

This study demonstrates targeting of blood-brain barrier disruption to deliver payloads at sites of neurological disease.
Abstract
Diseases that lead to blood-brain barrier (BBB) disruption will pathologically expose normally inaccessible brain extracellular matrix (ECM) to circulating blood components. Therefore, we hypothesized that brain ECM-targeting moieties could specifically target the disrupted BBB and potentially deliver therapies. Variable lymphocyte receptors (VLRs) that preferentially associate with brain ECM were identified from an immune VLR library via yeast surface display biopanning coupled with a moderate throughput ECM screen. Brain ECM binding of VLR clones to murine and human brain tissue sections was confirmed. After systemic administration, P1C10, the lead brain ECM-targeting VLR candidate, specifically accumulated in brains with mannitol-disrupted BBB and at disrupted BBB regions in two different intracranial glioblastoma models. We also demonstrate P1C10’s ability to deliver doxorubicin-loaded liposomes, leading to significantly improved survival in glioblastoma-bearing mice. Thus, VLRs can be used to selectively target pathologically exposed brain ECM and deliver drug payloads.
INTRODUCTION
Under healthy conditions, brain endothelial cells form the blood-brain barrier (BBB) that differentially segregates blood components from the central nervous system (CNS) (1). Because of the BBB’s specialized barrier properties, including tight junctions and drug efflux transport, CNS uptake of systemically administered therapeutics is often limited (1, 2). However, multiple events including acute insults such as trauma and stroke, chronic pathologies such as tumors and multiple sclerosis, and artificial methods such as high-frequency focused ultrasound and osmotic agents can disrupt BBB integrity (1, 3–7). Although the time course, extent and location of BBB disruption markedly differ with each condition, the consistent result is pathological exposure of normally sequestered brain extracellular matrix (ECM) (1, 2, 6, 8–10). Therefore, targeting brain regions with pathologically disrupted BBB could serve as a noncell-intrinsic method of delivering therapy to previously “difficult-to-access” CNS disease sites. This strategy is potentially superior to therapies directed against cell-intrinsic targets because normal brain ECM serves as the targeting ligand rather than cell-associated disease variants or markers that are often altered or lost after treatments, resulting in therapeutic resistance (11). Moreover, a single ECM-targeting ligand could be applied to many different CNS diseases or conditions and provide a versatile therapeutic platform, contrasting to other recent ECM-targeted approaches that selectively target injury or disease-associated ECM (4, 12).
To test the potential benefits of ECM-targeted drug delivery, we first applied the concept to the incurable brain cancer, glioblastoma (GBM). Better GBM therapies are sorely needed because patients have a uniformly poor prognosis with median survival of less than 2 years despite aggressive clinical treatments of surgery, chemotherapy, and radiation (13, 14). Recent data indicate that GBM targeting of immunotherapy through interleukin-13 (IL-13)–targeted chimeric antigen receptor (CAR) T cells could have marked effects for a short time in selected patients, and CAR T cells targeting chondroitin sulfate proteoglycan 4 recently demonstrated efficacy in a murine model (11, 12, 15). Thus, targeting of GBM and, specifically, ECM components could offer new treatment strategies. In addition, central or internal GBM regions exhibit disrupted BBB revealed by magnetic resonance imaging enhancement with gadolinium contrast, suggesting compatibility with ECM-targeted approaches (6, 16). Thus, we hypothesized that pathologically exposed brain ECM in GBM could be targeted for therapeutic purposes.
While standard peptides and antibodies could be used as ECM-targeting reagents, we chose to deploy lamprey antigen receptors known as variable lymphocyte receptors (VLRs) (17). VLRs are crescent-shaped, leucine-rich repeat proteins that recognize antigenic targets with specificity and affinity comparable to immunoglobulin (Ig)–based antibodies (1, 18, 19). Combining the unique binding site geometry with the approximately 500 million years of evolutionary distance between lamprey and mammals, VLRs could possibly recognize conserved proteins and glycans in ECM that may not be effectively targeted by mammalian antibodies (17–19). To identify VLRs that bind brain ECM, we screened an immunized lamprey VLR library in yeast surface display format by biopanning. The resultant enriched pool of ECM-binding clones was subsequently assayed for preferential binding to brain ECM using a moderate throughput, enzyme-linked immunosorbent assay (ELISA)–based screen. The lead clone demonstrated preferential accumulation at disrupted BBB sites in animals with osmotic BBB disruption or intracranial GBM tumors and also effectively targeted doxorubicin-loaded liposomes to improve survival.
RESULTS
Identification of brain ECM-binding VLRs
Brain ECM-binding VLRs were identified by mining a yeast surface display library of VLRs. The library was generated from the VLR repertoire of lamprey immunized with mechanically isolated murine brain microvessel plasma membrane preparations that contained associated brain ECM (20). The library was first enriched for ECM binders via two rounds of biopanning on decellularized ECM generated by cultured mouse brain endothelial cells (bEnd.3 cell line). Next, we sought to identify ECM-binding clones that preferentially bound bEnd.3 ECM compared to control mouse fibroblast ECM (3T3 cell line). For this analysis, we picked individual yeast clones into 96-well plates and then expanded and induced them for VLR display. To enable moderate throughput analysis of the clones and avoid cumbersome subcloning of many VLR clones, we removed the VLRs directly from yeast surface via reduction. After removal, we evaluated the comparative binding of VLRs to bEnd.3 and 3T3 ECM via ELISA screening (Fig. 1A). In total, 285 clones were assayed for differential binding, and observed binding signals were as high as ~5-fold preference for bEnd.3 ECM (Fig. 1B). Ten clones demonstrated at least a 2.5-fold preference for bEnd.3 ECM. To further validate the ELISA-based screening method, the individual VLR-displaying yeast clones were panned against both bEnd.3 and 3T3 ECM and imaged via bright-field microscopy. For example, yeast displaying clones P1C10 and P2C7 (ELISA fold change, 4.2 and 4.5, respectively) bound selectively to bEnd.3 ECM compared to 3T3 ECM, whereas a nonbinding VLR clone P1E9 showed very little ECM binding (Fig. 1C, P1E9).
Fig. 1. Identification of VLRs that bind brain ECM.
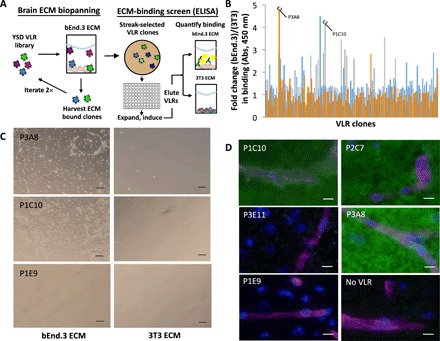
(A) A yeast surface display VLR library underwent two rounds of biopanning to enrich for clones that bind bEnd.3 ECM. Next, individual clones were grown, induced, and screened for comparative binding to bEnd.3 and 3T3 ECM. VLRs were released from the yeast surface using reducing agent, and VLR binding to ECM was measured by probing the anti–c-myc epitope tag in an ELISA format. (B) Fold changes in ECM binding. The ratio of bEnd.3 ECM/3T3 ECM ELISA signals is shown for 285 VLR clones. Lead clones P3A8 and P1C10 are noted. Abs, absorbance. (C) Preferential binding to bEnd.3 ECM was evaluated for selected clones by biopanning individual clones onto bEnd.3 and 3T3 ECM. Yeast binding was qualitatively analyzed by bright-field microscopy. Scale bars, 100 μm. (D) VLRs were reduced off the yeast surface and used in labeling murine brain sections and detected via an anti–c-myc epitope tag antibody (green). Microvessels (magenta) were labeled with IS-GB4 lectin, and cell nuclei (blue) were visualized with Hoechst 33342 stain. Scale bars, 20 μm.
Next, to confirm the in vivo relevance of ECM-binding VLR candidates, they were assayed for binding to murine brain sections. Eight of the top 10 VLR clones from the ELISA screen showed binding to murine brain sections (Fig. 1D). Two VLRs, including the P3E11 clone shown in Fig. 1D, did not show brain tissue labeling despite their binding to cell culture ECM. Each of the binding clones demonstrated a diffuse parenchymal brain ECM immunolabeling pattern, with no apparent vascular or cell type enrichment despite the fact that brain endothelial cell ECM was used as the biopanning substrate. Given their positive binding and substantial bEnd.3 ECM selectivity in the ELISA assay, P1C10 and P3A8 were subjected to more detailed evaluation.
