

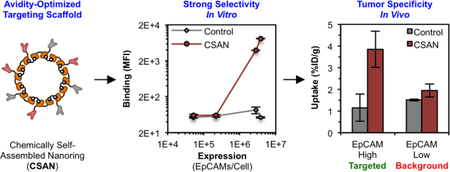
Abstract
Nature uses multivalency to govern many biological processes. The development of macromolecular and cellular therapies has largely been dependent on engineering similar polyvalent interactions to enable effective targeting. Such therapeutics typically utilize high-affinity binding domains that have the propensity to recognize both antigen-overexpressing tumors and normal-expressing tissues, leading to “on-target, off-tumor” toxicities. One strategy to improve these agents’ selectivity is to reduce the binding affinity, such that biologically relevant interactions between the therapeutic and target cell will only exist under conditions of high avidity. Pre-clinical studies have validated this principle of avidity optimization in the context of chimeric antigen receptor (CAR) T cells; however, a rigorous analysis of this approach in the context of soluble multivalent targeting scaffolds has yet to be undertaken. Using a modular protein nanoring capable of displaying ≤8 fibronectin domains with engineered specificity for a model antigen, epithelial cell adhesion molecule (EpCAM), this study demonstrates that binding affinity and ligand valency can be optimized to afford discrimination between EpCAMHigh (2.8 – 3.8 ×106 antigens/cell) and EpCAMLow (5.2×104 – 2.2×105 antigens/cell) tissues both in vitro and in vivo.
Graphical Abstract
INTRODUCTION
Many biological processes – including antibody-antigen, virus-cell, and cell-cell recognition – are governed by multivalent interactions between macromolecular species.1 Given the ability to enhance apparent affinity, polyvalency has also been employed to target various therapeutic agents to the desired tissue, including antibody drug conjugates (ADCs), drug-loaded nanoparticles, and even chimeric antigen receptor (CAR) T cells. While the affinities of these engineered proteins, particles, and cells can be high, their therapeutic efficacy is ultimately derived from their ability to recognize a specific antigen on the surface of a malignant cell. However, many of the antigens targeted by these agents are not exclusive to the neoplasm, and the recognition of healthy tissue that also expresses the antigen has led to adverse events known as “on-target, off-tumor” toxicities.2, 3 Such toxicities have both restricted and prevented the clinical use of otherwise promising treatments. For example, CAR T cells targeting the pan-B cell marker CD19 are remarkably efficacious for certain B cell neoplasms, but the recognition and eradication of healthy B cells leads to aplasia.4, 5 Furthermore, CAR T cells recognizing ErbB26, carbonic anhydrase IX (CAIX)7, and carcinoembryonic antigen (CEA)8, yielded potent solid organ toxicities that, in some cases, halted further development. Likewise, bispecific T cell engagers (BiTEs)9, 10 and antibodies11, 12 targeting epithelial cell adhesion molecule (EpCAM) produced dose-limiting colitis and hepatotoxicity, respectively.
Accordingly, many groups have sought strategies to mitigate on-target, off-tumor toxicities. One approach is to reduce the binding affinity of the targeting agent such that there is insufficient avidity to stably recognize the lower level of antigen expression on the healthy tissue.13, 14 Indeed, CAR T cells utilizing reduced affinity single chain variable fragments have exhibited reduced cytotoxicity towards antigenLow cell lines in vitro and increased therapeutic windows in murine xenograft models.13–17 Likewise, enhancements in selectivity have been observed for nanoparticles18–20 and dendrimers21–23 when either the affinity or valency of the binding domains are reduced. These studies suggest that avidity optimization may be an effective strategy to reduce on-target, off-tumor recognition by these highly multivalent agents.
Most protein-based scaffolds – such as antibodies and their various formats – present a unique challenge, as the small number of potential ligand/cell interactions limits their avidity for the target cell. Theoretical models24–31 and analytical experiments32–35 have indicated that avidity optimization could enhance the selectivity of these species as well; however, there is very limited work demonstrating this phenomenon in vitro36–38 and in vivo39, 40. Furthermore, none of these studies have directly examined all facets of avidity (i.e., binding affinity, ligand valency, and antigen expression density) nor have any used non-antibody protein topologies. Thus, the present study aims to translate the theoretical models into both in vitro and in vivo results using a soluble protein scaffold with variable ligand affinity and valency, plus a small panel of cell lines with different antigen expression densities.
For this, we utilized a protein scaffold previously developed by our group, called the chemically self-assembled nanoring (CSAN; Figure 1).41 CSANs are formed when bivalent dihydrofolate reductase (DHFR2) fusion proteins are spontaneously oligomerized by a chemical dimerizer, bis-methotrexate (bisMTX).41, 42 CSANs can be further functionalized by fusing various binding entities to the DHFR2 subunits.43–45 In this work, human tenth type III fibronectin (Fn3) domains with either no binding efficacy (i.e., a non-targeted control domain) or with engineered specificity for a model antigen, EpCAM46, were fused. By co-assembling stoichiometric ratios of these targeted and non-targeted subunits, CSAN populations with defined overall EpCAM-targeting valencies were formed. We hypothesized that by varying the affinity and valency of the Fn3 domains in the CSAN, we could prepare a scaffold capable of binding specifically to cells that overexpress EpCAM but with limited binding to cells expressing low quantities of EpCAM. Accordingly, we titrated a number of these avidity-modulated CSANs against various EpCAM-expressing cell lines, enabling us to quantitate the apparent affinity of the scaffold in the context of variable Fn3 affinities, valencies, and EpCAM-expression densities. The results of these experiments were then applied to the development of avidity-optimized CSANs capable of performing cellular discrimination both in vitro and in vivo.
Figure 1. Chemically Self-Assembled Nanorings (CSANs).
