

PES1 is a new component of telomerase assembly that regulates telomerase activity, telomere length, and cellular senescence.
Abstract
Telomerase defers the onset of telomere shortening and cellular senescence by adding telomeric repeat DNA to chromosome ends, and its activation contributes to carcinogenesis. Telomerase minimally consists of the telomerase reverse transcriptase (TERT) and the telomerase RNA (TR). However, how telomerase assembles is largely unknown. Here, we demonstrate that PES1 (Pescadillo), a protein overexpressed in many cancers, forms a complex with TERT and TR through direct interaction with TERT, regulating telomerase activity, telomere length maintenance, and senescence. PES1 does not interact with the previously reported telomerase components Reptin, Pontin, p23, and Hsp90. PES1 facilitates telomerase assembly by promoting direct interaction between TERT and TR without affecting TERT and TR levels. PES1 expression correlates positively with telomerase activity and negatively with senescence in patients with breast cancer. Thus, we identify a previously unknown telomerase complex, and targeting PES1 may open a new avenue for cancer therapy.
INTRODUCTION
Telomerase is a ribonucleoprotein (RNP) enzyme that adds telomeric repeat DNA to chromosome ends (1–3). This prevents progressive shortening of telomeres caused by the failure of the DNA replication machinery to duplicate the very end of each chromosome. Once telomeres are shortened to a certain length, cells enter replicative senescence or, alternatively, undergo apoptosis, a major tumor-suppressive mechanism. Telomerase, which is required for de novo telomeric repeat DNA synthesis and telomere maintenance, is expressed in approximately 90% of cancer cells but undetectable in the majority of normal somatic cells (4–6). Thus, telomerase is thought to be a relevant factor in distinguishing cancer cells from normal cells and has become a target for cancer therapy.
Telomerase is minimally composed of the telomerase reverse transcriptase (TERT) and the telomerase RNA (TR). Studies have shown in vitro assembly of active telomerase by combining the purified RNA component with the TERT synthesized in rabbit reticulocyte extract (7–9). A few accessory proteins have been identified to associate with the active telomerase RNP complex. The molecular chaperones p23 and Hsp90 bind to human TERT (hTERT), and chemical inhibition of Hsp90 decreases telomerase activity (10, 11). However, determining whether Hsp90 is required for active telomerase assembly is difficult because chemical inhibition of a key chaperone in human cells potentially has pleiotropic and indirect effects. Assembly of human TR (hTR) and hTERT into catalytically active telomerase is facilitated by the adenosine triphosphatases Reptin and Pontin (12). Pontin knockdown (KD) reduces telomerase activity and hTR levels. It is unclear whether the loss of telomerase activity after Pontin KD is caused by reduced hTR levels, disrupted telomerase assembly, or a combination of both.
To identify additional components of the human telomerase holoenzyme assembly, we purified hTERT complexes from human cancer cells by co-immunoprecipitation (co-IP) and examined hTERT-interacting partners with mass spectrometry (MS). We identify PES1 (also known as Pescadillo), which was shown to be up-regulated in many cancers and to regulate cell cycle progression, as well as the synthesis and maturation of ribosome (13–20), as a component of active telomerase complex. PES1 increases telomerase activity without affecting the expression of hTERT or hTR. Moreover, PES1 regulates telomere length and cellular senescence. Mechanistically, PES1 facilitates telomerase assembly by promotion of direct interaction between hTERT and hTR.
RESULTS
Identification and characterization of PES1 as an hTERT-interacting protein
We used co-IP combined with MS to identify Flag-tagged hTERT-interacting proteins. Besides the previously reported hTERT-interacting proteins Hsp90, GNL3L, and 14-3-3 (10, 21, 22), we identified PES1 as a potential hTERT interaction partner (Fig. 1A). Co-IP of exogenous proteins with Myc-tagged hTERT and Flag-tagged PES1 confirmed the hTERT/PES1 interaction (Fig. 1B). Ribonuclease (RNase) treatment did not alter the hTERT/PES1 interaction, whereas RNase treatment abolished the interaction of hTERT with DKC1 (dyskeratosis congenita 1, dyskerin), which was shown to associate with hTERT via hTR (12), indicating that the hTERT/PES1 interaction is not mediated by hTR. PES1, a breast cancer gene 1 (BRCA1) C-terminal (BRCT) domain–containing protein, associates with WD repeat domain 12 (WDR12) to control nucleolar localization and ribosome biogenesis (23). However, WDR12 and the BRCT domain–containing protein 53BP1 did not interact with hTERT (fig. S1A). PES1 also did not associate with the previously reported telomerase complex components Pontin, Reptin, p23, and Hsp90 (10–12), although these components interacted with hTERT (fig. S1, B and C). Endogenous hTERT associated with endogenous PES1 (Fig. 1C). Because of low abundance of hTERT (∼500 molecules per cell in cancer cells) and a dearth of antibodies that efficiently immunoprecipitate hTERT and PES1 (12), we established a stable human breast cancer MCF7 cell line expressing Flag-tagged PES1 protein similar to endogenous levels by lentiviral infection of Flag-PES1 in PES1-depleted cells (fig. S1D). We refer to this cell line as MCF7-Flag-PES1+shPES1. MCF7-Flag-PES1+shPES1 cells had similar behaviors as the control cells MCF7-Flag+shCon and parental MCF7 cells according to PES1 modulation of estrogen receptor α expression and cell proliferation (figs. S1, D and E). Substituting endogenous PES1 with Flag-PES1 allows for quantitative IP using well-characterized antibodies against Flag tag. It has been shown that the amount of hTERT bound to Pontin and Reptin peaks in S phase of the cell cycle (12). Unlike Pontin and Reptin, however, PES1 interacted with hTERT similarly throughout the cell cycle using the established cell lines (fig. S1F). Overexpression of PES1 did not change hTERT expression. These data suggest that PES1 is a component of another telomerase complex.
Fig. 1. PES1 directly interacts with hTERT.
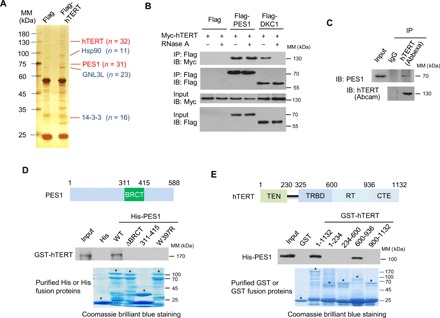
(A) Cell extracts from MCF7 cells stably expressing Flag (control) or Flag-tagged hTERT (Flag-hTERT) were immunopurified with anti-Flag affinity gel, and the purified complex was subject to SDS–polyacrylamide gel electrophoresis (PAGE) and silver-stained. The differential protein bands were retrieved and analyzed by MS. n, number of peptides identified by MS analysis; MM, molecular mass. (B) HEK293T cells transiently transfected with Myc-tagged hTERT (Myc-hTERT) and Flag-PES1 or Flag-DKC1 were treated with or without RNase A (0.1 mg/ml), and immunoprecipitated (IP) with anti-Flag, followed by immunoblotting (IB) with the indicated antibodies. (C) MCF7 cell lysates were immunoprecipitated with anti-hTERT from Abbexa and immunoblotted with anti-hTERT from Abcam and anti-PES1. IgG, immunoglobulin G. (D) Glutathione-Sepharose beads bound with GST-hTERT were incubated with purified His-tagged wild-type (WT) or mutated PES1. After washing the beads, the bound proteins were subjected to SDS-PAGE and immunoblotted with anti-GST antibody. Also shown on top of the graph is a schematic diagram of the PES1 protein, illustrating the location of the BRCT domain. Asterisks indicate the positions of the expected full-length fusion proteins. (E) Glutathione-Sepharose beads bound with WT or truncated hTERT were incubated with purified His-tagged PES1. After washing the beads, the bound proteins were examined by immunoblot with anti-His. Schematic diagram of the hTERT deletion constructs used is shown at the top. Asterisks indicate the positions of the expected full-length fusion proteins. TEN, telomerase N-terminal; TRBD, TR-binding domain; RT, reverse transcriptase; CTE, C-terminal extension.
The interaction between PES1 and hTERT is direct because histidine (His) or glutathione S-transferase (GST) pull-down experiments showed that purified His-tagged PES1 protein interacted with purified GST-hTERT, and purified GST-hTERT, but not GST alone, associated with His-PES1 (Fig. 1, D and E). To increase the expression level of full-length hTERT, we optimized the coding sequence of hTERT in the GST-hTERT construct (table S2) and efficiently purified the GST-hTERT protein (fig. S1G).
Although PES1 (311-415) containing the BRCT domain did not interact with hTERT, deletion of the BRCT domain or mutation of one of the highly conserved residues within the BRCT domain (W397R) abolished the PES1-hTERT interaction (Fig. 1D), suggesting that the BRCT domain is required for the PES1-hTERT interaction. Deletion of the BRCT domain or the W397R mutation abrogated the ability of PES1 to localize in the nucleolus (fig. S1H). On the other hand, hTERT (600-936) containing the RT domain, but not other regions, interacted with PES1 (Fig. 1E).
