

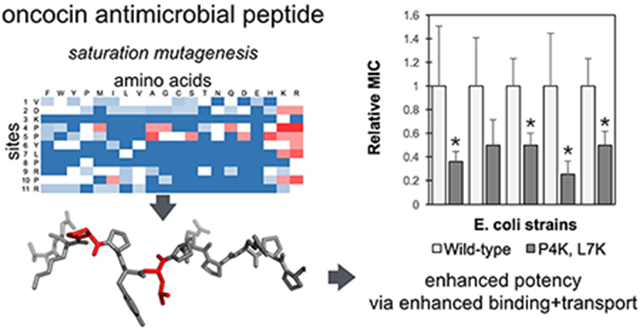
Abstract
Oncocin is a proline-rich antimicrobial peptide that inhibits protein synthesis by binding to the bacterial ribosome. In this work, the antimicrobial activity of oncocin was improved by systematic peptide mutagenesis and activity evaluation. We found that a pair of cationic substitutions (P4K and L7K/R) improves the activity by 2-4 fold (p<0.05) against multiple Gram-negative bacteria. An in vitro transcription / translation assay indicated that the increased activity was not because of stronger ribosome binding. Rather a cellular internalization assay revealed a higher internalization rate for the optimized analogs thereby suggesting a mechanism to increase potency. In addition, we found that the optimized peptides’ benefit is dependent upon nutrient-depleted media conditions. The molecular design and characterization strategies have broad potential for development of antimicrobial peptides.
Graphical Abstract
1. Introduction
Antibiotics are essential to modern medicine.1–5 However, the resistance to available antibiotics has become an urgent issue over the last two decades. Particularly, many patients are suffering from organisms such as vancomycin-resistant enterococci, methicillin-resistant Staphylococcus aureus (MRSA), multidrug and extensively drug-resistant Mycobacterium tuberculosis and Enterobacteriaceae.6 Moreover, a lack of investment has steadily slowed the discovery of new antibiotics.7
Antimicrobial peptides (AMPs) are a promising alternative to conventional antibiotics.8 AMPs are oligopeptides containing a varying number of amino acids, ranging in length from about 8-50 amino acids.9 Over 85% of AMPs are cationic peptides with an abundance of lysine and arginine residues analyzed from the antimicrobial peptide database (APD3).9
Proline-rich antimicrobial peptides (PrAMPs) have attracted attention in recent years.10–12 PrAMPs have specific intracellular targets rather than killing bacteria through membrane lytic mechanisms.10 PrAMPs have been identified in some insects, crustaceans and mammals.13 As their name states, these peptides are rich in proline, and they often have a common motif including arginine. In Gram-negative bacteria, most PrAMPs are transported into the cytoplasm through a specialized transporter such as SbmA on the bacterial membrane;14,15 however, the function and mechanism of SbmA-mediated transportation remains unclear. The intracellular target of PrAMPs was first identified as the heat-shock protein DnaK; later, the 70S ribosome was identified as the main target for binding.11,16,17 The binding of PrAMPs to the ribosome inhibits protein synthesis. This non-lytic mechanism reduces the likelihood of adverse effects in humans thereby providing a promising feature for drug design.
Oncocin (Onc18, VDKPPYLPRPRPPRRIYNR-NH2) is a PrAMP optimized from the milkweed bug (Oncopeltus fasciatus) to kill Gram-negative pathogens.18 Oncocin’s primary mechanism of action is assumed to have three major steps: passive diffusion aross the bacterial outer membrane, active transport through SbmA and maybe other receptors from periplasm to cytoplasm, and inhibition of protein synthesis by binding to the 70S ribosome.17,19 Recently, the crystal structure of the oncocin and ribosome bound complex has been determined.17 From the complex structure, it was shown that oncocin inhibits protein synthesis by blocking the peptidyl transferase center and the peptide-exit tunnel of the ribosome.
There have been studies to engineer improved oncocin activity. Different analogs of Onc18, with arginines replaced with non-natural amino acids, increase stability. For example, Onc72 (VDKPPYLPRPRPPROIYNO-NH2) and Onc112 (VDKPPYLPRPRPPRrIYNr-NH2) improved serum stability and antimicrobial activity compared to Onc18 by switching arginine with ornithine (O) and D-arginine (r), respectively.18,20 In addition, Onc110 (VDKPPYLPRPRPxRxjYNO-NH2) enhanced protease stability by incorporating trans-4-hydroxyproline (x) and tert-leucine (j).19 Moreover, computer simulations have been applied to rationally design stronger binding peptides to DnaK protein by substituting seven residues from the N-terminal region with four optimized residues selected from molecular modeling and docking. 21 Although six derivatives were found to bind more strongly to DnaK, the minimum inhibitory concentrations (MICs) against wild type E. coli were not improved. The use of array synthesis has been applied to optimize oncocin activity against Gram-positive Staphylococcus aureus and Pseudomonas aeruginosa, against which oncocin has rather weak activity.22 A total of 361 singly substituted analogs were produced to evaluate against a luminescent P. aeruginosa strain. The combination of two favorable substitutions (D2R/P12W) yielded an optimized peptide that was 10-fold and 100-fold more active against P. aeruginosa and against S. aureus, respectively.23 The same approach has been applied to apidaecin, another PrAMP, to improve the activity against P. aeruginosa and against S. aureus as well.24 There have been studies to investigate the importance of the C-terminal portion of oncocin to the antimicrobial activity.25 C-terminally truncated Onc112 ΔC7 and Onc112 ΔC9 derivatives, which lacked the last 7 and 9 amino acids, respectively, showed complete loss of antimicrobial activity. However, both truncated peptides displayed some inhibitory activity with an in vitro translation system, though with a reduced activity than that of full-length Onc112. It was concluded that the C terminus of Onc112 is essential for cell transport but not for ribosome binding. Nevertheless, it remains unknown if the N-terminal portion is impactful for internalization.
In the current study, we systematically evaluate oncocin mutants for growth inhibition of Gram-negative bacteria and elucidate the mechanistic basis of these mutational impacts. The first objective of the current work is to optimize the activity of oncocin by systematic variant analysis using array synthesis for efficient peptide production. From the binding structure of Onc112 on the ribosome, only the N-terminal part of oncocin binds to the peptide exit channel of the ribosome.17 A positional alanine-scan for oncocin showed that substitution of residues 3, 6-9 and 11 abolished the antimicrobial activity almost completely, while other positions only increased the MIC values slightly.19 This motivates further optimization of oncocin activity by mutagenesis for the first 11 residues. In the current work, a peptide array of the monosubstituted variants for the first 11 amino acids was screened to select more potent peptides. The most effective substitutions identified were combined to further improve the activity, and the most potent candidates were synthesized on resin and tested against multiple Gram-negative bacterial strains. The second objective is to study the mechanisms for the improved antimicrobial activities. We applied in vitro GFP translation inhibition as well as molecular dynamics (MD) simulations to investigate the optimized analogs and provide insights into the interactions with the ribosome. In addition, we developed and implemented a high-performance liquid chromatography (HPLC)-based kinetic assay to study peptide binding and transport.
2. Materials and Methods
2.1. SPOT Synthesis
While various strategies exist for peptide engineering, a major hurdle for large scale analysis is the inefficiency of conventional peptide synthesis on resin. SPOT synthesis is a powerful technique to efficiently synthesize numerous peptides, which can be used for functional analysis.26 It is based on synthesis of peptide arrays on cellulose, which is less costly than conventional solid state synthesis on resin. Efficient peptide production via array synthesis empowers engineering and elucidation of antimicrobial activity by systematic substitution analysis. All peptides synthesized on cellulose used in this work were purchased from Kinexus (Vancouver, Canada). Each peptide includes an additional glycine on the C-terminus for initial immobilization. The peptides were delivered as a set of small paper discs and then dissolved in 160 μL sterile water for each spot and stored at 4 °C. Randomly selected peptide spots were evaluated via analytical HPLC and matrix-associated laser-desorption time-of-flight mass spectrometry. The purity for monosubstituted and multisubstituted variants were 60.4 ± 10.0 %, and 63.9 ± 3.2 %, respectively (Table S1 and S2), which is consistent with published ranges for SPOT synthesis.27 The consistency in purity between peptide variants provides for effective comparison.