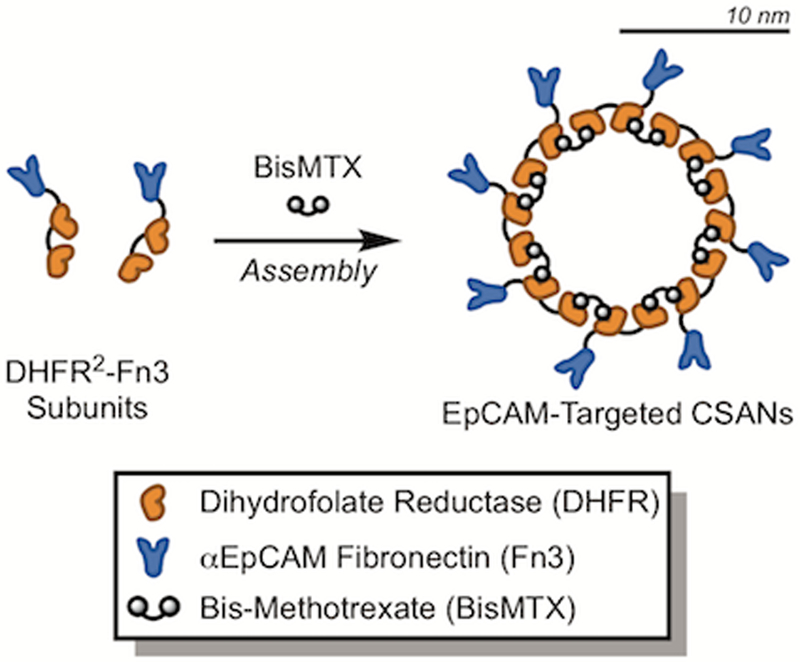
Bivalent DHFR2 proteins fused to a targeting domain are spontaneously oligomerized into CSANs via the chemical dimerizer, bisMTX. In this work, a fibronectin domain with engineered specificity for epithelial cell adhesion molecule (EpCAM)46 was fused, enabling the formation of EpCAM-targeted CSANs. While not drawn to scale, the CSAN scaffold as a whole is 19 ± 5 nm in diameter, as depicted by the rough scale bar.43
RESULTS AND DISCUSSION
Scaffold Apparent Affinity Varies Quantitatively as a Function of Ligand Valency
We previously reported the development of a panel Fn3 domains with engineered specificity for EpCAM46, which is overexpressed on a wide range of epithelial malignancies (i.e., carcinomas)47, 48 and certain cancer stem cell populations.49, 50 For this work, we fused two of these ligands – the “high-affinity” clone C5 (Kd = 17 ± 1 nM) and the “low-affinity” clone B22 (Kd = 1,100 ± 200 nM) – to our DHFR2 subunits, enabling the formation of EpCAM-targeted CSANs (Figure 1). Similarly, we fused a “non-targeted” Fn3 domain (termed clone NT) with no binding efficacy to serve as a negative control.
Due to the self-assembling nature of the CSANs, bispecific CSANs incorporating two distinct targeting domains can be formed by incubating mixtures of the desired DHFR2 fusion proteins with bisMTX.43–45 Furthermore, our previous characterization of the CSAN platform has demonstrated that, while a narrow distribution of species are formed during the self-assembly process, the most abundant41 and mass-average42 moiety is an octameric nanoring displaying eight targeting domains. Thus, we hypothesized that mixing stoichiometric ratios of an EpCAM-targeted DHFR2-Fn3 fusion protein with the non-targeted DHFR2-Fn3 fusion protein would enable the formation of CSANs exhibiting “reduced valency” of the EpCAM-binding Fn3s, given that the non-binding domains would now occupy a statistical number of positions within the predominantly octameric scaffold (Figure 2A). In turn, these reduced valency CSANs would exhibit a corresponding lower affinity for EpCAM-expressing cells, with a direct correlation between the CSAN’s overall apparent affinity and the valency of its EpCAM-binding domains.
Figure 2. Modular Assembly of CSANs Enables Affinity Tuning.

(A) CSANs with a reduced valency of EpCAM-binding Fn3 domains can be formed by co-assembling defined molar ratios of targeted and non-targeted DHFR2-Fn3 fusion proteins using the chemical dimerizer, bisMTX. This was done for both the “high-affinity” Fn3 clone C5 and the “low-affinity” Fn3 clone B22. (B&C) CSANs of distinct average valencies were titrated against MCF-7 cells via flow cytometry to determine their apparent affinity for cellular EpCAM, as previously described.43, 46 The apparent affinity of both scaffolds, (B) C5-targeted CSANs and (C) B22-targeted CSANs, varied directly with the proportion of binding domains in the CSAN. The legends in (B) and (C) give the ratio of targeted to non-targeted (T:NT) subunits incorporated into each CSAN. The x-axis provides the total concentration of DHFR2-Fn3 monomers that were oligomerized into CSANs using bisMTX. Each sample is normalized to the fitted maximum fluorescence intensity for that sample, enabling determination of the fraction of bound ligand at each concentration. Un-normalized binding isotherms are provided in Figure S4. Each titration was performed at least three independent times, and data is presented as the mean ± standard deviation of these trials.
To test this hypothesis, we assembled defined ratios of EpCAM-targeted DHFR2-Fn3 subunits (utilizing either clone C5 or B22) and non-targeted DHFR2-Fn3 subunits into CSANs with distinct EpCAM-binding valencies (Figure 2A). As in our previous work with DHFR2-Fn3 fusion proteins, complete CSAN oligomerization was observed within 30 min of the addition of bisMTX, as demonstrated by size exclusion chromatography (SEC; Figure S1).42, 51 Oligomerized CSANs were further characterized by dynamic light scattering (DLS; Figure S2) with results in close agreement to those previously reported for Fn3-targeted CSANs.43
We then titrated each of these CSAN species – with targeted:non-targeted (T:NT) domain ratios of either 8:0 (i.e., fully targeted), 4:4, 2:6, or 1:7 (i.e., a single targeting domain) – against the EpCAM-expressing cell line, MCF-7, using flow cytometry. As predicted, the apparent affinity of the CSAN scaffold varied directly with the proportion of EpCAM-binding Fn3 domains incorporated into the nanoring (Figures 2B–C). Indeed, this relationship was true for both of the Fn3 clones tested, C5 (Figure 2B) and B22 (Figure 2C). The apparent Kd,N values obtained for each Fn3 clone and valency tested are given in Table S1. For clarity, the abbreviation Kd,N will be used throughout the text to distinguish multivalent apparent affinities from true monomeric dissociation constants, depicted as the traditional Kd. That the Kd values for the monovalent 1:7 T:NT species are higher than those of the parent ligands suggests a thermodynamic penalty for fusing these ligands to DHFR2 domains and further organizing them into nanorings. Importantly, CSANs composed solely of the non-targeted subunits (i.e., T:NT ratio of 0:8) exhibited no binding to the EpCAM-positive cells (Figure S3).