PES1 forms an active complex with hTR via hTERT
Human telomerase minimally consists of hTERT and hTR. Since PES1 directly interacts with hTERT, we tested whether PES1 forms a complex with hTERT and hTR. Co-IP followed by re-IP indicated that PES1, hTERT, and hTR form a complex in human breast cancer MCF7 and human liver cancer HepG2 cells (Fig. 2A and fig. S2A). Consistent with previous results (12), protein fractionation experiments performed by fast protein liquid chromatography (FPLC) showed that the elution pattern of Pontin overlapped with that of hTERT and DKC proteins and hTR in a complex of more than 1 MDa (Fig. 2B and fig. S2D). The elution pattern of PES1 overlapped with that of hTERT and DKC proteins and hTR in another complex with a size of 0.5 to 1 MDa. PES1, hTERT, hTR, and telomerase activity coexist in this complex. p23, Hsp90, hTERT, hTR, and telomerase activity might be present in a complex of less than 0.5 MDa. Although the elution pattern of Hsp90 overlapped with PES1, Hsp90 did not bind to PES1 in co-IP experiments as mentioned above (see Discussion). PES1 interacted with hTR in hTERT-positive MCF7 and HepG2 cells but not hTERT-negative U2OS cells (Fig. 2C and fig. S2B). PES1 did not affect hTR level. Moreover, KD of endogenous hTERT in MCF7-Flag-PES1+shPES1 cells greatly attenuated the interaction of PES1 with hTR (Fig. 2D). In vitro RNA pull-down experiments showed that purified GST-hTERT, but not GST-PES1, bound to hTR, and GST-PES1 interacted with hTR in the presence of GST-hTERT (Fig. 2E). As mentioned above, DKC1 interacts with hTERT via hTR. We examined if PES1 associates with DKC1 through hTR. RNase treatment abolished the PES1-DKC1 interaction in MCF7 cells (fig. S2C), suggesting that the PES1-DKC1 interaction is hTR dependent. These results, combined with the data shown above, suggest that PES1 forms an active complex with hTR through hTERT.
Fig. 2. PES1 forms an active complex with telomerase.
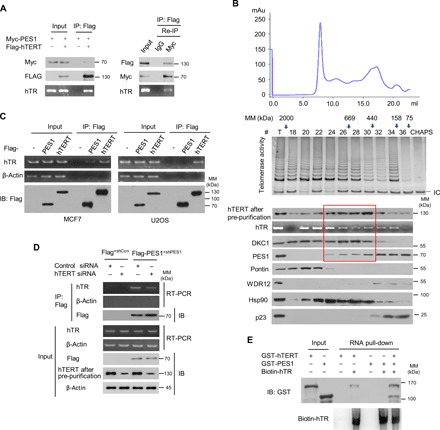
(A) MCF7 cells transfected with the indicated plasmids were immunoprecipitated with anti-Flag. The immune complexes were eluted with Flag peptide and re-IP using anti-Myc or normal IgG. The resulting precipitates were used for detection of Flag-hTERT and Myc-PES1 expression by immunoblot. hTR levels were determined by RT-PCR. (B) MCF7 cell extracts were fractionated on Superose 6 size exclusion columns. Fast protein liquid chromatography (FPLC) chromatographic elution profiles are indicated. The elution positions of calibration proteins with known molecular masses (kDa) are shown by arrows, and an equal volume from each chromatographic fraction was used for determination of protein expression by immunoblot with the indicated antibodies. To increase the specificity of hTERT detection, cells were immunoprecipitated with anti-hTERT from Abbexa, followed by immunoblot with anti-hTERT from Abcam (hTERT after pre-purification). hTR levels were determined by reverse transcription polymerase chain reaction (RT-PCR) and telomerase activity by telomerase repeat amplification protocol (TRAP). CHAPS buffer was used as a negative control for TRAP assay. IC, internal control; mAu, milli-Absorbance unit. Red frame indicates a potential complex with hTERT/hTR/DKC1/PES1. (C) MCF7 and U2OS cells were transfected with Flag-tagged PES1 or hTERT or empty vector, and immunoprecipitated with anti-Flag agarose. The resulting precipitates were used for assessment of Flag-PES1 or Flag-hTERT expression by immunoblot. hTR levels were determined by RT-PCR. β-Actin was used as a negative control for hTR determination. (D) MCF7-Flag-PES1+shPES1 cells or control (MCF7-Flag+shCon) cells were transfected with control small interfering RNA (siRNA) or hTERT siRNA as indicated and were analyzed as in (C). (E) Biotinylated hTR was incubated with purified GST-hTERT, GST-PES1, or GST-hTERT plus GST-PES1. The resulting complexes were subject to RNA pull-down assay. The amount of biotin-hTR was detected by the Chemiluminescent Nucleic Acid Detection Module Kit (Thermo Fisher Scientific).
PES1 promotes assembly of hTERT and hTR through enhancement of the hTERT-hTR interaction
Since PES1 forms an active telomerase complex, we investigated whether PES1 regulates the interaction between hTERT and hTR.To this end, we generated PES1 knockout (KO) MCF7 cells by CRISPR-Cas9. However, it was very difficult to obtain a single PES1 KO MCF7 clone due to growth arrest after a long period of culture. Thus, we used mixed clones for further study (fig. S3A). As disruption of PES1 gene results in early embryonic lethality in this study and a previous study (24), we prepared PES1 KO mouse embryonic fibroblasts (MEFs) by adding Cre recombinase adenovirus (Ad-Cre) to MEFs isolated from conditional PES1fl/fl mice (fig. S3B). PES1 KO MCF7 cells or MEFs showed decreased interaction between TERT and TR (Fig. 3A). The effect of PES1 KO on the TERT-TR interaction could be rescued by PES1 reexpression in PES1 KO MCF7 cells and MEFs. Since we could only passage the PES1 KO cells a few times due to the limited cell proliferation, we knocked down PES1 using small-interfering RNA (siRNA) in MCF7 and HepG2 cells. As expected, PES1 KD reduced the hTERT-hTR interaction (Fig. 3B). This effect could be rescued by reexpression of wild-type (WT) PES1, but not PES1 (ΔBRCT) or PES1 (W397R) that fails to interact with hTERT in the PES1 KD cells. Similar effects were observed in PES1 KD MEFs (Fig. 3C). Moreover, RNA pull-down showed that purified His-PES1, but not His-PES1 (ΔBRCT) or His-PES1 (W397R), enhanced binding of hTR to hTERT in vitro (Fig. 3D). Together, these data suggest that the direct interaction between PES1 and hTERT is required for PES1 promotion of hTERT and hTR assembly.
Fig. 3. PES1 enhances assembly of TERT and TR.
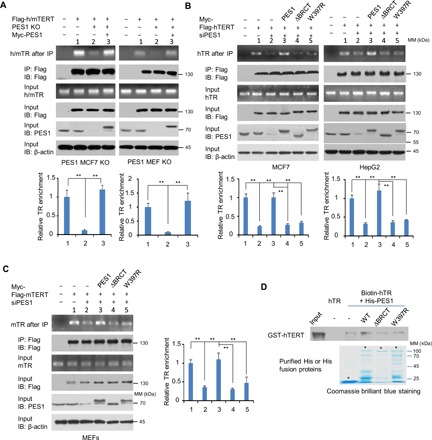
(A) PES1 WT or KO MCF7 cells (left) and MEFs (right) were transiently transfected with the indicated plasmids and immunoprecipitated with anti-Flag. The precipitates were used for assessment of Flag-h/mTERT, PES1, and β-actin expression by immunoblot. TR levels were determined by RT-PCR. Flag-h/mTERT, Flag-tagged human or mouse TERT. Flag-hTERT was used for MCF7 cells, and Flag-mTERT was used for MEFs. (B) siRNA-mediated PES1 KD MCF7 or HepG2 cells were transfected with Flag-hTERT and siRNA-resistant Myc-tagged PES1, PES1 (ΔBRCT), or PES1 (W397R) as indicated and were analyzed as in (A). (C) siRNA-mediated PES1 KD MEFs were transfected with Flag-mTERT and siRNA-resistant Myc-tagged PES1, PES1 (ΔBRCT), or PES1 (W397R) as indicated and were analyzed as in (A). Data shown are mean ± SD of three independent experiments (A to C). **P < 0.01. (D) Biotinylated hTR was incubated with purified GST-hTERT and WT or mutant His-PES1. The resulting complexes were subject to RNA pull-down assay. Unlabeled hTR was used as a negative control. Asterisks indicate the positions of the expected full-length fusion proteins.
PES1 enhances telomerase activity in mammalian cells and in vitro
Since PES1 facilitates assembly of hTERT and hTR, we investigated whether PES1 regulates telomerase activity. We performed direct telomerase activity assays with biotin to quantify telomerase activity. Flag-hTERT and hTR were transfected into PES1 KO MCF7 cells or MEFs. KO of PES1 in MCF7 cells or MEFs reduced telomerase activity (Fig. 4A). Reexpression of WT PES1, but not PES1 (ΔBRCT) or PES1 (W397R), in the PES1 KO cells rescued this effect. We also detected telomerase activity by telomerase repeat amplification protocol (TRAP) assay and found similar effects in these cells (fig. S4A) and in PES1 KD HepG2 cells (fig. S4B). Moreover, PES1 KO hepatocytes from liver-specific PES1fl/fl mice also revealed reduced telomerase activity (Fig. 4B). As expected, overexpression of PES1, but not WDR12 and 53BP1, which fail to interact with hTERT, increased telomerase activity in cancer cells (fig. S4C).
Fig. 4. PES1 regulates telomerase activity and telomere length.