2.2. Purified Peptides
All peptides synthesized on resin with C-terminal amides were purchased from United Biosystems (Herndon, USA). All purities were above 95 % as analyzed by reverse-phase HPLC (Fig. S1).
2.3. Microdilution Broth Assay
E. coli and Salmonella strains (listed in Table 1) were grown on lysogeny broth (LB, Fisher Scientific BP1427-2) with agar overnight and the colonies were added to 2 mL nutrient broth (RPI N15100) for liquid culture. 150 μL overnight bacterial culture diluted in 3 mL of 12.5 % Mueller Hinton Broth (MHB, Sigma-Aldrich 70192) was shaken at 37 °C for 6 h to reach exponential growth phase. The culture was diluted with 12.5 % MHB to reach a suspension of 5 × 105 colony-forming unit (cfu)/mL. The bacterial density was measured as the optical density at 600 nm (OD600). Aqueous peptide solutions were serially twofold diluted in sterile water from 12.8 to 0.1 mg/L. 10 μL of the peptide solution was transferred to a sterile 96-well plate and mixed with 90 μL diluted bacterial culture. Cultures were incubated for 18 ± 1 h at 37 °C. The MIC was defined as the lowest peptide concentration with no detectable bacterial growth measured by OD600 value (< 0.05) after incubation (see Figure S2). MIC values were also assessed in different concentrations of MHB and minimal M9 broth (Cat. No. M8000, TEKNOVA). The experiment was performed in triplicate.
Table 1.
Bacteria used in this study
| Bacterial strain | Description | Source |
|---|---|---|
| E. coli JW0013 | ΔdnaK734::kan, Δ(araD-araB)567, ΔlacZ4787(::rrnB-3),λ-, rph-1, Δ(rhaD-rhaB)568, hsdR514 | Keio Collection, GenoBase |
| E. coli O157:H7 472 | Common pathogenic species | University of Minnesota |
| E. coli MC1061 F’ | Plasmid-free, recA+, non-amber suppressor strain | Lucigen |
| E. coli J2210 | Common pathogenic species | University of Minnesota |
| E. coli JJ2487 | Common pathogenic species | Minneapolis VA hospital |
| Salmonella enterica serovar Enteritidis MH91989 | Common pathogenic species | University of Minnesota |
| Salmonella enterica serovar Typhimurium ATCC SL1344 | Common pathogenic species | University of Minnesota |
2.4. In vitro Translation Assay
The inhibition of cell free GFP synthesis by Onc18 and its mutants was evaluated with an E. coli lysate-based transcription-translation coupled assay (RTS100, 5Prime) as previously described.28 The GFP translation inhibition assay is intended to focus on oncocin’s inhibition of the ribosome in contrast to the overall growth inhibition assay. Briefly, transcription-translation reactions with or without oncocin variants were mixed following the manufacturer’s instructions with the exception that the final reaction volume was reduced from 50 μL to 6 μL. Each reaction contained 1.44 μL E. coli lysate, 1.2 μL reaction mix, 1.44 μL amino acids, 0.12 μL methionine, 0.6 μL reconstitution buffer, 0.6 μL GFPmut3 vector,29 and 0.6 μL peptides or control buffer. Reactions were incubated for 4 h at 30 °C in a CFX Connect Real-Time PCR Detection System instrument. GFP expression was quantified through the rate of production in the first 30 min (λExc. = 450-490 nm, λEm = 515-530 nm). The production rate was calculated by differentiation of fluorescent signals at different time points. All assays were conducted in triplicate.
2.5. Cellular Binding and Internalization Assay
Binding and internalization to E. coli were measured by incubating oncocin analogs in bacterial culture and measuring the reduction in free peptide over time via HPLC. E. coli strain JW0013 was grown in minimal M9 broth to provide low background for identification of oncocin analogs within the culture supernatant via HPLC. 0.5 mL of overnight culture was mixed with 125 μg/mL of peptide and 5 mL of M9 medium and incubated at 37 °C. At 0, 45, and 90 minutes, 1 mL of culture was centrifuged at 3000 × g for 3 min, and supernatant was filtered (0.2 μm). The filtrate was analyzed via reverse-phase HPLC using a Dionex UltiMate 3000 UHPLC (Dionex, Sunnyvale, CA, USA) with an XBridge Peptide BEH C18 column (Waters Corp., Milford, MA, USA) with a linear gradient from 5.5 to 35.5% aqueous acetonitrile for 17 min in the presence of 0.1 % trifluoroacetic acid as ion pair reagent. The absorbance was measured at 280 nm.
We analyze cellular association and internalization of the peptide as first order with respect to both peptide and cells.
| (1) |
where km is the rate constant for association and internalization and P and C are the concentrations of extracellular peptides and cells, respectively.
To reduce uncertainty associated with imperfect knowledge of cell concentration, internalization assays were performed with two different peptides simultaneously. Assuming sufficiently large cell surface area to prevent competition between peptides, the kinetic model of the two peptides becomes
| (2) |
| (3) |
which can be combined and rearranged to calculate the ratio of internalization rate constants of two peptides:
| (4) |
The experiment is designed to have the total number of oncocin peptides remain stoichiometrically low, by at least two orders of magnitude, as compared to transport receptors thereby avoiding competition between peptides.
2.6. SbmA Quantification
Nine milliliters of E. coli JW0013 in mid-log phase were pelleted by centrifugation at 3,200 × g for 10 min and resuspended in lysis buffer (9.38 g/L sodium phosphate dibasic heptahydrate, 2.07 g/L sodium phosphate monobasic monohydrate, 29.2 g/L NaCl, 3.1 g/L 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), 1.7 g/L imidazole, and 5 % glycerol in water). The samples were subjected to four freeze/thaw cycles. The lysed bacterial cells were spun at 12,000 g for 10 mins and supernatants were used for Western blotting. Protein concentrations were determined by a bicinchoninic acid (BCA) assay. Lysate was separated on a 4-12% SDS-PAGE gel and transferred onto a polyvinylidene difluoride membrane using a semidry transfer apparatus (Bio-Rad) at 30 V for 60 min.The blot was stained with Ponceau Red (59803S, Cell Signaling Technology) solution to note the efficiency of protein transfer. For detection, the blot was blocked for 1 hr at room temperature in 5% milk powder in Tris-buffered saline with Tween (TBST) solution (40 mM tris(hydroxymethyl)aminomethane-HCl, pH 7.5, 200 mM NaCl, 0.1% [vol/vol] Tween 20 (polysorbate 20)) and then incubated overnight with rabbit anti-SbmA antibody (HP6001, HycultBiotech) at a titer of 1:500 (in 5% bovine serum albumin (BSA) in TBST). The membrane was washed and incubated for 1 hr at room temperature with a goat anti-rabbit secondary antibody conjugated to horseradish peroxidase (7074S, Cell Signaling Technology) at a titer of 1:5,000. Detection was performed using a ChemiDoc system (Bio-Rad).
2.7. Molecular Modeling of the Binding Complex
To our knowledge, the crystal structure of oncocin-ribosome binding complex was available only for Thermus thermophilus (PDB: 4z8c).17 In order to obtain a structural model with oncocin bound to an E. coli ribosome, the crystal structure for E. coli ribosome (PDB: 4v7u)30 was aligned with the Thermus thermophilus ribosome using the Pymol align function (Fig. 1 A,B).31 The E. coli and Thermus thermophilus ribosomes exhibit 79 % and 71 % homology in their 16S RNA and 23S RNA sequences, respectively, calculated from BLAST.32
Fig. 1.