To quantitatively define the effect of a multivalent scaffold’s valency on its apparent affinity, the obtained Kd,N values were plotted against the number of targeting domains incorporated into the nanoring (Figure 3). This analysis yielded a consistent descriptor of how the CSAN’s apparent affinity changes as a function of both the singly-targeted ligand’s affinity (Kd,1, units of molarity) and the valency (N, unit-less number of ligands per scaffold) of the binding domains:
| (Eq. 1) |
Note that this equation form is based purely on empiric observations and may not represent the underlying mechanistic relationship. This data can also be considered in terms of the cooperativity parameter, α (equation 2), and the enhancement parameter, β (equation 3)1:
| (Eq. 2) |
2ion 3enhancement parameter, ∫n terms of the ressing cells when assessed by flow cytometry scaffold. to all cell lines, with) . 2ion 3enhancement parameter, ∫n terms of the ressing cells when assessed by flow cytometry scaffold. to all cell lines, with
| (Eq. 3) |
Figure 3. Apparent Affinity Varies with Binding Domain Valency.
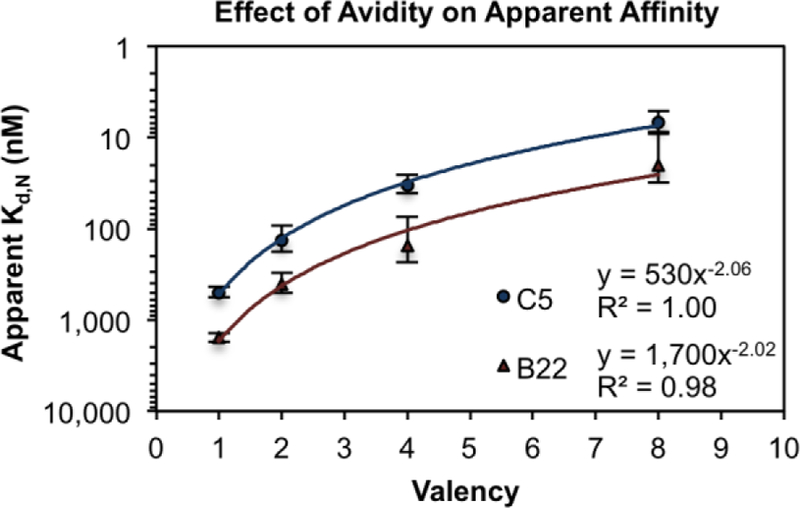
CSANs composed of different valencies of EpCAM-binding Fn3 domains (either clone C5 or B22) were titrated against EpCAM-expressing MCF-7 cells via flow cytometry. The resulting Kd,N values were plotted against the valency of the binding domains, and the correlation was fit to a power regression curve, as this model provided the best coefficient of determination (R2 value) for both Fn3 clones. Each titration was performed at least three times, and data is presented as the mean ± standard deviation of the Kd,N values resulting from these independent trials. Apparent dissociation constants are given in Table S1.
Multivalent interactions are characterized as positively cooperative when α > 1, non-cooperative when α = 1, and negatively cooperative when α < 1.21 Like other well-characterized multivalent targeting scaffolds1, 21, the CSANs exhibit values of α < 1, indicating that they, too, are negatively-cooperative (Table S1). This negative cooperativity could be a result of steric hindrance that impedes highly multivalent binding in spite of the increased local concentration that results from scaffold tethering to the cell surface. Furthermore, the β values are consistent with a 78-fold enhancement of the binding affinity for fully-targeted CSANs (Table S1).
Antigen Expression Density Impacts Scaffold Apparent Affinity
Cellular discrimination based upon antigen expression levels necessitates that antigen quantity be a predictive variable in a multivalent scaffold’s apparent affinity calculation. Therefore, we sought to more clearly define the role of EpCAM expression density on the CSANs’ cell-binding potential and incorporate this analysis into equation 1.
Using flow cytometry, EpCAM expression was quantitatively defined for a small panel of cell lines, including MDA-MB-231 (5.2 ± 0.1 ×104 EpCAMs/cell), SK-OV-3 (2.2 ± 0.4 ×105 EpCAMs/cell), LNCaP (2.8 ± 0.1 ×106 EpCAMs/cell), and MCF-7 (3.8 ± 0.9 ×106 EpCAMs/cell; Figure 4A–D, respectively). Fully-targeted CSANs (T:NT 8:0) utilizing the high-affinity Fn3 clone C5 were titrated against each of these cell lines via flow cytometry, yielding apparent Kd,N values of 27 ± 2.8 nM, 19 ± 3.4 nM and 7.2 ± 3.7 nM, and 6.9 ± 1.8 nM, respectively. Plotting these Kd,N values against the absolute EpCAM expression per cell afforded a local, yet descriptive, model of how the apparent affinity is affected by a reduction in the antigen expression density (Figure 4E) within this regime. As expected, minor reductions in expression – for instance, from 3.8×106 EpCAMs/cell (MCF-7) to 2.8×106 EpCAMs/cell (LNCaP) – have an insignificant impact on the CSAN’s apparent affinity (p > 0.8). However, larger differences in antigen density – from 3.8×106 EpCAMs/cell (MCF-7) to 5.2×104 EpCAMs/cell (MDA-MB-231) – showed a marked reduction (approximately four-fold; p < 0.001) in the apparent affinity (Figure 4E).
Figure 4. Apparent Affinity Varies with Antigen Expression Density.

(A–D) The absolute EpCAM expression of four cell lines was quantitated via flow cytometry by comparing the mean fluorescence intensity of the cellular samples to those of standard calibration beads. Values in parentheses represent mean numbers of EpCAM/cell. Experiments were performed in triplicate, and a representative histogram is shown for each cell line. (E) To isolate the effect of EpCAM expression density on the apparent affinity of the CSAN scaffold, fully-targeted CSANs utilizing clone C5 (8:0 C5:NT) were titrated against the same four EpCAM-expressing cell lines shown in panels A–D. The minimum apparent Kd,N value (6.9 ± 1.8 nM) was obtained when the CSANs were bound to the MCF-7 cells; therefore, this value was chosen as the baseline (i.e, value of 1.0) and is represented by the dashed grey line. The apparent Kd,N values obtained for the LNCaP (7.2 ± 3.7 nM), SK-OV-3 (19 ± 3.4 nM), and MDA-MB-231 (27 ± 2.8 nM) were scaled relative to the MCF-7 Kd,N value to provide the fold by which the apparent Kd,N had been increased. This “fold increase in Kd,N value” is plotted against the quantitated number of EpCAMs/cell, providing a local fit within the confines of this data set. The relationship was fit to a power regression curve, as this model provided the best coefficient of determination (R2 value). Each titration was performed at least three times, and data is presented as the mean ± standard deviation of the fold increase in the apparent Kd,N values resulting from these independent trials. The red lines and dot indicate the threshold number of EpCAMs/cell needed to permit theoretical octavalent CSAN binding to the cells, per the model proposed in the main text. (F) The theoretical number of EpCAMs accessible to the CSAN at each of the eight binding events. The black dashed line indicates the threshold number of accessible EpCAMs needed to permit octavalent CSAN binding to the cells. (G) The theoretical effective molarity of EpCAMs accessible to the CSAN at each of the eight binding events. See Figure S5 for additional details regarding the model used to produce panels (F–G) and the associated threshold values.
