgenic mouse experiments.124,130 Conditional deletion of microsomal b5 in liver creates a liver b5-null mouse, but these mice develop and breed normally and have no overt phenotype. In liver microsomes prepared from these animals, NADH-mediated metabolism was essentially abolished for most substrates, and the NADPH-dependent metabolism of many substrates was reduced by 50–90%131 (the NADH-b5 reductase/b5-supported reduction of P450s can be reconstituted in vitro, despite the unfavorable redox potential of b5 for reducing ferric P450s (step 2 in Scheme 1)123,132). The results indicate that microsomal b5 can play a major role in the in vivo metabolism of certain drugs and chemicals but in a rather P450- and substrate-dependent manner.130 The extent to which metabolism was significantly affected by the (hepatic) absence of b5 is substrate-dependent.131 In mice treated with the carcinogen benzo[a]pyrene, in vivo DNA adduct levels were significantly higher (7-fold) in the livers of (liver) NADPH-P450 reductase knockout mice than wild-type mice.124 In the same study, no significant difference in DNA adduct formation was observed in liver between NADH-b5 reductase-null and wild-type mice. Thus, NADPH- P450 reductase and b5 both appear to modulate P450-mediated activation of benzo[a]pyrene in vitro but hepatic P450 enzymes appear to be more important for benzo[a]pyrene detoxication than its activation in vivo.
What are the Limits of P450 Catalysis?
Recent work by Arnold and associates133–137 and also by the Fasan group138–145 has pushed the limits of what P450s—and other heme proteins—can do in the realm of catalysis. Arnold’s work in directed evolution, which was recognized by a share of the 2018 Nobel Prize in Chemistry, includes ≥ 47 papers with P450s and their derivatives.
This area developed largely from work with bacterial P450 102A1 (P450bm3), which has advantages over most other P450s in that it is self-sufficient (i.e., it has a reductase domain that binds NADPH and delivers electrons to the heme domain, eliminating the need to co-express a separate reductase), high catalytic activity towards fatty acids (turnover number ~ 200 s−1), and high expression levels.146 Directed evolution (or, more appropriately “molecular breeding”), with high throughput selection methods, was used to develop modified P450 102A1 catalysts that can catalyze various transformations of interest.137 Some of the reactions involve drugs, and batteries of mutated P450 102A1 catalysts can be used to screen for their abilities to synthesize drug metabolites and new lead molecules.147
Further studies led to the ability to catalyze propane 1-hydroxylation, a dramatic departure from the classical ω−1, ω−2, … hydroxylation of fatty acids.148 Another development was the synthesis of anti-Markovnikov carbonyl products from styrenes by selected P450 102A1 mutants (Scheme 5A),135 although it should be noted that such reactions had been demonstrated earlier with rat P450 2B168 and with P450 biomimetic models.149
Scheme 5.

Some unusual reactions catalyzed by modified P450 catalysts.135,144,145 Ts: tosyl.
Further molecular breeding, incorporating random and site-directed mutagenesis and unnatural amino acid mutagenesis, has yielded P450-derived enzymes that can catalyze unusual reactions, e.g. cyclopropanation and nitrene transfer (Scheme 5B,5C).133,134,143 These reactions do not involve the normal P450 chemistry (Scheme 1), and Arnold’s group found that substitution of the conserved cysteine heme ligand by serine led to higher activity.137 These catalysts are no longer P450s, by definition (no thiolate ligand or classical FeII-CO spectrum) and have been termed P411 by Arnold.137,150 Nevertheless, some of these P411 variants are capable of highly stereoselective catalysis.134
The nitrene transfer mechanism (Scheme 6),144 with roots in earlier work by White151 and Dawson and Breslow,152 is rather unrelated to the normal catalytic cycle (Scheme 1). Consistent with the P411 catalysis, these reactions can also be catalyzed by myoglobin derivatives.140
Scheme 6.

A proposed mechanism of nitrene transfer for P450s.144
Further Unresolved Issues
Exactly How Does Structure Drive Function?
At least 822 P450 structures (from > 146 P450s) are available in the Protein Data Bank, and a large fraction contain bound ligands. In many cases, the atom(s) on the substrate that is oxidized is positioned closest to the heme iron and, presumably, the oxygen of the FeO3+ complex (Compound I). Such structures can explain many P450 reactions, and it is certainly easier to understand reactions with enzyme structures than without any.
However, there are some gaps in the field, aside from the difficulty in crystallizing some of the P450s. As pointed out earlier, there is a paucity of structures of complexes with accessory proteins. Examples are with ferredoxin-P450 fusion proteins, one a mitochondrial P450 (11A1)153 (PDB 3N9Y, 3NA1, 3N9Z, 3NA0, 4JWS, 4JWU, 4JX1, 3W9C) and two others with bacterial P450 101A1.79,154 The only other case involves the separated heme and flavin domains of bacterial P450 102A1 (1BVY).155
Another issue is that the structures can explain major sites of reaction, but structures explaining minor products are not generally seen due to the energetic differences in binding in the less favorable conformations. In this regard, the computational prediction software may even be more generally useful than structural models.
Another issue is that there are some examples of explaining natural human (or other) genetic variants at a structural level but not many (e.g., PDB 5X23, 5X24).156 For instance, it would be interesting (and useful) to understand why the human P450 2D6*53 variant has higher activity than wild-type P450 2D6.157 Two recent examples from our own group exemplify the problems. Zebrafish P450 17A1 and P450 17A2 both catalyze steroid 17α-hydroxylations (progesterone and pregnenolone) but only P450 17A1 will catalyze the second (lyase) reaction, i.e., cleaving the C17-C20 bond (vide supra).158 However, the X-ray structure showed nearly identical active sites,158 and site-directed mutagenesis of all five possible perturbations (four near the heme periphery) did not impart lyase activity to P450 17A2.62 In the case of human P450 21A2, > 100 single amino acid substitutions have been reported, with clinical deficiencies. Although having the structure of wild-type P450 21A2 is useful in understanding the low-activity variants,159–161 we have been unsuccessful in obtaining diffractable crystals of any of the variants.
The difficulty of relating structure to function can be appreciated by considering the implications of the Eyring equation:
| (4) |
, in which a difference of 1.3 kcal mol−1, less than a typical single hydrogen bond, translates to a 10-fold difference in reaction rate at 37 °C (Kb is the Boltzmann constant, h is Planck’s constant, and T is the absolute temperature). Further, a free energy difference of 6.4 kcal mol−1 is associated with a 50,000-fold change in the rate. Thus, small differences in structure (binding) can lead to major changes in catalysis. This, of course, is an issue not only for P450s but for all efforts at understanding enzyme structure-function relationships.
Prediction of Catalysis.
As mentioned earlier, structural models have the capability of understanding regioselectivity of oxidation reactions and even predicting sites with new ligands. The experience to date with AutoDock programs162,163 is not better than with programs such as MetaSite164–166 that are based on past history with similar molecules, in the absence of structural constraints. Success rates of ~ 80% in predicting the top three sites of oxidation are reasonable and competitive with predictions by trained drug metabolism experts.167,168
There are still anomalies that would not be predicted. For instance, testosterone and the important androgenic derivative dihydrotestosterone differ only in a double bond and twists in the (steroid) A-ring. However, P450 3A4 hydroxylates testosterone at the 6β position (plus lesser amounts of hydroxylation at the 2β, 1β, and 15β positions)71,169 but dihydrotestosterone is hydroxylated primarily at the thermodynamically unfavored C18 methyl group.72
Another deficiency, especially in the field of drug metabolism, is that we do not really have the ability to predict rates of oxidation of compounds, even when we can predict sites. This can realistically only be done in situations where Hammett analysis (with substituted aryl groups)170,171 or some other well-defined linear free energy or another quantitative structural-activity paradigm can be applied. Beyond such cases we have limited predictably with new substrates, as well as with predicting the effects of most single-amino acid substitutions in P450s. As noted above, the problem can be appreciated when the Erying relationship is considered.