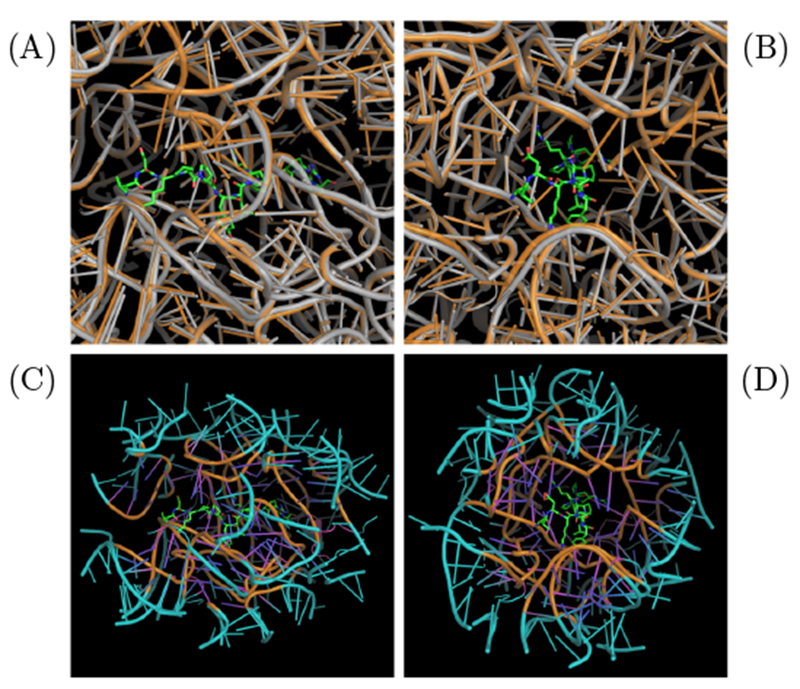
System construction for MD simulation. (A) and (B) are orthogonal views of the E. coli (PDB: 4v7u, gray) and Thermus thermophilus (PDB: 4z8c, orange) ribosome structures aligned via Pymol align with oncocin bound (shown in sticks). (C) and (D) illustrate the reduced E. coli ribosome with oncocin bound. The reduced region contains all residues within 20 Å of the oncocin. A buffer region between 15 Å to 20 Å from the oncocin (shown in cyan) is confined with position restraints.
Molecular dynamics simulation and free energy calculations for the entire ribosome are computationally expensive. We propose that a reduced system can be used to estimate binding energies because binding affinity is determined primarily from local interactions. Residues with any atom within 20 Å of the oncocin were included in the reduced system (Figure 1 C,D). The reduced system was stabilized by defining a buffer region between 15 Å to 20 Å of the oncocin. The heavy atoms in the buffer region (15-20 Å) were subjected to position restraints over the course of the simulation to stabilize the reduced system. In contrast, all the atoms within 15 Å of oncocin were allowed to move freely. The buffer dimensions were chosen to include the predominant short-range interactions (typically within 12 Å), while limiting the computational cost associated with a larger volume. A similar reduced system approach was implemented to calculate the binding free energy of sparsomycin to the ribosomalpeptidyl-transferase P-site.33 The predicted binding free energy was in reasonable agreement with the experimental value. The reduced system was solvated with water molecules with a thickness of 10 Å on each side. As long as the water layer is large enough to prevent the interactions of the peptide-ribosome system with its periodic image, the choice of thickness does not affect the binding free energy. This is evidenced by the similar binding free energy of peptide p41 bound to the SH3 domain of tyrosine kinase Abl.34,35 In addition, magnesium and chloride ions were added to neutralize the system.
2.8. Molecular Dynamics Simulation Details
All MD simulations in this work were performed with NAMD2.1236 with a time step of 2 fs. The CHARMM36 force field was used for proteins and RNAs. 37 TIP3P model was used for water molecules.38 Energy minimization was performed for 2000 steps. Constant temperature and pressure simulations were performed with harmonic restraints enforced on all the protein and RNA heavy atoms to relax the system at 300 K and 1 bar for 500 ps. Subsequently, the restraints on oncocin were removed and ran for 1 ns. Finally, all the restraints except in the buffer region were relaxed for full production. The pressure coupling at 1 atm was accomplished using the Langevin piston,39,40 while temperature control was achieved via the Langevin thermostat.41 The oscillation period and damping time for pressure control were 50 ps and 25 ps, respectively. The damping coefficient for temperature control was set to 1 ps−1. The electrostatic interactions were calculated by the particle-mesh Ewald (PME) method.42 The grid spacing for the PME method was set to 1 Å. The van der Waals interactions were smoothly turned off between 10 and 12 Å with the force switching method. 43
2.9. Relative Binding Free energy Calculations
The relative binding free energy calculations were based on our previous work.44 Briefly, the relative binding free energy (ΔΔGbind) between the wild-type and the mutant was based on free energy perturbation.35,45 Instead of calculating the absolute binding free energy for the wild-type and for mutants directly and independently, the free energy change due to a mutation was estimated in bulk and in bound phases, and respectively. The alchemical transformation for a mutation is carried out with the dual topology approach.46,47 Both the initial state (λ = 0) and the final states (λ = 1) are defined concurrently. As the MD simulation progresses, the potential energy is properly scaled based on the λ value. For non-alanine mutations, the double-alanine-transformation scheme was applied. For instance, the relative binding free energy between leucine (L) and lysine (K) in this method is equivalent to ΔΔG(L → K) = ΔΔG(L → A)+ΔΔG(A → K) = ΔΔG(L → A) − ΔΔG(K → A). This gave a better convergence than transforming directly from L → K, according to our previous study.44
The system setup for MD simulations is summarized here. The system for the bound phase was the same as that described in the previous section. The bulk phase contained onc18 analogs with waters at least 20 Å on each side. The free energy perturbation transformation was evenly divided into 80 windows. MD simulation was performed for each window for 50,000 production steps after 5,000 equilibration steps. The simulation was carried out bi-directionally (forward and backward) in both aqueous solution and binding site. The free energy change and statistical uncertainty were calculated by means of the Bennett acceptance ratio (BAR),48 combining statistical data assessed from both forward and backward directions using the ParseFEP tool.49
2.10. Statistical analysis of MIC values
MIC, the lowest tested peptide concentration that shows activity, was determined using two-fold serial dilutions of peptide. The true MIC value is less than or equal to the last dilution inhibiting growth and greater than the first dilution not inhibiting growth. We developed a statistical analysis based on censoring statistics, where the exact values of the outcome are not observed and only the boundaries or the intervals are known.50 The noise of the average MIC was assumed to be normally distributed; applying a binned filter results in the following piecewise probability between two values a and b from a normal distribution
| (5) |
| (6) |
where μ is the MIC value, σ is the standard deviation and X is the experimental data. To compute the marginal distribution of μ independent of σ contingent on the observed data, P(μ, σ|X) was integrated over the domain of σ:
| (7) |
Each set of experimental measurements will have a distribution of μ. The MIC value is defined as the mode.
A hypothesis test was performed to evaluate statistical significance of differences between the MIC values of the mutants and the wild-type oncocin. The null hypothesis is that both peptides have the same MIC values μα = μβ.
Three random values were drawn according to the cumulative probability distributions of μα and μβ. The mean of the μα set was compared to the mean of the μβ set. This process was repeated 5000 times. If 95% of the trials were μα < μβ (or 95% of trials were μα > μβ), their p-value is <0.05; if so, the null hypothesis was rejected. The MATLAB script used to perform the statistical analysis for our proposed method is provided in the SI.
