

Abstract
Top-down mass spectrometry and direct dissociation of gas phase intact proteins have been demonstrated to be a powerful platform for identifying proteins from complex mixtures and for elucidating post-translational modifications (PTMs). Fragmentation of proteins in the atmospheric pressure/vacuum interface of the electrospray ionization mass spectrometer is an effective dissociation technique that can be utilized for on-line HPLC top-down analysis. We demonstrate the capability to perform intact protein identifications in a single-stage time-of- flight (TOF) mass spectrometer in a data independent (DIA) acquisition fashion by rapidly switching the in-source dissociation (ISD) energy during protein elution from a liquid chromatography (LC) column. The intact protein and product ion masses obtained at low and high ISD energies, respectively, were measured using a TOF mass analyzer. By coupling on-line protein separations to dissociation in the atmospheric pressure/vacuum interface region of the mass spectrometer, we identified proteins in simple complexity mixtures, including subunits from the human 20S proteasome complex, and PTMs such as phosphorylation and N-terminal acetylation events. This proof-of-principle study demonstrates that a data-independent pseudo- MS/MS method could be a relatively in-expensive platform for top-down MS.
Keywords: liquid chromatography, electrospray ionization, top-down mass spectrometry, proteasome, protein sequencing, data independent acquisition
1. Introduction
Mass spectrometry (MS)-based proteomics is a powerful tool available to analyze comprehensive sets of proteins in simple to complex sample mixtures. Two major routes that are commonly used in MS-based proteomics analysis are top-down and bottom-up methodologies. Bottom-up methods utilize proteases that cleave the protein at specific sites to generate smaller fragments that are then searched against sequence databases to identify the protein. Top-down MS (TDMS) strategies provide the intact mass of the molecule being studied and indicate the presence of any isoforms and/or proteoforms [1–3].
Technologies for TDMS and top-down proteomics (TDP) have advanced greatly over the past several decades [4]. Shortly after the development of electrospray ionization (ESI) and the measurement of large proteins by ESI-MS, fragmentation of large polypeptides to greater than 100 kDa by collisionally activated dissociation (CAD) to yield sequence-relatable product ions was demonstrated [5, 6]. With the efficient coupling of ESI to high mass accuracy and high resolving power mass analyzers, such as the Fourier transform ion cyclotron resonance (FTICR), quadrupole time-of-flight (QTOF), and orbitrap analyzers, and the development of ion activation/dissociation methods such as electron capture/transfer dissociation (ECD, ETD) [7] and ultraviolet photodissociation (UVPD) [8], top-down MS/proteomics is emerging as a powerful tool for not only large scale protein identification, but also proteoform elucidation that can impact basic science and translational medical research. Combined with improvements in intact protein separation methods [9], on-line liquid chromatography (LC)-MS/MS TDP protein and proteoform identification is becoming routine in more laboratories [10].
However, to perform top-down MS/proteomics experiments does not necessarily require the most up-to-date and advanced instrument platform and it depends on the goals of the study. In its simplest form, TDMS requires a means to measure the masses of each protein, dissociate the gas phase proteins into products, and measure these products with sufficient accuracy to yield information on sequence, origin (identification), and proteoform identity. Resolving power to unambiguously determine product ion charge could be a requirement for any TDMS platform, but other means to assign product ion mass could be considered, as in the early days of TDMS [11].
Most online TDMS strategies use collisionally activated dissociation (CAD) in a data dependent acquisition (DDA) mode with precursor ion selection [12]. However, with ESI, proteins are multiply charged and their signal is distributed over many channels, i.e., charge states. Often the three most abundant charge states are chosen by the DDA charge state selection algorithm for MS/MS, although there is no rigorous rule in choosing the precursor ion(s). But different precursor charge states of the same protein can yield different MS/MS sequence coverage [13, 14]. DDA of multiple precursor ions can yield lower duty cycle analysis. A data independent acquisition (DIA) approach, in which no precursor ion(s) is selected, i.e., all ions are fragmented, and in-source dissociation (ISD) may be an alternative method.
ISD, referred to as “nozzle-skimmer” dissociation (NSD) in earlier studies, was first reported for peptide and protein analysis by Loo et al. in 1988 [15]; the report showed that the energy bias in the atmosphere-vacuum region of the ESI interface dramatically changed the charge distribution of multiply charged proteins through CAD processes. Later this effect was found to be advantageous for polypeptide sequencing [5]. Since then, many research groups have used this technique for characterizing intact proteins by direct sample infusion [16]. Vellaichamy et al. employed ISD in a LTQ-FT instrument for on-line top-down identification of proteins in yeast cell lysates [17]; as intact proteins eluted from a polymeric column, the ISD voltage was lowered to aid the detection of intact mass and increased to promote protein fragmentation. Since then instruments with dual mass analyzers such as LTQ-FTs [18, 19] and orbitraps [20–22] have been used for on-line top-down protein identification studies, where typically two mass analyzers are utilized to collect the intact and fragment ion data, and in these experiments, CAD in the ion trap or ISD in the source region is used to fragment the protein. The fragmentation obtained by ISD is comparable to CAD and the dissociation in the source region can be used as an additional MS/MS stage to perform a pseudo-MS3 experiment in non-trapping instruments, e.g., triple quadrupole [11, 23] and quadrupole time-of-flight (QTOF) instruments [24].