Multiple Conformations.
One of the reasons why structural predictions are not better is the presence of multiple conformations of P450 proteins. There is considerable evidence for their existence, both from the structures of proteins bound to different ligands30,96,172 and from the existence of multiple crosspeaks in NMR spectra.173 In principle, NMR methods would be more useful in revealing multiple conformations but solving structures is formidable for such large proteins. Through-space spectroscopic methods such as fluorescence and EPR tagging are qualitatively useful but also deficient in revealing total individual structures (and necessarily require perturbation of the protein). Molecular dynamics simulations, although more theoretical than experimental, can also address questions about multiple configurations.
The existence of multiple conformations of P450s, or at the very least multiple docking models, is self-evident in considering multiplicity of reaction products from a single enzyme and substrate. There is also extensive evidence for substrate-induced conformational changes upon binding, as clearly evidenced in the crystal structures30 (and NMR measurements125,173).
One question is whether these changes involve induced fit or conformational selection models (Scheme 7), a general issue in enzymology.174 In the induced fit model, the binding of the substrate leads to a conformation change. In contrast, in the conformational selection model, multiple states of the unbound protein coexist, one of which has substrate complementarity and binds (Scheme 7). Which of the two mechanisms is dominant with P450s is unclear, although some of our own kinetic studies with P450 3A4 led to a preference for an induced fit model.175 The two phenomena are not mutually exclusive, and we have utilized two forms of both free and bound P450 to obtain fits of ligand binding kinetic data.97,102,175,176 However, the improved fitting seen with adding more steps may be misleading, in that the most minimal kinetic models are generally the safest even if not the most complete.103
Scheme 7.

Induced fit vs. conformational selection hypotheses to explain multiplicity of enzyme conformations involved in catalysis.
We have explored the effects of substrate binding on circular dichroism spectra. However, any changes seen are probably too weak to be useful, in that changes in helicity are needed.
Another anomaly of substrate binding is the slow changes in heme perturbation spectra seen with some P450s. Bacterial P450 101A1 binds its substrate (camphor) rapidly (k= 106 M−1s−1)177 and so do several human P450s.102,121,122,159 However, under the same conditions several mammalian P450s show slow changes in heme perturbation spectra upon mixing with substrate.92,175,176,178,179 In some cases the rates of initial contact of the substrate/ligand with the P450 has been shown to be fast, as judged by fluorescence kinetics of interaction of ligands with the protein.102,175,179 The slow heme perturbation results are currently rationalized in models that involve diffusion- limited encounters of a substrate with a ligand followed by conformational changes and “worming” of the ligand into the active site, moving near the heme iron.179 However, it is presently unclear why some P450s exhibit such behavior and others do not, as well as whether an induced fit or a conformational selection model is more relevant.
Conclusions
P450 is a mature field, in that the basic catalytic mechanism is understood in the context of electronic changes leading to oxygen activation and the oxidation of a substrate (Scheme 1). Questions still exist about some of the electronics of Compound I and possibly other active species. More questions abound about the role of the protein structure in driving catalytic selectivity, including internal motion and conformational changes. Prediction of altered catalytic activity due to small changes is problematic. Finally, there is a paucity of detailed information about interactions with accessory proteins.
Acknowledgments.
The author thanks Drs. F. K. Yoshimoto and M. J. Reddish for helpful comments and K. Trisler for assistance in preparation of the manuscript.
(Grant support). This work was supported in part by National Institutes of Health Grant R01 GM118122.
Footnotes
Conflict of Interest. The author declares no conflict of interest.
References
- (1).Fisher HF; Conn EE; Vennesland B; Westheimer FH The Enzymatic Transfer of Hydrogen. I. The Reaction Catalyzed by Alcohol Dehydrogenase. J Biol. Chem 1953, 202, 687–697. [PubMed] [Google Scholar]
- (2).Walsh C Enzymatic Reaction Mechanisms, W. H. Freeman Co: San Francisco, 1979. [Google Scholar]
- (3).Rendic S; Guengerich FP Survey of Human Oxidoreductases and Cytochrome P450 Enzymes Involved in the Metabolism of Xenobiotic and Natural Chemicals. Chem. Res. Toxicol 2015, 28, 38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hayaishi O; Katagiri M; Rothberg S Studies on Oxygenases; Pyrocatechase. J Bio. Chem 1957, 229, 905–920. [PubMed] [Google Scholar]
- (5).Bugg TDH Dioxygenase Enzymes: Catalytic Mechanisms and Chemical Models. Tetrahedron 2003, 59, 7075–7101. [Google Scholar]
- (6).Guengerich FP; Yoshimoto FK Formation and Cleavage of C-C Bonds by Enzymatic Oxidation-Reduction Reactions. Chem. Rev 2018, 118, 6573–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hernandez-Ortega A; Quesne MG; Bui S; Heyes DJ; Steiner RA; Scrutton NS; de Visser SP Catalytic Mechanism of Cofactor-Free Dioxygenases and How They Circumvent Spin-Forbidden Oxygenation of Their Substrates. J Am. Chem. Soc 2015, 137, 7474–7487. [DOI] [PubMed] [Google Scholar]
- (8).Guengerich FP Common and Uncommon Cytochrome P450 Reactions Related to Metabolism and Chemical Toxicity. Chem. Res. Toxicol 2001, 14, 611–650. [DOI] [PubMed] [Google Scholar]
- (9).Omura T; Sato R A New Cytochrome in Liver Microsomes. J Bio. Chem 1962, 237; 1375–1376. [PubMed] [Google Scholar]
- (10).Omura T; Sato R The Carbon Monoxide-Binding Pigment of Liver Microsomes.Evidence for Its Hemoprotein Nature. J Bio. Chem 1964, 239, 2370–2378. [PubMed] [Google Scholar]
- (11).Omura T; Sato R The Carbon Monoxide-Binding Pigment of Liver Microsomes. Solubilization, Purification, and Properties. J Biol. Chem 1964, 239, 2379–2385. [PubMed] [Google Scholar]
- (12).Hayaishi O; Hashimoto K Pyrocatecase. A New Enzyme Catalizing Oxidative Breakdown of Pyrocatechin. J Biochem. (Tokyo) 1950, 37, 371–374. [Google Scholar]