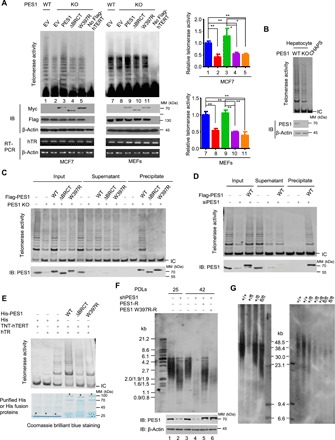
(A) Direct telomerase assay in PES1 KO MCF7 cells or MEFs transfected with Flag-hTERT, hTR, and PES1 or its mutants. Transfected cells were immunoprecipitated with anti-Flag, followed by telomerase activity detection. Parental cells without Flag-hTERT transfection were used as a negative control. Values shown are mean ± SD of three independent experiments. *P < 0.05, **P < 0.01. (B) PES1 WT and KO mouse hepatocytes were isolated from liver-specific PES1 KO mice generated by mating conditional PES1 KO mice with Alb-Cre transgenic mice and were analyzed by TRAP assay. (C) IP-TRAP analysis of PES1 KO MCF7 cells transfected with Flag-tagged WT or mutant PES1. When the binding of Flag-tagged proteins to anti-Flag agarose beads was complete, the reaction mixtures were centrifuged and the supernatants were collected for TRAP assay. The precipitates were washed, lysed, and used for TRAP assay. Flag-tagged PES1 or endogenous PES1 levels were analyzed by immunoblot with anti-PES1. (D) IP-TRAP analysis of siRNA-mediated PES1 KD HepG2 cells transfected with Flag-PES1 or empty vector. (E) TRAP assay was performed by mixing in vitro transcribed hTR and in vitro translated hTERT with purified WT and mutated His-PES1. (F) Representative MCF7 monoclones stably expressing PES1 shRNA or control shRNA and WT or mutant PES1 were harvested at indicated PDLs. Reexpression of WT or mutant PES1 was performed after 25 PDLs by splitting the same clone. Telomere lengths were measured using terminal restriction fragment (TRF) analysis. Representative immunoblot reveals PES1 expression. PES1-R, shRNA-resistant PES1. PES1 W397R-R, shRNA-resistant PES1 W397R. (G) PES1 WT and KO mouse hepatocytes isolated from liver-specific PES1 KO mice at 3 months were subjected to TRF analysis. Left and right panels indicate two nest mice. fl, flox.
To test to what extent PES1 associates with telomerase activity, we determined telomerase activity on lysates before and after immunodepletion (supernatants and precipitates, respectively) in PES1 KO or KD cancer cells expressing exogenous Flag-tagged PES1 similar to the endogenous level. Analysis of the relative telomerase activity in the supernatant and precipitate after volume normalization showed that the activity of the precipitate was a little higher than that of the supernatant. Considering that efficiencies of IP and elution were not 100%, these data indicate that PES1 associates with more than 50% telomerase activity in cancer cells (Fig. 4, C and D). However, the precipitates from PES1 KO cells expressing exogenous Flag-tagged PES1 (ΔBRCT) or PES1 (W397R) did not show telomerase activity, and the supernatants from these cells had similar activity to PES1 KO cells (Fig. 4C). These results suggest that PES1 associates with the majority of telomerase activity in cancer cells.
Next, we tested whether PES1 activates telomerase activity in vitro. Since we have successfully purified GST-hTERT protein from Escherichia coli, we attempted to assemble active telomerase by combining purified hTR with the purified hTERT. However, the purified hTERT did not reveal telomerase activity (fig. S4D). This might be due to lack of posttranslational modification of the purified hTERT from E. coli. Thus, we still used hTERT synthesized in rabbit reticulocyte extract as previously reported (12). hTERT synthesized in rabbit reticulocyte extract had telomerase activity (Fig. 4E and fig. S4D). The purified WT PES1, but not PES1 (ΔBRCT) or PES1 (W397R), markedly increased hTERT-mediated telomerase activity (Fig. 4E).
PES1 affects telomere length maintenance in cancer cells and in mice
Telomerase activity is critical for telomere length maintenance (1, 2). The observation that PES1 regulates telomerase activity prompted us to examine the effect of PES1 on telomere length maintenance. Telomere length, as determined by telomere restriction fragment (TRF) assay, did not change with increasing population doubling levels (PDLs) in telomerase-positive MCF7 and HepG2 cells (from 25 to 42 PDLs in MCF7 cells and from 35 to 66 PDLs in HepG2 cells; Fig. 4F and fig. S5A). Stable PES1 KD reduced telomere length in MCF7 and HepG2 cells at 25 and 35 PDLs, respectively. In contrast to control cells, stable PES1 KD cells showed decreased telomere length with the increasing PDLs. Reexpression of PES1, but not PES1 (W397R), in the stable PES1 KD MCF7 cells almost rescued this effect at 42 PDLs (Fig. 4F). Both transient and stable PES1 KD in telomerase-negative human osteosarcoma U2OS cells had no effect on telomere length (fig. S5B). Moreover, PES1 KO mouse hepatocytes isolated from liver-specific PES1 KO mice revealed decreased telomere length compared to WT cells (Fig. 4G).
PES1 regulates senescence in cultured cells and in vivo
Telomere shortening is associated with cell division in normal cells. Replicative senescence is the result of telomere shortening (3). Cellular senescence induced by telomere shortening is characterized by typical alterations in cell morphology (flattened cells with enlarged cytoplasm) and senescence-associated β-galactosidase (SA-β-gal) activity (25). In addition, senescent cells fail to proliferate. Since PES1 regulates telomere length maintenance, we investigated whether PES1 modulates senescence. Consistent with the results of the telomere length experiments, senescence did not change with increasing PDLs in MCF7 and HepG2 cells stably transfected with control short hairpin RNA (shRNA) in terms of morphology and SA-β-gal activity (Fig. 5A and fig. S6A). Stable PES1 KD increased senescence in MCF7 and HepG2 cells at 25 and 35 PDLs, respectively. In contrast to control cells, stable PES1 KD cells revealed increased senescence with the increasing PDLs. Reexpression of PES1, but not PES1 (W397R), in the stable PES1 KD MCF7 cells almost rescued this effect at 42 PDLs (Fig. 5A). However, stable PES1 KD in telomerase-negative U2OS cells had no effect on senescence (fig. S6B). Furthermore, PES1 KO mouse hepatocytes isolated from liver-specific PES1 KO mice showed increased senescence compared to WT cells (Fig. 5B).
Fig. 5. PES1 KD or KO triggers senescence.
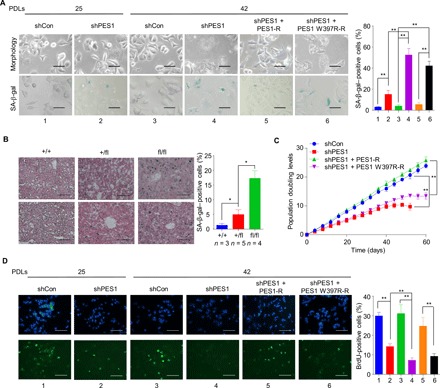
(A) Cells from Fig. 4F were captured by microscopy (top) and subjected to SA-β-gal staining (bottom). The percentage of SA-β-gal–positive cells on the right was calculated from five randomly chosen fields. At least 150 cells were analyzed per experiment. Scale bars, 50 μm. Data shown are mean ± SD of three independent experiments. **P < 0.01. (B) Representative frozen liver sections from liver-specific PES1 KO mice at 3 months were subjected to SA-β-gal staining followed by eosin staining. The PES1+/+ and PES1+/f mice were littermates of the PES1f/f mice. Scale bars, 100 μm. *P < 0.05. (C) Growth curves of representative MCF7 monoclones stably expressing PES1 shRNA (shPES1) or control shRNA (shCon) and shRNA-resistant WT or mutated PES1 from 34 PDLs. (D) Cells from Fig. 4F were labeled with BrdU and stained with anti-BrdU. Scale bars, 100 μm. Data shown are mean ± SD of three independent experiments. **P < 0.01.
In agreement with the results of the cellular senescence, stable PES1 KD MCF7 and HepG2 cells had a smaller number of PDLs than control cells during the same periods (e.g., 40 to 80 days; Fig. 5C and fig. S6C), and the change in the amplitude of PDLs in telomerase-negative U2OS cells was smaller than that in telomerase-positive MCF7 and HepG2 cells (fig. S5D). PES1 KD led to growth arrest in MCF7 and HepG2 cells, but not in U2OS cells, approximately 50 (MCF7) and 70 (HepG2) days after cell growth (Fig. 5C and fig. S6, C and D). Reexpression of PES1, but not PES1 (W397R), in the stable PES1 KD MCF7 cells rescued this effect (Fig. 5C). Moreover, the proportion of cells undergoing DNA synthesis, as measured by bromodeoxyuridine (BrdU) incorporation, did not change with increasing PDLs in MCF7 and HepG2 cells stably transfected with control shRNA (Fig. 5D and fig. S6E). Stable PES1 KD decreased the percentage of BrdU-positive cells in MCF7 and HepG2 cells at 25 and 35 PDLs, respectively. In contrast to control cells, stable PES1 KD cells showed decreased percentage of BrdU-positive cells with the increasing PDLs. Reexpression of PES1, but not PES1 (W397R), in the stable PES1 KD MCF7 cells almost rescued this effect at 42 PDLs (Fig. 5D). However, stable PES1 KD in telomerase-negative U2OS cells had less effect on BrdU incorporation than that in telomerase-positive MCF7 and HepG2 cells (fig. S6F).
To determine whether PES1 depletion leads to senescence and proliferation defects through regulation of telomerase, PES1 KD MCF7 cells were stably transfected with hTERT and hTR and subjected to SA-β-gal staining and cell proliferation detection. Overexpression of hTERT and hTR largely rescued cell senescence and proliferation defects (fig. S6, G and H), suggesting that PES1 regulates cell senescence and proliferation largely through telomerase.
Correlation of PES1 expression with telomerase activity and cellular senescence in patients with breast cancer
To explore the clinical significance of PES1 in regulation of telomerase and senescence, we conducted immunohistochemistry (IHC), TRAP, TRF, and SA-β-gal staining in human breast cancer tissue samples. We have previously verified the specificity of the anti-PES1 antibody (15). PES1 expression correlated positively with telomerase activity but negatively with senescence (Fig. 6, A and B). However, PES1 did not associate with telomere length (Fig. 6A), suggesting that other factor(s) may also be involved in regulation of telomere length in breast cancer tissues we used.