FTMS instruments, i.e., FT-ICR and Orbritrap systems, are ideal for TDMS because of their high mass accuracy and resolving power. However, other instrument platforms have been used in the past for TDMS, including triple quadrupole [15], quadrupole ion trap [25], and QTOF systems [26]. TOF instruments have only one mass analyzer without a means for precursor ion selection. However, with ISD, ESI-TOF has been used to characterize large proteins, such as light and heavy chain subunits from monoclonal antibodies using NSD in a TOF instrument [27].
We demonstrate an on-line LC-ESI-TOF approach for TDMS using DIA and ISD for protein dissociation. Intact proteins separated by HPLC elute from the column and are subjected to ISD by switching the ISD voltage from “low” to “high” every second; both the intact and product ion masses obtained at low and high voltages, respectively, are measured using the TOF analyzer. This approach is similar to the label-free DIA quantification strategy known as ‘MSE’ in the QTOF instruments for quantifying absolute amounts of peptides in shotgun proteomics experiments [28]. MSE is performed without any precursor ion selection and the energy in the collision cell continuously alternates between a low-energy mode for peptide precursor identification and an elevated-energy mode for generation of peptide fragmentation data. Previously we demonstrated DIA top-down MS using ISD with a high resolving power FT FTICR analyzer [29]. Here we show similar capabilities using a lower performing, yet sufficient resolving power TOF analyzer.
2. Materials and methods
2.1. Materials and sample preparation
Most peptide and protein standards were purchased from Sigma-Aldrich (St. Louis, MO USA) unless otherwise stated. LC-MS grade water was purchased from Baker-VWR (Radnor, PA, USA) and acetonitrile (ACN) was purchased from EMD (Billerica, MA, USA). Solutions of albumin from bovine, porcine, rabbit, and sheep were prepared at 1 mg/mL concentration in MS grade water and diluted to 1.5 µM with 0.1% formic acid (FA). 10 µL of the sample was injected into the Agilent dual ESI source.
Standard proteins (bradykinin, neurotensin, ubiquitin, ribonuclease A, transferrin, bovine serum albumin, and carbonic anhydrase) were made at 1 mg/mL concentration in MS grade water. An equimolar mixture of these proteins (10 pmol each) was prepared by diluting in 0.1% FA. The human 20S (h20S) proteasome complex was purchased from Boston Biochem (Cambridge, MA USA). The h20S proteasome sample (10 pmol) was denatured with 0.5% FA and loaded onto the HPLC column. The HPLC and MS parameters were the same for the analyses of standard proteins and h20S proteasome sample.
2.2. LC-MS parameters
5 µL of the standard protein mix was injected into the Agilent C3 column (pore size 300 Ǻ) with dimensions 2.1 mm × 150 mm. LC-MS was performed with an Agilent 1200 LC system connected on-line with an Agilent 6220 ESI-TOF mass spectrometer (Santa Clara, CA USA) [30]. A resolving power of ca. 15,000 (FWHM) at m/z of 1000 was routinely achieved by this instrument and the mass window can go as high as 3200 m/z in the extended dynamic range mode. Solvent A was 99.9% H2O/0.1% FA and solvent B was 90% ACN/9.9% H2O/0.1% FA. The flow rate was 0.2 mL/min and the following gradient was used: isocratic hold at 5% B for 3 min followed by three steps of linear increases to 25% B at 4 min, 55% B at 34 min, 80% B at 36 min, followed by an isocratic hold at 80% B for 9 min. The column was finally equilibrated with 5% B for 10 min prior to the next run. The column temperature was maintained at 60° C throughout the run.
The MS source parameters were as follows: ESI capillary voltage 4300 V, drying gas temperature 350° C, drying gas flow rate 10 L/min, nebulizer pressure 25 psig, and fragmentor voltage 250 V. To induce ISD during LC run, the fragmentor voltage was altered between 250 V and 350 V every second during the elution window. The data was acquired at the rate of 1 spectrum/sec and the acquisition window was set from m/z 100 to 3000.
The peaks in the total ion chromatogram (TIC) were manually integrated and the mass spectra at different fragmentor voltages were obtained using the Agilent MassHunter Qualitative Analysis software. The multiple charge state distributions of intact proteins were deconvoluted using the Maximum Entropy deconvolution algorithm. The fragment ion spectra were manually analyzed to find the sequence coverage of each protein.
3. Results and discussion
3.1. Optimization of in-source dissociation
ISD in the Agilent ESI-TOF instrument is the fragmentation that occurs as the ions are transported from the back end of the capillary inlet towards the skimmer. This region is under higher pressure than the high vacuum TOF region and it provides higher ion transfer efficiency for large multiply charged product ions. Generally, small molecules and peptides require low fragmentor voltages (175 V) and proteins need higher voltages (225 V) for intact mass analysis. We optimized the fragmentor voltage for protein ISD by comparing the fragmentation efficiency observed at different voltages ranging from 250–375 V using 66 kDa bovine serum albumin (BSA). We found that the signal intensity and number of product ions observed increased up to 350 V and further increase in the voltage did not generate new fragment ions. Therefore, we decided to use 250 V for intact protein analysis and 350 V for fragmentation (Figure 1).
Figure 1.