- (13).Mason HS Mechanisms of Oxygen Metabolism. Science 1957, 125, 1185–1190. [DOI] [PubMed] [Google Scholar]
- (14).Witkop B Remembering Heinrich Wieland (1877–1957). Portrait of an Organic Chemist and Founder of Modern Biochemistry. Med. Res. Rev 1992, 12, 195–274. [DOI] [PubMed] [Google Scholar]
- (15).Cooper DY; Levine S; Narasimhulu S; Rosenthal O; Estabrook RW Photochemical Action Spectrum of the Terminal Oxidase of Mixed Function Oxidase Systems. Science 1965, 147, 400–402. [DOI] [PubMed] [Google Scholar]
- (16).Hamilton GA Oxidation by Molecular Oxygen. I. Reactions of a Possible Model System for Mixed-Function Oxidases. J Am. Chem. Soc 1964, 86, 3391–3392. [Google Scholar]
- (17).Strobel HW; Coon MJ Effect of Superoxide Generation and Dismutation on Hydroxylation Reactions Catalyzed by Liver Microsomal Cytochrome P-450. J Biol. Chem 1971, 246, 7826–7829. [PubMed] [Google Scholar]
- (18).Martinez S; Hausinger RP Catalytic Mechanisms of FeII- and 2-Oxoglutarate- Dependent Oxygenases. J Bio. Chem 2015, 290, 20702–20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ortiz de Montellano PR Substrate Oxidation. In Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th ed; Ortiz de Montellano PR, Ed.; Springer: New York, 2015, pp 111–176. [Google Scholar]
- (20).Theorell H; Ehrenberg A; Chance B Electronic Structure of the Peroxidase- Peroxide Complexes. Arch. Biochem. Biophys 1952, 37, 237–239. [DOI] [PubMed] [Google Scholar]
- (21).Groves JT; Nemo TE; Myers RS Hydroxylation and Epoxidation Catalyzed by Iron-Porphine Complexes. Oxygen Transfer from Iodosylbenzene. J Am. Chem. Soc 1979, 101, 1032–1033. [Google Scholar]
- (22).Groves JT Models and Mechanisms of Cytochrome P450 Action. In Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd ed; Ortiz de Montellano PR, Ed.; Kluwer Academic/Plenum Publishers: New York, 2005, pp 1–43. [Google Scholar]
- (23).Yosca TH; Rittle J; Krest CM; Onderko EL; Silakov A; Calixto JC; Behan RK; Green MT Iron(IV) hydroxide pKa and the Role of Thiolate Ligation in C-H Oxidation by Cytochrome P450. Scene 2013, 342, 825–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Krest CM; Onderko EL; Yosca TH; Calixto JC; Karp RF; Livada J; Rittle J; Green MT Reactive Intermediates in Cytochrome P450 Catalysis. J Biol. Chem 2013, 288, 17074–17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Rittle J; Green MT Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics. Science 2010, 330, 933–937.d [DOI] [PubMed] [Google Scholar]
- (26).Grant JL; Hsieh CH; Makris TM Decarboxylation of Fatty Acids to Terminal Alkenes by Cytochrome P450 Compound I. J Am. Chem. Soc 2015, 137, 4940–4943. [DOI] [PubMed] [Google Scholar]
- (27).Davydov R; Makris TM; Kofman V; Werst DE; Sligar SG; Hoffman BM Hydroxylation of Camphor by Reduced Oxy-Cytochrome P450cam: Mechanistic Implications of EPR and ENDOR Studies of Catalytic Intermediates in Native and Mutant Enzymes. J Am. Chem. Soc 2001, 123, 1403–1415. [DOI] [PubMed] [Google Scholar]
- (28).Davydov R; Gilep AA; Strushkevich NV; Usanov SA; Hoffman BM Compound I Is the Reactive Intermediate in the First Monooxygenation Step during Conversion of Cholesterol to Pregnenolone by Cytochrome P450scc: EPR/ENDOR/Cryoreduction/Annealing Studies. J Am. Chem. Soc 2012, 134, 17149–17156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Davydov R; Strushkevich N; Smil D; Yantsevich A; Gilep A; Usanov S; Hoffman BM Evidence That Compound I Is the Active Species in Both the Hydroxylase and Lyase Steps by which P450scc Converts Cholesterol to Pregnenolone: EPR/ENDOR/Cryoreduction/Annealing Studies. Biochemistry 2015, 54, 7089–7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Poulos TL; Johnson EF Structures of Cytochrome P450 Enzymes. In Cytochrome P450: Structure, Function, and Biochemistry, Ortiz de Montellano PR, Ed.; Springer: New York, 2015, pp 3–32. [Google Scholar]
- (31).Schlichting I; Berendzen J; Chu K; Stock AM; Maves SA; Benson DE; Sweet BM; Ringe D; Petsko GA; Sligar SG The Catalytic Pathway of Cytochrome P450cam at Atomic Resolution. Science 2000, 287, 1615–1622. [DOI] [PubMed] [Google Scholar]
- (32).Nagano S; Cupp-Vickery JR; Poulos TL Crystal Structures of the Ferrous Dioxygen Complex of Wild-Type Cytochrome P450eryF and Its Mutants, A245S and A245T: Investigation of the Proton Transfer System in P450eryF. J Bioi. Chem 2005, 280, 22102–22107.d [DOI] [PubMed] [Google Scholar]
- (33).Nagano S; Poulos TL Crystallographic Study on the Dioxygen Complex of Wild-type and Mutant Cytochrome P450cam. Implications for the Dioxygen Activation Mechanism. J Bioi. Chem 2005, 280, 31659–31663. [DOI] [PubMed] [Google Scholar]
- (34).Augusto O; Beilan HS; Ortiz de Montellano PR The Catalytic Mechanism of Cytochrome P-450: Spin-Trapping Evidence for One-Electron Substrate Oxidation. J Biol. Chem 1982, 257, 11288–11295. [PubMed] [Google Scholar]
- (35).Macdonald TL; Zirvi K; Burka LT; Peyman P; Guengerich FP Mechanism of Cytochrome P-450 Inhibition by Cyclopropylamines. J Am. Chem. Soc 1982, 104, 2050–2052. [Google Scholar]
- (36).Guengerich FP; Yun CH; Macdonald TL Evidence for a 1-Electron Oxidation Mechanism in A-Dealkylation of N,N-Dialkylanilines by Cytochrome P450 2B1. Kinetic Hydrogen Isotope Effects, Linear Free Energy Relationships, Comparisons with Horseradish Peroxidase, and Studies with Oxygen Surrogates. J Biol. Chem 1996, 271, 27321–27329. [DOI] [PubMed] [Google Scholar]
- (37).Stearns RA; Ortiz de Montellano PR Cytochrome P-450 Catalyzed Oxidation of Quadricyclane. Evidence for a Radical Cation Intermediate. J Am. Chem. Soc 1985, 107, 4081–4082. [Google Scholar]
- (38).Sato H; Guengerich FP Oxidation of 1,2,4,5-Tetramethoxybenzene to a Cation Radical by Cytochrome P450. J Am. Chem. Soc 2000, 122, 8099–8100. [Google Scholar]
- (39).Bondon A; Macdonald TL; Harris TM; Guengerich FP Oxidation of Cycloalkylamines by Cytochrome P-450. Mechanism-based Inactivation, Adduct Formation, Ring Expansion, and Nitrone Formation. J Biol. Chem 1989, 264, 1988–1997. [PubMed] [Google Scholar]
- (40).Macdonald TL; Gutheim WG; Martin RB; Guengerich FP Oxidation of Substituted N,N-Dimethylanilines by Cytochrome P-450: Estimation of the Effective Oxidation-Reduction Potential of Cytochrome P-450. Biochemistry 1989, 28, 2071–2077. [DOI] [PubMed] [Google Scholar]