Fig. 6. PES1 correlates with telomerase activity and cellular senescence in breast cancer samples.
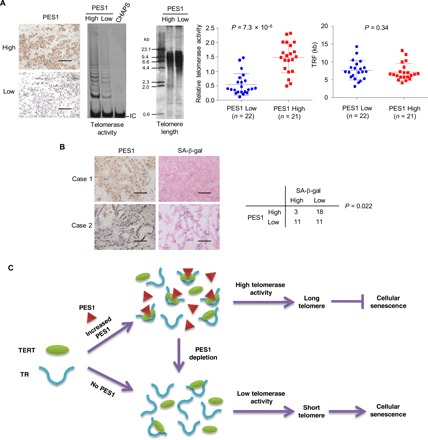
(A) Representative immunohistochemical staining of PES1, telomerase activity, and telomere length in 43 human breast cancer samples. Scale bars, 100 μm. Correlation of PES1 levels with relative telomerase activity and telomere length was generated by Mann-Whitney test. (B) Representative immunohistochemical staining of PES1 and SA-β-gal in frozen breast cancer tissue sections from (A). Scale bars, 100 μm. Correlation between PES1 expression and SA-β-gal staining was generated by Fisher’s exact test. (C) A proposed model underlying the role of PES1 in regulation of telomerase and senescence. Telomerase minimally comprises TERT and TR. When PES1 expression levels are elevated in some cases (e.g., cancer), it facilitates assembly of TERT and TR by forming a complex with TERT and TR, resulting in high telomerase activity, long telomere, and inhibition of senescence. PES1 depletion has opposite effects.
Effects of PES1 on regulation of noncanonical functions of hTERT
It has been reported that TERT exerts noncanonical functions, such as inflammation and transcriptional activation, through interactions with nuclear factor κB (NF-κB) and wingless/integrated (Wnt) signaling. hTR is required for TERT regulation of NF-κB signaling but not Wnt signaling (26, 27). Since PES1 interacts with hTERT directly, we investigated whether PES1 regulates NF-κB and Wnt signaling. As expected, tumor necrosis factor–α (TNFα), a cytokine critical for inflammation, stimulated NF-κB–dependent reporter activity, and overexpression of hTERT together with hTR further increased the reporter activity. However, PES1 overexpression failed to regulate the reporter activity (fig. S7A). Similar effects were observed with NF-κB–dependent gene transcription in response to TNFα (fig. S7B). In addition, LiCl induced Wnt reporter activity and hTERT overexpression further enhanced the reporter activity (fig. S7C). Again, overexpression of PES1 could not modulate the reporter activity.
TERT has also been shown to regulate cell proliferation, apoptosis, tRNA expression, DNA damage response, and c-Myc stability, independent of its telomere function (28–32). Next, we investigated whether PES1 might be involved in these processes. Ectopic PES1 expression promoted cell proliferation of MCF7 cells, accompanied by increased c-Myc expression. These effects were greatly attenuated in TERT KD cells (fig. S7D), suggesting that PES1 regulates cell proliferation and c-Myc expression largely through TERT. KD of PES1 failed to regulate apoptosis, tRNA expression, and the level of γH2AX, a marker of DNA damage (fig. S7, E to G), whereas TERT KD induced apoptosis, reduced tRNA expression, and decreased γH2AX level, as previously reported (29–31). Together, these data suggest that PES1 may be responsible for some noncanonical functions of TERT.
DISCUSSION
The catalytic core of human telomerase consists of hTERT and hTR.Assembly of hTERT and hTR into catalytically active telomerase is facilitated by other cellular proteins. It has been reported that the affinity-purified telomerase complex has a molecular mass of ∼550 kDa from HeLa cells with antisense oligonucleotides complementary to the template region of hTR, although the components, except for hTERT and hTR, remain unclear (33). Glycerol gradient sedimentation analyses using extracts from HeLa cells stably expressing Flag-hTERT have shown that telomerase resides in a large complex of 1 to 2 MDa, with components containing Pontin and Reptin (12). Pontin and Reptin are critical for telomerase activity and for increased hTR levels. However, Pontin and Reptin coimmunoprecipitate a substantial pool of hTERT that yields only low enzymatic activity. The molecular chaperones p23 and Hsp90 have been shown to bind to hTERT and are associated with active telomerase in cells (10, 11). How p23 and Hsp90 regulate telomerase assembly and whether p23, Hsp90, and hTERT form a complex are still unclear. In this study, we used co-IP combined with MS to identify hTERT-interacting proteins from MCF7 cells stably expressing Flag-hTERT and found that PES1 associated with hTERT. Further experiments indicate that PES1 interacts with hTERT directly and PES1 forms an active complex with hTR via hTERT. To characterize the size of the telomerase holoenzyme complex, we performed FPLC analyses using extracts from MCF7 cells. Consistent with a previous report, Pontin, hTERT, hTR, and telomerase activity reside in a large complex of over 1 MDa (12). PES1, hTERT, hTR, and telomerase activity coexist in a complex of 0.5 to 1 MDa. p23, hTERT, hTR, and telomerase activity seem to be present in a complex of less than 0.5 MDa. The observation that the elution pattern of Hsp90 overlaps with PES1 may be due to limited resolution of the FPLC used or other unknown reasons because Hsp90 binds to hTERT, but not PES1, in co-IP experiments. In addition, Pontin and p23 fail to interact with PES1. Since Hsp90 was shown to associate with p23 (34, 35), our data, combined with previous results, indicate that hTERT protein exists in at least three different complexes (hTERT/hTR/Pontin/Reptin, hTERT/hTR/Hsp90/p23, and hTERT/hTR/PES1). However, PES1 was not identified in the recently reported human telomerase holoenzyme complex (36). The reason why PES1 was not found may be that different cell lines, transfection of different constructs, and different purification methods were used. They used human embryonic kidney (HEK) 293T cells transfected with TERT plus hTR instead of MCF7 cells transfected with TERT alone in this study. They purified the complex with streptavidin agarose resin prebound to a 5′-biotinylated oligonucleotide instead of Flag agarose in this report. Thus, we identify a previously unknown complex for telomerase assembly, which regulates telomerase activity, telomere length, and senescence (Fig. 6C). Mechanistically, PES1 facilitates telomerase assembly by enhancement of direct interaction between hTERT and hTR. High telomerase activity in cancer cells is usually thought to result from increased expression of hTERT and hTR. Using PES1 KO or KD cancer cell lines, PES1 KO MEFs, PES1 KO mouse hepatocytes, and tumor samples, we show that PES1 stimulates telomerase activity without affecting the expression of hTERT or hTR. PES1 associates with more than 50% telomerase activity in mammalian cells. Our data indicate that PES1 is a key cofactor for telomerase assembly and also reveal a causal role for PES1 in regulation of telomerase activity.
Telomerase activity is important for telomere length maintenance (37). Although Pontin and Reptin, and p23 and Hsp90, the components of the telomerase holoenzyme, can regulate telomerase activity, whether they change telomere length is unclear. In this study, we show that stable PES1 KD telomerase-positive cancer cells, but not telomerase-negative cancer cells, or PES1 KO mouse hepatocytes reveal decreased telomere length. In normal human cells, telomerase is often repressed, causing telomere shortening. However, elevated telomerase activity is observed in more than 90% of human cancers and is critical for telomere maintenance and tumor cell growth. In contrast to hTERT, mouse TERT (mTERT) is widely expressed in primary cells and mouse tissues. Telomere shortening can trigger senescence. Recently, KD of the ribosomal RNA (rRNA)–processing factors Dkc1, rRNA processing 5 (Rrp5), and ribosome biogenesis regulatory protein homolog 1 (Rrs1) has been shown to induce cellular senescence in MEFs and MCF-7 cells through the p53-p21 pathway (38). As a control, transient KD of PES1, another rRNA-processing factor, also stimulates senescence in MCF-7 cells, although the underlying mechanism remains unclear. In this study, we show that PES1 induces senescence through regulation of telomerase activity and telomere length.
Because telomerase is intimately associated with human cancer, it has long been considered as an attractive target for cancer therapy (39–41). Blocking telomerase activity is becoming a strategy for limiting the life span of telomerase-expressing cancer cells. Some telomerase inhibitors have been shown to cause telomerase inhibition and subsequent telomere shortening and senescence in cancer cells. However, the issue of successful inhibition of telomerase in cancer therapy remains to be resolved. Cellular senescence is now thought to be a tumor-suppressive mechanism used for cancer treatment (42–46). Senescence limits tumor development and progression and determines the clinical outcome of conventional cancer therapies. To date, plenty of effective chemotherapeutic drugs were exploited to trigger cellular senescence in cancer therapy through different mechanisms and pathways. We show that PES1 regulates not only telomerase activity but also senescence. Previous studies indicate that PES1 is overexpressed in many cancers, including breast cancer, gastric cancer, colon cancer, neuroblastoma, head and neck squamous cell carcinoma, and astrocytomas (13–20). High PES1 expression in patients with cancer is associated with worse overall and relapse-free survival. PES1 KD inhibits cancer cell growth in vitro and in nude mice. Thus, targeting PES1 may open a new avenue for cancer treatment.