Analogous to biparatopic antibodies26, 52, 53, this phenomenon is supported by a biophysical model of how CSANs might bind to cell membranes decorated with target antigens (Figure S5). Briefly, the known radius of the CSANs43, 44 permits calculation of the cell surface area and extracellular volume accessible to the nanoring. Having quantitated the EpCAM expression of the various cell lines, both the number (Figure 4F) and effective molarity (Figure 4G) of EpCAM moieties present in the accessible area and volume, respectively, can be calculated. As shown, the model of EpCAM accessibility supports the trend in cell binding efficacy observed in Figure 4E, where similar Kd,N values are observed for the high-expressing cell lines (MCF-7 and LNCaP) and reduced affinity is observed for the low-expressing cell lines (MDA-MB-231 and SK-OV-3). Additional details regarding this model (including assumptions and numerical values) are discussed in the Supporting Information.
Combining our observations with the proposed model, we sought to estimate the minimum EpCAM density required to enable fully octavalent binding of a CSAN to a cell membrane. Using equation 1, the observed Kd,N values for the fully-targeted CSANs against the MCF-7, LnCAP, SK-OV-3, and MDA-MB-231 cells were converted to estimated binding valencies of N = 8, 8, 5, and 4, respectively. This suggested that the CSANs could bind octavalently to the MCF-7 and LNCaP cells, but only pentavalently to the SK-OV-3 cells and tetravalently to the MDA-MB-231 cells. Per the data in Figure 4F, this pattern of binding is most consistent with a threshold number of accessible antigens between 0.042 – 0.057 EpCAMs, as this “cut off” permits eight binding events for the MCF-7 and LNCaP cells, but only five and three for the SK-OV-3 and MDA-MB-231 cells, respectively. Thus, the threshold is estimated as the midpoint of this range, 0.049 EpCAMs, and that value is represented by the horizontal black dashed line in Figure 4F. This corresponds to 7.7×105 EpCAMs/cell, as indicated by the red point in Figure 4E; likewise, the upper and lower bounds of this range are represented by the projected red dashed lines. (See page S9 of the Supporting Information for further details.) Collectively, this data suggests that CSANs are unlikely to discriminate between cells expressing ≥7.7×105 antigens/cell, as there is sufficient antigen density to support maximum octavalent binding to these membranes. Below this threshold, however, the CSANs are no longer capable of all eight binding interactions with the cell surface, thus lowering the avidity for the target cell and decreasing the apparent Kd,N value. However, we note that the specific threshold value is likely related to the inherent design and geometry of the CSAN; other multivalent scaffolds of different sizes and conformational flexibilities may demonstrate deterioration of the apparent affinity at a different antigen density threshold.
Incorporating the antigen expression (R, number of receptors/cell) analysis into equation 1 provided an updated view of the holistic relationship:
| (Eq. 4) |
Of note, equation 4 is merely a local empirical model of how the Kd,N value changes within this regime of antigen expression densities (R). This is highlighted by the fact that, as R approaches infinity, the fold-change in Kd,N (and thus Kd,N itself) approaches zero. Thus, the model may overestimate the apparent affinities of multivalent ligands towards cells with very high numbers of antigens/cell; however, we note such densities are unlikely to be encountered in physiologic settings. For these studies, the Kd,N value for the MCF-7 cells was set as the reference point (i.e., fold-change = 1.0) as it was the highest-expressing cell line in our panel, and because the next highest-expressing cell line (LNCaP) provided a Kd,N value that was not significantly different, suggesting the physiologic maximum Kd,N had been reached (Figure 4E). Finally, because the antigen expression term of equation 4 was constructed using data from only the fully-targeted CSANs (T:NT 8:0) displaying clone C5, we tested the predictive capacity of the equation by calculating the apparent Kd,N values for reduced-valency CSANs against the various cell lines. As shown in Table S2, equation 4 predicts Kd,N values that are within reasonable agreement with the experimentally-determined dissociation constants, suggesting that this is an appropriate model for predicting CSAN apparent affinities based upon the variables that compose the avidity of the scaffold/cell interaction.
Affinity and Valency Tuning Enables Target Discrimination In Vitro
We next explored whether modulating the CSAN’s avidity could reduce binding to EpCAMLow cells while maintaining binding to EpCAMHigh cells. EpCAM-targeted CSANs of different T:NT ratios (either 8:0 or 4:4) were titrated against each of the EpCAM-expressing cell lines in parallel via flow cytometry. A fully-targeted CSAN utilizing the high-affinity Fn3 clone C5 (8:0 C5:NT) bound appreciably to all cell lines (Figure 5E–H). However, reducing the valency of the Fn3 in the CSAN (4:4 C5:NT), yielded a construct that still bound well to the MCF-7, LNCaP, and SK-OV-3 cells but exhibited greatly diminished binding to the MDA-MB-231 cells (Figure 5I–L). These observations suggest that valency reduction may be a viable strategy for enabling cellular discrimination.
Figure 5. Avidity Modulation Enables Cellular Discrimination In Vitro.