- (41).Groves JT; McClusky GA; White RE; Coon MJ Aliphatic Hydroxylation by Highly Purified Liver Microsomal Cytochrome P-450: Evidence for a Carbon Radical Intermediate. Biochem. Biophys. Res. Commun 1978, 81, 154–160. [DOI] [PubMed] [Google Scholar]
- (42).Frey PA Radicals in Enzymatic Reactions. Curr. Opin. Chem Biol 1997, 1, 347–356. [DOI] [PubMed] [Google Scholar]
- (43).Auclair K; Hu Z; Little DM; Ortiz de Montellano PR; Groves JT Revisiting the Mechanism of P450 Enzymes with the Radical Clocks Norcarane and Spiro[2,5]octane. J Am. Chem. Soc 2002, 124, 6020–6027. [DOI] [PubMed] [Google Scholar]
- (44).Gorsky LD; Koop DR; Coon MJ On the Stoichiometry of the Oxidase and Monooxygenase Reactions Catalyzed by Liver Microsomal Cytochrome P-450: Products of Oxygen Reduction. J Biol. Chem 1984, 259, 6812–6817. [PubMed] [Google Scholar]
- (45).Jung C; Schunemann V; Lendzian F; Trautwein AX; Contzen J; Galander M; Bottger LH; Richter M; Barra AL Spectroscopic Characterization of the Iron-oxo Intermediate in Cytochrome P450. Biol. Chem 2005, 386, 1043–1053. [DOI] [PubMed] [Google Scholar]
- (46).Newcomb M; Zhang R; Chandrasena RE; Halgrimson JA; Horner JH; Makris TM; Sligar SG Cytochrome P450 Compound I. J Am. Chem. Soc 2006, 128, 4580–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Kellner DG; Hung SC; Weiss KE; Sligar SG Kinetic Characterization of Compound I Formation in the Thermostable Cytochrome P450 CYP119. J Biol. Chem 2002, 277, 9641–9644. [DOI] [PubMed] [Google Scholar]
- (48).Jung C; de Vries S; Schunemann V Spectroscopic Characterization of Cytochrome P450 Compound I. Arch. Biochem. Biophys 2011, 507, 44–55. [DOI] [PubMed] [Google Scholar]
- (49).Isin EM; Guengerich FP Complex Reactions Catalyzed by Cytochrome P450 Enzymes. Biochim. Biophys. Acta 2007, 1770, 314–329. [DOI] [PubMed] [Google Scholar]
- (50).Guengerich FP; Isin EM Unusual Metabolic Reactions and Pathways. In Handbook of Metabolic Pathways of Xenobiotics, Lee P; Aizawa H; Gau L; Prakash C; Zhong D, Eds.; John Wiley & Sons: Chichester, UK, 2014; Vol 1, pp 147–197. [Google Scholar]
- (51).Ortiz de Montellano PR Oxygen Activation and Reactivity. In Cytochrome P450: Structure, Mechanism, and Biochemistry, 2nd ed; Ortiz de Montellano PR, Ed.; Plenum Press: New York, 1995, pp 245–303. [Google Scholar]
- (52).Yoshimoto FK; Gonzalez E; Auchus RJ; Guengerich FP Mechanism of 17α,20-Lyase and New Hydroxylation Reactions of Human Cytochrome P450 17A1: 18O Labeling and Oxygen Surrogate Evidence for a Role of a Perferryl Oxygen. J. Bioi. Chem 2016, 291, 17143–17164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Akhtar M; Corina D; Pratt J; Smith T Studies on the Removal of C-19 in Oestrogen Biosynthesis using 18O2. J. Chem. Soc, Chem. Commun 1976, 854–856. [Google Scholar]
- (54).Akhtar M; Calder MR; Corina DL; Wright JN Mechanistic Studies on C-19 Demethylation in Oestrogen Biosynthesis. Biochem. J 1982, 201, 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Caspi E; Arunachalam T; Nelson PA Biosynthesis of Estrogens: Aromatization of (19R)-, (19S)-, and (19S)-[19–3H,2H,1H]-3β-Hydroxyandrost-5- en-17-ones by Human Placental Aromatase. J Am. Chem. Soc 1986, 108, 1847–1852. [Google Scholar]
- (56).Akhtar M; Lee-Robichaud P; Akhtar ME; Wright JN The Impact of Aromatase Mechanism on Other P450s. J Steroid Biochem. Mol. Biol 1997, 61, 127–132. [PubMed] [Google Scholar]
- (57).Yoshimoto FK; Guengerich FP Mechanism of the Third Oxidative Step in the Conversion of Androgens to Estrogens by Cytochrome P450 19A1 Steroid Aromatase. J Am. Chem. Soc 2014, 136, 15016–15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Akhtar M; Corina D; Miller S; Shyadehi AZ; Wright JN Mechanism of the Acyl-carbon Cleavage and Related Reactions Catalyzed by Multifunctional P- 450s: Studies on Cytochrome P45017a. Biochemistry 1994, 33, 4410–4418. [DOI] [PubMed] [Google Scholar]
- (59).Mak PJ; Gregory MC; Denisov IG; Sligar SG; Kincaid JR Unveiling the Crucial Intermediates in Androgen Production. Proc. Natl. Acad. Sci. U S. A 2015, 112, 15856–15861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Gregory M; Mak PJ; Sligar SG; Kincaid JR Differential Hydrogen Bonding in Human CYP17 Dictates Hydroxylation Versus Lyase Chemistry. Angew. Chem. Int. Ed 2013, 52, 5342–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Mak PJ; Duggal R; Denisov IG; Gregory MC; Sligar SG; Kincaid JR Human Cytochrome CYP17A1: The Structural Basis for Compromised Lyase Activity with 17-Hydroxyprogesterone. J Am. Chem. Soc 2018, 140, 7324–7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Gonzalez E; Johnson KM; Pallan PS; Phan TTN; Zhang W; Lei L; Wawrzak Z; Yoshimoto FK; Egli M; Guengerich FP Inherent Steroid 17 α,20-Lyase Activity in Defunct Cytochrome P450 17A Enzymes. J Biol. Chem 2018, 293, 541–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Shyadehi AZ; Lamb DC; Kelly SL; Kelly DE; Schunck WH; Wright JN; Corina D; Akhtar M Mechanism of the Acyl-carbon bond Cleavage Reaction Catalyzed by Recombinant Sterol 14 α -Demethylase of Candida albicans (Other Names Are: Lanosterol 14 α -Demethylase, P-45014dm, and CYP51). J Biol. Chem 1996, 271, 12445–12450. [DOI] [PubMed] [Google Scholar]
- (64).Guengerich FP; Munro AW Unusual Cytochrome P450 Enzymes and Reactions. J Biol. Chem 2013, 288, 17065–17073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Varfaj F; Zulkifli SNA; Park H-G; Challinor VL; De Voss JJ; Ortiz de Montellano PR Carbon-Carbon Bond Cleavage in Activation of the Prodrug Nabumetone. Drug Metab. Dispos 2014, 42, 828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Shaik S; Cohen S; Wang Y; Chen H; Kumar D; Thiel W P450 Enzymes: Their Structure, Reactivity, and Selectivity-Modeled by QM/MM Calculations. Chem. Rev 2010, 110, 949–1017. [DOI] [PubMed] [Google Scholar]
- (67).Miller RE; Guengerich FP Oxidation of Trichloroethylene by Liver Microsomal Cytochrome P-450: Evidence for Chlorine Migration in a Transition State Not Involving Trichloroethylene Oxide. Biochemistry 1982, 21, 1090–1097. [DOI] [PubMed] [Google Scholar]
- (68).Liebler DC; Guengerich FP Olefin Oxidation by Cytochrome P-450: Evidence for Group Migration in Catalytic Intermediates Formed with Vinylidene Chloride and trans-1-Phenyl-1-butene. Biochemistry 1983, 22, 5482–5489. [DOI] [PubMed] [Google Scholar]
- (69).Ortiz de Montellano PR; Mangold BLK; Wheeler C; Kunze KL; Reich NO Stereochemistry of Cytochrome P-450-Catalyzed Epoxidation and Prosthetic Heme Alkylation. J Biol. Chem 1983, 258, 4208–4213. [PubMed] [Google Scholar]