MATERIALS AND METHODS
Plasmids, siRNAs, shRNAs, and reagents
The eukaryotic expression vectors encoding Flag- or Myc-tagged proteins or untagged proteins were generated by inserting polymerase chain reaction (PCR)–amplified fragments into pcDNA3 (Invitrogen). Prokaryotic plasmids encoding GST- or His-fusion proteins were constructed in pGEX-KG (Amersham Pharmacia Biotech) and pET32a (Novagen), respectively. The target sequences of siRNAs and/or shRNAs for PES1 and hTERT were listed in table S1. Because of the extremely low expression level of GST-hTERT protein in E. coli, we optimized the coding sequence of hTERT (table S2) and cloned the optimized sequence into the pGEX-KG vector. The Wnt signaling reporter plasmids TOPFlash and FOPFlash were gifts from X. Yan (Nanchang University). The NF-κB-Luc reporter was a gift from X. Yu (Beijing Institute of Basic Medicine). Lentiviral vectors for gene overexpression were obtained by inserting PCR-amplified gene fragments into pCDH-EF1-MCS-T2A-Puro (System Biosciences). Lentiviral shRNA vectors were constructed by cloning shRNA fragments into pSIH-H1-Puro (System Biosciences). Lentiviruses were produced by cotransfection of HEK293T cells with recombinant lentivirus vectors and pPACK Packaging Plasmid Mix (System Biosciences) using MegaTran reagent (Origene) and were used to infect target cells according to the manufacturers’ instructions.
Anti-Myc (sc-40HRP), β-actin (sc-47778HRP), and anti-PES1 for immunoblot (sc-166300) were purchased from Santa Cruz Biotechnology; anti-Flag (A8592) and anti-Flag M2 agarose (A2220) were obtained from Sigma-Aldrich; anti-GST (RPN1236) and anti-His (27471001) antibodies were purchased from GE Healthcare Life Sciences; anti-PES1 for IHC (A300-902A) was purchased from Bethyl Laboratories; anti-hTERT (abx120550) was purchased from Abbexa; anti-hTERT (ab32020), anti-DKC1, and anti-WDR12 (ab95070) were purchased from Abcam; anti-Hsp90 (60318-1-Ig) and anti-p23 (15216-1-AP) were purchased from Proteintech; anti–c-Myc (D84C12) was purchased from Cell Signaling Technology.
PES1 conditional KO mice
To generate PES1 conditional KO mice, a 16.03-kb genomic fragment, comprising the LoxP-floxed exons 5 to 12, was cloned into the Pfrt1 vector containing an FRT-floxed neomycin resistance cassette and a TK cassette. The neomycin cassette was inserted 621 base pairs (bp) upstream of exon 5, and the other LoxP was inserted 1275 bp downstream of exon 12. Not I–linearized targeting vector was electroporated into TC1 mouse embryonic stem (ES) cells. The chimeric mice were generated by microinjection of targeted ES cells into C57BL/6J blastocysts. Alb-Cre transgenic mice have been reported previously (47). The following PCR primers were used for genotyping: PES1 forward, 5′-GTGTTTAGAGCCACAAGAGGT-3′; PES1 reverse, 5′-GGTCTTGTCTTCCCGTTTCTC-3′; Alb-Cre forward, 5′-ATGAAATGCGAGGTAAGTATGG-3′; Alb-Cre reverse, 5′-CGCCGCATAACCAGTGAAAC-3′. All experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health after securing the approval of the Committee of Animal Care of the Beijing Institute of Biotechnology. MEFs were isolated from PES1fl/fl mouse embryos at day 14.5 of gestation. Briefly, embryos were surgically removed and separated from maternal tissues and the yolk sac. The bodies were minced finely and then incubated in trypsin-EDTA solution with shaking at 37°C for 15 to 30 min. The solution was allowed to settle for 2 min, and the supernatant was centrifuged for 3 min at 1000g. The resulting pellet was resuspended in culture medium and MEFs were obtained. Cre-expressing adenovirus AD-Cre-EGFP (BioWit Technologies) was added to MEFs to KO PES1. PES1 KO hepatocytes were isolated from 3- to 4-month-old PES1fl/fl Alb-Cre mice by a two-step collagenase perfusion method. Briefly, livers were first perfused with 20 to 30 ml of calcium and magnesium-free Hanks’ balanced salt solution (HBSS); the second perfusion was performed with 40 ml of collagenase solution (0.5 g/liter). The liver was excised and the cells were dispersed in HBSS. Cells were then centrifuged at 50g for 1 min. The resulting pellet was resuspended in 20 ml of L15 medium containing 5% fetal bovine serum, 1 mM dexamethasone, and 1 mM insulin, and centrifuged at 50g for 1 min. This step was repeated three times. The resulting hepatocyte pellets were collected.
PES1 KO MCF7 cells
PES1 KO MCF7 cells were generated by CRISPR-Cas9. MCF7-Cas9 stable cells were constructed by transfection with lentivirus expressing Cas9, which was packaged in 293T cells by coexpression of lentiCas9-Blast (Addgene #52962), psPAX2 (Addgene #12260), and pCMV-VSV-G (Addgene #8454). The single guide RNA sequence GTTCGTCCGGAAGCTCCGGA was cloned into the lentiGuide-Puro (Addgene #52963) and packaged into lentivirus in 293T cells, followed by transfection into MCF7-Cas9 cells. After selection with puromycin, cells were collected, and genomic DNA was extracted and amplified by PCR with the following primers: forward, 5′-AGGAGGCATGAGAGTCCTTC-3′; reverse, 5′-CCGTTCCTTGATGATGTGGTC-3′. The PCR products were sequenced using the forward primer. The PES1 expression levels were examined by Western blot.
Cell culture and transfection
Human breast cancer MCF7 cells, human liver cancer HepG2 cells, and HEK293T cells were purchased from the American Type Culture Collection and have been previously tested for mycoplasma contamination. Cells were routinely cultured in Dulbecco’s modified Eagle’s medium (Invitrogen) containing 10% fetal bovine serum (Hyclone). Normal human mammary epithelial cell–TERT cells stably expressing hTERT were a gift from Y. Zhao (Beijing Institute of Genomics) and cultured in Medium 171 with mammary epithelial growth supplement (Invitrogen). Lipofectamine 2000 reagents (Invitrogen) were used for HEK293T cell transfection and Lipofectamine LTX reagents (Invitrogen) were used for cancer cell transfection. For MEF transfection, the vectors were introduced into MEFs using a Nucleofector 2b device (Lonza) with program N-024 according to the manufacturer’s instructions. Luciferase reporter assays were performed according to the manufacturer’s instructions (Promega). Briefly, cells were transfected with NF-κB or Wnt reporter, PES1 or hTERT/TR constructs, and β-gal reporter (an internal control).
Mass spectrometry
The Flag-tagged hTERT complex was obtained by co-IP with anti-Flag from 5 × 108 MCF7 cells stably expressing Flag-hTERT. Cells were lysed in high-salt IP buffer [20 mM tris-HCl at pH 8.0, 1 M NaCl, 1.5% Nonidet P-40 (NP40), 5 mM EDTA, and 5 mM β-mercaptoethanol supplemented with protease inhibitors] and immunoprecipitated with anti-Flag M2 agarose. After washing four times with the high stringent lysis buffer, the immunoprecipitants were resolved on gradient SDS–polyacrylamide gel electrophoresis (PAGE), silver-stained, and subjected to MS sequencing and data analysis. In-solution and in-gel digestion were performed according to a previously published method (48). Briefly, gel bands were minced and destained with 50% acetonitrile in 50 mM ammonium bicarbonate. Proteins were reduced with 10 mM dithiothreitol (DTT) at 56°C, followed by alkylation with 55 mM iodoacetamide at room temperature in the dark. Trypsin digestion was performed overnight at 37°C with gentle shaking. Peptides were extracted using 1% trifluoroacetic acid in 50% acetonitrile. Samples were vacuum-dried and reconstituted in 0.1% formic acid for subsequent MS analysis. The treated samples were examined by nano–LC-MS/MS (nanoACQUITY UPLC and SYNAPT G2 HD mass spectrometer, Waters). MS/MS data were obtained with data-dependent analysis mode and processed with PLGS 2.4 software (Waters), and the resulting peak list was searched against the National Center for Biotechnology Information database with the MASCOT search engine.
Co-immunoprecipitation
Cells transfected with indicated vectors were lysed in NP40 lysis buffer (25 mM Hepes-KOH at pH 7.5, 1.5 mM MgCl2, 150 mM KCl, 0.5% NP40, 10% glycerol, and 5 mM β-mercaptoethanol supplemented with protease inhibitors) for 30 min on ice and disrupted by brief sonication at 4°C. Extracts were clarified by centrifugation at 12,000 rpm for 15 min and immunoprecipitated with anti-Flag M2 agarose for 4 hours at 4°C. Where indicated, RNase A was included in the co-IP experiments at 0.1 mg/ml. Agarose was then washed four times for 10 min each with 0.7 ml of lysis buffer. The immunoprecipitants were subjected to standard Western blot/immunoblot analysis with indicated antibodies.
GST/His pull-down assay
GST or His fusion proteins were expressed in E. coli, purified, and eluted according to the manufacturers’ instructions (Amersham Pharmacia and QIAGEN). For hTERT fusion protein expression, 0.3 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 16°C for 20 hours was used to stimulate the expression. For other fusion proteins, bacteria were cultured for 20 hours at 20°C with 0.1 mM IPTG.Purified fusion proteins with His tag or GST tag were incubated with GST protein bound to GST beads or His protein bound to His beads for 4 hours at 4°C. After washing, the precipitated components were analyzed by immunoblot.
Fast protein liquid chromatography
MCF7 cells (2 × 108) were suspended in nuclear/cytoplasmic separation buffer (20 mM tris-HCl at pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, and 0.1 mM EDTA supplemented with RNase inhibitors and protease inhibitors) and lysed by a dounce homogenizer. After centrifugation at 1000 rpm for 10 min, nuclear pellets were resuspended in a buffer containing 25 mM tris at pH 7.6, 0.2 mM EDTA, 5 mM MgCl2, 0.1 M KCl, 10% glycerol, 0.5 mM DTT, 1 mM benzamidine, 0.2 mM phenylmethylsulfonyl fluoride (PMSF), and RNase inhibitors. Nuclear extracts were applied to a Superose 6 gel filtration column and an FPLC system (GE Healthcare) and calibrated with protein standards (blue dextran, 2000 kDa; thyroglobulin, 669 kDa; ferritin, 440 kDa; catalase, 158 kDa; bovine serum albumin, 75 kDa; all from Amersham Biosciences). The column was eluted at a flow rate of 0.5 ml/min and fractions were collected. Samples were subjected to TRAP, Western blot, and reverse transcription PCR analysis.