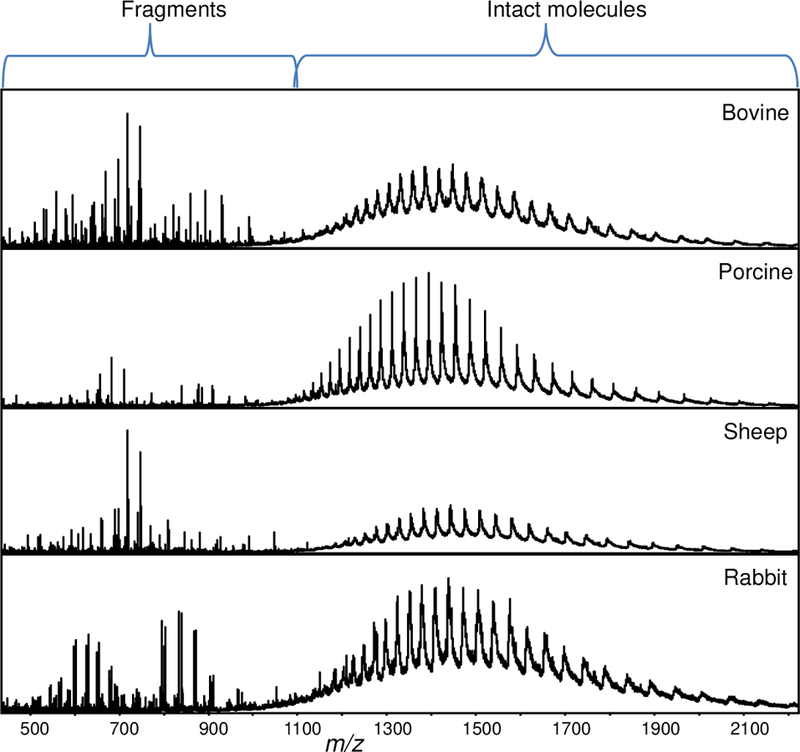
Mass spectra of albumins from different species acquired at high ISD voltage (350 V).
Also, we evaluated the fragmentation of albumin proteins from different species. Sequence alignment of the N-terminal 40 amino acid residues of albumins from bovine, sheep, porcine, and rabbit show that 26 out of 40 residues are conserved among all four species (Figure 2, top). ISD can be a powerful and convenient means to generate CAD-based product ions for TDMS. The mass spectra obtained at 350 V from these different albumin species shows differences in the fragment ions obtained and sequence tags (short stretches of continuous amino acids) that are specific for each albumin species (Figure 2).
Figure 2.

(top) Homology between the N-terminal 40 residues in albumins from different species. Shown in red are the conserved residues and the residues in blue are similar in two or three species. (bottom) Mass spectra of albumins acquired at 350 V ISD voltage. The b-ions pertaining to the N-terminal 30 amino acids obtained at different charge states (5+ in red, 4+ in green, 3+ in blue, and 2+ in purple) are shown. For porcine albumin, a y-ion sequence tag (in black) was also observed.
Loo et al. extensively analyzed albumins from a variety of species using ISD and CAD using a low resolving power triple quadrupole instrument and they reported sequence specific differences from each species [5]. The ESI-TOF data is consistent with the earlier report, showing abundant b-product ions (at 2+ to 6+ charge states) originating from the N-terminal region from all albumin species and a series of y-product ions only for porcine albumin. Moreover, Loo et al. had noticed an error in the published (at that time) rabbit albumin sequence at position 20 and the fragment ions they observed suggested the presence of Leu/Ile instead of Lys. The b193+ and b203+ ions that were observed in the ESI-TOF data confirm their finding.
The mass measurement accuracy of the product ions is sufficient to distinguish lysine (‘K’, incremental mass = 128.094) from glutamine (‘Q’, incremental mass = 128.058). This is illustrated in Table 1 for sheep albumin, which has a ‘Q’ at position 20 from the N-terminus. The measured m/z of fragment ions is compared with the calculated m/z when ‘Q’ is present and if ‘Q’ is replaced by ‘K’. To distinguish K from Q at m/z 2400 (which is the mass of the peptides that originate from residue 20), the error on mass measurement accuracy should be less than 16 ppm. The actual errors are well within 5–6 ppm, which suggests that K and Q can be accurately distinguished with this analyzer. We found that ISD is sufficiently efficient to measure sequence information from the N-terminal 40 residues of all albumins and can distinguish sequence variations among albumins from different species.
Table 1.
Error in mass accuracy between calculated & experimental m/z when ‘Q’ is present at position 20 in sheep albumin is compared to the error in accuracy when ‘K’ is supposed to replace ‘Q’. The deviation in the mass at m/z of 800 is within 6 ppm, which is sufficient to differentiate ‘K’ from ‘Q’.
| Residue position from N-terminus |
Amino acid | Experimental b3+ |
Calculateda b3+ |
Calculatedb b3+ |
Error (ppm) between Calc’da and Expt’l b3+ |
Error (ppm) between Calc’db and Expt’l b3+ |
|---|---|---|---|---|---|---|
| 20 | Q | 790.3648 | 790.3686 | 790.3807 | 4.7488 | 20.0578 |
| 21 | G | 809.3720 | 809.3757 | 809.3878 | 4.5550 | 19.5456 |
| 22 | L | 847.0655 | 847.0704 | 847.0825 | 5.7689 | 20.0925 |
| 23 | V | 880.0877 | 880.0932 | 880.1053 | 6.2342 | 20.0203 |
| 24 | L | 917.7826 | 917.7879 | 917.8000 | 5.7602 | 18.9439 |
b3+ if ‘Q’ is present
b3+ if ‘K’ is present
3.2. DIA of standard protein mixture
To test the ability of DIA/ISD with LC-TOF-MS for protein identification, we measured a 7-protein mixture. The proteins were separated on a reversed-phase C3 column; as the proteins are eluted from the column, the fragmentor voltage was altered from low (250 V) to high (350 V) every second. The intact mass spectra obtained at 250 V and fragmentation spectra generated at 350 V were analyzed to identify the proteins and each peak in the TIC was monitored for the corresponding protein (Figure 3). For small peptides such as bradykinin, 100% sequence coverage was obtained (Figure 4A). For larger proteins such as BSA and 77 kDa transferrin, 7–8 amino acid sequence tags mostly from the ends of the protein were identified (Figures 4B and 4C).