- (70).Guengerich FP; Macdonald TL Chemical Mechanisms of Catalysis by Cytochromes P-450: A Unified View. Acct. Chem. Res 1984, 17, 9–16. [Google Scholar]
- (71).Krauser JA; Guengerich FP Cytochrome P450 3A4-Catalyzed Testosterone 6β-Hydroxylation Stereochemistry, Kinetic Deuterium Isotope Effects, and Rate- limiting Steps. J Biol. Chem 2005, 280, 19496–19506. [DOI] [PubMed] [Google Scholar]
- (72).Cheng Q; Sohl CD; Yoshimoto FK; Guengerich FP Oxidation of Dihydrotestosterone by Human Cytochromes P450 19A1 and 3A4. J Bioi. Chem 2012, 287, 29554–29567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Poulos TL; Finzel BC; Howard AJ High-resolution Crystal Structure of Cytochrome P450cam. J Moi. Bioi 1987, 195, 687–700. [DOI] [PubMed] [Google Scholar]
- (74).Raag R; Poulos TL Crystal Structure of the Carbon Monoxide-Substrate- Cytochrome P-450CAM Ternary Complex. Biochemistry 1989, 28, 7586–7592. [DOI] [PubMed] [Google Scholar]
- (75).Raag R; Li H; Jones BC; Poulos TL Inhibitor-induced Conformational Change in Cytochrome P-450 CAM. Biochemistry 1993, 32, 4571–4578. [DOI] [PubMed] [Google Scholar]
- (76).Liou SH; Mahomed M; Lee YT; Goodin DB Effector Roles of Putidaredoxin on Cytochrome P450cam Conformational States. J Am. Chem. Soc 2016, 138, 10163–10172. [DOI] [PubMed] [Google Scholar]
- (77).Conrad HE; Lieb K; Gunsalus IC Mixed Function Oxidation. 3. An Electron Transport Complex in Camphor Ketolactonization. J Biol. Chem 1965, 240, 4029–4037. [PubMed] [Google Scholar]
- (78).Tyson CA; Lipscomb JD; Gunsalus IC The Roles of Putidaredoxin and P450cam in Methylene Hydroxylation. J Biol. Chem 1972, 247, 5777–5784. [PubMed] [Google Scholar]
- (79).Tripathi S; Li H; Poulos TL Structural Basis for Effector Control and Redox Partner Recognition in Cytochrome P450. Science 2013, 340, 1227–1230. [DOI] [PubMed] [Google Scholar]
- (80).Myers WK; Lee YT; Britt RD; Goodin DB The Conformation of P450cam in Complex with Putidaredoxin Is Dependent on Oxidation State. J Am. Chem. Soc 2013, 135, 11732–11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Skinner SP; Liu W-M; Hiruma Y; Timmer M; Blok A; Hass MAS; Ubbink M Delicate Conformational Balance of the Redox Enzyme Cytochrome P450cam. Proc. Nall. Acad. Sci. U. S. A 2015, 112, 9022–9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Liou SH; Myers WK; Oswald JD; Britt RD; Goodin DB Putidaredoxin Binds to the Same Site on Cytochrome P450cam in the Open and Closed Conformation. Biochemistry 2017, 56, 4371–4378. [DOI] [PubMed] [Google Scholar]
- (83).Rettie AE; Rettenmeier AW; Howald WN; Baillie TA Cytochrome P-450 Catalyzed Formation of δ4-VPA, a Toxic Metabolite of Valproic Acid. Science 1987, 235, 890–893. [DOI] [PubMed] [Google Scholar]
- (84).Wang RW; Kari PH; Lu AY; Thomas PE; Guengerich FP; Vyas KP Biotransformation of Lovastatin. IV. Identification of Cytochrome P450 3A Proteins as the Major Enzymes Responsible for the Oxidative Metabolism of Lovastatin in Rat and Human Liver Microsomes. Arch. Biochem. Biophys 1991, 290, 355–361. [DOI] [PubMed] [Google Scholar]
- (85).Guengerich FP; Kim DH Enzymatic Oxidation of Ethyl Carbamate to Vinyl Carbamate and Its Role as an Intermediate in the Formation of 1,N6 Ethenoadenosine. Chem. Res. Toxicol 1991, 4, 413–421. [DOI] [PubMed] [Google Scholar]
- (86).Cooper HLR; Mishra G; Huang XY; Pender-Cudlip M; Austin RN; Shanklin J; Groves JT Parallel and Competitive Pathways for Substrate Desaturation, Hydroxylation, and Radical Rearrangement by the Non-heme Diiron Hydroxylase AlkB. J Am. Chem. Soc 2012, 134, 20365–20375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Morikawa T; Mizutani M; Aoki N; Watanabe B; Saga H; Saito S; Oikawa A; Suzuki H; Sakurai N; Shibata D et al. Cytochrome P450 CYP710A encodes the sterol C-22 Desaturase in Arabidopsis and Tomato. Plant Cell 2006, 18, 1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Arnqvist L; Persson M; Jonsson L; Dutta PC; Sitbon F Overexpression of CYP710A1 and CYP710A4 in Transgenic Arabidopsis Plants Increases the Level of Stigmasterol at the Expense of Sitosterol. Planta 2008, 227, 309–317. [DOI] [PubMed] [Google Scholar]
- (89).Skaggs BA; Alexander JF; Pierson CA; Schweitzer KS; Chun KT; Koegel C; Barbuch R; Bard M Cloning and characterization of the Saccharomyces cerevisiae C-22 Sterol Desaturase Gene, Encoding a Second Cytochrome P-450 Involved in Ergosterol Biosynthesis. Gene 1996, 169, 105–109. [DOI] [PubMed] [Google Scholar]
- (90).Kelly SL; Lamb DC; Baldwin BC; Corran AJ; Kelly DE Characterization of Saccharomyces cerevisiae CYP61, Sterol Δ22-Desaturase, and Inhibition by Azole Antifungal Agents. J Biol. Chem 1997, 272, 9986–9988. [DOI] [PubMed] [Google Scholar]
- (91).Kramlinger VM; Nagy LD; Fujiwara R; Johnson KM; Phan TT; Xiao Y; Enright JM; Toomey MB; Corbo JC; Guengerich FP Human Cytochrome P450 27C1 Catalyzes 3,4-Desaturation of Retinoids. FEBS Lett 2016, 590, 1304–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Johnson KM; Phan TTN; Albertolle ME; Guengerich FP Human Mitochondrial Cytochrome P450 27C1 is Localized in Skin and Preferentially Desaturates tan-Retinol to 3,4-Dehydroretinol. J Biol. Chem 2017, 292, 13672–13687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Dunham NP; Chang WC; Mitchell AJ; Martinie RJ; Zhang B; Bergman JA; Rajakovich LJ; Wang B; Silakov A; Krebs C et al. Two Distinct Mechanisms for C-C Desaturation by Iron(II)- and 2-(Oxo)glutarate-Dependent Oxygenases: Importance of α -Heteroatom Assistance. J Am. Chem. Soc 2018, 140, 7116–7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Guy JE; Abreu IA; Moche M; Lindqvist Y; Whittle E; Shanklin J A Single Mutation in the Castor Δ9-18:0-Desaturase Changes Reaction Partitioning from Desaturation to Oxidase Chemistry. Proc. Natl. Acad. Sci. U S. A 2006, 103, 17220–17224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Wojdyla Z; Borowski T On How the Binding Cavity of AsqJ Dioxygenase Controls the Desaturation Reaction Regioselectivity: A QM/MM Study. J Biol.inorg. Chem 2018, 23, 795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Petrunak EM; Rogers SA; Aube J; Scott EE Structural and Functional Evaluation of Clinically Relevant Inhibitors of Steroidogenic Cytochrome P450 17A1. Drug Metab. Dispos 2017, 45, 635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Gonzalez E; Guengerich FP Kinetic Processivity of the Two-step Oxidations of Progesterone and Pregnenolone to Androgens by Human Cytochrome P450 17A1. J Biol. Chem 2017, 292, 13168–13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Imai T; Yamazaki T; Kominami S Kinetic Studies on Bovine Cytochrome P45011b Catalyzing Successive Reactions from Deoxycorticosterone to Aldosterone. Biochemistry 1998, 37, 8097–8104. [DOI] [PubMed] [Google Scholar]