Immunofluorescence
Immunofluorescence was performed as previously described (15). Briefly, cells grown on glass coverslips were fixed, permeabilized, and blocked in normal goat serum. The coverslips were then incubated with primary antibodies, followed by incubation with corresponding secondary antibodies. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Confocal images were collected using an LSM 780 confocal microscope (Zeiss).
Cell cycle synchronization
MCF7 cells at G1 phase were obtained by treating cells with aphidicolin (10 μg/ml) for 14 hours. After extensive washing by phosphate-buffered saline (PBS), cells were released from G1 phase by adding fresh medium, and S phase cells were collected 3 hours later. For G2-M phase cells, cells were treated with nocodazole (50 ng/ml) for 14 hours and subjected to mechanical shake-off. Detached cells were M phase cells while rest cells were G2 cells. The cell phases were examined by flow cytometry.
RNA binding protein IP assay
Cultured cells were exposed to UVC (400 mJ/cm2) and lysed in NP40 lysis buffer (50 mM Hepes-KOH at pH 7.5, 150 mM KCl, 2 mM EDTA, 0.5% NP40, and RNase inhibitors and protease inhibitors). Lysates were immunoprecipitated by anti-Flag M2 agarose for 4 hours at 4°C and washed twice with stringent buffer (100 mM tris-HCl at pH 7.4, 500 mM LiCl, 0.1% Triton X-100, 1 mM DTT, and RNase inhibitors and protease inhibitors) and twice with the NP40 lysis buffer. The RNA in the precipitates was assessed by RT-PCR analysis. For re-IP assay, the precipitates containing RNA were eluted with Flag peptide and reimmunoprecipitated with immunoglobulin G (IgG) or anti-Myc antibody and protein A/G. The re-IP complex was then washed and subjected to Western blot and RT-PCR analysis.
RNA pull-down assay
Linearized pCDNA3-hTR was used as template to transcribe biotinylated RNA using Riboprobe In Vitro Transcription Systems (Promega) according to the manufacturer’s instructions. One-half microgram of the biotinylated RNA was incubated at room temperature for 1 hour with 5 μg of indicated recombinant proteins in biotin pull-down assay buffer (50 mM tris-HCl at pH 7.9, 10% glycerol, 100 mM KCl, 5 mM MgCl2, 10 mM β-mercaptoethanol, and 0.1% NP-40). Twenty microliters of washed streptavidin agarose beads (Invitrogen) was added to each binding reaction and further incubated at room temperature for 30 min. Beads were washed briefly five times and boiled in SDS loading buffer, the retrieved proteins were detected by standard Western blot analysis, and the recovered biotin-hTR was examined by the Chemiluminescent Nucleic Acid Detection Module Kit (Thermo Fisher Scientific).
Quantitative RT-PCR
Total RNA was extracted using TRIzol reagent according to the manufacturer’s instructions (Invitrogen). First-strand cDNA was reverse-transcribed with random primers using Moloney murine leukemia virus reverse transcriptase (Promega). The first-strand cDNA was used for PCR amplification with the following primers: hTR forward, 5′-GCCTGCCGCCTTCCACCGTTCATT-3′; hTR reverse, 5′-GACTCGCTCCGTTCCTCTTCCTG-3′; human β-actin forward, 5′-AGAAGAGCTACGAGCTGCCTGA-3′; human β-actin reverse, 5′-CAATGATCTTGATCTTCATTGTGCT-3′; mouse TR forward, 5′-ACCTAACCCTGATTTTCATTAGC-3′; mouse TR reverse, 5′-GCTCTTCGCGGCGGCAGC-3′; mouse β-actin forward, 5′-ATGGATGACGATATCGCTGCG-3′; mouse β-actin reverse, 5′-CCTTCTGACCCATTCCCACC-3′; interleukin-6 (IL-6) forward, 5′-AGGAGAAGATTCCAAAGATGTAGCCGCCC-3′; IL-6 reverse, 5′-TCTGCCAGTGCCTCTTTGCTGCT-3′; MCP1 forward, 5′-CAGCATGAAAGTCTCTGCCGCCCT-3′; MCP1 reverse, 5′-TAGCTCGCGAGCCTCTGCACT-3′; interferon-β (IFN-β) forward, 5′-AATTGAATGGGAGGCTTGAATACTGCCTCAAGG-3′; IFN-β reverse, 5′-GTCTCATTCCAGCCAGTGCTAGATGAATCTTGT-3′; IκBα forward, 5′-TCGGAGCCCTGGAAGCAGCA-3′; IκBα reverse, 5′-AGTCTGCTGCAGGTTGTTCTGGAAGTTG-3′; human pre-tRNA-Leu forward, 5′-GTCAGGATGGCCGAGTGGTCTAAG-3′; human pre-tRNA-Leu reverse, 5′-CCACGCCTCCATACGGAGAACCAGAAGACCC-3′; human pre-tRNA-Arg forward, 5′-GGCTCTGTGGCGCAATGGATA-3′; human pre-tRNA-Arg reverse, 5′-TTCGAACCCACAACCTTTGAATTGCTC-3′; human pre-tRNA-Tyr forward, 5′-CCTTCGATAGCTCAGCTGGTAGAGCGGAGG-3′; human pre-tRNA-Tyr reverse, 5′-CGGAATTGAACCAGCGACCTAAGGATGTCC-3′. β-Actin was used as an internal control. The relative expression was calculated by the comparative Ct method.
TRAP assay
The TRAP assay was performed to determine telomerase activity by using the TRAPeze Telomerase Detection kit (Millipore). Briefly, cultured cell pellets were lysed with 1× CHAPS lysis buffer for 30 min on ice, and breast cancer tissues were ground with mortar and pestle in liquid nitrogen before CHAPS buffer lysis. After centrifugation at 12,000 rpm for 20 min, the concentration of supernatant proteins was determined by the Bio-Rad Protein Assay Kit. Twenty to fifty nanograms of cultured cell lysates, 50 to 100 ng of breast cancer tissue lysates, or 200 to 500 ng of mouse cell lysates was used for TRAP assays. The final reaction products were mixed with GelRed loading buffer (Generay Biotech) and run on a 10% polyacrylamide gel. The gel was exposed by Gel Image System (Tanon). For in vitro reconstitution of telomerase, Flag-hTERT was transcribed and translated using the TNT Quick Coupled Transcription/Translation System (Promega), GST-hTERT was purified from E. coli according to the manufacturer’s instructions (Amersham Pharmacia Biotech), and hTR was transcribed using Riboprobe In Vitro Transcription Systems (Promega) according to the manufacturer’s instructions. Eight microliters of TNT-hTERT or 5 μg of GST-hTERT was mixed with 1 μg of hTR and incubated at 30°C for 90 min, followed by TRAP analysis.
IP-TRAP
For estimation of efficiency of PES1 association with telomerase activity, cells were lysed in 200 μl of NP40 buffer (25 mM Hepes-KOH at pH 7.5, 1.5 mM MgCl2, 150 mM KCl, 0.5% NP40, 10% glycerol, and 5 mM β-mercaptoethanol supplemented with protease inhibitors and RNase inhibitors), and 2 μl was used for TRAP assay and 2 μl was used for immunoblot. The remaining cell lysates (add NP40 buffer to 500 μl) were subjected to IP with anti-Flag agarose. After centrifugation, supernatants were collected, and 5 μl out of 500 μl of the supernatants was used for TRAP assay. The immunoprecipitates were eluted with 100 μl of Flag peptides, and 1 μl of eluate was subjected to TRAP assay.
Direct telomerase activity assay
The direct telomerase activity (DTA) assay was performed to quantitate telomerase activity under the guidance of published methods with minor modifications (49, 50). Briefly, 1 × 107 to 5 × 107 cells were lysed with 1× DTA CHAPS lysis buffer (10 mM tris-HCl at pH 7.5, 1 mM MgCl2, 1 mM EGTA, 0.5% CHAPS, 10% glycerol, 300 mM KCl, 1 mM PMSF, and 1 mM DTT) for 1 hour. After centrifugation at 12,000 rpm for 30 min, the supernatants were transferred to a new tube, mixed with 20 μg of anti-hTERT antibody (abx120550, Abbexa), and rotated in a cold room for 2 hours. Twenty microliters of Protein G agarose (Santa Cruz Biotechnology) was added and rotated for 1 hour. For cells expressing exogenous Flag-tagged hTERT, cell lysates were immunoprecipitated with anti-Flag M2 agarose for 2 hours. After washing of the immunoprecipitates with 5 ml of DTA CHAPS lysis buffer, telomerase was eluted with telomerase peptide antigen (N-ARPAEEATSLEGALSGTRH-C) or Flag peptide. The eluted endogenous telomerase or Flag-hTERT was incubated with Bio-L-18GGG [5′-biotin-CTAGACCTGTCATCA(TTAGGG)3-3′] in reaction buffer (20 mM Hepes-KOH buffer at pH 8.0, 2 mM MgCl2, 200 mM KCl, 1 mM spermidine, 0.1% v/v Triton X-100, 10 mM DTT, 1 mM deoxyadenosine triphosphate, 1 mM deoxythymidine triphosphate, and 1 mM deoxyguanosine triphosphate) at 37°C overnight. The DNA products were immunoprecipitated with Dynabeads M-280 Streptavidin (Thermo Fisher Scientific), eluted with 5 mM d-biotin, and resolved in formamide. After treatment at 80°C for 10 min, telomere DNA was separated with 8 M urea and 10% polyacrylamide gels and transferred to a nylon membrane. The biotin signal was detected by the Chemiluminescent Nucleic Acid Detection Module Kit (89880, Thermo Fisher Scientific).