3. Results and Discussion
3.1. Activity of Monosubstituted Oncocin Analogs
The antimicrobial activity of 209 Onc18 variants – representing all 19 natural amino acid variants at each of the first 11 sites – was evaluated against E. coli. Strain JW0013 was chosen because it lacks the secondary Onc18 target heat shock protein DnaK, which enables us to focus on ribosome binding and transport. The Onc18 variants were obtained from SPOT synthesis, which is an array-based synthesis strategy that provides time- and cost-efficient production of peptides. The antimicrobial activity of each analog was assessed by the MIC against JW0013. The greatest decrease in MIC relative to wild-type (2.8-fold to 1.7 ± 0.8 μg/mL, p < 0.05) was achieved via mutation of proline at site 4 to basic lysine or arginine (Figure 2). Arginine mutations at L7 and P10 reduced the MIC twofold while lysine mutations at these sites were nominally beneficial. Mutation of the proline at site 5 to mildly cationic histidine, as well as neutral alanine, reduced the MIC twofold. A host of other mutations, including Y6R, yielded modest nominal reductions in the MIC. Beyond specific beneficial mutations, much can be learned from the relative tolerance of each site to mutation. Five of the 11 tested sites (V1, D2, P4, P5, and R9) were generally tolerant of mutation with at least 12 mutant amino acids providing activity >50% of wild-type. Conversely, K3, L7, and P8 were especially restrictive with 0, 2, and 1 mutations, respectively, that did not substantially hinder activity. Other sites exhibited intermediate tolerance. Notably, mutational tolerance at a given site did not correlate with diversity at that site in natural homologs (Figure S3).
Fig. 2.
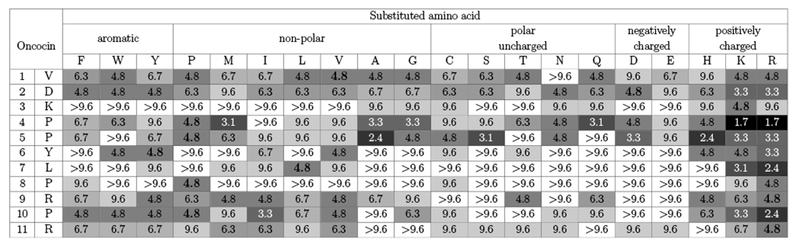
E. coli inhibition from monosubstituted Onc18. The first two columns denote the site and amino acid of the first 11 residues. Each subsequent column represents the minimum inhibitory concentration (MIC, in μg/mL) of an amino acid replacement at each position. The values are the mean MIC (n = 3) evaluated by dilution test against E. coli strain JW0013. Boxes are color coded: darker colors indicate lower MIC values and higher activity, and white color represents no activity at the highest concentration. The wild-type Onc18 residues are emboldened.
Of 20 monosubstituted mutants that showed improved activity compared to the wild-type, 11 (55%) were to positively charged lysine or arginine substitutions, which – considering that only 9% of all mutants were cationic – suggests the importance of the cationic amino acids to the improved activity. In addition, evaluation of activity as a function of peptide charge reveals that as the net charge increased from 3e to 7e, peptides became more active, though with dispersed MIC values (Figure S4). An analogous work using SPOT synthesis to improve oncocin activity against Gram-positive pathogen was studied by Daniel et al.23 The authors performed systematic mutagenesis of the entire Onc18 against P. aeruginosa reporter strain H1001 and also found that lysine and arginine substitutions were more favorable. Most of the favorable substitutions were on the C-terminal region (P12 to R19) for Gram-positive bacteria, which were not included for substitutions in this work. It would be informative to extend mutagenesis to the oncocin C-terminal region against Gram-negative bacteria as a future study for comparison.
3.2. Activity of Multisubstituted Oncocin Analogs
From the results of the monosubstituted analogs, we combined several favorable substitutions to design multisubstitution analogs for further evaluation (Table 2). Because the positively charged substitutions displayed better improvement, we focused on the combinations of histidine, lysine and arginine. Since P4K and P4R mutants showed strong improvement, the proline was replaced with either lysine or arginine residue. For other positions, the wild-type amino acid was also included. The histidine substitution at position 5 was included with other lysine and/or arginine substitutions at position 6, 7 and 10. While additional mutations that exhibited modest improvement could have been included, we desired to keep combinatorial diversity constrained for experimental efficiency. The resultant 72 multisubstituted peptides were SPOT synthesized and evaluated for inhibition of E. coli strain JW0013 (Table 3 and Table S3).
Table 2.
Amino acid substitutions in Onc18 allowed in the multisubstitution library based on improved MIC values against E. coli strain JW0013 as monosubstitutions. The proline with asterisk at position 4 is completely replaced with lysine and arginine residues
| Position | Original amino acid (first 11 residues) | Favorable substitution | Tested variants |
|---|---|---|---|
| 1 | V | 1 | |
| 2 | D | 1 | |
| 3 | K | 1 | |
| 4 | P* | K,R | 2 |
| 5 | P | H | 2 |
| 6 | Y | R | 2 |
| 7 | L | K,R | 3 |
| 8 | P | 1 | |
| 9 | R | 1 | |
| 10 | P | K,R | 3 |
| 11 | R | 1 | |
| total | 72 | ||
Table 3.
Antimicrobial activity of select multisubstituted oncocin derivatives from SPOT synthesis against E. coli strain JW0013. The net charge of histidine is assigned 0.1. The MIC were averaged from at least three samples at different days. The values indicate the mode ± the average difference between the mode and the limits of the 68% confidence interval. * denotes p<0.05. The results of other peptides are in Table S3
| Sequence (First 11 amino acids) | Net Charge | MIC [μg/mL] | Factor of improvement |
|---|---|---|---|
| VDKPPYLPRPR | +5 | 3.1±1.3 | – |
| P4K | +6 | 1.2±0.3 | 2.6* |
| P4K, P10K | +7 | 1.2±0.3 | 2.6* |
| P4K, L7K | +7 | 0.6±0.1 | 5.2* |
| P4K, L7K, P10R | +8 | 1.2±0.3 | 2.6* |
| P4K, L7R | +7 | 0.4±0.2 | 7.8* |
| P4K, P5H, L7K | +7.1 | 0.8±0.4 | 3.9* |
| P4R | +6 | 1.7±0.8 | 1.8 |
| P4R, L7K | +7 | 0.8±0.4 | 3.9* |
| P4R, L7R | +7 | 1.2±0.3 | 2.6* |
| P4R, P5H, L7R | +7.1 | 0.8±0.4 | 3.9* |
In this iteration, the P4K analog was nominally more active than the P4R analog. This might result from slightly different peptide purity or yield of the first and second SPOT synthesis. The additional substitutions of L7R or L7K combined with the P4K analog further improved the activity to 7.8- or 5.2-fold, respectively, stronger than the wild-type (p < 0.05 for both). These were the most active analogs among all multisubstituted peptides (MIC = 0.4 ± 0.2 and 0.6 ± 0.1 ng/mL). Similarly, lysine or arginine substitutions at position 7 within the P4R context also further improved the activity. The addition of the P5H substitution to other mutants showed no significant improvement in activity. Moreover, substitution to lysine or arginine at position 10 also had no improvement.
While multiple neutral-to-cationic mutations – including several combinations – provided benefit, improved activity required more than an increased net charge. Rather, the particular site and cationic amino acid impacted performance. Evaluation of activity as a function of peptide charge (Figure 3) reveals that as the net charge increased from 5e (wild-type) to 6e or 7e, peptides became more active but with MIC values dispersed from 0.4 to 2.4 μg/mL. This indicates that the net charge was not the only determining factor for antimicrobial activity. As the net charge further increased, peptide activities decreased significantly.
Fig. 3.