Figure 3.
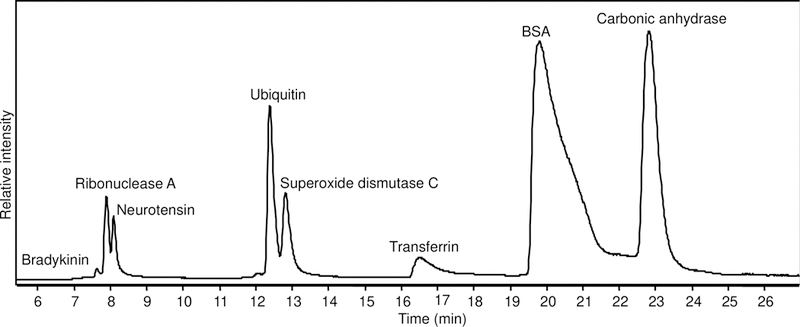
TIC (LC-MS) of standard protein mix run through a C3 column. The peaks are labeled with the corresponding protein eluting after analyzing the intact and fragment ions generated from it. Superoxide dismutase C was a contaminant present in carbonic anhydrase II sample.
Figure 4.
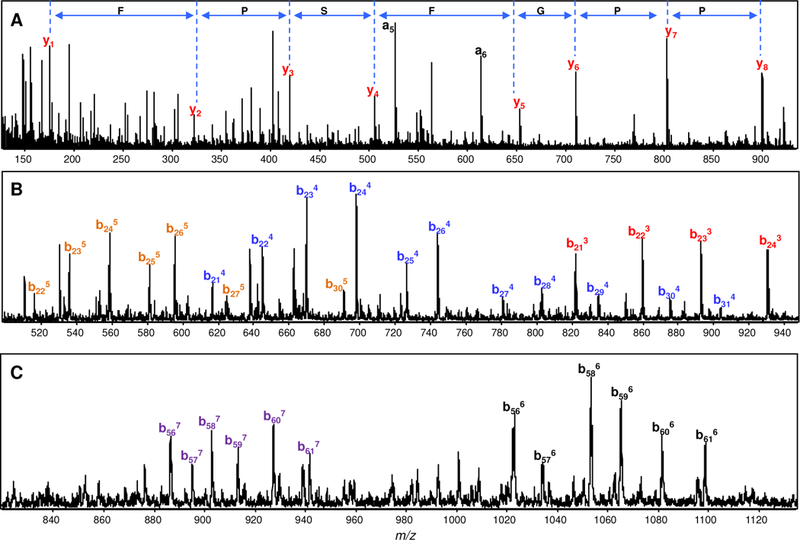
Fragmentation of proteins in protein mixture at high ISD voltage during elution from the column. Mass spectra of (A) bradykinin [1RPPGFSPFR9] showing the y-series sequence tag, (B) BSA showing the b-series ions obtained at different charge states 5+ (orange), 4+ (blue), and 3+ (red) from the N-terminal 21–31 amino acids, and (C) human transferrin showing the b-series ions obtained at 6+ (black) and 7+ (purple) charge states from the amino-terminal 56–61 amino acids.
The bovine carbonic anhydrase II sample contained bovine superoxide dismutase C (SOD C) as a contaminant and we found this by measuring both the intact mass and the mass of product ions obtained by ISD (Figure 5). SOD C has three cysteine residues, with one of them in the reduced state (Cys6) and the remaining two are disulfide bonded (Cys56 - Cys145). The measured average mass of 15591.34 matches with a deviation of 3 ppm to the theoretical mass of 15591.39 calculated with two cysteines disulfide bonded and one free cysteine. Furthermore, the sequence tag 2TKAVCVL8 of singly charged fragment ions from the amino-terminus of SOD C was obtained and this confirms the presence of a free cysteine at this position from the amino acid incremental mass obtained. Since the sequence information within the disulfide bonded regions is seldom obtained, the dissociation of backbone carbonyl bonds at position 6 supports that it is a free cysteine. Although the information about the nature of the cysteine residues has already been reported in the UniProt database, the intact mass and product ion masses from the SOD C data show that the state of cysteines can be obtained by this method.
Figure 5.

(A) Intact mass spectrum of superoxide dismutase C with the inset showing the deconvoluted intact mass, and (B) fragment ion spectrum of superoxide dismutase C showing the fragment ion assignments and the sequence tag obtained.
3.3. LC-MS and DIA of 20S proteasome proteins
The proteasome complex is a supra-macromolecular assembly of different polypeptides that function to degrade a majority of intracellular proteins [31]. The two main components of the 26S proteasome complex are the core particle, or the 20S proteasome complex where the proteolysis occurs, and the 19S regulatory particle that controls protein entry and has ATPase subunits that power the protein degradation. These two components function together to perform nonspecific protein degradation by allowing the entry of the substrate protein, proteolytic cleavage of the protein at specific sites followed by exit of the short peptide chains, which are then cleaved into constituent amino acids and finally salvaged into various biosynthetic pathways. This mechanism, along with ubiquitin as the intra-cellular molecular tag to label proteins for degradation, is known as the “Ubiquitin-Proteasomal System” of protein degradation [32].