TRF assay
TRF assay was performed to determine telomere length by using the TeloTAGGG Telomere Length Assay Kit (Roche). Briefly, genomic DNA was purified using SDS and proteinase K digestion, followed by phenol-chloroform extraction methods. DNA samples were digested with Hinf I/Rsa I (New England Biolabs). The digested genomic DNA was run on 0.8% gels in 1× tris-acetate-EDTA buffer (6 hours, 5 V/cm). For mouse hepatocyte telomere length analysis, DNA was separated by pulsed-field gel electrophoresis (PFGE, 0.5 to 1.5 s, 6 V/cm) for 20 hours with the λ DNA-Mono Cut Mix (New England Biolabs) as a marker. After electrophoresis, the gels were treated with 0.2 M HCl, denature buffer (0.5 M NaOH and 1.5 M NaCl), and neutralization buffer (0.5 M tris-HCl at pH 8.0 and 3 M NaCl). The DNA were then transferred to nylon membrane by capillary transfer using 20× SSC transfer buffer, hybridized with digoxigenin-telomere probe, and subjected to chemiluminescence detection. The mean TRF length of breast cancer tissues was then calculated as ∑(ODi)/∑(ODi/Li), where ODi is total chemiluminescence above background in interval i and Li is the average length of i in base pairs according to the manufacturer’s instructions.
SA-β-gal staining
SA-β-gal staining was carried out according to the manufacturer’s instructions (Cell Signaling Technology). Briefly, mouse liver and human breast cancer tissues were flash-frozen in optimal cutting temperature and 5-μm sections were cut. Cells or frozen tissue sections were washed once with PBS and fixed with 0.5% glutaraldehyde in PBS at pH 7.2 for 15 min. After washing in PBS, cells were stained in X-gal solution [100 mM sodium phosphate, 2 mM MgCl2, 150 mM NaCl, 0.01% sodium deoxycholate, 0.02% NP40, 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, and X-gal (1 mg/ml) at pH 6.0] overnight at 37°C. Tissue sections were then stained with eosin.
BrdU incorporation assay
Cells grown on coverslips were incubated with 5-BrdU for 1 to 4 h and stained with anti-BrdU antibody according to the manufacturer’s instructions (BD Pharmingen). Briefly, cells were fixed in 3% paraformaldehyde for 15 min and permeabilized with Triton X-100. After DNA was denatured in 2 M HCl for 10 min, cells were incubated with fluorescein-conjugated mouse anti-BrdU antibody for 30 min. The coverslips were washed four times, incubated with DAPI (0.1 mg/ml) for 5 min, washed, and mounted on glass slides.
Population doubling levels
Cells were maintained continuously in culture, plated (100,000 cells per 6-cm dish), and split every 4 days. At each passage, the cells were counted by a hemocytometer and seeded at the same number of cells at each subculture. PDLs were calculated using the equation PDL = log (Nf/N0)/log2, where Nf is the number of final cells and N0 is the number of initial cells. Cumulative PDLs were calculated by summing the PDLs from all passages.
Cell proliferation assay
MCF7 cells transfected with indicated plasmids were seeded onto 96-well plates (3 × 103 cells per well). Cell proliferation was monitored using CCK8 according to the manufacturer’s instructions (Dojindo Molecular Technologies).
Apoptosis analysis
Annexin V–fluorescein isothiocyanate (FITC) Apoptosis Detection Kit I (BD Pharmingen) was used for apoptosis analysis according to the manufacturer’s instructions. Briefly, cells were washed with PBS and resuspended in 1× binding buffer at a concentration of 1 × 106 cells/ml. Next, 5 μl of FITC–annexin V and 5 μl of propidium iodide were added to 100 μl of the cell suspension, and the samples were incubated for 15 min in the dark. After incubation, 400 μl of 1× binding buffer was added. Apoptosis was analyzed by flow cytometry (Becton Dickinson). The cells undergoing apoptosis were annexin V–FITC–positive/PI–negative (early apoptotic stage) plus annexin V–FITC–positive/PI–positive (late apoptotic or necrotic).
Immunohistochemistry
Breast cancer samples were obtained from the Chinese PLA General Hospital, with the informed consent of patients and with institutional approval for experiments from the Chinese PLA General Hospital and Beijing Institute of Biotechnology. Immunohistochemical staining was performed as described previously (49) using rabbit anti-PES1 (A300-902A, Bethyl Laboratories) as primary antibody. Each specimen was assigned a score according to the intensity of the nuclear staining (no staining = 0, weak staining = 1, moderate staining = 2, strong staining = 3) and the extent of stained cells (0% = 0, 1 to 24% = 1, 25 to 49% = 2, 50 to 74% = 3, 75 to 100% = 4). The final immune reactive score was determined by multiplying the intensity score with the extent score, ranging from 0 (the minimum score) to 12 (the maximum score). The optimal cutoff value of the IHC scores was estimated using receiver operating characteristic curve analysis. We defined score 0 to 4 as low PES1 and score 5 to 12 as high PES1.
Statistical analysis
Trial experiments or similar experiments done previously were used to assess sample size with adequate statistical power. Statistical significance in SA-β-gal staining, PDLs, and BrdU incorporation assay was assessed by two-tailed Student’s t test. The correlation of PES1 expression with telomerase activity and telomere length was determined using the Mann-Whitney test. The correlation between PES1 expression and SA-β-gal staining in clinical samples was determined using Fisher’s exact test. All statistical tests were two-sided. Statistical calculations were performed using SPSS 18.0. In all assays, P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Y. Zhu for assistance with the PFGE experiments. Funding: Q.Y. was supported by the National Natural Science Foundation of China (81330053, 81630067, and 81872246) and the National Key Research and Development Program of China (2017YFA0505602). L.C. was supported by the National Natural Science Foundation of China (81572382). S.G. was supported by the National Natural Science Foundation of China (81472827) and Hundred-Talent Program of Chinese Academy of Sciences. C.N. was supported by the Beijing Natural Science Foundation (5182002). Author contributions: Q.Y. conceived the project, designed the experiments, and analyzed the data. S.G. designed the experiments and analyzed the data. Xiao Yang was responsible for establishment of PES1 conditional KO mice. L.C. designed and performed the experiments. B.Y., S.Y., and C.N. performed the experiments aided by H.M., X.G., Xiaohui Yang, J.H., R.J., E.W., C.Z. and D.X. J.L. collected clinical samples. J.W., B.L., and S.C. performed the FPLC experiments. Y.T., N.H., and X.C. generated PES1 conditional KO mice. L.C. drafted Materials and Methods and the figure legends, and Q.Y. wrote other parts of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaav1090/DC1
Fig. S1. PES1 specifically interacts with hTERT.
Fig. S2. PES1 forms a complex with hTERT and hTR.
Fig. S3. Identification of PES1 KO MCF7 cell line and PES1 KO mice.
Fig. S4. PES1 modulates telomerase activity and telomere length.
Fig. S5. PES1 modulates telomere length.
Fig. S6. PES1 KD induces cellular senescence.
Fig. S7. Effects of PES1 on modulation of noncanonical functions of TERT.
Table S1. siRNAs and shRNAs for PES1 and hTERT.
Table S2. Optimized coding sequence of hTERT.