- (99).Yamazaki T; Ohno T; Sakaki T; Akiyoshi-Shibata M; Yabusaki Y; Imai T; Kominami S Kinetic Anaylsis of Successive Reactions Catalyzed by Bovine Cytochrome P45017a,lyase. Biochemistry 1998, 37, 2800–2806. [DOI] [PubMed] [Google Scholar]
- (100).Tagashira H; Kominami S; Takemori S Kinetic Studies of Cytochrome P45017α,lyase dependent Androstenedione Formation from Progesterone. Biochemistry 1995, 34, 10939–10945. [DOI] [PubMed] [Google Scholar]
- (101).Higuchi A; Kominami S; Takemori S Kinetic Control of Seroidogenesis by Steroid Concentration in Guinea Pig Adrenal Microsomes. Biochim. Biophys. Acta 1991, 1084, 240–246. [DOI] [PubMed] [Google Scholar]
- (102).Sohl CD; Guengerich FP Kinetic Analysis of the Three-step Steroid Aromatase Reaction of Human Cytochrome P450 19A1. J Biol. Chem 2010, 285, 17734–17743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Johnson KA Introduction to Kinetic Analysis of Enzyme Systems. In Kinetic Analysis of Macromolecules. A Practical Approach, Johnson KA, Ed.; Oxford University Press: Oxford, UK, 2003; pp 1–18. [Google Scholar]
- (104).Johnson KA Binding Equilibria. 12th New Enzymology Kinetics Workshop, KinTek, Austin, TX, 2017, p 23. [Google Scholar]
- (105).Bell-Parikh LC; Guengerich FP Kinetics of Cytochrome P450 2E1-catalyzed Oxidation of Ethanol to Acetic Acid via Acetaldehyde. J Biol. Chem 1999, 274, 23833–23840. [DOI] [PubMed] [Google Scholar]
- (106).Chowdhury G; Calcutt MW; Nagy LD; Guengerich FP Oxidation of Methyl and Ethyl Nitrosamines by Cytochrome P450 2E1 and 2B1. Biochemistry 2012, 51, 9995–10007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Chowdhury G; Calcutt MW; Guengerich FP Oxidation of N Nitrosoalkylamines by Human Cytochrome P450 2A6: Sequential Oxidation to Aldehydes and Carboxylic Acids and Analysis of Reaction Steps. J Biol. Chem 2010, 285, 8031–8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Hildebrandt A; Estabrook RW Evidence for the Participation of Cytochrome b5 in Hepatic Microsomal Mixed-Function Oxidation Reactions. Arch. Biochem. Biophys 1971, 143, 66–79. [DOI] [PubMed] [Google Scholar]
- (109).Canova-Davis E; Chiang JYL; Waskell L Obligatory Role of Cytochrome b5 in the Microsomal Metabolism of Methoxyflurane. Biochem. Pharmacoi 1985, 34, 1907–1912. [DOI] [PubMed] [Google Scholar]
- (110).Gorsky LD; Coon MJ Effects of Conditions for Reconstitution with Cytochrome b5 on the Formation of Products in Cytochrome P-450-Catalyzed Reactions. Drug Metab. Dispos 1986, 14, 89–96. [PubMed] [Google Scholar]
- (111).Jansson I; Schenkman JB Influence of Cytochrome b5 on the Stoichiometry of the Different Oxidative Reactions Catalyzed by Liver Microsomal Cytochrome P- 450. Drug Metab. Dispos 1987, 15, 344–348. [PubMed] [Google Scholar]
- (112).Bart AG; Scott EE Structural and Functional Effects of Cytochrome b5 Interactions with Human Cytochrome P450 Enzymes. J Biol. Chem 2017, 292, 20818–20833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Katagiri M; Kagawa N; Waterman MR The Role of Cytochrome b5 in the Biosynthesis of Androgens by Human P450c17. Arch. Biochem. Biophys 1995, 317, 343–347. [DOI] [PubMed] [Google Scholar]
- (114).Yamazaki H; Johnson WW; Ueng YF; Shimada T; Guengerich FP Lack of Electron Transfer from Cytochrome b5 in Stimulation of Catalytic Activities of Cytochrome P450 3A4. Characterization of a Reconstituted Cytochrome P450 3A4/NADPH-Cytochrome P450 Reductase System and Studies with Apo-Cytochrome b5. J Biol. Chem 1996, 271, 27438–27444. [DOI] [PubMed] [Google Scholar]
- (115).Lee-Robichaud P; Akhtar ME; Akhtar M Control of Androgen Biosynthesis in the Human Through the Interaction of Arg347 and Arg358 of CYP17 with Cytochrome b5. Biochem. J 1998, 332, 293–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Auchus RJ; Lee TC; Miller WL Cytochrome b5 Augments the 17,20-Lyase Activity of Human P450c17 Without Direct Electron Transfer. J Biol. Chem 1998, 273, 3158–3165. [DOI] [PubMed] [Google Scholar]
- (117).Yamazaki H; Nakamura M; Komatsu T; Ohyama K; Hatanaka N; Asahi S; Shimada N; Guengerich FP; Shimada T; Nakajima M et al. Roles of NADPH-P450 Reductase and Apo- and Holo-Cytochrome b5 on Xenobiotic Oxidations Catalyzed by 12 Recombinant Human Cytochrome P450s Expressed in Membranes of Escherichia coli. Prot. Express. Purif 2002, 24, 329–337. [DOI] [PubMed] [Google Scholar]
- (118).Guryev OL; Gilep AA; Usanov SA; Estabrook RW Interaction of Apo-Cytochrome b5 with Cytochromes P4503A4 and P45017A: Relevance of Heme Transfer Reactions. Biochemistry 2001, 40, 5018–5031. [DOI] [PubMed] [Google Scholar]
- (119).Yamazaki H; Shimada T; Martin MV; Guengerich FP Stimulation of Cytochrome P450 Reactions by Apo-Cytochrome b5 Evidence Against Transferof Heme from Cytochrome P450 3A4 to Apo-Cytochrome b5 or Heme Oxygenase. J Biol. Chem 2001, 276, 30885–30891. [DOI] [PubMed] [Google Scholar]
- (120).Pearl NM; Wilcoxen J; Im S; Kunz R; Darty J; Britt RD; Ragsdale SW; Waskell L Protonation of the Hydroperoxo Intermediate of Cytochrome P450 2B4 Is Slower in the Presence of Cytochrome P450 Reductase Than in the Presence of Cytochrome b5. Biochemistry 2016, 55, 6558–6567. [DOI] [PubMed] [Google Scholar]
- (121).Yun CH; Kim KH; Calcutt MW; Guengerich FP Kinetic Analysis of Oxidation of Coumarins by Human Cytochrome P450 2A6. J Biol. Chem 2005, 280, 12279–12291. [DOI] [PubMed] [Google Scholar]