The proteins that constitute the 20S complex form a hollow cylinder shaped structure that can be composed of only two distinct subunits, as in prokaryotes [33], to 14 unique subunits as in eukaryotes [34]. In humans, the 20S proteasome complex is made up of four stacked rings, and each ring is a heptamer of 7 different α subunits or 7 different β subunits. The α subunits form the two outer rings and the β subunits form the two inner rings such that the whole 20S proteasome is arranged as a α7β7α7β7 barrel [35].
The first detailed proteomic characterization of the human 26S proteasome complex was done by Wang et al. using bottom-up proteomic approaches [36]. Subunits in the 19S and 20S particles along with the N-terminal modification on all subunits and post-translational modifications on few of them were detected. Later in 2008, Uttenweiler-Joseph et al. used a combination of proteomic approaches and measured the intact mass of all the subunits in the h20S complex by eluting proteins intact from native two-dimensional gels followed by analysis by high resolution FTICR MS [37]. However, this method is time consuming and requires eluting intact proteins from 2D gels, a low efficiency method using current protocols, in sufficient amounts to be analyzed by top-down MS methods.
Using our DIA top-down LC-MS approach, the h20S proteins were separated on the reversed-phase C3 column and sequence tags were measured by inducing dissociation through ISD. All the proteins eluted between 20 and 32 min as shown in the chromatogram (Figure 6). Alternating the ISD voltage generated both intact and fragment mass spectra for all the proteins eluting within this timeframe. With the intact mass and the sequence tags generated from the fragment mass spectra, the proteins in the h20S complex were identified, and one such representative identification is shown for the ß6 subunit (Figure 7). The list of all the proteins identified along with the sequence tag that was obtained by ISD is shown in Table 2.
Figure 6.
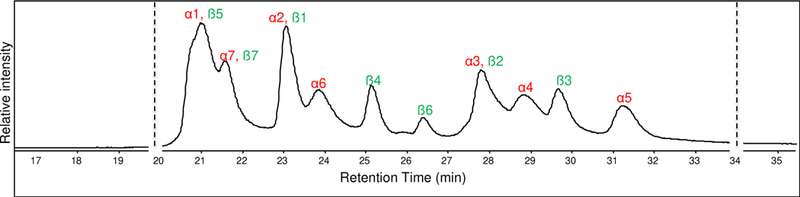
TIC of h20S proteins showing the peaks labeled with protein(s) being eluted. Α and ß subunits are labeled in red and green, respectively.
Figure 7.

(A) Intact mass spectrum of ß6 subunit measured at low ISD voltage, and (B) fragment mass spectrum obtained at high ISD voltage. The deconvoluted mass is shown in the inset of the intact mass spectrum and amino acid sequence tag deduced by mapping the fragment ions is given in the fragment ion spectrum. From the intact and fragment mass, this protein was identified as ß6 subunit.
Table 2.
List of h20S proteasome subunits identified by LC separation followed by ISD. The average theoretical and experimental MW, deviation in the measured mass, sequence tags obtained, and the modifications observed for each protein are tabulated.
| UniProt ID | Subunit | Average Theoretical MW (Da) |
Average Experimental MW (Da) |
Error in mass accuracy (ppm) |
Sequence tag obtained from NSD |
Modification |
|---|---|---|---|---|---|---|
| P25786 | α1 | 29597.7 | 29597.8 | 3.38 |
249PADEPA254 260PME262 3RNQ5 |
+Nacetyl Metini |
| P25787 | α2 | 25809.4 | 25809.39 | 0.39 |
5YSFSLTT11 8SLTTFS13 |
−Metini +Nacetyl Ala1 |
| P25788 | α3 | 28424.1 | 28423.9 | 7.04 | Co-eluting protein |
−Metini +Nacetyl Ser1 +[PO3]− |
| P25789 | α4 | 29394.7 | 29395.04 | 11.57 |
33CLGI36 239HEEEEA244 |
−Metini +Nacetyl Ser1 |
| P28066 | α5 | 26453.1 | 26452.8 | 11.34 |
13NTFSPEGR20 23QVEYAIEAI31 36TAIGIQT42 39GIQT42 218ATVQP222 |
+Nacetyl Metini |
| P60900 | α6 | 27310.3 | 27310.32 | 0.73 |
2RGSSAG7 10RHITIFS16 12ITIFS16 238LVAL241 235DAH237 219VTVENP224 |
−Metini +Nacetyl Ser1 |
| O14818 | α7 | 27797.7 | 27798.1 | 14.39 | Co-eluting protein |
−Metini +Nacetyl Ser1 |
| P20618 | β1 | 23549.0 | 23548.3 | 29.73 | Co-eluting protein | No modification |
| P49721 | β2 | 22878.3 | 22878.32 | 0.87 |
2EYLIGI7 195SFPK198 |
+Nacetyl Metini |
| P49720 | β3 | 22859.8 | 22859.81 | 0.44 |
146YGMCESLWEP155 185IVH187 186VHIIEKDK193 |
−Metini +Nacetyl Ser1 |
| P28070 | β4 | 24391.8 | 24391.6 | 8.20 | 2QNPMVT7 | No modification |
| P28074 | β5 | 22458.4 | 22458.3 | 4.45 | Co-eluting protein | No modification |
| P28072 | β6 | 21903.9 | 21903.7 | 9.13 | 2TIMAVQF8 | No modification |
| Q99436 | β7 | 25295.04 | 25295.05 | 0.4 | 229QTM231 | No modification |
Chromatographically, 6 of the subunits were well resolved from the other proteins and the other 8 subunits overlapped with another protein (Figure 6). Overlapping charge state distributions in the intact mass spectra were found for proteins that co-eluted. Most of the proteins were identified with sequence tags composed of 6–7 residues (Table 2). From the fragment ion data acquired at high ISD voltage, 9 out of the 14 proteins were found to be N- terminally acetylated (Table 2). The other 5 subunits with unmodified N-termini belonged to the β hepatmeric ring. The resolving power of TOF instrument was sufficient to assign charge states for multiply charged product ions up to 6+ charge, and a mass measurement accuracy of ±10 ppm on average for intact protein masses and ±7 ppm for product ions was sufficient for protein identification for these proteins.