Flow cytometry histogram plots depicting the binding of various constructs to EpCAM-expressing cell lines. (A–D) Qualitative EpCAM expression of indicated cell lines using a commercially-available antibody-fluorophore conjugate. The same cells were labeled with Fn3-targeted CSANs (50 nM) composed of T:NT ratios of (E–H) 8:0 C5:NT, (I–L) 4:4 C5:NT, (M–P) 8:0 B22:NT, or (Q–T) 4:4 B22:NT. In all panels, the background fluorescence of the negative control sample is shown by the gray histograms, (A–D) binding of the positive-control antibody is shown by the green histograms, (E–L) binding of CSANs utilizing clone C5 is shown by the blue histograms, and (M–T) binding of Fn3 CSANs utilizing clone B22 is shown by the red histograms. Triplicate experiments were performed as described in the methods, and the histogram plots for one representative trial are shown. Histograms for the full-range of CSAN concentrations tested (i.e., 5, 50, and 500 nM) are shown in Figure S6. (U) Quantitative depiction of avidity-modulated CSANs (50 nM) binding to EpCAM+ cells, as in panels A–T. Median fluorescence intensity values are plotted against the EpCAM expression of each of the four cell lines. Data is presented as the mean ± standard deviation of three independent trials.
Likewise, a fully-targeted CSAN utilizing the low-affinity Fn3 clone B22 (8:0 B22:NT) bound strongly to both the EpCAMHigh MCF-7 and LNCaP cells; however, binding was undetectable against the EpCAMLow SK-OV-3 and MDA-MB-231 cells (Figure 5M–P), suggesting that affinity reduction – in this case, by a factor of approximately of 65-fold between the parent Fn3 clones – may be sufficient to enable target cell discrimination, at least in vitro. As expected, reducing the valency of Fn3 clone B22 in the CSAN (4:4 B22:NT) marginally reduced the binding to MCF-7 and LNCaP cells, while binding to the SK-OV-3 and MDA-MB-231 cells remained undetectable (Figure 5Q–T). Data shown in Figure 5 represents CSAN binding at 50 nM, though the same trends were observed at all CSAN concentrations tested (i.e., 5, 50, and 500 nM; Figure S6).
Notably, the CSANs utilizing the low-affinity Fn3 clone B22 exhibited “switch-like” discrimination between the EpCAMLow (5.2×104 – 2.2×105 EpCAMs/cell) and EpCAMHigh (2.8×106 – 3.8×106 EpCAMs/cell) cell lines, with essentially no binding efficacy towards the MDA-MB-231 and SK-OV-3 cells but robust binding to the LNCaP and MCF-7 cells (Figure 5U). This phenomenon was diminished in the CSANs utilizing the high-affinity Fn3 clone C5, and entirely absent in the positive control samples where the antibody bound very well to all cell lines. Theoretical models of “super selectivity” have postulated that this on-off binding pattern can be achieved by multivalent nanoparticles when the affinity of the individual ligand-receptor interactions is low because the number of distinct binding arrangements increases in a non-linear way with respect to antigen density.25 Indeed, the experimental results in Figure 5 are in direct agreement with this model.
Collectively, these results demonstrate that the avidity of a multivalent targeting scaffold can be tuned to enable discrimination between cell populations expressing distinct quantities of the cognate antigen. To this end, reducing either the affinity of the binding ligand, the valency of the binding domains, or both are all viable approaches.
Avidity Optimization Yields Tunable Biodistribution In Vivo
Lastly, we evaluated whether the principles of avidity modulation determined in vitro would translate to an in vivo setting. Four week old, male, athymic nude (NU/J) mice were xenografted with an EpCAMHigh MCF-7 tumor (3.8×106 EpCAMs/cell) on the left flank and an EpCAMLow MDA-MB-231 (5.2×104 EpCAMs/cell) tumor on the right flank. We hypothesized that high-avidity CSANs (i.e, high affinity and/or valency) would distribute to both tumors regardless of EpCAM expression while low-avidity CSANs (i.e., reduced affinity and/or valency) would preferentially distribute to the high EpCAM-expressing tumor (i.e, MCF-7 tumor).
After two weeks, when tumors had grown to 8–12 mm in diameter, mice were randomly assigned to one of five groups (n = 3 mice/group): (1) 0:8 T:NT CSANs; (2) 8:0 C5:NT CSANs; (3) 4:4 C5:NT CSANs; (4) 8:0 B22:NT CSANs; and (5) 4:4 B22:NT CSANs. For this experiment, the CSANs were oligomerized with a previously-synthesized bisMTX analog that incorporates the copper-64 (64Cu) radiochelator, DOTA51; as with our previous work using bisMTX analogs43, 51, 54, all DHFR2-Fn3 monomers completely oligomerized into CSANs within 30 min of the addition of the DOTA-bisMTX (Figure S7).
Two weeks after tumor engraftment, mice in each group were given an intravenous (tail vein) injection of the indicated CSAN radiolabeled with 64Cu. After 18 h, mice were euthanized and tissues were collected for gamma counting to assess the in vivo biodistribution of the CSANs (Figure S8). The non-targeted control CSAN (0:8 T:NT) distributed to both tumors equally (Figure 6A–B) at a level commensurate with background tissue uptake (Figure S8). The highest avidity CSAN (8:0 C5:NT) exhibited significantly greater uptake in both tumors relative to the control (p ≤ 0.02). Further, it was unable to effectively discriminate between the tumors, exhibiting similar uptake in both the MCF-7 tumor (5.0 ± 1.8 %ID/g) and MDA-MB-231 tumor (5.3 ± 1.4 %ID/g) regardless of EpCAM expression (Figure 6A); this provided an MCF-7:MDA-MB-231 tumor uptake ratio of 0.94 ± 0.15, further reflecting the equivalent targeting to both tumors (Figure 6B). Reducing the valency of the C5 domain (4:4 C5:NT) did not affect uptake in the MCF-7 tumor (4.2 ± 1.1 %ID/g); however, uptake by the MDA-MB-231 tumor was reduced (2.5 ± 0.4 %ID/g; p < 0.03) though not ablated, with a tumor ratio of 1.74 ± 0.76 (Figure 6B). Though trending towards a selective preference for the MCF-7 xenograft, targeting to the respective tumors was not significantly different (p = 0.07) for this construct (Figure 6A). Reducing only the affinity of the CSAN (8:0 B22:NT) exhibited a similar pattern, again with unchanged MCF-7 tumor uptake (5.0 ± 1.5 %ID/g) and reduced MDA-MB-231 tumor uptake (2.6 ± 0.2 %ID/g; p < 0.03). In this case, however, discrimination between the two tumors did reach statistical significance, albeit barely (p < 0.05; Figure 6A), and this is reflected in the tumor uptake ratio of 1.94 ± 0.46 (Figure 6B). Finally, reducing both the affinity and valency of the CSAN (4:4 B22:NT) provided sustained MCF-7 tumor uptake (3.9 ± 0.8 %ID/g) and statistically insignificant uptake in the MDA-MB-231 tumor (2.0 ± 0.3 %ID/g; p > 0.07 relative to control; Figure 6A). Accordingly, there was a significant preference for the MCF-7 tumor over the MDA-MB-231 tumor (p < 0.03), collectively indicating targeting specificity with a tumor uptake ratio of 1.97 ± 0.21 (Figure 6B).