- (122).Kim D; Cha GS; Nagy LD; Yun CH; Guengerich FP Kinetic Analysis of Lauric Acid Hydroxylation by Human Cytochrome P450 4A11. Biochemistry 2014, 53, 6161–6172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Stiborova M; Indra R; Moserova M; Frei E; Schmeiser HH; Kopka K; Philips DH; Arlt VM NADH:Cytochrome b5 Reductase and Cytochrome b5 Can Act as Sole Electron Donors to Human Cytochrome P450 1A1-Mediated Oxidation and DNA Adduct Formation by Benzo[a]pyrene. Chem. Res. Toxicol 2016, 29, 1325–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Reed L; Mrizova I; Barta F; Indra R; Moserova M; Kopka K; Schmeiser HH; Wolf CR; Henderson CJ; Stiborova M et al. Cytochrome b5 Impacts on Cytochrome P450-Mediated Metabolism of Benzo[a]pyrene and Its DNA Adduct Formation: Studies in Hepatic Cytochrome b5/P450 Reductase Null (HBRN) mice. Arch. Toxicol 2018, 92, 1625–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Estrada DF; Laurence JS; Scott EE Substrate-modulated Cytochrome P450 17A1 and Cytochrome b5 Interactions Revealed by NMR. J Biol. Chem 2013, 288, 17008–17018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Peng HM; Liu J; Forsberg SE; Tran HT; Anderson SM; Auchus RJ Catalytically Relevant Electrostatic Interactions of Cytochrome P450c17 (CYP17A1) and Cytochrome b5 J Biol. Chem 2014, 289, 33838–33849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Yablokov E; Florinskaya A; Medvedev A; Sergeev G; Strushkevich N; Luschik A; Shkel T; Haidukevich I; Gilep A; Usanov S et al. Thermodynamicsof Interactions Between Mammalian Cytochromes P450 and b5. Arch. Biochem. Biophys 2017, 619, 10–15. [DOI] [PubMed] [Google Scholar]
- (128).Shimada T; Mernaugh RL; Guengerich FP Interactions of Mammalian Cytochrome P450, NADPH-Cytochrome P450 Reductase, and Cytochrome b5 enzymes. Arch. Biochem. Biophys 2005, 435, 207–216. [DOI] [PubMed] [Google Scholar]
- (129).Zhang H; Hamdane D; Im SC; Waskell L Cytochrome b5 Inhibits Electron Transfer from NADPH-Cytochrome P450 Reductase to Ferric Cytochrome P450 2B4. J Biol. Chem 2008, 283, 5217–5225. [DOI] [PubMed] [Google Scholar]
- (130).Finn RD; McLaughlin LA; Ronseaux S; Rosewell I; Houston JB; Henderson CJ; Wolf CR Defining the in Vivo Role for Cytochrome b5 in Cytochrome P450 Function through the Conditional Hepatic Deletion of Microsomal Cytochrome b5. J Biol. Chem 2008, 283, 31385–31393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Henderson CJ; McLaughlin LA; Finn RD; Ronseaux S; Kapelyukh Y; Wolf CR A Role for Cytochrome b5 in the In Vivo Disposition of Anticancer and Cytochrome P450 Probe Drugs in Mice. Drug Metab. Dispos 2014, 42, 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).West SB; Levin W; Ryan D; Vore M; Lu AYH Liver microsomal electron transport systems. II. The Involvement of Cytochrome b5 in the NADH-Dependent Hydroxylation of 3,4-Benzpyrene by a Reconstituted Cytochrome P-448- Containing System. Biochem. Biophys. Res. Commun 1974, 58, 516–522. [DOI] [PubMed] [Google Scholar]
- (133).Arnold FH Directed Evolution: Bringing New Chemistry to Life. Angew. Chem. Int. Ed 2017, 57, 4143–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Knight AM; Kan SBJ; Lewis RD; Brandenberg OF; Chen K; Arnold FH Diverse Engineered Heme Proteins Enable Stereodivergent Cyclopropanation of Unactivated Alkenes. ACS Cent. Sci 2018, 4, 372–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Hammer SC; Kubik G; Watkins E; Huang S; Minges H; Arnold FH Anti- Markovnikov Alkene Oxidation by Metal-oxo-mediated Enzyme Catalysis. Science 2017, 358, 215–218. [DOI] [PubMed] [Google Scholar]
- (136).Renata H; Lewis RD; Sweredoski MJ; Moradian A; Hess S; Wang ZJ; Arnold FH Identification of Mechanism-Based Inactivation in P450-Catalyzed Cyclopropanation Facilitates Engineering of Improved Enzymes. J Am. Chem. Soc 2016, 138, 12527–12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Renata H; Wang ZJ; Arnold FH Expanding the Enzyme Universe: Accessing Non-Natural Reactions by Mechanism-Guided Directed Evolution. Angew. Chem. Int. Ed 2015, 54, 3351–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Rentmeister A; Arnold FH; Fasan R Chemo-enzymatic Fluorination of Unactivated Organic Compounds. Nat. Chem. Biol 2009, 5, 26–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Wei Y; Tinoco A; Steck V; Fasan R; Zhang Y Cyclopropanations via Heme Carbenes: Basic Mechanism and Effects of Carbene Substituent, Protein Axial Ligand, and Porphyrin Substitution. J Am. Chem. Soc 2018, 140, 1649–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Vargas DA; Tinoco A; Tyagi V; Fasan R Myoglobin-Catalyzed C-H Functionalization of Unprotected Indoles. Angew. Chem Int. Ed 2018, 57, 9911–9915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Sreenilayam G; Moore EJ; Steck V; Fasan R Metal Substitution Modulates the Reactivity and Extends the Reaction Scope of Myoglobin Carbene Transfer Catalysts. Adv. Synth. Catal 2017, 359, 2076–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Singh R; Bordeaux M; Fasan R P450-Catalyzed Intramolecular sp3 C-H Amination with Arylsulfonyl Azide Substrates. ACS Catal 2014, 4, 546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Kolev JN; Zaengle JM; Ravikumar R; Fasan R Enhancing the Efficiency and Regioselectivity of P450 Oxidation Catalysts by Unnatural Amino Acid Mutagenesis. Chembiochem 2014, 15, 1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Giovani S; Alwaseem H; Fasan R Aldehyde and Ketone Synthesis by P450- Catalyzed Oxidative Deamination of Alkyl Azides. ChemCatChem 2016, 8, 2609–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Fasan R Enzymatic Catalysis: New Functional Twists for P450s. Nat. Chem 2017, 9, 609–611. [DOI] [PubMed] [Google Scholar]
- (146).Ravichandran KG; Boddupalli SS; Hasemann CA; Peterson JA; Deisenhofer J Crystal Structure of Hemoprotein Domain of P450 BM-3, a Prototype for Microsomal P450’s. Science 1993, 261, 731–736. [DOI] [PubMed] [Google Scholar]
- (147).Sawayama AM; Chen MM; Kulanthaivel P; Kuo MS; Hemmerle H; Arnold FH A Panel of Cytochrome P450 BM3 Variants to Produce Drug Metabolites and Diversify Lead Compounds. Chemistry 2009, 15, 11723–11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Fasan R; Meharenna YT; Snow CD; Poulos TL; Arnold FH Evolutionary History of a Specialized P450 Propane Monooxygenase. J Moi. Bioi 2008, 383, 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Groves JT; Myers RS Catalytic Asymmetric Epoxidations with Chiral Iron Porphyrins. J Am. Chem. Soc 1983, 105, 5791–5796. [Google Scholar]
- (150).Brandenberg OF; Prier CK; Chen K; Knight AM; Wu Z; Arnold FH Stereoselective Enzymatic Synthesis of Heteroatom-Substituted Cyclopropanes. ACS Catal 2018, 8, 2629–2634. [Google Scholar]
- (151).White RE; McCarthy MB Aliphatic Hydroxylation by Cytochrome P-450. Evidence for Rapid Hydrolysis of an Intermediate Iron-Nitrene Complex. J Am. Chem. Soc 1984, 106, 4922–4926. [Google Scholar]
- (152).Svastits EW; Dawson JH; Breslow R; Gellman SH Functionalized Nitrogen Atom Transfer Catalyzed by Cytochrome P-450. J Am. Chem. Soc 1985, 107, 6427–6428. [Google Scholar]