P1C10 displays brain-selective ECM binding
P1C10 and P3A8 were cloned into our previously described intein fusion yeast display system that allows site-specific modification of the VLR at the C terminus (21). In this way, P1C10 and P3A8 were functionalized with Cy5 fluorescent dye (22). Direct immunolabeling of murine tissues using VLR-Cy5 conjugates indicated that P1C10-Cy5 had substantial brain ECM selectivity compared to kidney (10.1-fold increase), heart (20.2-fold increase), and liver (5.7-fold increase) tissues (Fig. 2A). In contrast, P3A8-Cy5 bound both brain and liver ECM with similar intensity but, like P1C10-Cy5, also did not show binding to kidney and heart tissues (fig. S1). Next, we tested the ability of P1C10 to cross-react with human brain ECM using human brain cryosections. P1C10-Cy5 bound human brain ECM compared with controls, with a pattern similar to that observed in murine brain sections (Fig. 2B). Furthermore, P1C10-Cy5 also bound to ECM in cryosections of freshly resected human GBM specimens (Fig. 2C). Given the desirable attributes of brain selectivity and murine-human cross-reactivity, P1C10 affinity was measured and the monomeric dissociation constant (Kd) for binding to bEnd.3 ECM was 48.38 ± 6.05 nM (Fig. 2D).
Fig. 2. Binding characterization of P1C10.
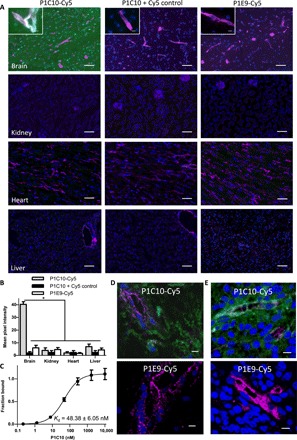
(A) P1C10-Cy5 (green) was used to probe murine tissues. GS-IB4 lectin labels microvessels (magenta), and cell nuclei are stained with Hoechst 33342 (blue). The P1C10 + Cy5 control column of images are labeled as in the left column except the P1C10 VLR and Cy5 that are not chemically conjugated. Images in the right column are labeled with nonbinding VLR, P1E9-Cy5. Scale bars, 100 μm. Inset images are higher magnification of brain labeling. Scale bars, 20 μm. (B) Quantification of VLR signal from images derived from groups presented in (A) [*P < 0.01, analysis of variance (ANOVA)]. (C) P1C10 VLR demonstrates a monomeric affinity of 48.38 ± 6.05 nM for bEnd.3 ECM. Three replicates (means ± SD) fit to a monomeric equilibrium binding model are plotted. (D) Freshly resected, snap-frozen, histologically normal human brain section labeled with either P1C10-Cy5 (green) or negative control P1E9-Cy5. Anti-Cd31 monoclonal antibody (mAb) labels microvessels (magenta), and cell nuclei are stained with Hoechst 33342 (blue). (E) A patient-derived GBM specimen was freshly resected, snap-frozen, and sectioned for labeling with P1C10-Cy5 (green) and compared to negative control, P1E9-Cy5. Anti-Cd31 mAb labels microvessels (magenta), and cell nuclei are stained with Hoechst 33342 (blue). Scale bars, 20 μm (D).
Brain ECM-binding VLR is retained within brain after BBB disruption
We next evaluated whether P1C10 would accumulate at sites of BBB disruption by using hyperosmolar mannitol for transient global BBB disruption in murine brain (23). P1C10 or RBC36 modified with IR800 near-infrared dye was administered intravenously at 1 mg/kg to healthy C57BL/6 mice. RBC36 is a VLR that recognizes human H antigen trisaccharide (18) and was used as an isotype control. Next, mannitol was administered intravenously to transiently open the BBB. With the BBB still disrupted, mice were perfused to remove unbound VLR. We resected and imaged whole brains for retained IR800 signal (Fig. 3). Mice treated with P1C10-IR800 had a significant, 3.3-fold increase in accumulated brain fluorescence compared to RBC36-IR800 and a 7.6-fold increase compared to saline-treated animals. Thus, P1C10 selectively accumulates in normal brains after transient BBB disruption.
Fig. 3. Brain retention of VLRs after BBB disruption with mannitol.
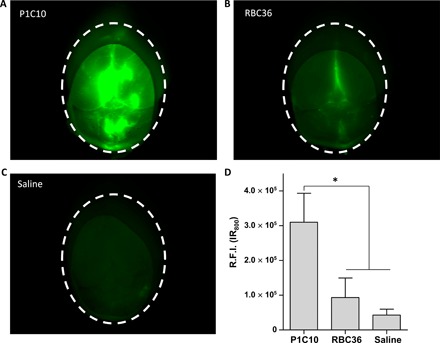
C57BL/6 mice were intravenously administered VLR-IR800, followed by hyperosmolar mannitol to transiently disrupt the BBB. While the BBB was still disrupted, mice were perfused to remove unbound VLR. Representative, whole-brain images of retained IR800 signal from mice administered (A) P1C10-IR800, (B) RBC36-IR800, or (C) saline animals are presented and quantified in (D). Dotted line outlines total brain area that was quantified in each image (means ± SD, n = 3 mice per group; R.F.I., relative fluorescent intensity; *P < 0.01, ANOVA).
Brain ECM-binding VLRs selectively accumulate within intracranial GBM
We used two different intracranial GBM models to test the hypothesis that pathological exposure of brain ECM can also be targeted by the identified VLRs. Intracranial implantation of murine GL261 or human U87 GBM cells results in brain tumors that exhibit chaotic vasculature and regional BBB disruption (24–26). First, P1C10 or RBC36 (1 mg/kg) isotype control (both modified with IR800 near-infrared dye) were intravenously administered to C57BL/6 mice bearing syngeneic, intracranial GL261 GBM. VLRs circulated for 30 min before mice were perfused, brain resected, and imaged for IR800 signal. The mean fluorescence intensity of the GL261 tumor region for mice treated with P1C10-IR800 was 112-fold higher than that of the contralateral brain region (Fig. 4A). By contrast, the fluorescence intensity of the GL261 tumor region for mice treated with control RBC36-IR800 was only increased ninefold compared with contralateral brain. Moreover, the accumulated P1C10-IR800 in tumor was 13-fold higher than that observed for control RBC36-IR800. Together, these data indicate that P1C10-IR800 selectively accumulated within GL261 orthotopic tumors. Next, given the capability to target pathologically exposed ECM in mouse tumors, we also tested whether P1C10 could target ECM in orthotopically implanted human tumors. VLR uptake was monitored after VLRs were administered as above to severe combined immunodeficient (SCID) mice bearing intracranial U87 GBM tumors. In addition to P1C10, which exhibits diffuse binding to brain ECM, we also evaluated another VLR (192, identified in our laboratory), which binds much more selectively to the basolateral side of the brain vasculature in addition to kidney and liver vessels and ECM (fig. S2) (20). P1C10, 192, and RBC36 were fused to a rabbit Fc region (VLR-Fc) to facilitate avidity and detection while minimizing renal clearance. VLR-Fc (3 mg/kg each) was administered intravenously and allowed to circulate for 30 min. After full-body perfusion, organs were removed, sectioned, and imaged for the presence of VLR-Fc. Both VLR clones P1C10-Fc and 192-Fc accumulated within the margins of the GBM, with P1C10-Fc being distributed throughout the tumor ECM (Fig. 4B). While 192-Fc exhibited some parenchymal distribution, it was more concentrated outside of large tumor vessels (Fig. 4B), recapitulating the perivascular binding pattern observed in tissue section labeling assays. Neither VLR appreciably accumulated in the normal contralateral brain hemisphere from the same mouse, and isotype control RBC36-Fc was not found in tumor or normal brain regions (Fig. 4B). P1C10-Fc did not preferentially accumulate above background in treated mouse kidney, heart, or liver sections. In contrast, we found 192-Fc signal proximal to blood vessels of renal glomeruli and liver sinusoidal blood vessels (Fig. 4B). Quantitatively, P1C10-Fc demonstrated a 21.2-fold increase in U87 tumor compared to contralateral controls, as well as increases of 21.2-fold compared to kidney, 15.9-fold compared to liver, and 29.6-fold compared to heart. Both P1C10-Fc (25.4-fold) and 192-Fc (11.9-fold) yielded increased accumulation in U87 tumor regions compared to the RBC36-Fc control. Both distributions mirrored those suggested by tissue section labeling (Fig. 2 and fig. S2). Together, both P1C10-Fc and 192-Fc specifically target and preferentially accumulate at regions of vascular disruption in GBM, with P1C10-Fc displaying more brain specificity.