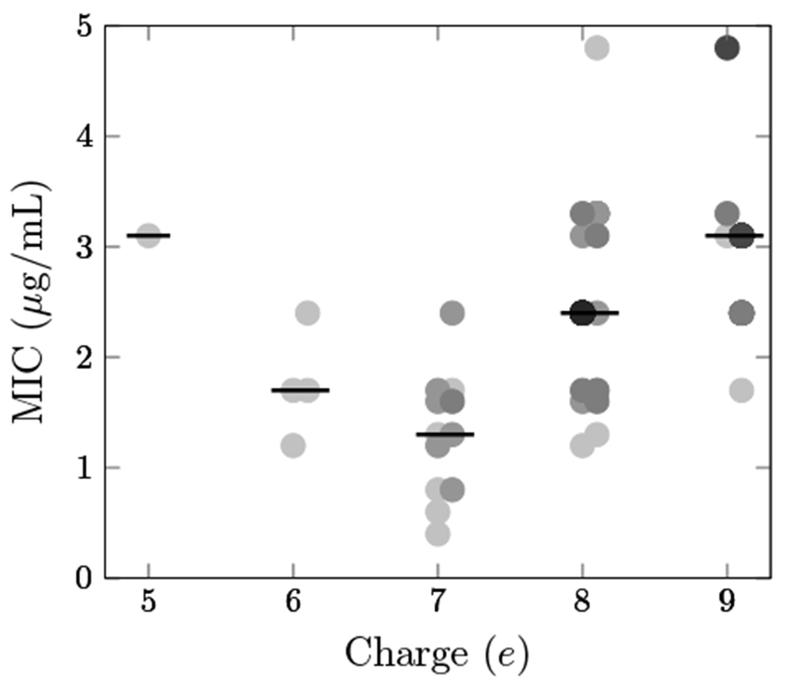
Peptide activity varies with net charge. MIC on E. coli JW0013 is plotted against net charge for 72 multisubstituted variants of Onc18 produced via SPOT synthesis. The darkness of each data point is proportional to the number of overlapping peptides at that MIC/charge combination. Lines indicate median values.
3.3. Activity of Optimized Peptides
Peptide activity was then evaluated, across multiple E. coli and Salmonella species, for a subset of peptides in a more purified form (Table 4). Given their efficacy in the previous activity assay, all combinations of lysine and arginine substitutions of proline at position 4 and leucine at position 7 were evaluated, as well as the wild-type Onc18. It is worth noting that the sequence of the purified peptides are slightly different from the SPOT synthesized peptides: the former has a C-terminal amidation and the latter has an additional glycine at the C-terminus without amidation. Consistent with SPOT synthesis results, three of the four double-cationic variants at sites 4 and 7 (P4K/L7K, P4K/L7R, and P4R/L7R) substantially aid activity (2.1 - 2.8-fold). P4K showed nominally improved activity against E. coli JW0013 while the arginine counterpart did not show any improvement. Both lysine and arginine substitutions at position 7 were nominally more active. Comparing the results from Table 3 and Table 4, the enhanced activity of the mutants was maintained in this form. Also the amidated forms were about 3.8-fold more active (from 3.0 μg/mL to 0.8 μg/mL) for the wild-type against E. coli JW0013. Moreover, the peptides P4K, P4R, P4K/L7K, were approximately 2-fold more active for the C-amidated form. The difference in activity may result from differences in purity and yield or increased uptake stability, or activity due to amidation. The C-amidated form was found more active against E. coli for wild-type oncocin.18 Our results were consistent with their observations.
Table 4.
Antimicrobial activity of purified oncocin derivatives against multiple Gram-negative bacteria. The substituted residues are emboldened. The values indicate the mode ± the average difference between the mode and the limits of the 68% confidence interval. The MIC values that are at least 2 times smaller than the wild-type are shown in bold. * denotes p<0.05
| MIC (μg/mL) |
|||||||
|---|---|---|---|---|---|---|---|
| peptides | E. coli JW0013 | E. coli O157:H7 | E. coli MC1061 | E. coli J2210 | E. coli JJ2487 | Salmonella MH91989 | Salmonella SL1344 |
| VDKPPYLPRPRPPRRIYNR-NH2 | 0.83 ± 0.42 | 3.14 ± 1.28 | 0.30 ± 0.07 | 0.79 ± 0.35 | 0.60 ± 0.14 | 0.60 ± 0.14 | 0.60 ± 0.14 |
| VDKKPYLPRPRPPRRIYNR-NH2 | 0.60 ± 0.14 | 2.40 ± 0.56 | 0.30 ± 0.07 | 0.39 ± 0.18 | 0.60 ± 0.14 | 0.60 ± 0.14 | 0.60 ± 0.14 |
| VDKRPYLPRPRPPRRIYNR-NH2 | 0.83 ± 0.42 | 2.40 ± 0.56 | 0.30 ± 0.07 | 0.60 ± 0.14 | 0.39 ± 0.18 | 0.60 ± 0.14 | 0.60 ± 0.14 |
| VDKPPYKPRPRPPRRIYNR-NH2 | 0.60 ± 0.14 | 2.40 ± 0.56 | 0.15 ± 0.03* | 0.30 ± 0.07* | 0.79 ± 0.35 | 0.60 ± 0.14 | 0.60 ± 0.14 |
| VDKPPYRPRPRPPRRIYNR-NH2 | 0.42 ± 0.21 | 2.40 ± 0.56 | 0.20 ± 0.09 | 0.39 ± 0.18 | 0.39 ± 0.18 | 0.60 ± 0.14 | 0.42 ± 0.21 |
| VDKKPYKPRPRPPRRIYNR-NH2 | 0.30 ± 0.07* | 1.57 ± 0.68 | 0.15 ± 0.03* | 0.20 ± 0.09* | 0.30 ± 0.07* | 0.42 ± 0.21 | 0.42 ± 0.21 |
| VDKKPYRPRPRPPRRIYNR-NH2 | 0.39 ± 0.18 | 1.57 ± 0.68 | 0.15 ± 0.03* | 0.30 ± 0.07* | 0.30 ± 0.07* | 0.39 ± 0.18 | 0.39 ± 0.18 |
| VDKRPYKPRPRPPRRIYNR-NH2 | 0.83 ± 0.42 | 2.40 ± 0.56 | 0.15 ± 0.03* | 0.30 ± 0.07* | 0.39 ± 0.18 | 0.39 ± 0.18 | 0.39 ± 0.18 |
| VDKRPYRPRPRPPRRIYNR-NH2 | 0.39 ± 0.18 | 1.66 ± 0.82 | 0.15 ± 0.03* | 0.30 ± 0.07* | 0.42 ± 0.21 | 0.42 ± 0.21 | 0.39 ± 0.18 |
In addition to the DnaK knockout E. coli strain, we also tested the antimicrobial activities of the purified peptides against other Gram-negative E. coli strains. The aforementioned double substituted variants P4K/L7K, P4K/L7R and P4R/L7R analogs were generally the most potent, with 2.0-3.9-fold increase in potency. In evaluation against additional Gram-negative species, the optimized peptides showed only slight improvement against Salmonella MH91989 and Salmonella SL1344.
3.4. In vitro GFP Translation Inhibition
To investigate the mechanism for improved antimicrobial activities, the optimized peptides were evaluated for translational inhibition potency. In vitro transcription/translation of GFP was performed in the presence or absence of Onc18 variants, titrated from 2 - 200 μg/mL (Fig. 4). Onc18 and its variants inhibit protein expression presumably via ribosomal binding. The IC50 values were in the range of 20.5 to 34.5 μg/mL. This indicated their binding affinity to the ribosome was approximately the same. This showed that the increased antimicrobial activity might not result from stronger binding to the ribosome. Wild-type Onc18 had the nominally lowest IC50 value compared to other optimized peptides. Although the first 11 residues were selected for optimization because of their presence in the binding pocket, there are seemingly other factors that contribute to improved antimicrobial activities.
Fig. 4.