Although many of the subunits in the 20S proteasomal core have been reported to be phosphorylated [38], only the α3 subunit was found to be phosphorylated; this was identified by the presence of the unmodified and singly phosphorylated forms of this subunit, differing by 80 Da (mass of the PO3− group) in the deconvoluted mass spectrum (Figure 8). The result is consistent with our previous TDMS study using FTICR MS [29]. The abundance of the phosphorylated form was higher than the non-phosphorylated counterpart, also consistent with a report using two dimensional gel electrophoresis [39].
Figure 8.
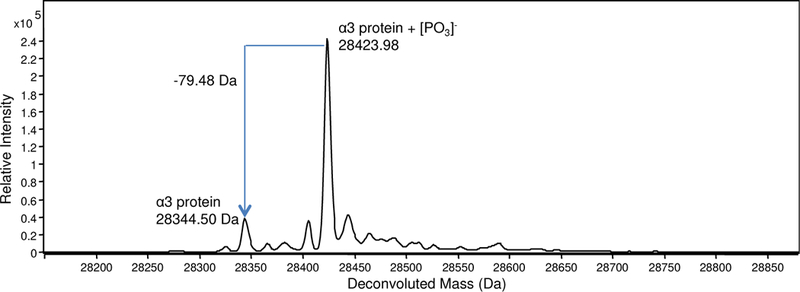
Deconvoluted mass spectrum of the α3 subunit of h20S proteasome showing the presence of both singly phosphorylated and unmodified forms of the protein.
4. Conclusion
By taking advantage of the resolution and mass measurement accuracy available with a TOF mass analyzer, we have combined it with in-source dissociation for proteins eluting on-line from a LC column to demonstrate that it is possible to utilize a stand-alone TOF instrument for top-down protein identification for samples of moderate complexity. This proof-of-principle study presents a relatively in-expensive solution for TDMS. The highlight of the method is that the ISD voltage can be switched from low to high every second enabling both intact and fragment ion mass measurements in the same run. With the resolving power available in this instrument, fragment ions up to 6+ charges were unambiguously assigned. Furthermore, an advantage of using the TOF instrument is that the data acquisition speed is sufficiently high to preserve the chromatographic resolution.
While this method is highly useful for identification of proteins in samples with moderate complexity, the upper limit on the number of proteins identified using this method is dependent on the peak capacity of the LC column. Co-eluting proteins often present a significant problem in reversed phase LC and when a data-independent technique such as ISD dissociation is chosen for fragmentation, product ions from the co-eluting proteins are present together in the mass spectrum, posing additional problems during data interpretation. For these situations where sample complexity can overwhelm the chromatography, additional up-front fractionation techniques such as solution phase electrophoresis [40–42] and on-line orthogonal multidimensional separation methods [43] can help to alleviate the complexity issue [44].
Highlights.
Top-down proteomics with an LC-TOF analyzer is demonstrated
Data-independent acquisition (DIA) for top-down MS is shown
All subunits and several PTMs for the h20S proteasome are identified
Acknowledgements
Support from the US National Institutes of Health (R01GM103479) and the US Department of Energy (UCLA Institute of Genomics and Proteomics; DE-FC03–02ER63421) are acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5. References
- [1].Armirotti A, Damonte G, Achievements and perspectives of top-down proteomics, Proteomics 10 (2010) 3566–3576. [DOI] [PubMed] [Google Scholar]
- [2].Casado-Vela J, Cebrian A, Gomez del Pulgar MT, Sanchez-Lopez E, Vilaseca M, Menchen L, Diema C, Selles-Marchart S, Martinez-Esteso MJ, Yubero N, Bru-Martinez R, Lacal JC, Lights and shadows of proteomic technologies for the study of protein species including isoforms, splicing variants and protein post-translational modifications, Proteomics 11 (2011) 590–603. [DOI] [PubMed] [Google Scholar]
- [3].McLafferty FW, Breuker K, Jin M, Han X, Infusini G, Jiang H, Kong X, Begley TP, Top-down MS, a powerful complement to the high capabilities of proteolysis proteomics, The FEBS journal 274 (2007) 6256–6268. [DOI] [PubMed] [Google Scholar]
- [4].Zhou H, Ning Z, Starr AE, Abu-Farha M, Figeys D, Advancements in top-down proteomics, Analytical chemistry 84 (2011) 720–734. [DOI] [PubMed] [Google Scholar]
- [5].Loo JA, Edmonds CG, Smith RD, Tandem mass spectrometry of very large molecules: serum albumin sequence information from multiply charged ions formed by electrospray ionization, Analytical chemistry 63 (1991) 2488–2499. [DOI] [PubMed] [Google Scholar]
- [6].Han X, Jin M, Breuker K, McLafferty FW, Extending top-down mass spectrometry to proteins with masses greater than 200 kilodaltons, Science 314 (2006) 109–112. [DOI] [PubMed] [Google Scholar]
- [7].McLafferty FW, Horn DM, Breuker K, Ge Y, Lewis MA, Cerda B, Zubarev RA, Carpenter BK, Electron capture dissociation of gaseous multiply charged ions by Fourier-transform ion cyclotron resonance, J. Am. Soc. Mass Spectrom 12 (2001) 245–249. [DOI] [PubMed] [Google Scholar]