Fig. 4. Tissue accumulation of VLRs after systemic administration in GBM models.
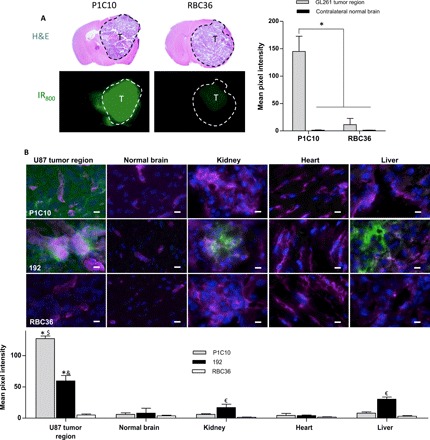
(A) C57BL/6 mice, bearing GL261 intracranial tumors, were intravenously administered VLR-IR800 (1 mg/kg). After 30 min, mice were perfused and the brains resected and imaged for IR800 signal (green). H&E images of coronal brain sections are depicted above the IR800 signal for representative mice, T represents the tumor region in each image and the tumor region is outlined. Signal from GL261 tumor region and normal contralateral brain was quantified (means ± SD, n = 3 mice per group; *P < 0.01, ANOVA). (B) VLR-Fc fusions (3 mg/kg) were administered via tail vein injection to SCID mice bearing intracranial U87 GBM and allowed to circulate for 30 min. After full-body perfusion, organs were harvested, sectioned, and probed for Fc region (VLR, green), blood vessels (GS-IB4, magenta), and nuclei (Hoechst 33342, blue). Columns from left to right are U87 tumor region, normal contralateral brain, kidney (focused on a glomerulus), heart, and liver. Scale bars, 20 μm. Accumulated VLR-Fc fluorescence signal intensity is quantified below the images for each tissue (*P < 0.01, comparing VLR, U87 tumor signal for either P1C10-Fc or 192-Fc to RBC36-Fc, t test. $P < 0.01 for P1C10-Fc U87 tumor signal compared to all other tissues, ANOVA. &P < 0.05 for 192-Fc U87 tumor signal compared to all other tissues, ANOVA. €P < 0.05 for 192 VLR signal within a tissue type compared to P1C10 or RBC36, ANOVA).
VLRs can be conjugated to doxorubicin-loaded liposomes
Given the targeting specificity of P1C10, we wished to test the effectiveness of ECM targeting for therapeutic delivery. Thus, we next used VLRs to decorate doxorubicin-loaded liposomes as a proof-of-principle exploration of pathologically exposed ECM for therapeutic targeting in GBM. Doxorubicin was chosen as the test drug because it is easily visualized due to intrinsic fluorescence, there is abundant literature describing liposome loading and tumor targeting, and doxorubicin is a substrate for drug efflux transporters in cancer and at the BBB (24, 25, 27). Pegylated liposomes were loaded with doxorubicin as previously described (28). Resultant liposomes had a median diameter of 94.2 nm and contained doxorubicin (1 to 2 mg/ml; fig. S3). Next, we prepared VLR-intein fusion protein for attachment by installing an azide group at the C terminus of the VLR as previously described (fig. S3) (22). The azide-functionalized VLRs were next attached to the liposome surface by reacting with a strained cyclooctyne (DBCO) that was doped into the liposome formulation (DBCO-PEG2000-DSPE). Specific VLR attachment was confirmed by measuring a significant increase in protein concentration in liposomal formulations having the VLR-azide (fig. S3). Last, VLRs retained their binding activity after immobilization on the liposome surface as demonstrated by a 4.5-fold increase in doxorubicin signal for P1C10-targeted liposomes binding to murine brain sections compared to RBC36-targeted control liposomes (Fig. 5). To demonstrate the feasibility of performing treatment studies with VLR-targeted doxorubicin-loaded liposomes, we incubated U87 GBM cells cultured on bEnd.3 ECM with P1C10- or RBC36-targeted doxorubicin-loaded liposomes in vitro (Fig. 5). We observed an enhanced cell killing with P1C10-targeted liposomes with a median effective concentration (EC50) of 199.0 ± 1.7 nM compared with an EC50 of 3312.0 ± 2.6 nM for RBC36-targeted liposomes.
Fig. 5. Characterization of P1C10-targeted doxorubicin-loaded liposomes.
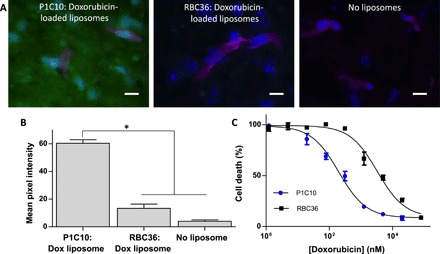
(A) VLR-targeted doxorubicin-loaded liposomes were used in a binding assay with murine brain tissue sections and imaged for doxorubicin fluorescence (green). GS-IB4 lectin labels microvessels (magenta), and cell nuclei are stained with Hoechst 33342 (blue). (B) Doxorubicin fluorescent signal, from groups presented in (A), is quantified for each group (*P < 0.01, ANOVA). (C) U87 cell viability as a function of doxorubicin concentration provided by VLR-conjugated doxorubicin-loaded liposomes bound to bEnd.3 ECM. The EC50 was determined by seeding U87 cells onto bEnd.3-derived brain ECM, incubating with VLR-conjugated doxorubicin labeled liposomes, washing unbound liposomes, and then culturing for 72 hours at which point the cell viability was measured. All dilutions were performed in triplicate. EC50 for P1C10 = 199.0 ± 1.7 nM and RBC36 = 3312.0 ± 2.6 nM (P < 0.05, Student’s t test).
P1C10-targeted liposomes significantly extend survival of GBM-bearing mice
To demonstrate the potential therapeutic utility of targeting pathologically exposed brain ECM, we treated SCID mice bearing intracranial U87 GBM with P1C10-, 192-, or RBC36-targeted doxorubicin-loaded liposomes. For initial accumulation studies, tumor-bearing mice were administered one intraperitoneal dose of VLR-targeted doxorubicin liposomes, perfused, and then imaged for accumulated doxorubicin fluorescence. Whole-brain images of doxorubicin signal from mice treated with either P1C10- or RBC36-targeted doxorubicin liposomes show enhanced doxorubicin uptake with P1C10-targeted liposomes (Fig. 6, A and B). Hematoxylin and eosin (H&E) staining of brain sections adjacent to sections used for fluorescent imaging demonstrates that the elevated doxorubicin signal was localized within tumor margins rather than contralateral normal brain. Higher-resolution fluorescent images indicate that P1C10 mediates an elevated doxorubicin accumulation within the tumor region compared to control contralateral brain tissue (7.6-fold increase) or the signal observed within the tumor region after exposure to control RBC36 (7.9-fold increase) (Fig. 6, C to E). These data correlate with the VLR brain distribution data (Fig. 4), indicating that P1C10 mediates doxorubicin accumulation at the tumor site. To test therapeutic efficacy, we administered VLR-targeted liposomes to mice having detectable tumor burden 1 week after implantation (fig. S4). Four weekly therapeutic doses of 12 mg doxorubicin/kg of doxorubicin-loaded liposomes targeted by P1C10, 192, or RBC36 were administered by intraperitoneal injection at days 7, 14, 21, and 28. There were no significant differences in weight between groups (fig. S4C). Median survivals after tumor implantation were 43, 30, and 28 days for P1C10-, 192-, and RBC36-targeted groups, respectively. The P1C10-targeted group demonstrated significantly improved survival compared to both the 192 and RBC36 control groups. Neither the 192 nor the RBC groups offered any survival benefit over a no-treatment control. These data indicate that targeting of pathologically exposed brain ECM with P1C10 can offer a therapeutic advantage versus perivascular targeting (192) or no targeting (RBC36).