- (153).Strushkevich N; MacKenzie F; Cherkesova T; Grabovec I; Usanov S; Park HW Structural Basis for Pregnenolone Biosynthesis by the Mitochondrial Monooxygenase System. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 10139–10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Hiruma Y; Hass MAS; Kikui Y; Liu W-M; Olmez B; Skinner SP; Blok A; Kloosterman A; Koteishi H; Loehr F et al. The Structure of the Cytochrome P450cam-Putidaredoxin Complex Determined by Paramagnetic NMR Spectroscopy and Crystallography. J Mol. Bio 2013, 425, 4353–4365. [DOI] [PubMed] [Google Scholar]
- (155).Sevrioukova IF; Li H; Zhang H; Peterson JA; Poulos TL Structure of a Cytochrome P450-Redox Partner Electron-Transfer Complex. Proc. Nat. Acad. Sci. U. S. A 1999, 96, 1863–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Maekawa K; Adachi M; Matsuzawa Y; Zhang Q; Kuroki R; Saito Y; Shah MB Structural Basis of Single-Nucleotide Polymorphisms in Cytochrome P450 2C9. Biochemistry 2017, 56, 5476–5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Muroi Y; Saito T; Takahashi M; Sakuyama K; Niinuma Y; Ito M; Tsukada C; Ohta K; Endo Y; Oda A et al. Functional Characterization of Wild-type and 49 CYP2D6 Allelic Variants for N-Desmethyltamoxifen 4-Hydroxylation Activity. Drug Metab. Pharmacokin 2014, 29, 360–366. [DOI] [PubMed] [Google Scholar]
- (158).Pallan PS; Nagy LD; Lei L; Gonzalez E; Kramlinger VM; Azumaya CM; Wawrzak Z; Waterman MR; Guengerich FP; Egli M Structural and Kinetic Basis of Steroid 17 α,20-Lyase Activity in Teleost Fish Cytochrome P450 17A1 and Its Absence in Cytochrome P450 17A2. J Biol. Chem 2015, 290, 3248–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Pallan PS; Wang C; Lei L; Yoshimoto FK; Auchus RJ; Waterman MR; Guengerich FP; Egli M Human Cytochrome P450 21A2, the Major Steroid 21- Hydroxylase: Structure of the Enzyme-Progesterone Substrate Complex and Rate- Limiting C-H Bond Cleavage. J Biol. Chem 2015, 290, 13128–13143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Pallan PS; Lei L; Wang C; Waterman MR; Guengerich FP; Egli M Research Resource: Correlating Human Cytochrome P450 21A2 Crystal Structure and Phenotypes of Mutations in Congenital Adrenal Hyperplasia. Mol. Endocrinol 2015, 29, 1375–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Wang C; Pallan PS; Zhang W; Lei L; Yoshimoto FK; Waterman MR; Egli M; Guengerich FP Functional Analysis of Human Cytochrome P450 21A2 Variants Involved in Congenital Adrenal Hyperplasia. J Biol. Chem 2017, 292, 10767–10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Roberts AG; Yang J; Halpert JR; Nelson SD; Thummel KT; Atkins WM The Structural Basis for Homotropic and Heterotropic Cooperativity of Midazolam Metabolism by Human Cytochrome P450 3A4. Biochemistry 2011, 50, 10804–10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Livezey M; Nagy LD; Diffenderfer LE; Arthur EJ; Hsi DJ; Holton JM; Furge LL Molecular Analysis and Modeling of Inactivation of Human CYP2D6 by Four Mechanism Based Inactivators. Drug Metab. Lett 2012, 6, 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Cruciani G; Carosati E; De Boeck B; Ethirajulu K; Mackie C; Howe T; Vianello R MetaSite: Understanding Metabolism in Human Cytochromes from the Perspective of the Chemist. J Med. Chem 2005, 48, 6970–6979. [DOI] [PubMed] [Google Scholar]
- (165).Trunzer M; Faller B; Zimmerlin A Metabolic Soft Spot Identification and Compound Optimization in Early Discovery Phases Using MetaSite and LC- MS/MS Validation. J Med. Chem 2009, 52, 329–335. [DOI] [PubMed] [Google Scholar]
- (166).Shin YG; Le H; Khojasteh C; Hop CE Comparison of Metabolic Soft Spot Predictions of CYP3A4, CYP2C9 and CYP2D6 Substrates Using MetaSite and StarDrop. Comb. Chem. High Throughput Screen 2011, 14, 811–823. [DOI] [PubMed] [Google Scholar]
- (167).Boyer S; Arnby CH; Carlsson L; Smith J; Stein V; Glen RC Reaction Site Mapping of Xenobiotic Biotransformations. J Chem. info. Model 2007, 47, 583–590. [DOI] [PubMed] [Google Scholar]
- (168).Guengerich FP New Horizons in Predictive Toxicology: Current Status and Application. In Metabolism-Based Toxicity Prediction, Wilson AEG, Ed.; Royal Soc. Chem.: Cambridge, 2012, pp 542–562. [Google Scholar]
- (169).Krauser JA; Voehler M; Tseng LH; Schefer AB; Godejohann M; Guengerich FP Testosterone 1β-Hydroxylation by Human Cytochrome P450 3A4. Eur. J. Biochem 2004, 271, 3962–3969. [DOI] [PubMed] [Google Scholar]
- (170).Hammett LP The Effect of Structure upon the Reactions of Organic Compounds. Benzene Derivatives. J Am. Chem. Soc 1937, 59, 96–103. [Google Scholar]
- (171).Smith MB; March J Quantitative Treatments of the Effect of Structure on Reactivity. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure; 6th ed; Wiley-Interscience: New York, 2007, pp 401–412. [Google Scholar]
- (172).Ekroos M; Sjögren T Structural Basis for Ligand Promiscuity in Cytochrome P450 3A4. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 13682–13687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Estrada DF; Skinner AL; Laurence JS; Scott EE Human Cytochrome P450 17A1 Conformational Selection: Modulation by Ligand and Cytochrome £5. J Biol. Chem 2014, 289, 14310–14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Johnson KA Role of Induced Fit in Enzyme Specificity: A Molecular Forward/Reverse Switch. J Biol. Chem 2008, 283, 26297–26301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Isin EM; Guengerich FP Kinetics and Thermodynamics of Ligand Binding by Cytochrome P450 3A4. J Biol. Chem 2006, 281, 9127–9136. [DOI] [PubMed] [Google Scholar]
- (176).Isin EM; Guengerich FP Multiple Sequential Steps Involved in the Binding of Inhibitors to Cytochrome P450 3A4. J Biol. Chem 2007, 282, 6863–6874. [DOI] [PubMed] [Google Scholar]
- (177).Griffin BW; Peterson JA Camphor Binding by Pseudomonas putida Cytochrome P-450. Kinetics and Thermodynamics of the Reaction. Biochemistry 1972, 11, 4740–4746. [DOI] [PubMed] [Google Scholar]
- (178).Sevrioukova IF; Poulos TL Dissecting Cytochrome P450 3A4-Ligand Interactions Using Ritonavir Analogues. Biochemistry 2013, 52, 4474–4481. [DOI] [PubMed] [Google Scholar]
- (179).Sohl CD; Isin EM; Eoff RL; Marsch GA; Stec DF; Guengerich FP Cooperativity in Oxidation Reactions Catalyzed by Cytochrome P450 1A2: Highly Cooperative Pyrene Hydroxylation and Multiphasic Kinetics of Ligand Binding. J Biol. Chem 2008, 283, 7293–7308. [DOI] [PubMed] [Google Scholar]