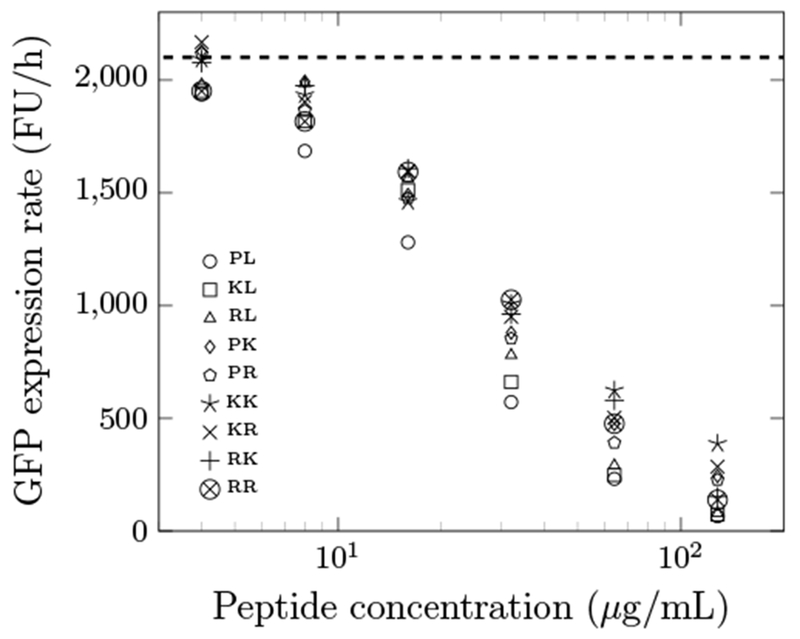
GFP expression rate of the optimized peptides at different concentrations. FU/h indicates relative fluorescence units per hour, which was computed as the slope of the fluorescence signal versus time. The dashed line is the negative control without any AMPs. The data are the average from a triplicate. The average standard deviation is 13% of the average. The maximum standard deviation is 24% of the average. The first and second letters of the legend indicate residues at position 4 and 7, respectively.
3.5. Relative Binding Free Energy from MD Simulations
Relative binding free energy calculations from MD simulations have been widely applied to ligand-receptor binding system and drug discovery.51,52 It is instructive to assess if the computational protocol is applicable to PrAMPs and the ribosome binding complex. We performed free energy perturbation analysis using MD simulations to calculate free energy changes upon binding (Table 5).
Table 5.
Relative binding free energy of the optimized peptides compared to the wild-type oncocin from MD simulations. The free energy change and uncertainty were calculated from the Bennett acceptance ratio of the forward and backward transformations. Negative ΔΔG indicates more favorable binding
| ΔΔG (kcal/mol) | |
|---|---|
| P4K | −0.7 ± 0.2 |
| P4R | −3.2 ± 0.2 |
| L7K | −2.2 ± 0.2 |
| L7R | −5.8 ± 0.2 |
| P4K/L7K | 1.6 ± 0.3 |
| P4K/L7R | −2.7 ± 0.3 |
| P4R/L7K | −2.8 ± 0.2 |
| P4R/L7R | −3.9 ± 0.3 |
Except for P4K that showed little change and P4K/L7K that was computed to be an unfavorable mutation, optimized peptides were predicted to bind the ribosome more strongly than the WT. These calculations did not agree with the results from GFP translation experiment (Fig. 4), which indicated comparable potency. There could be several reasons for the discrepancy between simulations and experiment. One possible reason is that the CHARMM36 force fields might not describe the interactions between peptides and RNA with sufficient accuracy. The DNA and RNA force fields were optimized separately from protein force fields to have better conformation sampling.53,54 Comparison of other force fields for the interactions between peptides and nucleic acids may be required to improve accuracy. Another reason might be that the free energy perturbation protocol assumes that the mutants are already at the binding site without explicitly considering the insertion pathway. The additional positive charges on the mutants may interfere with the ribosomal insertion. Moreover, the additional charges may drive the mutants to associate with other intracellular molecules to induce off-target effects. Thus, care must be taken if one intends to optimize PrAMPs based on this MD approach.
3.6. Internalization Rate of the Optimized Peptides
We hypothesized that enhanced cell association and internalization could be a mechanism for the observed increase in potency of the Onc18 variants especially considering the potential for cations to interact with negatively charged moieties on the cell surface. Thus, we devised an assay to measure cell association and internalization. Peptide was incubated with E. coli cells, and at various times cell-associated peptide was removed via centrifugation. Cell-free peptide was quantified via HPLC, and a cell association/internalization rate constant was computed assuming first-order kinetics. WT Onc18 exhibits slow association/internalization whereas the P4K/L7K mutant exhibits 7.7 ± 0.6-fold faster dynamics (τ1/2 = 33.3 ± 19.4 min, Fig. 5AB). On the contrary, in 100% M9 broth, the association/internalization rate for both peptides were slower and the rate constant for the P4K/L7K mutant was only 2.4 ± 0.3 times higher than the WT (p<0.05, Fig. 5CD).
Fig. 5.
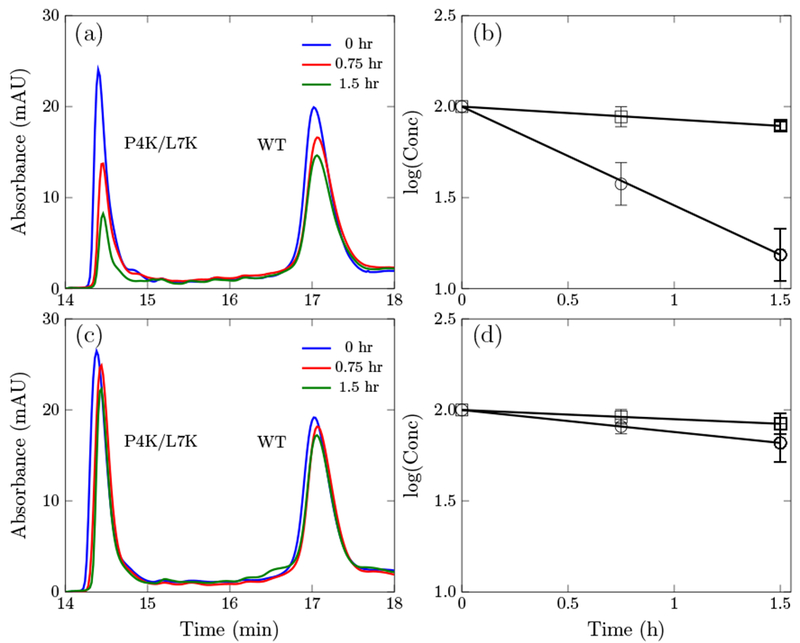
Absorbance at 280 nm of the wild-type oncocin (WT) and the P4K/L7K analog at different time point and the corresponding kinetic curve. (a) and (b) are in 20% M9 growth media. (c) and (d) are in 100% M9 growth media.
To ensure the decrease of peptides in the supernatant were not because of degradation by protease secreted from cells, the mutant was added to a cell-free supernatant. The peptides remained intact after two hours indicating no degradation occurred outside of cells (Figure S5). Moreover, the full protocol was also designed with the ability to observe degraded peptide, but no degraded product was detected during the time course of the assay.
3.7. Growth Media Dependence of the Antimicrobial Activity
Antimicrobial activity is known to be dependent on the growth medium.55 The commonly used media for oncocin studies are 1% tryptic soy broth (TSB) or 12.5% MHB.18,21 It is informative to evaluate if oncocin and the optimized peptides remain potent in the minimal M9 broth, which was used in the internalization and kinetic assay. Table 6 shows the MIC values of the oncocin and the two most potent mutants P4K/L7K and P4K/L7R in 20%, 60% and 100% of MHB and minimal M9 broth. The peptides were less active in concentrated media relative to that in diluted media. The two mutants showed equal potency as the wild-type in the 100% MHB broth (2.40 μg/mL), and they were 4-fold less potent in the 100% M9 broth (9.60 μg/mL, p<0.05 vs. wild-type). However, the mutants became more potent than the wild-type in both 20% MHB and M9 broths.