Fig. 6. VLR targeting of doxorubicin-loaded liposomes in a GBM model.
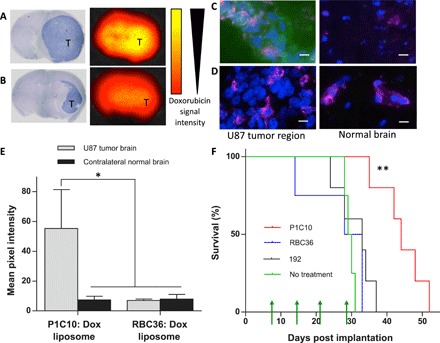
SCID mice bearing intracranial U87 tumors were intraperitoneally administered 12 mg doxorubicin/kg of VLR-targeted doxorubicin-loaded liposomes. (A) Accumulation of P1C10-targeted doxorubicin-loaded liposomes after 30 min of circulation. Representative H&E-stained murine brain bearing U87 glioma with the tumor region, dark blue and denoted with “T.” The paired fluorescence image depicts doxorubicin fluorescence signal in an adjacent serial brain section. Yellow represents a stronger signal. (B) Accumulation of RBC36-targeted doxorubicin-loaded liposomes. Stained and imaged as in (A). (C) Accumulation of P1C10 or (D) RBC36-targeted doxorubicin-loaded liposomes was evaluated using doxorubicin fluorescence (green). GS-IB4 lectin labels microvessels (magenta), and cell nuclei are stained with Hoechst 33342 (blue). Doxorubicin accumulation was evaluated in the U87 tumor region and the contralateral normal brain. Scale bars, 20 μm. (E) Doxorubicin fluorescence signal, from groups presented in (C) and (D), is quantified for each group (*P < 0.05, ANOVA). (F) P1C10-, 192-, or RBC36-targeted doxorubicin-loaded liposomes were administered once per week for 4 weeks (green arrows). The percentage of surviving mice was plotted for each group using Kaplan-Meier curves. P1C10 (n = 5) targeting conferred a significant survival benefit compared to 192 (n = 5), RBC36 (n = 4), and nontreated (n = 4) groups (**P < 0.01, log-rank tests).
DISCUSSION
This study presents an approach for identifying VLRs that accumulate in pathologically exposed brain ECM for therapeutic targeting of brain diseases. We used VLRs rather than conventional targeting molecules such as immunoglobulins, peptides, or DARPins (29–31). As described earlier, VLRs have a leucine repeat structure, are smaller than immunoglobulins, have a large evolutionary distance from mammals, and have additional propensity to bind glycostructures (17–19, 32). All four of these features are desirable for identifying targeting ligands that accumulate in brain ECM.Whereas the unique structure and evolutionary distance expand the potential epitope range, the small size- and sugar-binding properties of VLRs could prove to be beneficial for accumulation and penetration into the tightly packed, glycosylated environment of brain ECM (27, 33). The novel, moderate throughput screening assay described in this study was suitable for identification of VLRs that specifically bind brain ECM. In particular, coupling an ECM biopanning protocol with a differential ECM-binding ELISA that simply used VLRs cleaved directly from the yeast surface enabled rapid identification of brain ECM-specific binders. In addition, the principles of this novel screening assay could be applied to identify targeting ligands that bind ECM derived from other organs or disease states.
Unlike typical therapeutic strategies that are designed to target disease-specific, cell-intrinsic markers (24), we show that targeting of pathologically exposed ECM could be effective in neurological diseases that exhibit differential BBB permeability such as GBM (6, 8, 9). Another advantage is that there are available techniques to artificially expose sequestered ECM via local BBB disruption such as targeted high-frequency focused ultrasound. To our knowledge, only two other recent studies have suggested targeting ECM as a strategy to treat neurological diseases. The first study identified a peptide that homed to traumatic brain injury (TBI) injury sites and appeared to selectively bind brain ECM associated with the TBI injury site (4). The second study generated CAR T cells targeted to chondroitin sulfate proteoglycan 4, which is reportedly overexpressed in brain tumor ECM (12). Both studies rely on targeting diseased ECM and are therefore likely best suited for treating diseases demonstrating these target-specific biomarkers. In contrast, the VLRs identified in our work bind normal brain ECM and strategically use the pathological exposure of normal ECM for targeting specificity. This approach could therefore obviate the need to identify a disease-specific target, and the ECM-targeting VLRs could be customized for any neurological disease that exhibits BBB disruption (or via artificial BBB disruption) by simply altering the therapeutic payload. Last, the initial immunohistochemical and biodistribution analyses with VLR clone P1C10 demonstrated high brain ECM specificity compared to peripheral tissues, indicating that brain ECM can serve as a “specific” target when coupled with pathological disruption of BBB properties. Thus, this strategy represents a significant departure from existing platforms that focus on targeting disease-related cell-intrinsic markers and/or schemes to circumvent the BBB.Specific targeting of BBB disruption could potentially reduce off-target effects compared with ligands that target overexpressed receptors as these proteins can also be expressed elsewhere in the body, although this would require further study.
This study used two different intracranial GBM murine models to demonstrate the functional relevance of targeting pathologically permeable BBB to deliver therapeutics. Implanting GL261 intracranially into C57BL/6 mice is a widely used immunocompetent syngeneic mouse GBM model for immunotherapy-related studies (26). In addition, implantable human-derived U87 GBM is a commonly used preclinical model, although the specific patient origin is unclear (24, 25, 34). Benefits include that it is a human tumor, has controlled tumor initiation, and can be implanted intracranially in SCID mice. Previous studies have demonstrated that treating U87 intracranial glioma with IL-13– or 2C5 nucleosome–recognizing, antibody-targeted, and doxorubicin-loaded liposomes significantly extended survival, with the IL-13 targeting leading to a partial cure (24, 25). Similarly, P1C10 targeting resulted in doxorubicin accumulation within GBM xenografts, reduced in vitro EC50, and significantly improved survival. In contrast to the IL-13 study, we observed no cured animals after doxorubicin treatment. However, the unoptimized doxorubicin dosing and administration in our study is different from the previous IL-13 work, and IL-13 may also result in tumor cell endocytosis (35), which may confer superior efficacy compared to an ECM-targeted doxorubicin payload. We administered all treatment groups in this study equal amounts of VLR-conjugated, doxorubicin-loaded liposomes. Thus, the therapeutic benefit observed with P1C10 VLR is directly related to its specific targeting of disrupted BBB regions, and the biodistribution data suggest that the therapeutic benefit results from enhanced accumulation of doxorubicin selectively within the GBM. The survival benefit was only observed with P1C10, which displays diffuse parenchymal brain ECM binding, and not with the perivascular accumulating VLR 192. Additional studies comparing the P1C10 and 192 VLRs, as well as possible use of mass spectrometry or glycan arrays or other methods to identify the ECM antigenic components recognized by these VLRs, could offer insights into possible differences in therapeutic efficacy that may be based on actual delivered dose or differential localization.
Although GBM was used as a proof-of-concept neurological disease in this study since these tumors exhibit pathological BBB disruption and ECM exposure, it is important to note that recent studies demonstrate GBM tumors exhibit only partially disrupted BBB (6). The invasive tumor margins generally reside behind an intact BBB.Therefore, targeting pathologically permeabilized BBB with VLRs appears to be an effective strategy for delivering drug to the tumor core, but further work is needed to combine these VLRs with a therapeutic platform that is capable of spreading and inclusively targeting the clinically important invasive margin. Alternatively, techniques such as targeted high-frequency focused ultrasound or stereotactic radiation could further disrupt the BBB, potentially allowing brain ECM-binding VLRs to deliver therapeutics to the tumor margins. In addition, rather than targeting doxorubicin as in this proof-of-principle study, P1C10 VLR could be used to target and deliver more clinically relevant anti-GBM treatments such as temozolomide-loaded liposomes or immune checkpoint inhibitors (36, 37). To gather evidence for broad application and versatility of our approach, we envision extending our paradigm of delivering therapies via brain ECM targeting to additional disease models that show pathological exposure of brain ECM, including stroke, trauma, and neurodegenerative conditions. In addition, given the observed P1C10 accumulation after BBB disruption via hyperosmolar mannitol, it would be conceivable to use a combination of ECM-targeting VLRs and localized anatomical BBB disruption techniques such as high-frequency focused ultrasound to specifically target therapies to diseased brain regions that are associated with an intact BBB (27, 38). Although relatively low VLR immunogenicity has been reported in recent murine studies, VLRs would likely require humanization for future clinical applications, potentially by grafting antigen-binding domains onto human proteins that have a structurally homologous leucine repeat structure (39, 40). Last, with the potential of VLRs to target and deliver many different therapeutic payloads having various mechanisms of action, detailed toxicity testing for each VLR-payload combination will be necessary.