- [8].Brodbelt JS, Photodissociation mass spectrometry: new tools for characterization of biological molecules, Chem. Soc. Rev 43 (2014) 2757–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Capriotti AL, Cavaliere C, Foglia P, Samperi R, Lagana A, Intact protein separation by chromatographic and/or electrophoretic techniques for top-down proteomics, Journal of chromatography 1218 (2011) 8760–8776. [DOI] [PubMed] [Google Scholar]
- [10].Parks BA, Jiang L, Thomas PM, Wenger CD, Roth MJ, Boyne MT 2nd, Burke PV, Kwast KE, Kelleher NL, Top-down proteomics on a chromatographic time scale using linear ion trap fourier transform hybrid mass spectrometers, Analytical chemistry 79 (2007) 7984–7991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Loo JA, Edmonds CG, Smith RD, Primary sequence information from intact proteins by electrospray ionization tandem mass spectrometry, Science 248 (1990) 201–204. [DOI] [PubMed] [Google Scholar]
- [12].Lee JE, Kellie JF, Tran JC, Tipton JD, Catherman AD, Thomas HM, Ahlf DR, Durbin KR,Vellaichamy A, Ntai I, Marshall AG, Kelleher NL, A robust two-dimensional separation for top-down tandem mass spectrometry of the low-mass proteome, J. Am. Soc. Mass Spectrom 20 (2009) 2183–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chanthamontri C, Liu J, McLuckey SA, Charge State Dependent Fragmentation of Gaseous alpha- Synuclein Cations via Ion Trap and Beam-Type Collisional Activation, Int. J. Mass Spectrom 283 (2009) 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Scherperel G, Reid GE, Emerging methods in proteomics: top-down protein characterization by multistage tandem mass spectrometry, Analyst 132 (2007) 500–506 [DOI] [PubMed] [Google Scholar]
- [15].Loo JA, Udseth HR, Smith RD, Collisional Effects on the Charge Distribution of Ions from Large Molecules, Formed by Electrospray-ionization Mass Spectrometry, Rapid Commun. Mass Spectrom 2 (1988) 207–210. [Google Scholar]
- [16].Thevis M, Ogorzalek Loo RR, Loo JA, Mass Spectrometric Characterization of Transferrins and their Fragments Derived by Reduction of Disulfide Bonds, J. Am. Soc. Mass Spectrom 14 (2003) 635–647. [DOI] [PubMed] [Google Scholar]
- [17].Vellaichamy A, Tran JC, Catherman AD, Lee JE, Kellie JF, Sweet SM, Zamdborg L, Thomas PM, Ahlf DR, Durbin KR, Valaskovic GA, Kelleher NL, Size-sorting combined with improved nanocapillary liquid chromatography-mass spectrometry for identification of intact proteins up to 80 kDa, Analytical chemistry 82 (2010) 1234–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tipton JD, Tran JC, Catherman AD, Ahlf DR, Durbin KR, Lee JE, Kellie JF, Kelleher NL,Hendrickson CL, Marshall AG, Nano-LC FTICR tandem mass spectrometry for top-down proteomics: routine baseline unit mass resolution of whole cell lysate proteins up to 72 kDa., Analytical chemistry 84 (2012) 2111–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, Wu C, Sweet SM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL, Mapping intact protein isoforms in discovery mode using top-down proteomics, Nature 480 (2011) 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ahlf DR, Compton PD, Tran JC, Early BP, Thomas PM, Kelleher NL, Evaluation of the compact high-field orbitrap for top-down proteomics of human cells, Journal of proteome research 11 (2012) 4308–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Michalski A, Damoc E, Lange O, Denisov E, Nolting D, Muller M, Viner R, Schwartz J, Remes P, Belford M, Dunyach JJ, Cox J, Horning S, Mann M, Makarov A, Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes, Mol. Cell. Proteomics 11 (2011) O111 013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mohr J, Swart R, Samonig M, Bohm G, Huber CG, High-efficiency nano- and micro-HPLC-- high-resolution Orbitrap-MS platform for top-down proteomics, Proteomics 10 (2010) 3598– 3609. [DOI] [PubMed] [Google Scholar]
- [23].Bure C, Lange C, Comparison of Dissociation of Ions in an Electrospray Source, or a Collision Cell in Tandem Mass Spectrometry, Curr. Org. Chem 7 (2003) 1613–1624. [Google Scholar]
- [24].Chen J, Shiyanov P, Schlager JJ, Green KB, A pseudo MS3 approach for identification of disulfide-bonded proteins: uncommon product ions and database search., J. Am. Soc. Mass Spectrom 23 (2012) 225–243. [DOI] [PubMed] [Google Scholar]
- [25].Mekecha TT, Amunugama R, McLuckey SA, Ion trap collision-induced dissociation of human hemoglobin alpha-chain cations, J. Am. Soc. Mass Spectrom 17 (2006) 923–931. [DOI] [PubMed] [Google Scholar]
- [26].Armirotti A, Benatti U, Damonte G, Top-down proteomics with a quadrupole time-of-flight mass spectrometer and collision-induced dissociation, Rapid Commun. Mass Spectrom 23 (2009) 661– 666. [DOI] [PubMed] [Google Scholar]