REFERENCES AND NOTES
- 1.De Vitis M., Berardinelli F., Sgura A., Telomere length maintenance in cancer:At the crossroad between telomerase and alternative lengthening of telomeres (ALT). Int. J. Mol. Sci. 19, E606 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu R. A., Upton H. E., Vogan J. M., Collins K., Telomerase mechanism of telomere synthesis. Annu. Rev. Biochem. 86, 439–460 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deng Y., Chan S. S., Chang S., Telomere dysfunction and tumour suppression: The senescence connection. Nat. Rev. Cancer 8, 450–458 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arndt G. M., MacKenzie K. L., New prospects for targeting telomerase beyond the telomere. Nat. Rev. Cancer 16, 508–524 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Ganesan K., Xu B., Telomerase inhibitors from natural products and their anticancer potential. Int. J. Mol. Sci. 19, E13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ouellette M. M., Wright W. E., Shay J. W., Targeting telomerase-expressing cancer cells. J. Cell. Mol. Med. 15, 1433–1442 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt J. C., Cech T. R., Human telomerase: Biogenesis, trafficking, recruitment, and activation. Genes Dev. 29, 1095–1105 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gómez D. L. M., Farina H. G., Gómez D. E., Telomerase regulation: A key to inhibition? Int. J. Oncol. 43, 1351–1356 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Egan E. D., Collins K., Biogenesis of telomerase ribonucleoproteins. RNA 18, 1747–1759 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holt S. E., Aisner D. L., Baur J., Tesmer V. M., Dy M., Ouellette M., Trager J. B., Morin G. B., Toft D. O., Shay J. W., Wright W. E., White M. A., Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev. 13, 817–826 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Forsythe H. L., Jarvis J. L., Turner J. W., Elmore L. W., Holt S. E., Stable association of hsp90 and p23, but not hsp70, with active human telomerase. J. Biol. Chem. 276, 15571–15574 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Venteicher A. S., Meng Z., Mason P. J., Veenstra T. D., Artandi S. E., Identification of ATPases pontin and reptin as telomerase components essential for holoenzyme assembly. Cell 132, 945–957 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kinoshita Y., Jarell A. D., Flaman J. M., Foltz G., Schuster J., Sopher B. L., Irvin D. K., Kanning K., Kornblum H. I., Nelson P. S., Hieter P., Morrison R. S., Pescadillo, a novel cell cycle regulatory protein abnormally expressed in malignant cells. J. Biol. Chem. 276, 6656–6665 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Lapik Y. R., Fernandes C. J., Lau L. F., Pestov D. G., Physical and functional interaction between Pes1 and Bop1 in mammalian ribosome biogenesis. Mol. Cell 15, 17–29 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Cheng L., Li J., Han Y., Lin J., Niu C., Zhou Z., Yuan B., Huang K., Li J., Jiang K., Zhang H., Ding L., Xu X., Ye Q., PES1 promotes breast cancer by differentially regulatingERα and ERβ. J. Clin. Invest. 122, 2857–2870 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J., Yu L., Zhang H., Wu J., Yuan J., Li X., Li M., Down-regulation of pescadillo inhibits proliferation and tumorigenicity of breast cancer cells. Cancer Sci. 100, 2255–2260 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J., Zhou X., Lan X., Zeng G., Jiang X., Huang Z., Repression of PES1 expression inhibits growth of gastric cancer. Tumour Biol. 37, 3043–3049 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Xie W., Feng Q., Su Y., Dong B., Wu J., Meng L., Qu L., Shou C., Transcriptional regulation of PES1 expression by c-Jun in colon cancer. PLOS ONE 7, e42253 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakaguro M., Kiyonari S., Kishida S., Cao D., Murakami-Tonami Y., Ichikawa H., Takeuchi I., Nakamura S., Kadomatsu K., Nucleolar protein PES1 is a marker of neuroblastoma outcome and is associated with neuroblastoma differentiation. Cancer Sci. 106, 237–243 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weber A., Hengge U. R., Stricker I., Tischoff I., Markwart A., Anhalt K., Dietz A., Wittekind C., Tannapfel A., Protein microarrays for the detection of biomarkers in head and neck squamous cell carcinomas. Hum. Pathol. 38, 228–238 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Okamoto N., Yasukawa M., Nguyen C., Kasim V., Maida Y., Possemato R., Shibata T., Ligon K. L., Fukami K., Hahn W. C., Masutomi K., Maintenance of tumor initiating cells of defined genetic composition by nucleostemin. Proc. Natl. Acad. Sci. U.S.A. 108, 20388–20393 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seimiya H., Sawada H., Muramatsu Y., Shimizu M., Ohko K., Yamane K., Tsuruo T., Involvement of 14-3-3 proteins in nuclear localization of telomerase. EMBO J. 19, 2652–2661 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hölzel M., Rohrmoser M., Schlee M., Grimm T., Harasim T., Malamoussi A., Gruber-Eber A., Kremmer E., Hiddemann W., Bornkamm G. W., Eick D., Mammalian WDR12 is a novel member of the Pes1-Bop1 complex and is required for ribosome biogenesis and cell proliferation. J. Cell Biol. 170, 367–378 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lerch-Gaggl A., Haque J., Li J., Ning G., Traktman P., Duncan S. A., Pescadillo is essential for nucleolar assembly, ribosome biogenesis, and mammalian cell proliferation. J. Biol. Chem. 277, 45347–45355 (2002). [DOI] [PubMed] [Google Scholar]
- 25.He S., Sharpless N. E., Senescence in health and disease. Cell 169, 1000–1011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh A., Saginc G., Leow S. C., Khattar E., Shin E. M., Yan T. D., Wong M., Zhang Z., Li G., Sung W.-K., Zhou J., Chng W. J., Li S., Liu E., Tergaonkar V., Telomerase directly regulates NF-κB-dependent transcription. Nat. Cell Biol. 14, 1270–1281 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Park J.-I., Venteicher A. S., Hong J. Y., Choi J., Jun S., Shkreli M., Chang W., Meng Z., Cheung P., Ji H., McLaughlin M., Veenstra T. D., Nusse R., McCrea P. D., Artandi S. E., Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 460, 66–72 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y., Tergaonkar V., Noncanonical functions of telomerase: Implications in telomerase-targeted cancer therapies. Cancer Res. 74, 1639–1644 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Cao Y., Li H., Deb S., Liu J.-P., TERT regulates cell survival independent of telomerase enzymatic activity. Oncogene 21, 3130–3138 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Khattar E., Kumar P., Liu C. Y., Akıncılar S. C., Raju A., Lakshmanan M., Maury J. J. P., Qiang Y., Li S., Tan E. Y., Hui K. M., Shi M., Loh Y. H., Tergaonkar V., Telomerase reverse transcriptase promotes cancer cell proliferation by augmenting tRNA expression. J. Clin. Invest. 126, 4045–4060 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Masutomi K., Possemato R., Wong J. M. Y., Currier J. L., Tothova Z., Manola J. B., Ganesan S., Lansdorp P. M., Collins K., Hahn W. C., The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc. Natl. Acad. Sci. U.S.A. 102, 8222–8227 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koh C. M., Khattar E., Leow S. C., Liu C. Y., Muller J., Ang W. X., Li Y., Franzoso G., Li S., Guccione E., Tergaonkar V., Telomerase regulates Myc-driven oncogenesis independent of its reverse transcriptase activity. J. Clin. Invest. 125, 2109–2122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schnapp G., Rodi H. P., Rettig W. J., Schnapp A., Damm K., One-step affinity purification protocol for human telomerase. Nucleic Acids Res. 26, 3311–3313 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ali M. M. U., Roe S. M., Vaughan C. K., Meyer P., Panaretou B., Piper P. W., Prodromou C., Pearl L. H., Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 440, 1013–1017 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karagöz G. E., Duarte A. M. S., Ippel H., Uetrecht C., Sinnige T., van Rosmalen M., Hausmann J., Heck A. J. R., Boelens R., Rüdiger S. G. D., N-terminal domain of human Hsp90 triggers binding to the cochaperone p23. Proc. Natl. Acad. Sci. U.S.A. 108, 580–585 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen T. H. D., Tam J., Wu R. A., Greber B. J., Toso D., Nogales E., Collins K., Cryo-EM structure of substrate-bound human telomerase holoenzyme. Nature 557, 190–195 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y., Snow B. E., Hande M. P., Yeung D., Erdmann N. J., Wakeham A., Itie A., Siderovski D. P., Lansdorp P. M., Robinson M. O., Harrington L., The telomerase reverse transcriptase is limiting and necessary for telomerase function in vivo. Curr. Biol. 10, 1459–1462 (2000). [DOI] [PubMed] [Google Scholar]
- 38.Nishimura K., Kumazawa T., Kuroda T., Katagiri N., Tsuchiya M., Goto N., Furumai R., Murayama A., Yanagisawa J., Kimura K., Perturbation of ribosome biogenesis drives cells into senescence through 5S RNP-mediated p53 activation. Cell Rep. 10, 1310–1323 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Crees Z., Girard J., Rios Z., Botting G. M., Harrington K., Shearrow C., Wojdyla L., Stone A. L., Uppada S. B., Devito J. T., Puri N., Oligonucleotides and G-quadruplex stabilizers: Targeting telomeres and telomerase in cancer therapy. Curr. Pharm. Des. 20, 6422–6437 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Shay J. W., Wright W. E., Telomerase therapeutics for cancer: Challenges and new directions. Nat. Rev. Drug Discov. 5, 577–584 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Harley C. B., Telomerase and cancer therapeutics. Nat. Rev. Cancer 8, 167–179 (2008). [DOI] [PubMed] [Google Scholar]
- 42.Cairney C. J., Bilsland A. E., Evans T. R. J., Roffey J., Bennett D. C., Narita M., Torrance C. J., Keith W. N., Cancer cell senescence: A new frontier in drug development. Drug Discov. Today 17, 269–276 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Acosta J. C., Gil J., Senescence: A new weapon for cancer therapy. Trends Cell Biol. 22, 211–219 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Nardella C., Clohessy J. G., Alimonti A., Pandolfi P. P., Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 11, 503–511 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Collado M., Blasco M. A., Serrano M., Cellular senescence in cancer and aging. Cell 130, 223–233 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Shay J. W., Roninson I. B., Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene 23, 2919–2933 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Ye H., Zhang C., Wang B.-J., Tan X.-H., Zhang W.-P., Teng Y., Yang X., Synergistic function of Kras mutation and HBx in initiation and progression of hepatocellular carcinoma in mice. Oncogene 33, 5133–5138 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Jin B.-F., He K., Wang H.-X., Wang J., Zhou T., Lan Y., Hu M.-R., Wei K.-H., Yang S. C., Shen B.-F., Zhang X.-M., Proteomic analysis of ubiquitin-proteasome effects: Insight into the function of eukaryotic initiation factor 5A. Oncogene 22, 4819–4830 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Cohen S. B., Reddel R. R., A sensitive direct human telomerase activity assay. Nat. Methods 5, 355–360 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Tomlinson C. G., Sasaki N., Jurczyluk J., Bryan T. M., Cohen S. B., Quantitative assays for measuring human telomerase activity and DNA binding properties. Methods 114, 85–95 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaav1090/DC1
Fig. S1. PES1 specifically interacts with hTERT.
Fig. S2. PES1 forms a complex with hTERT and hTR.
Fig. S3. Identification of PES1 KO MCF7 cell line and PES1 KO mice.
Fig. S4. PES1 modulates telomerase activity and telomere length.
Fig. S5. PES1 modulates telomere length.
Fig. S6. PES1 KD induces cellular senescence.
Fig. S7. Effects of PES1 on modulation of noncanonical functions of TERT.
Table S1. siRNAs and shRNAs for PES1 and hTERT.
Table S2. Optimized coding sequence of hTERT.