In conclusion, we present a proof-of-principle study for exploiting the pathological exposure of brain ECM for therapeutic benefit. Identification of VLRs and other targeting reagents that can target the pathological exposure of brain ECM and be combined with compatible therapeutic payloads may provide a new approach for treating many debilitating neurological diseases that currently lack effective treatments, including incurable GBM.
MATERIALS AND METHODS
Experimental design
Sample size
Sample sizes were dependent on the assay and are presented within the methods section for each assay.
Rules for stopping data collection
Data were collected at a predefined endpoint for each in vitro assay based on manufacturer’s protocol or recommendations from literature. With murine survival studies, mice were euthanized in accordance with the Institutional Animal Care and Use Committee (IACUC) guidelines.
Data inclusion/exclusion criteria
Data generated for all assays were presented.
Outliers
No outliers were removed in this study.
Selection of endpoints
Study endpoints were predefined on the basis of the assay (please see methods sections below) and were not changed on the basis of outcomes.
Replicates
All in vitro assays were performed in multiple replicates as described within the methods section. Murine studies were powered as described below to measure statistically significant differences between groups.
Research objectives
The overall goal of this proof-of-concept study was to identify VLRs that bind normal brain ECM and demonstrate the functional ability of these VLRs to treat neurological pathologies that present with pathologic exposure of brain ECM.
Research subjects or units of investigation
NOD-SCID and C57BL/6 mice, bEnd.3 and 3T3 tissue-cultured cells, U87 and GL261 GBM cell lines, murine tissue sections (multiple organs), human brain sections, and human GBM sections were used in this study.
Experimental design
This study was a controlled laboratory experiment. Initial measurements were made using quantitative ELISA-based assays. Clones were validated using qualitative and quantitative in vitro assays and qualitative and quantitative murine studies.
Randomization
Mice were randomized into groups after intracranial implantation of U87 or GL261 GBM cells.
Blinding
Researchers were not blinded in this study given the limited number of individuals approved to perform murine assays making blinding impractical for this proof-of-concept study.
Cell culture
bEnd.3 cells [American Type Culture Collection (ATCC CRL-2299)] were cultured in Dulbecco’s modified Eagle’s medium (DMEM):F12 (1:1) supplemented with 10% fetal bovine serum (FBS). NIH/3T3 cells (ATCC CRL-1658) were cultured in DMEM supplemented with 10% FBS. Cultures were decellularized to expose ECM using nonenzymatic cell removal techniques. Namely, versene treatment (three washes at 37°C for 10 min each) or three 10-min washes with 10 mM tris, 10 mM EDTA, and 1% Triton X-100 at 4°C, followed by 30-min incubation with deoxyribonuclease (0.2 mg/ml) in phosphate-buffered saline (PBS) with calcium and magnesium were used for decellularization (4, 41). Following decellularization, all plates were stored for a maximum of 6 months at 4°C in PBS + 1% bovine serum albumin (BSA) until use. U87-MG (ATCC HTB-14) cells were grown in DMEM supplemented with 10% FBS and passaged using accutase.
Yeast culture
For VLR surface display assays, EBY100 yeast were grown in SD-CAA medium [dextrose (20.0 g/liter), yeast nitrogen base (6.7 g/liter), casamino acids (5.0 g/liter), Na2HPO4·7 H2O (10.19 g/liter), and NaH2HPO4·H2O (8.56 g/liter)] as previously described and induced when the culture reached an optical density at 600 nm (OD600nm) between 0.8 and 0.9 using SG-CAA medium (21). Yeast was induced for 48 hours at room temperature before VLR or VLR-intein was harvested. For VLR secretion, YVH10 yeast transformed with pRS316–VLR-intein was grown in SD-2XSCAA + Trp [dextrose (20 g/liter), yeast nitrogenous base (6.7 g/liter), Na2HPO4·7H2O (10.19 g/liter), NaH2HPO4·H2O (8.56 g/liter), Arg (190 mg/liter), Met (108 mg/liter), Tyr (52 mg/liter), Ile (290 mg/liter), Lys (440 mg/liter), Phe (200 mg/liter), Glu (1260 mg/liter), Asp (400 mg/liter), Val (480 mg/liter), Thr (220 mg/liter), Gly (130 mg/liter), and Trp (40 mg/liter), lacking leucine and uracil]. Cultures were initiated, and then, cell density reset the following day to an OD600nm = 0.1 and grown for 72 hours at 30°C. Yeast were induced by replacing the medium with an equivalent volume of SG-2XSCAA + Trp [galactose replacing dextrose (20 g/liter)] containing 0.1% (w/v) BSA and culturing the cells for 72 hours at 20°C. Yeast supernants were harvested, filtered through 0.22-μm PES (polyethersulfone) membranes, and dialyzed against tris-buffered saline (TBS) [25 mM tris, 300 mM NaCl, 2 mM KCl (pH 7.9)]. Imidazole (5 mM) plus 200 ml of the dialyzed supernant was gently mixed overnight at 4°C with 1 ml of cobalt HisPur resin (Thermo Fisher Scientific). Beads were collected, washed with TBS plus 10 mM imidazole, and eluted with TBS plus 250 mM imidazole. Elutants were then buffer-exchanged into 50 mM HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid] (pH 7.2) (removing the imidazole and neutralizing pH) using 10 K MWCO (10 kDa molecular weight cut-off) filters. Of note, regenerated cellulose membranes contain high amounts of thiols that induce nondesirable intein cleavage during isolation. We recommend using a PES membrane available through Pierce and Pall for buffer exchanges with secreted VLR-intein fusions before expressed protein ligation (EPL).
Combining biopanning with moderate throughput ELISA-based screen
The VLR YSD library underwent two rounds of biopanning against bEnd.3 ECM to enrich for brain ECM-binding clones as previously described (20, 42). For the ELISA-based screen, selected biopanning clones were streaked out, and individual clones were picked into 96-well polypropylene plates. Clones were expanded at 30°C overnight in SD-CAA medium and split into two plates (one plate for screening and one plate for frozen stock). The screening plate was induced at 20°C using SG-CAA medium for 24 hours. VLR displaying yeast were washed with 50 mM HEPES (pH 7.2), and VLR was reduced off of the yeast surface using 20 μl of 50 mM MESNA solution in 50 mM HEPES (pH 7.2) for 45 min. Supernatants containing VLR were then diluted 1:10 with 50 mM HEPES (pH 7.2). Plates containing decellularized bEnd.3 or 3T3 ECM were blocked with 1% BSA and 1.5% goat serum in PBS and incubated with the HEPES-diluted VLRs for 1 hour at 37°C. Wells were washed five times with PBS + 0.05% Tween 20. Next, an anti-myc antibody (9E10, BioLegend) modified with horseradish peroxidase (HRP) (1:1000) was added to each well and incubated for 45 min at room temperature. Wells were then washed seven times with PBS + 0.05% Tween 20, 1 min per wash. VLR binding was detected by incubation with one-step 3,3′,5,5′-tetramethylbenzidine (TMB) substrate for 15 to 30 min to develop signal. The reaction was stopped via acidification with 1 M HCl and quantified using absorbance signal at 450 nm. A blank well was included on every plate to establish background signal.
Affinity measurement
To characterize VLR affinity for bEnd.3 ECM, dilutions of purified VLR were incubated with ECM. P1C10 VLR was diluted using a sixfold dilution scheme from 10 μM to 0.21 nM and then incubated with bEnd.3 ECM. Wells were washed five times with PBS + 0.05% Tween 20 and then inubcated with anti-myc 9E10, 1:750, (BioLegend) and goat anti-mouse IgG modified with HRP. Wells were washed seven times with PBS + 0.05% Tween 20 and then incubated with TMB substrate. Reaction was stopped after 15 min by acidification with 1 M HCl. Absorbance at 450 nm was quantified, and data were fit to a one-site equilibrium binding model to determine Kd as previously described (43). The amino acid sequence for VLR P1C10 is ACPSQCSCDQTTVKCHSRRLTSVPAGIPTTTKILRLYSNQITKLEPGVFDHLVNLEKLYISWNQLSALPVGVFDKLTKLTHLSLGYNQLKSVPRGAFDNLKSLTHIWLLNNPWDCECSDILYLKNWIVQHASIVNLQGHGGVDNVKCSGTNTPVRAVTEASTSPSKCP.