Figure 6. Xenograft Tumor Distribution of Avidity-Modulated CSANs.

(A) EpCAMHigh MCF-7 and EpCAMLow MDA-MB-231 xenograft tumors were resected, weighed, and counted for activity 18 h after IV infusion of avidity modulated CSANs. Activity values were time-corrected to 18 h to account for continued decay during the weighing/counting process. Data is presented as the mean ± standard deviation of the mice (n = 3) in each treatment group. Unless specified otherwise, statistical relevance marks comparison of the indicated sample to the 0:8 T:NT sample for that tumor, as determined by a two-tailed Student’s t-test. For clarity, intertumoral comparisons that failed to reach statistical significance are not marked. (B) The ratio of CSAN uptake in the MCF-7 tumors to MDA-MB-231 tumors is plotted on the primary y-axis (bar graph, linear scale). The corresponding MCF-7:MDA-MB-231 ratio of median fluorescence intensity values obtained in the in vitro studies (Fig. 5) are plotted along the secondary y-axis (line graph, log scale). Data is presented as the mean ratio ± standard deviation for three mice (in vivo) or independent trials (in vitro).
All targeted CSANs exhibited significant uptake by the EpCAMHigh MCF-7 tumor, regardless of their avidity (p < 0.03 for all species relative to control). This is similar to the in vitro flow cytometry observations where all constructs exhibited robust MCF-7 cell binding (Figure 5U). Likewise, a 50% reduction in CSAN valency alone (4:4 C5:NT) was insufficient to ablate binding to the relatively EpCAMLow MDA-MB-231 cells both in vivo (p < 0.01) and in vitro. In contrast, affinity reduction alone (8:0 B22:NT) only eliminated MDA-MB-231 cell binding in vitro, whereas the construct still exhibited significant MDA-MB-231 tumor uptake in vivo (p < 0.001) to a level similar to the 4:4 C5:NT CSAN. Only reduction of both affinity and valency (4:4 B22:NT) yielded durable cellular discrimination both in vitro and in vivo. The minor discrepancies between the in vitro and in vivo results indicate that, not unsurprisingly, there are additional factors in vivo that will influence the biodistribution of these scaffolds. This is highlighted in Figure 6B, where the in vitro cell binding ratio (orange line) is compared to the in vivo xenograft uptake ratio (colored bars). Little correlation is seen between the MFI ratios and the %ID/g ratios, suggesting that a scaffold’s ability to display super selective binding25 across a range of antigen expression densities is more important than the quantitative difference in binding potential. That is, all constructs yielded MCF-7:MDA-MB-231 MFI ratios ≥100 (Figure 6B), but only the 8:0 B22:NT and 4:4 B22:NT species that exhibited switch-like discrimination between EpCAMHigh and EpCAMLow cell lines in vitro (Figure 5U) were able to provide convincing tumor selectivity in vivo. Thus, while in vitro work can help design candidate scaffolds, a small panel of prioritized molecules should be evaluated in vivo to elucidate idiosyncrasies and identify the most promising targeting construct(s).
Interestingly, the in vivo tumor targeting profile did correlate with the apparent affinity of the scaffolds. The highest affinity construct (8:0 C5:NT) has a Kd,N value on the order of 10−9 M and exhibited equivalent uptake by both tumors. The two scaffolds with Kd,N values of 10−8 M (4:4 C5:NT and 8:0 B22:NT) demonstrated a degree of selectivity, with diminished targeting to the MDA-MB-231 tumor. Finally, the lowest-affinity scaffold with a Kd,N of 10−7 M (4:4 B22:NT) targeted the high-expressing MCF-7 tumor specifically. This result is consistent with other work highlighting the benefit of low-nanomolar (or subnanomolar) affinities for robust tumor targeting across a range of antigen expression levels.55–59
As suggested by the in vitro analyses (equation 4), it is possible that further valency reduction alone – e.g., to 2:6 C5:NT – could have also afforded complete tumor discrimination in vivo. Indeed, the 2:6 C5:NT CSANs exhibited a similar apparent affinity to the 4:4 B22:NT CSANs when titrated against the MCF-7 cells (140 ± 40 nM vs. 150 ± 80 nM, respectively; Figure 3). In the same manner, a further reduction in affinity alone – via the fusion of an even lower affinity parent Fn3 clone (e.g., B17 with a Kd of 3.1 ± 0.8 µM46) – may afford discrimination without the need for valency reduction. Finally, while the MDA-MB-231 cells express a relatively low level of EpCAM, it is still a malignant breast cancer cell line that may aberrantly express EpCAM relative to normal, healthy tissue. Therefore, CSANs that distributed to this tumor may or may not be taken up by healthy tissue. However, the use of such a cell line in this discrimination study (and others) may be highly valuable as a “built-in” safety margin, as a lack of binding to the MDA-MB-231 xenograft could be highly predictive of a lack of recognition of normal epithelium. The results of ongoing studies addressing these numerous possibilities will be reported in due course.
CONCLUSIONS
Using a modular self-assembling protein nanoring, this work demonstrates that the overall, apparent affinity of multivalent targeting scaffolds varies as a function of monomeric ligand affinity, binding domain valency, and cellular antigen expression density. The relationship between these variables was elucidated in vitro and then translated in vivo to identify scaffolds capable of specifically targeting cells that overexpress the target antigen. Thus, this work provides insight into the future design of multivalent scaffolds that could be used for a host of applications, including therapeutic drug conjugates and diagnostic imaging agents.
The model antigen in these studies, EpCAM, has been extensively explored as a clinical candidate, as its overexpression on carcinomas and presence on several types of CSCs make it a potentially useful target. Unfortunately, on-target, off-tumor toxicities – primarily in colonic and hepatic epithelia9 – have prevented the FDA approval of EpCAM-directed therapies. Currently, the only clinically-used EpCAM-targeted agent is catumaxomab, a rat/mouse hybrid monoclonal antibody approved in the European Union (EU) for localized treatment of malignant ascites.11 However, this work suggests that avidity optimization of the binding scaffold may be a viable strategy to enable the clinical targeting of EpCAM and other promising, yet promiscuous, antigens.