Table 6.
Antimicrobial activity of the optimized peptides against E. coli strain JW0013 in different concentrations of Mueller-Hinton broth (MHB) and in minimal M9 broth. The values indicate the mode ± the average difference between the mode and the limits of the 68% confidence interval. * denotes p<0.05
| MIC (μg/mL) |
||||||
|---|---|---|---|---|---|---|
| MHB | M9 | |||||
| peptides | 20% | 60% | 100% | 20% | 60% | 100% |
| VDKPPYLPRPRPPRRIYNR-NH2 | 0.79 ± 0.35 | 1.20 ± 0.28 | 2.40 ± 0.56 | 0.60 ± 0.14 | 1.20 ± 0.28 | 2.40 ± 0.56 |
| VDKKPYKPRPRPPRRIYNR-NH2 | 0.20 ± 0.09* | 0.79 ± 0.35 | 2.40 ± 0.56 | 0.30 ± 0.09* | 1.57 ± 0.68 | 9.60 ± 2.00* |
| VDKKPYRPRPRPPRRIYNR-NH2 | 0.20 ± 0.09* | 0.60 ± 0.14* | 2.40 ± 0.56 | 0.39 ± 0.18 | 1.20 ± 0.28 | 9.60 ± 2.00* |
The difference in transportation rate observed in the internalization assay may provide explanation to the media dependent potency for the peptides. In rich media, the internalization for the peptides were markedly slow, and the potency of the AMPs were hampered by transportation inefficiency. As a result, the peptides were less potent in rich media. On the contrary, the increased internalization rate in poor media facilitated peptide transportation and improved activity. The relative rate constant of the P4K/L7K mutant to the wild-type increased from 2.4 to 7.7 times from 100% to 20% M9 broth. The difference in transportation could be due to a varying number of receptor expression. In rich media, the cells need less receptors for the uptake of nutrients and consequently the internalization rate for the peptides was reduced. In poor media, the cells strive to obtain more nutrients, and the number of transport receptors may increase thereby increasing internalization.
It is reported that there are at least two inner membrane receptors facilitating PrAMP transportation. The inner membrane proteins YgdD and SbmA are essential for the complete susceptibility of E. coli.56 We reason that in low nutrient media, one of the receptors may be overexpressed which favors the association and transportation of the P4K/L7K mutant.
To investigate the expression level of receptors in different growth media, we carried out Western blot analysis to quantify SbmA in 20% and in 100% M9 media (Fig 6). SbmA expression was increased by 83% (p = 0.004) in the reduced medium relative to the rich medium. This supports our hypothesis that the enhanced antimicrobial activity for P4K/L7K and P4K/L7R mutants benefited from increasing SbmA receptors in poor media. Unfortunately, the atomic structure and the function of the SbmA receptors are still unknown. Future work is needed to study the underlying mechanisms of the transportation through these receptors. Additional factors for media-dependent antimicrobial activities include AMP binding to media components and the impact of osmolarity. Further experiments are needed to assess these hypotheses.
Fig. 6.
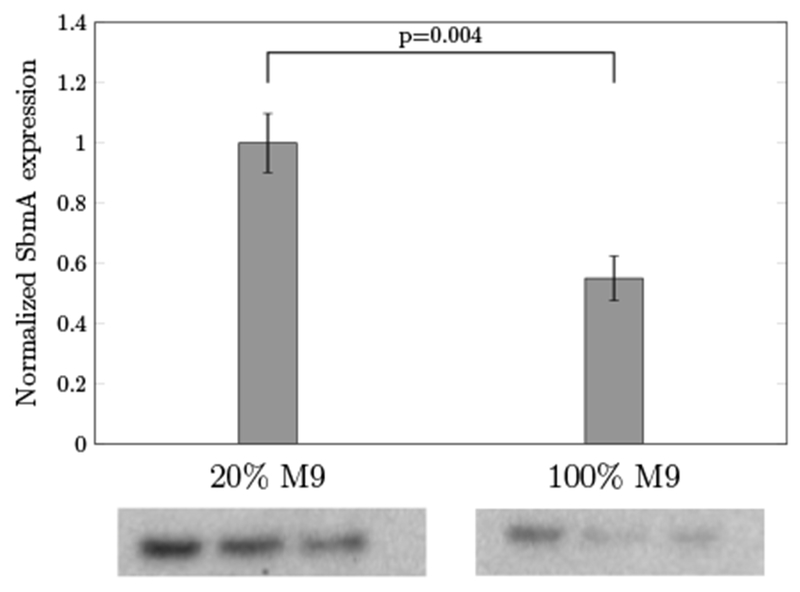
Quantification of SbmA grown in 20% and in 100% of M9 media by Western blot analysis. Values represent the mean ± the standard deviation of triplicate samples.
4. Conclusions
In this work, we performed systematic mutagenesis of the N-terminal 11 sites of Onc18 antimicrobial peptide. Positively charged lysine and arginine substitutions increased potency at several positions. Combinations of these favorable substitutions revealed that replacement of the proline at position 4 and leucine at position 7 with lysine or arginine could further increase the activity. The optimized sequences were 2-4 fold more active than the wild-type oncocin against a wide range of Gram-negative bacteria. An in vitro GFP transcription/translation inhibition assay indicated that the increased activity might not be attributed to stronger binding to the ribosome. An HPLC-based cell association and internalization kinetic assay revealed dramatically faster membrane binding and transport of the oncocin variants compared to the wild type that may be a key factor for higher antimicrobial activity. The dual potency of the oncocin variants in different growth media conditions provide new insights into the mechanisms of antimicrobial transport. This work provided a systematic approach to design and engineer AMPs to optimize their functions and can be applied to other PrAMPs.
Supplementary Material
Acknowledgements
This work was supported by grants from the National Institutes of Health (R01GM121777 and R01GM111358) and by a grant from the National Science Foundation (CBET-1412283). We acknowledge computational support from the Minnesota Supercomputing Institute and from the Extreme Science and Engineering Discovery Environment, which is supported by National Science Foundation ACI-10535753.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/b000000x/
Conflicts of interest
Y. Kaznessis is the founder and president of General Probiotics, Inc., a startup company that aims to commercialize technologies based on antimicrobial probiotics. K. Geldart is a current employee of General Probiotics, Inc. This interest was reviewed and managed by the University of Minnesota in accordance with its Conflict of Interest policies.