Manufacturing Cys-PEG3-azide EPL handle
Two hundred µM equivalents of Fmoc-Cys (SASRINTM resin)–OH (Bachem) were swollen in N,N′-dimethylformamide (DMF) using two 30-min washes. Azido-PEG3-amine (1 mM; Santa Cruz Biotechnology), dissolved in DMF, was added, followed by NMM (4-N-methyl-morpholine) and HCTU [2-(6-Chloro-1H-benzotriazol-1-yl)-N,N,N′,N′-tetramethylaminium hexafluorophosphate] for the activation of the amine. The reaction proceeded for 3 hours at room temperature. Resin was then washed nine times with DMF, two times with 20% piperidine for 15 min per wash, six times with DMF, and nine times with dichloromethane, dried for 10 min under N2, and transferred to a desiccator overnight. The peptide was cleaved off the resin using 94% trifluoroacetic acid (TFA) with 2% TIPs (triisopropylsilane) for 3 hours. TFA was evaporated, and the peptide precipitated with cold ether. The Cys-PEG3-azide peptide sequence was confirmed with mass spectrometry (fig. S5) and purified to >90% using high-performance liquid chromatography.
Intein EPL
To facilitate intein-mediated EPL of yeast-displayed VLRs, VLRs were cloned into the pCTre vector and fused to the engineered 202-08 intein using previously described methods (21, 22). For surface display experiments, 50 ml of yeast displaying VLR-intein fusion were pelleted, washed twice with 50 mM HEPES (pH 7.2), and then incubated with 200 mM MESNA in 50 mM HEPES (pH 7.2) (900 μl). This procedure reduces VLRs off the yeast surface and triggers the intein to form an unstable thioester. Immediately following MESNA addition, 5 mM 2mer peptide containing N-terminal cys and styrene handle was added to the yeast slurry (final volume, 1000 μl) (22). The mixture was incubated for 45 min at room temperature, and then, the yeast was pelleted. The supernatant was recovered and incubated at room temperature with shaking for an additional 2 to 18 hours to facilitate EPL. Buffer exchange of the VLR solution against PBS using 10 kDa MWCO filters removed MESNA and excess EPL ligand. Last, the tetrazine-Cy5 was incubated with VLR-styrene to facilitate covalent C-terminal modification of VLR and create VLR-Cy5 fusions. For secretion of intein-VLR fusions, VLRs were instead cloned into the pRS316 vector and fused to the 202-08 intein and produced and purified as noted in “Yeast culture” section above. Next, 200 mM MESNA was added to purified VLR-intein, followed by the addition of 5 mM Cys-PEG3-azide to produce VLR-azide fusions for liposome attachment. The mixture was incubated overnight at room temperature with gentle shaking. MESNA and excess EPL ligand were removed before use by buffer exchange with 10 kDa MWCO filters as described above. Western blots were used to characterize VLR-azide production. VLR samples were resolved on 4 to 12% bis-tris acrylamide and transferred to nitrocellulose using manufactures’ protocols. Blots were probed using anti-FLAG antibody (M1 murine monoclonal, Sigma) to detect the N-terminal FLAG tag.
VLR-IR800 fusions
VLR-IR800 was generated by creating VLR-azide using the intein-EPL reaction described above. After buffer exchange to remove MESNA and excess Cys-PEG3-azide, 5 mM DBCO-IR800 (LI-COR) was added to VLR fraction. Protein was shaken at 250 rpm, 30°C for 90 min, to facilitate the bioorthogonal azide-DBCO reaction and yield VLR-IR800. For GL261 imaging experiments, VLR-IR800 (1 mg/kg ) was intravenously administered to mice and allowed to circulate for 30 min before mice were perfused via cardiac puncture. Brains were resected, fixed in 4% paraformaldehyde, bisected, and imaged using a LiCor scanner. Serial sections were cut and stained with H&E to identify the tumor margins. n = 3 per group was generated over two independent experimental days.
Mannitol administration for temporary BBB disruption
Mannitol disruption was performed as previously described (23) with slight modification for this study. Briefly, C57BL/6 mice were anesthetized and then intravenously administered VLR-IR800 (1 mg/kg). Five minutes after injection, 200 μl of 25% mannitol solution in water was administered intravenously (~2 mg/g mannitol). Exactly 9 min after mannitol administration, while the BBB was still permeable (23), mice were perfused with 20 ml of PBS via cardiac puncture. Brains were removed and imaged for retained IR800 signal using a LiCor scanner. n = 3 per group was generated over two independent experimental days.
VLR rabbit Fc fusions
VLRs were cloned into a pIRES vector and fused to rabbit Fc region (VLR-Fc). 293F cells (Life Technologies) were transfected with pIRES-VLR-Fc expression plasmids using 293fectin (Life Technologies), according to the manufacturer’s protocol. Transfected 293F cells were grown for 3 days in FreeStyle (Life Technologies) media. VLR-Fc dimers were purified using protein A/G beads according to the manufacturer’s protocol (Pierce).
Fluorescence microscopy of murine specimens
Murine immunoflourescence techniques were used in two formats. First, VLRs directly reduced off of yeast surface were reoxidized and incubated with murine tissue sections, snap-frozen, and cut on a cryostat for 1 hour at room temperature. Sections were washed three times with PBS + 1% BSA, 1.5% goat serum and incubated with a master mix containing anti–c-myc (rabbit) antibody (1:500; BioLegend), goat anti-rabbit AF555 antibody (1:1000; Life Technologies), and Isolectin GS-IB4 AF488 (1:400; Thermo Fisher Scientific) for 1 hour at room temperature. Sections were incubated with Hoechst 33342 (1:800) for the final 15 min of labeling. Sections were washed three times, fixed with 4% paraformaldehyde, and mounted for imaging. The second application was identical to the first except VLRs are directly labeled with Cy5 using intein EPL described above (22). Images were collected on a Zeiss Imager Z2 upright fluorescent microscope. For quantification, a minimum of three fields per group were quantified to determine VLR channel mean pixel intensity using ImageJ. Data are presented as mean pixel intensity ± SD.
Fluorescence microscopy of human specimens
Human brain samples were obtained from surgical resections as approved by the University of Wisconsin Institutional Review Board. Samples were snap-frozen and sectioned on a cryostat. For VLR staining, VLR-Cy5 was incubated with human brain sections (1:100; 1 mg/ml) for 1 hour at 37°C. Anti-CD31 (1:100; Cell Sciences) and goat anti-mouse AF488 (1:200; Thermo Fisher Scientific) were added to illuminate microvessels. Last, nuclei were stained with Hoechst 33342 (1:800). All human sections were imaged using a Leica SP8 3X STED microscope using confocal settings.
Generation and characterization of VLR-targeted doxorubicin-loaded liposomes
Doxorubicin containing stealth liposomes decorated with VLR were produced. A lipid film containing 65% phosphatidylcholine, 32% cholesterol, 2.5% phosphatidylethanolamine-PEG2000, and 0.64% 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[dibenzocyclooctyl(polyethylene glycol)-2000] (DSPE-PEG2000-DBCO) (all lipids were from Avanti Polar Lipids) was created by dissolving lipids in 1:1 mixture of chloroform and methanol and removing solvent as previously described (28). Films were rehydrated in 155 mM (NH4)2SO4 (pH 5.5) via vortexing and heating to >65°C, sonicated for 1 hour, and extruded through double-stacked 100-nm filters. Liposomes were buffer-exchanged into 123 mM Na citrate using PD-10 desalting columns and immediately mixed with 30 mg of doxorubicin/100 mg of lipid, at >65°C, to facilitate postformation loading. After ~1 hour at >65°C, liposomes were transferred to 4°C overnight. The next day, free doxorubicin was removed using PD-10 desalting columns. Formation and size of liposomes were confirmed using dynamic light scattering (Malvern Zetasizer). To facilitate VLR decoration, VLR-azide was incubated with DBCO-containing liposomes for 90 min at 30°C with shaking. VLR covalently bound to the liposome surface was detected by BCA (bicinchoninic acid assay) in nondoxorubicin-loaded liposomes (because doxorubicin contains a free amine) using the manufacturer’s protocol (Pierce). Encapsulated doxorubicin was quantified by boiling liposomes with 1% Triton X-100 and measuring doxorubicin absorbance at 480 nm compared to a standard curve.