Pre-clinical work with CAR T cells has demonstrated that avidity optimization is a viable strategy for reducing on-target, off-tumor toxicities in that platform. Not unsurprisingly, the same variables identified in those studies – binding affinity, ligand expression/valency, and target density – have been found here to be crucial for soluble, multivalent targeting scaffolds as well. Arcangeli et al. suggested that CAR T cells using ligand affinities in the range of 10−7 M could offer a better safety profile compared to higher affinity constructs17; here, the only CSAN capable of completely discriminating between the EpCAMHigh and EpCAMLow xenograft tumors had an apparent affinity in this range (Kd,N = 1.5×10−7 M), while the higher-affinity constructs exhibited measurable uptake by both tumors (Kd,N values of 3.3×10−8 – 6.9×10−9 M). Whether this shared 10−7 M affinity threshold is merely a coincidence or represents a meaningful barrier for off-tumor toxicities remains to be seen.
A limitation of this work is that it was completed using a single scaffold, the CSAN, whose self-assembling nature drives the formation of heterogeneous populations of nanorings. While overall targeting potential of each population is distinct, as determined by the stoichiometric ratio of targeted and non-targeted subunits, the existence of a statistical distribution of species hinders the precise analysis of the binding characteristics. Importantly, the self-assembled populations yield reproducible results across trials, suggesting that the distribution of species is also recapitulated with each assembly. Furthermore, the antigen expression data obtained using flow cytometry demonstrates that, expectedly, there is a distribution of EpCAM expression within each cell population. It’s conceivable that the mean estimates of EpCAM density may not drive binding to cells that fall at either extreme of the distribution. In such cases, fluorescence microscopy techniques can be valuable for examining the relative levels of antigen expression and ligand binding, and can provide qualitative correlation with the flow cytometry results.60 The use of both techniques in parallel will be an important facet of future work in this area when more a more detailed examination of population binding mechanisms is required.
Finally, while the fundamental relationship between affinity, valency, and expression are likely to translate to other scaffolds, we caution that the specific numerical values (i.e., the exponents and numerator constant) in equation 4 may be different for other geometries, such as multi-specific antibodies and linear fusions of tandem alternative protein topologies (e.g., Fn3s, affibodies, designed ankyrin protein repeats, etc.). As other groups with specialties in these ligands and formats explore avidity optimization in their scaffolds, we are enthusiastic that the field as a whole will develop a collective set of design principles and thresholds that will facilitate the clinical use of these unique constructs for the targeting of difficult yet promising tumor associated antigens.
MATERIALS AND METHODS
Cells and Cell Culture
The MCF-7, LNCaP, SK-OV-3, and MDA-MB-231 cell lines were previously purchased from the American Type Culture Collection (ATCC). MCF-7 and MDA-MB-231 cells were grown at 37 ºC in a humidified atmosphere with 5% CO2 in Dulbecco’s Modified Eagle’s Medium (DMEM) with 4.5 g/L glucose, L-glutamine, and supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 µg/mL streptomycin. LNCaP and SK-OV-3 cells were grown at 37 ºC in a humidified atmosphere with 5% CO2 in Roswell Park Memorial Institute (RPMI) media with L-glutamine and supplemented with 10% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin. For passaging, cells were detached via trypsin. Cell count and viability were determined via trypan blue exclusion using a Bio Rad TC20 automated cell counter (Bio Rad Laboratories, Inc.).
Expression Plasmids
A gBlock Gene Fragment coding for the DHFR2-Fn3 clone C5 fusion protein was ordered from Integrated DNA Technologies (IDT) and cloned into the Novagen pET28a(+) vector (EMD Millipore, Cat: 69864–3) via NcoI and XhoI restriction sites. Separately, Fn3 clones B22 and NT replaced clone C5 using designed NheI and BamHI restriction sites flanking the Fn3 sequence region. Notably, the DHFR2-Fn3 proteins contain an N-terminal MYC epitope tag and C-terminal polyhistidine tag to facilitate detection via flow cytometry and purification via immobilized metal affinity chromatography (IMAC), respectively.
Protein Expression and Purification
DHFR2-Fn3 fusion proteins were expressed in Escherichia coli (E. coli) and purified from the soluble fraction of the cell lysates by IMAC.43 Purified proteins were analyzed by SEC on a Superdex 200 Increase 10/300 gel filtration column (GE Healthcare Life Sciences, Cat: 28990944) in phosphate buffered saline (PBS, pH 7.4) running buffer. Fusion protein retention times were compared to those of commercial molecular weight standards (Sigma Aldrich, Cat: MWGF1000–1KT).
CSAN Formation and Characterization
CSANs were formed by adding a 3-fold molar excess of the desired chemical dimerizer (either bisMTX or DOTA-bisMTX) to a solution of DHFR2-Fn3 fusion protein monomers in PBS. Consistent with our previous studies, CSAN oligomerization occured within 30 minutes of adding the dimerizer.41 Oligomerized CSANs were analyzed by SEC (as described above) and dynamic light scattering (DLS). For DLS, 60 µL of CSANs in PBS was loaded into a cuvette and analyzed on a Punk DLS unit (Unchained Labs). Hydrodynamic diameter values represent the mean ± standard deviation of at least three measurements. SEC and DLS results were consistent with our previous analyses of Fn3-targeted CSANs, including cryo-electron microscopy (cryo-EM) evidence of nanoring formation.43, 44
Apparent Affinity Determination of Fn3-Targeted CSANs
The apparent affinity of the Fn3-targeted CSANs was determined by flow cytometry, as previously described.46 Briefly, EpCAM-expressing cells (MCF-7, LNCaP, MDA-MB-231 lines) were cultured to approximately 80% confluency, detached, and counted, as described above. Aliquots of 5×104 cells were washed with PBSA (PBS with 0.1% w/v bovine serum albumin) and labeled with varying concentrations of Fn3-targeted CSANs for ≥90 min at 4 ºC. Cells were then pelleted (8,000 g, 30 s, 4 ºC) and resuspended in 50 µL anti-His antibody (clone 4E3D10H2/E3) Alexa Fluor 647 conjugate (Thermo Fisher Scientific, Cat: MA1–135-A647; 50 µg/mL in PBSA). After incubating at 4 ºC for ≥30 min in the dark, cells were washed thrice with 1 mL cold PBSA before the fluorescence was analyzed on an LSR II flow cytometer (BD Biosciences). Maximum fluorescence and Kd,N values were determined by minimizing the sum of squared errors.