- [27].Ren D, Pipes GD, Hambly D, Bondarenko PV, Treuheit MJ, Gadgil HS, Top-down N-terminal sequencing of Immunoglobulin subunits with electrospray ionization time of flight mass spectrometry, Analytical biochemistry 384 (2009) 42–48. [DOI] [PubMed] [Google Scholar]
- [28].Geromanos SJ, Vissers JP, Silva JC, Dorschel CA, Li GZ, Gorenstein MV, Bateman RH, Langridge JI, The detection, correlation, and comparison of peptide precursor and product ions from data independent LC-MS with data dependant LC-MS/MS, Proteomics 9 (2009) 1683–1695. [DOI] [PubMed] [Google Scholar]
- [29].Lakshmanan R, Wolff J, Alvarado R, Loo JA, Top-Down Protein Identification of Proteasome Proteins with nanoLC FT-ICR MS Employing Data-Independent Fragmentation Methods, Proteomics 14 (2014) 1271–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nshanian M, Lakshmanan R, Chen H, Ogorzalek-Loo RR, Loo JA, Enhancing sensitivity of liquid chromatography–mass spectrometry of peptides and proteins using supercharging agents, Int. J. Mass Spectrom 427 (2018) 157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bedford L, Paine S, Sheppard PW, Mayer RJ, Roelofs J, Assembly, structure, and function of the 26S proteasome, Trends in cell biology 20 (2010) 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wong E, Cuervo AM, Integration of clearance mechanisms: the proteasome and autophagy, Cold Spring Harbor perspectives in biology 2 (2010) a006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Loo JA, Berhane B, Kaddis CS, Wooding KM, Xie Y, Kaufman SL, Chernushevich IV, Electrospray ionization mass spectrometry and ion mobility analysis of the 20S proteasome complex, J. Am. Soc. Mass Spectrom 16 (2005) 998–1008. [DOI] [PubMed] [Google Scholar]
- [34].Gallastegui N, Groll M, The 26S proteasome: assembly and function of a destructive machine, Trends in biochemical sciences 35 (2010) 634–642. [DOI] [PubMed] [Google Scholar]
- [35].Loo JA, Benchaar SA, Zhang J, Advanced in Mass Spectrometry: Integrating Native Mass Spectrometry and Top-Down MS for Defining Protein Interactions Important in Biology and Medicine, Mass Spectrom 2 (2013) S0013 (0017 pp). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang X, Chen CF, Baker PR, Chen PL, Kaiser P, Huang L, Mass spectrometric characterization of the affinity-purified human 26S proteasome complex, Biochemistry 46 (2007) 3553–3565. [DOI] [PubMed] [Google Scholar]
- [37].Uttenweiler-Joseph S, Claverol S, Sylvius L, Bousquet-Dubouch MP, Burlet-Schiltz O, Monsarrat B, Toward a full characterization of the human 20S proteasome subunits and their isoforms by a combination of proteomic approaches, Methods in molecular biology (Clifton, N.J 484 (2008) 111–130. [DOI] [PubMed] [Google Scholar]
- [38].Lu H, Zong C, Wang Y, Young GW, Deng N, Souda P, Li X, Whitelegge J, Drews O, Yang PY, Ping P, Revealing the dynamics of the 20S proteasome phosphoproteome: a combined CID and electron transfer dissociation approach, Mol. Cell. Proteomics 7 (2008) 2073–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Claverol S, Burlet-Schiltz O, Girbal-Neuhauser E, Gairin JE, Monsarrat B, Mapping and structural dissection of human 20 S proteasome using proteomic approaches, Mol. Cell. Proteomics 1 (2002) 567–578. [DOI] [PubMed] [Google Scholar]
- [40].Ouvry-Patat SA, Torres MP, Quek HH, Gelfand CA, O’Mullan P, Nissum M, Schroeder GK, Han J, Elliott M, Dryhurst D, Ausio J, Wolfenden R, Borchers CH, Free-flow electrophoresis for top-down proteomics by Fourier transform ion cyclotron resonance mass spectrometry, Proteomics 8 (2008) 2798–2808. [DOI] [PubMed] [Google Scholar]
- [41].Tran JC, Doucette AA, Gel-eluted liquid fraction entrapment electrophoresis: an electrophoretic method for broad molecular weight range proteome separation, Analytical chemistry 80 (2008) 1568–1573. [DOI] [PubMed] [Google Scholar]
- [42].Witkowski C, Harkins J, Using the GELFREE 8100 Fractionation System for molecular weight- based fractionation with liquid phase recovery, J. Vis. Exp (2009). [DOI] [PMC free article] [PubMed]
- [43].Wu Q, Yuan H, Zhang L, Zhang Y, Recent advances on multidimensional liquid chromatography- mass spectrometry for proteomics: from qualitative to quantitative analysis--a review, Analytica chimica acta 731 (2012) 1–10. [DOI] [PubMed] [Google Scholar]
- [44].Kellie JF, Catherman AD, Durbin KR, Tran JC, Tipton JD, Norris JL, Witkowski CE 2nd, Thomas PM, Kelleher NL, Robust analysis of the yeast proteome under 50 kDa by molecular- mass-based fractionation and top-down mass spectrometry, Analytical chemistry 84 (2012) 209– 215. [DOI] [PMC free article] [PubMed] [Google Scholar]