In vitro U87 cytotoxicity assay
bEnd.3 cells were grown to confluence in 96-well plates. Wells were decellularized to leave bEnd.3 ECM, as described above. Next, 1000 U87 cells per well were replated onto the U87 ECM and allowed to attach overnight. Wells were washed once with Opti-MEM and then incubated with a dilution series of P1C10- or RBC36-targeted doxorubicin-loaded liposomes. Liposomes were applied to the cell-ECM mixture in a dilution series from 80 μM to 0.3 nM, using fourfold dilutions (concentration calculated from doxorubicin 480-nm absorbance signal), using Opti-MEM as a diluent and incubated for 2 hours. Wells were washed two times with complete media and incubated for an additional 72 hours. CellTiter-Glo (Promega) was used to quantify the number of live cells, according to the manufacturer’s protocol. EC50 calculations were made using GraphPad software suite. To directly quantify VLR-doxorubicin binding to murine sections, P1C10-RBC36, or nontargeted doxorubicin-loaded liposomes were incubated with murine sections, washed and then imaged on a Zeiss Imager Z2 upright fluorescent microscope. A minimum of three fields per group were quantified to determine the VLR channel mean pixel intensity using ImageJ. Data are presented as mean pixel intensity ± SD.
U87 delivery and survival studies
All animal studies were approved by IACUC at the University of Wisconsin-Madison. Nonobese diabetic–severe combined immunodeficient mice underwent intracranial implantation of U87 cells expressing luciferase as previously described (44). Tumor engraftment was determined by quantifying luminescence using luciferin and an IVIS imager, according to manufacturer’s protocols. For accumulation studies, tumors were grown until mice demonstrated tumor burden by displaying neurologic symptoms or hunched posture. For VLR-Fc studies, protein (3 mg/kg) was administered intravenously and allowed to circulate for 30 min. Mice were then perfused, and the organs were harvested. Organs were snap-frozen, embedded in OCT (optimal cutting temperature), and cut using a cryostat. Sections were stained with Isolectin GS-IB4 (1:400), Hoechst 33342 (1:800), and goat anti-rabbit AF555 (1:500) and then imaged on an upright Zeiss Imager Z2 fluorescent microscope. For doxorubicin accumulation studies, mice bearing U87 intracranial tumors received a single injection of P1C10- or RBC36-targeted doxorubicin-loaded liposomes intraperitoneally (12 mg doxorubicin/kg). Drug was allowed to circulate for 30 min, and then, mice were perfused with PBS and euthanized. Whole brains were bisected at the tumor site, and coronal images for doxorubicin fluorescence were taken using an IVIS imager (excitation, 465 nm; emission, 600 nm). Slices adjacent to the brain bisection used to expose the tumor were stained with H&E to define tumor margins and Isolectin GS-IB4 + Hoechst 33342 (1:400 and 1:800, respectively) for imaging on an upright Zeiss Imager Z2 fluorescent microscope. A minimum of three fields per group were quantified to determine the VLR channel mean pixel intensity using ImageJ.Data are presented as mean pixel intensity ± SD. For survival studies, after confirming tumor engraftment by luminescence, mice were intraperitoneally administered 12 mg/kg doxorubicin of P1C10-targeted (n = 5), 192-targeted (n = 5), or RBC36-targeted (n = 4) doxorubicin-loaded liposomes weekly for four cycles. Mice were monitored for neurological symptoms, hunched posture, and weight loss throughout the study and euthanized in accordance with guidelines of the IACUC protocol. The proportion of surviving mice over time was plotted using a Kaplan-Meier plot, and statistically significant differences were determined with log-rank tests.
Statistical analysis
Statistical methods used in each assay are defined in detail within each respective figure legend. In general, data are presented and means ± SD. The number of replicates used to generate these data is described in each method section. For murine survival studies, an n of 5 was chosen to power the study to observe a 30% survival difference between groups with 95% confidence.
Supplementary Material
Acknowledgments
Funding: This work was supported by NIH grants NS091851 (to E.V.S. and B.R.H.) and NS099158 (to E.V.S.), a Falk Medical Research Trust Catalyst award (to E.V.S. and J.S.K.), and a Defense Threat Reduction Agency grant HDTRA1-15-1-0012 (to E.V.S.). Author contributions: B.J.U., P.A.C., J.M.L., J.V.G., and S.B. performed experiments. B.J.U., P.A.C., B.R.H., J.S.K., and E.V.S. designed the experiments, evaluated the results, and wrote the manuscript. Competing interests: B.R.H. is a founder and shareholder of Novab Inc., a company developing commercial applications of lamprey VLRs. E.V.S., B.J.U., B.R.H., P.A.C., and J.S.K. are inventors on a provisional U.S. patent related to this work filed by the Wisconsin Alumni Research Foundation (no. 62783475, filed 21 December 2018). The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaau4245/DC1
Fig. S1. P3A8 binding to murine tissue sections.
Fig. S2. VLR 192 binding to tissue sections.
Fig. S3. Characterization of doxorubicin-loaded liposomes.
Fig. S4. Luminescence and weights from U87 tumor survival experiment.
Fig. S5. Mass spectrometry traces for the manufacture of Cys-PEG3-azide.
REFERENCES AND NOTES
- 1.Abbott N. J., Patabendige A. A. K., Dolman D. E. M., Yusof S. R., Begley D. J., Structure and function of the blood–brain barrier. Neurobiol. Dis. 37, 13–25 (2010). [DOI] [PubMed] [Google Scholar]
- 2.McCaffrey G., Davis T. P., Physiology and pathophysiology of the blood-brain barrier: P-glycoprotein and occludin trafficking as therapeutic targets to optimize central nervous system drug delivery. J. Invest. Med. 60, 1131–1140 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Minagar A., Alexander J. S., Blood-brain barrier disruption in multiple sclerosis. Mult. Scler. 9, 540–549 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Mann A. P., Scodeller P., Hussain S., Joo J., Kwon E., Braun G. B., Mölder T., She Z.-G., Kotamraju V. R., Ranscht B., Krajewski S., Teesalu T., Bhatia S., Sailor M. J., Ruoslahti E., A peptide for targeted, systemic delivery of imaging and therapeutic compounds into acute brain injuries. Nat. Commun. 7, 11980 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holodinsky J. K., Yu A. Y. X., Assis Z. A., Al Sultan A. S., Menon B. K., Demchuk A. M., Goyal M., Hill M. D., History, evolution, and importance of emergency endovascular treatment of acute ischemic stroke. Curr. Neurol. Neurosci. Rep. 16, 42 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Lockman P. R., Mittapalli R. K., Taskar K. S., Rudraraju V., Gril B., Bohn K. A., Adkins C. E., Roberts A., Thorsheim H. R., Gaasch J. A., Huang S., Palmieri D., Steeg P. S., Smith Q. R., Heterogeneous blood–tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin. Cancer Res. 16, 5664–5678 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lamsam L., Johnson E., Connolly I. D., Wintermark M., Hayden Gephart M., A review of potential applications of MR-guided focused ultrasound for targeting brain tumor therapy. Neurosurg. Focus 44, E10 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Nico B., Ribatti D., Morphofunctional aspects of the blood-brain barrier. Curr. Drug Metab. 13, 50–60 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Obermeier B., Verma A., Ransohoff R. M., The blood–brain barrier. Handb. Clin. Neurol. 133, 39–59 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Groothuis D. R., The blood-brain and blood-tumor barriers: A review of strategies for increasing drug delivery. Neuro Oncol. 2, 45–59 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown C. E., Badie B., Barish M. E., Weng L., Ostberg J. R., Chang W.-C., Naranjo A., Starr R., Wagner J., Wright C., Zhai Y., Bading J. R., Ressler J. A., Portnow J., D'Apuzzo M., Forman S. J., Jensen M. C., Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res. 21, 4062–4072 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pellegatta S., Savoldo B., Di Ianni N., Corbetta C., Chen Y., Patané M., Sun C., Pollo B., Ferrone S., DiMeco F., Finocchiaro G., Dotti G., Constitutive and TNFα-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: Implications for CAR-T cell therapy. Sci. Transl. Med. 10, eaao2731 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Siegel R. L., Miller K. D., Jemal A., Cancer statistics, 2015. CA Cancer J. Clin. 65, 5–29 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Ostrom Q. T., Gittleman H., Fulop J., Liu M., Blanda R., Kromer C., Wolinsky Y., Kruchko C., Barnholtz-Sloan J. S., CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the united states in 2008–2012. Neuro Oncol. 