References
- 1.Sengupta S, Chattopadhyay MK and Grossart H-P, Front Microbiol, 2013, 4, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gould IM and Bal AM, Virulence, 2013, 4, 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wright G, Nature, 2015, 517, 442. [DOI] [PubMed] [Google Scholar]
- 4.Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, Mueller A, Schäberle TF, Hughes DE, Epstein S et al. , Nature, 2015, 517, 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boumehira AZ, El-Enshasy HA, Hacène H, Elsayed EA, Aziz R and Park EY, Biotechnol. Bioprocess Eng, 2016, 21, 199–223. [Google Scholar]
- 6.World Health Organization, Antimicrobial resistance: global report on surveillance, World Health Organization, 2014. [Google Scholar]
- 7.Simpkin VL, Renwick MJ, Kelly R and Mossialos E, J. Antibiot, 2017, 70, 1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moual H, Thomassin J and Brannon J, J Clin Cell Immunol, 2013, 13, 2. [Google Scholar]
- 9.Wang G, Li X and Wang Z, Nucleic Acids Res, 2015, 44, D1087–D1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scocchi M, Tossi A and Gennaro R, Cell. Mol. Life Sci, 2011, 68, 2317–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krizsan A, Volke D, Weinert S, Sträter N, Knappe D and Hoffmann R, Angew. Chem. Int. Ed. Engl, 2014, 53, 12236–12239. [DOI] [PubMed] [Google Scholar]
- 12.Li W, Tailhades J, O’Brien-Simpson NM, Separovic F, Otvos L, Hossain MA and Wade JD, Amino Acids, 2014, 46, 2287–2294. [DOI] [PubMed] [Google Scholar]
- 13.Graf M, Mardirossian M, Nguyen F, Seefeldt AC, Guichard G, Scocchi M, Innis CA and Wilson DN, Nat. Prod. Rep, 2017, 34, 702–711. [DOI] [PubMed] [Google Scholar]
- 14.Mattiuzzo M, Bandiera A, Gennaro R, Benincasa M, Pacor S, Antcheva N and Scocchi M, Mol. Microbiol, 2007, 66, 151–163. [DOI] [PubMed] [Google Scholar]
- 15.Guida F, Benincasa M, Zahariev S, Scocchi M, Berti F, Gennaro R and Tossi A, J. Med. Chem, 2015, 58, 1195–1204. [DOI] [PubMed] [Google Scholar]
- 16.Zahn M, Kieslich B, Berthold N, Knappe D, Hoffmann R and Strater N, Protein Pept. Lett, 2014, 21, 407–412. [DOI] [PubMed] [Google Scholar]
- 17.Roy RN, Lomakin IB, Gagnon MG and Steitz TA, Nat. Struct. Mol. Biol, 2015, 22, 466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knappe D, Piantavigna S, Hansen A, Mechler A, Binas A, Nolte O, Martin LL and Hoffmann R, J. Med. Chem, 2010, 53, 5240–5247. [DOI] [PubMed] [Google Scholar]
- 19.Knappe D, Zahn M, Sauer U, Schiffer G, Sträter N and Hoffmann R, Chembiochem, 2011, 12, 874–876. [DOI] [PubMed] [Google Scholar]
- 20.Knappe D, Kabankov N and Hoffmann R, Int J Antimicrob Agents, 2011, 37, 166–170. [DOI] [PubMed] [Google Scholar]
- 21.Knappe D, Goldbach T, PD Hatfield M, Y Palermo N, Weinert S, Sträter N, Hoffmann R and Lovas S, Protein Pept. Lett, 2016, 23, 1061–1071. [DOI] [PubMed] [Google Scholar]
- 22.López-Pérez PM, Grimsey E, Bourne L, Mikut R and Hilpert K, Front. Chem, 2017, 5, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knappe D, Ruden S, Langanke S, Tikkoo T, Ritzer J, Mikut R, Martin LL, Hoffmann R and Hilpert K, Amino Acids, 2016, 48, 269–280. [DOI] [PubMed] [Google Scholar]
- 24.Hoffmann R, Knappe D, Hilpert K, Mikut R and Ruden S, Modified Apidaecin Derivatives as Antibiotic Peptides, 2012, United States Patent Application 14/346,624.
- 25.Seefeldt AC, Nguyen F, Antunes S, PÃl’rÃl’baskine N, Graf M, Arenz S, Inampudi KK, Douat C, Guichard G, Wilson DN and Innis CA, Nat. Struct. Mol. Biol, 2015, 22, 470–475. [DOI] [PubMed] [Google Scholar]
- 26.Hilpert K, Winkler DF and Hancock RE, Nat. Protoc, 2007, 2, 1333. [DOI] [PubMed] [Google Scholar]
- 27.Portwich M, Keller S, Strauss HM, Mahrenholz CC, Kretzschmar I, Kramer A and Volkmer R, Angew. Chem. Int. Ed. Engl, 2007, 46, 1654–1657. [DOI] [PubMed] [Google Scholar]
- 28.Dinos G, Wilson DN, Teraoka Y, Szaflarski W, Fucini P, Kalpaxis D and Nierhaus KH, Mol. Cell, 2004, 13, 113–124. [DOI] [PubMed] [Google Scholar]
- 29.Cormack BP, Valdivia RH and Falkow S, Gene, 1996, 173, 33–38. [DOI] [PubMed] [Google Scholar]
- 30.Dunkle JA, Xiong L, Mankin AS and Cate JHD, Proc. Natl. Acad. Sci. U.S.A, 2010, 107, 17152–17157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC. [Google Scholar]
- 32.Basic Local Alignment Search Tool (BLAST), https://blast.ncbi.nlm.nih.gov/Blast.cgi, [Online; accessed 12-June-2018].
- 33.Ge X and Roux B, J. Mol. Recognit, 2010, 23, 128–141. [DOI] [PubMed] [Google Scholar]
- 34.Gumbart J, Roux B and Chipot C, Protein: ligand standard binding free energies: A tutorial for alchemical and geometrical transformations, https://www.ks.uiuc.edu/Training/Tutorials/namd/PLB/tutorial-protein-ligand.pdf, [Online; accessed 03-October-2018].
- 35.Gumbart JC, Roux B and Chipot C, J. Chem. Theory Comput, 2012, 9, 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L and Schulten K, J. Comput. Chem, 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klauda JB, Venable RM, Freites JA, OŠConnor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD Jr and Pastor RW, J. Phys. Chem. B, 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW and Klein ML, J. Chem. Phys, 1983, 79, 926–935. [Google Scholar]
- 39.Martyna GJ, Tobias DJ and Klein ML, J. Chem. Phys, 1994, 101, 4177–4189. [Google Scholar]
- 40.Feller SE, Zhang Y, Pastor RW and Brooks BR, J. Chem. Phys, 1995, 103, 4613–4621. [Google Scholar]
- 41.Izaguirre JA, Catarello DP, Wozniak JM and Skeel RD, J. Chem. Phys, 2001, 114, 2090–2098. [Google Scholar]
- 42.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H and Pedersen LG, J. Chem. Phys, 1995, 103, 8577–8593. [Google Scholar]
- 43.Steinbach PJ and Brooks BR, J. Comput. Chem, 1994, 15, 667–683. [Google Scholar]
- 44.Lai P-K and Kaznessis YN, J. Chem. Theory Comput, 2017, 13, 3413–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gumbart JC, Roux B and Chipot C, J. Chem. Theory Comput, 2013, 9, 3789–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pearlman DA, J. Phys. Chem, 1994, 98, 1487–1493. [Google Scholar]
- 47.Gao J, Kuczera K, Tidor B and Karplus M, Science, 1989, 244, 1069–1072. [DOI] [PubMed] [Google Scholar]
- 48.Bennett CH, J. Comput. Phys, 1976, 22, 245–268. [Google Scholar]
- 49.Liu P, Dehez F. o., Cai W and Chipot C, J. Chem. Theory Comput, 2012, 8, 2606–2616. [DOI] [PubMed] [Google Scholar]
- 50.van de Kassteele J, van Santen-Verheuvel MG, Koedijk FDH, van Dam AP, van der Sande MAB and de Neeling AJ, Antimicrob. Agents Chemother, 2012, 56, 1557–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abel R, Wang L, Harder ED, Berne BJ and Friesner RA, Acc. Chem. Res, 2017, 50, 1625–1632. [DOI] [PubMed] [Google Scholar]
- 52.Cournia Z, Allen B and Sherman W, J. Chem. Inf. Model, 2017, 57, 2911–2937. [DOI] [PubMed] [Google Scholar]
- 53.Hart K, Foloppe N, Baker CM, Denning EJ, Nilsson L and MacKerell AD Jr, J. Chem. Theory Comput, 2011, 8, 348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Denning EJ, Priyakumar UD, Nilsson L and Mackerell AD, J. Comput. Chem, 2011, 32, 1929–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang G, Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies, CABI, 2017. [Google Scholar]
- 56.Paulsen VS, Mardirossian M, Blencke H-M, Benincasa M, Runti G, Nepa M, Haug T, Stensvåg K and Scocchi M, Microbiology, 2016, 162, 601–609. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.