17 Suppl 4, iv1–iv62 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown C. E., Alizadeh D., Starr R., Weng L., Wagner J. R., Naranjo A., Ostberg J. R., Blanchard M. S., Kilpatrick J., Simpson J., Kurien A., Priceman S. J., Wang X., Harshbarger T. L., D’Apuzzo M., Ressler J. A., Jensen M. C., Barish M. E., Chen M., Portnow J., Forman S. J., Badie B., Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 375, 2561–2569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woodworth G. F., Dunn G. P., Nance E. A., Hanes J., Brem H., Emerging insights into barriers to effective brain tumor therapeutics. Front. Oncol. 4, 126 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herrin B. R., Cooper M. D., Alternative adaptive immunity in jawless vertebrates. J. Immunol. 185, 1367–1374 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Han B. W., Herrin B. R., Cooper M. D., Wilson I. A., Antigen recognition by variable lymphocyte receptors. Science 321, 1834–1837 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo P., Hirano M., Herrin B. R., Li J., Yu C., Sadlonova A., Cooper M. D., Dual nature of the adaptive immune system in lampreys. Nature 459, 796–801 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.J. M. Lajoie, “Application of yeast surface display screening methods to antibody discovery and proteomics of the blood-brain barrier,” thesis, University of Wisconsin-Madison (2016). [Google Scholar]
- 21.Marshall C. J., Grosskopf V. A., Moehling T. J., Tillotson B. J., Wiepz G. J., Abbott N. L., Raines R. T., Shusta E. V., An evolved Mxe GyrA intein for enhanced production of fusion proteins. ACS Chem. Biol. 10, 527–538 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Umlauf B. J., Mix K. A., Grosskopf V. A., Raines R. T., Shusta E. V., Site-specific antibody functionalization using tetrazine–styrene cycloaddition. Bioconjug. Chem. 29, 1605–1613 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCarty D. M., DiRosario J., Gulaid K., Muenzer J., Fu H., Mannitol-facilitated CNS entry of rAAV2 vector significantly delayed the neurological disease progression in MPS IIIB mice. Gene Ther. 16, 1340–1352 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Madhankumar A. B., Slagle-Webb B., Wang X., Yang Q. X., Antonetti D. A., Miller P. A., Sheehan J. M., Connor J. R., Efficacy of interleukin-13 receptor–targeted liposomal doxorubicin in the intracranial brain tumor model. Mol. Cancer Ther. 8, 648–654 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Gupta B., Torchilin V. P., Monoclonal antibody 2C5-modified doxorubicin-loaded liposomes with significantly enhanced therapeutic activity against intracranial human brain U-87 MG tumor xenografts in nude mice. Cancer Immunol. Immunother. 56, 1215–1223 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oh T., Fakurnejad S., Sayegh E. T., Clark A. J., Ivan M. E., Sun M. Z., Safaee M., Bloch O., James C. D., Parsa A. T., Immunocompetent murine models for the study of glioblastoma immunotherapy. J. Transl. Med. 12, 107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nance E., Timbie K., Miller G. W., Song J., Louttit C., Klibanov A. L., Shih T.-Y., Swaminathan G., Tamargo R. J., Woodworth G. F., Hanes J., Price R. J., Non-invasive delivery of stealth, brain-penetrating nanoparticles across the blood-brain barrier using MRI-guided focused ultrasound. J. Control. Release 189, 123–132 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gray B. P., McGuire M. J., Brown K. C., A liposomal drug platform overrides peptide ligand targeting to a cancer biomarker, irrespective of ligand affinity or density. PLOS ONE 8, e72938 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winter G., Griffiths A. D., Hawkins R. E., Hoogenboom H. R., Making antibodies by phage display technology. Annu. Rev. Immunol. 12, 433–455 (1994). [DOI] [PubMed] [Google Scholar]
- 30.Steiner D., Forrer P., Plückthun A., Efficient selection of DARPins with sub-nanomolar affinities using SRP phage display. J. Mol. Biol. 382, 1211–1227 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Brown K. C., Peptidic tumor targeting agents: The road from phage display peptide selections to clinical applications. Curr. Pharm. Des. 16, 1040–1054 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hong X., Ma M. Z., Gildersleeve J. C., Chowdhury S., Barchi J. J. Jr., Mariuzza R. A., Murphy M. B., Mao L., Pancer Z., Sugar-binding proteins from fish: Selection of high affinity “lambodies” that recognize biomedically relevant glycans. ACS Chem. Biol. 8, 152–160 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolak D. J., Thorne R. G., Diffusion of macromolecules in the brain: Implications for drug delivery. Mol. Pharm. 10, 1492–1504 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allen M., Bjerke M., Edlund H., Nelander S., Westermark B., Origin of the U87MG glioma cell line: Good news and bad news. Sci. Transl. Med. 8, 354re3 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Kuznetsov V. A., Puri R. K., Kinetic analysis of high affinity forms of interleukin (IL)-13 receptors: Suppression of IL-13 binding by IL-2 receptor gamma chain. Biophys. J. 77, 154–172 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Preusser M., Lim M., Hafler D. A., Reardon D. A., Sampson J. H., Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat. Rev. Neurol. 11, 504–514 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim S.-S., Rait A., Kim E., DeMarco J., Pirollo K. F., Chang E. H., Encapsulation of temozolomide in a tumor-targeting nanocomplex enhances anti-cancer efficacy and reduces toxicity in a mouse model of glioblastoma. Cancer Lett. 369, 250–258 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Etame A. B., Diaz R. J., Smith C. A., Mainprize T. G., Hynynen K., Rutka J. T., Focused ultrasound disruption of the blood brain barrier: A new frontier for therapeutic delivery in molecular neuro-oncology. Neurosurg. Focus 1, E3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee J.-j., Kim H. J., Yang C.-S., Kyeong H.-H., Choi J.-M., Hwang D.-E., Yuk J.-M., Park K., Kim Y. J., Lee S.-G., Kim D., Jo E.-K., Cheong H.-K., Kim H.-S., A high-affinity protein binder that blocks the IL-6/STAT3 signaling pathway effectively suppresses non-small cell lung cancer. Mol. Ther. 22, 1254–1265 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hwang D.-E., Ryou J.-H., Oh J. R., Han J. W., Park T. K., Kim H.-S., Anti-human VEGF repebody effectively suppresses choroidal neovascularization and vascular leakage. PLOS ONE 11, e0152522 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crapo P. M., Gilbert T. W., Badylak S. F., An overview of tissue and whole organ decellularization processes. Biomaterials 32, 3233–3243 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X. X., Shusta E. V., The use of scFv-displaying yeast in mammalian cell surface selections. J. Immunol. Methods 304, 30–42 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Burns M. L., Malott T. M., Metcalf K. J., Hackel B. J., Chan J. R., Shusta E. V., Directed evolution of brain-derived neurotrophic factor for improved folding and expression in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 80, 5732–5742 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swanson K. I., Clark P. A., Zhang R. R., Kandela I. K., Farhoud M., Weichert J. P., Kuo J. S., Fluorescent cancer-selective alkylphosphocholine analogs for intraoperative glioma detection. Neurosurgery 76, 115–124 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/5/eaau4245/DC1
Fig. S1. P3A8 binding to murine tissue sections.
Fig. S2. VLR 192 binding to tissue sections.
Fig. S3. Characterization of doxorubicin-loaded liposomes.
Fig. S4. Luminescence and weights from U87 tumor survival experiment.
Fig. S5. Mass spectrometry traces for the manufacture of Cys-PEG3-azide.