Quantification of EpCAM Expression for Mammalian Cell Lines
Cells were grown to approximately 80% confluence and detached as described above. Cells were aliquoted at 1×106 cells/sample, pelleted (8,000 g, 30 sec, 4 ºC), and washed with 1 mL ice cold PBSA. Cells were labeled with mouse anti-human EpCAM monoclonal antibody (clone 9C4) Alexa Fluor 647 conjugate (BioLegend, Cat: 324212) at 4 ºC for 30 min in the dark. Cells were then washed three times with 1 mL PBSA before immediate analysis on a BD LSR II. To quantify the absolute expression of EpCAM, the fluorescence was compared to a calibration curve prepared from anti-mouse IgG beads (Bangs Laboratories, Inc., Cat: 815). Polystyrene beads with known quantities of immobilized monoclonal anti-mouse IgG were labeled with the same anti-EpCAM Alexa Fluor 647 conjugate as the cells under the same conditions. Beads were washed once with PBSA and pelleted at 2,500g for 2.5 min. The beads were then washed thrice with 1 mL PBSA and analyzed on the flow cytometer in parallel with the cells. The standard curve generated from the bead populations was compared to the fluorescence of the mammalian cells that were labeled with the same mouse anti-EpCAM antibody and used to calculate the cellular EpCAM expression.
Copper Chelation and Purification of 64Cu-Labeled CSANs
64CuCl2 (University of Wisconsin, Madison) was diluted with 100 μL of 100 µM sodium acetate buffer, pH 7.0 and pH adjusted using 1M HCl/NaOH. Approximately 7.9 mCi of the 64Cu solution was added to 40 μL of DOTA-bisMTX (2 mM) in DMSO and the solution was incubated at 50 ºC for 30 min. After cooling to room temperature, the chelated DOTA-bisMTX was combined with the desired mixture of DHFR2-Fn3 monomers at a 3:1 molar ratio in PBS, and the resulting solutions were incubated at room temperature for 30 min to allow for CSAN oligomerization. The 64Cu-labeled CSANs were then purified using a PD-10 column equilibrated PBS, pH 7.4, to remove excess DOTA-bisMTX and unchelated 64Cu.
Tumor Inoculation
All procedures performed in studies involving animals were in accordance with the ethical standards of the University of Minnesota and approved by the Institutional Animal Care and Use Committee (IACUC). Four-week-old, male, athymic nude (NU/J) mice (Jackson Laboratory) were anesthetized with isofluorane (induced at 5% and maintained at 2% in 1 mL/min O2) and inoculated with 2.5×106 EpCAMHigh MCF-7 cells on the left shoulder and 2.5×106 EpCAMLow MDA-MB-231 cells on the right shoulder, each as a 50% v/v suspension in Matrigel Matrix (Corning, Cat: 354248). Xenografts were allowed to grow for two weeks, yielding tumors of 8–12 mm in diameter. To support MCF-7 cell growth, the mice’s water supply was supplemented with 500 µM 17-β-estradiol valerate.
Biodistribution Analysis
All procedures performed in studies involving animals were in accordance with the ethical standards of the University of Minnesota and approved by the Institutional Animal Care and Use Committee (IACUC). Mice were anesthetized with isofluorane (induced at 5% and maintained at 2% in 1 mL/min O2) and tail vein injected with approximately 70–100 µCi of 64Cu-labeled CSANs, as measured by an Atomlab 100 Dosimeter with a setting of 50.2. After 18 h, mice were euthanized via gentle CO2 asphyxiation. Blood, bone, brain, heart, intestine, kidneys, liver, lungs, muscle, pancreas, skin, spleen, stomach, tail and tumors were resected, weighed, and had their radioactivity measured by a Hidex Automated Gamma Counter (Hidex) with counts averaged over 60 sec.
Statistical Considerations
Unless otherwise stated, experiments were performed in triplicate and data is presented as the mean ± standard deviation of three independent trials. Differences between means are compared using a two-tailed Student’s t-test or a one-way analysis of variance (ANOVA), as appropriate. A p-value <0.05 is deemed to be statistically significant, and is denoted in graphics with an (*), p<0.01 is denoted with (**), and p<0.001 is denoted with (***).
Supplementary Material
ACKNOWLDEGEMENTS
This work was supported by the National Institutes of Health (NIH) R21 CA185627 (CRW), F30 CA210345 (CMC), T32 GM008244 (CMC), and the University of Minnesota, including an endowed fellowship on behalf of Dr. and Mrs. Bighley (CMC). Flow cytometry work was performed in the University Flow Cytometry Resource core facility at the University of Minnesota. The biodistribution work was performed in the University Imaging Center at the University of Minnesota.
Footnotes
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS Publications Website at https://pubs.acs.org.
Supplementary figures and tables, sequences and characterization of protein constructs, theoretical models and assumptions, references
REFERENCES
- 1.Mammen M; Choi SK; Whitesides GM, Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors. Angew Chem Int Ed Engl 1998, 37 (20), 2754–2794. [DOI] [PubMed] [Google Scholar]
- 2.Jackson HJ; Rafiq S; Brentjens RJ, Driving CAR T-cells forward. Nat Rev Clin Oncol 2016, 13 (6), 370–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fesnak AD; June CH; Levine BL, Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer 2016, 16 (9), 566–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maude SL; Frey N; Shaw PA; Aplenc R; Barrett DM; Bunin NJ; Chew A; Gonzalez VE; Zheng Z; Lacey SF; Mahnke YD; Melenhorst JJ; Rheingold SR; Shen A; Teachey DT; Levine BL; June CH; Porter DL; Grupp SA, Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. New England Journal of Medicine 2014, 371 (16), 1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude SL; Laetsch TW; Buechner J; Rives S; Boyer M; Bittencourt H; Bader P; Verneris MR; Stefanski HE; Myers GD; Qayed M; De Moerloose B; Hiramatsu H; Schlis K; Davis KL; Martin PL; Nemecek ER; Yanik GA; Peters C; Baruchel A; Boissel N; Mechinaud F; Balduzzi A; Krueger J; June CH; Levine BL; Wood P; Taran T; Leung M; Mueller KT; Zhang Y; Sen K; Lebwohl D; Pulsipher MA; Grupp SA, Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 2018, 378 (5), 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan RA; Yang JC; Kitano M; Dudley ME; Laurencot CM; Rosenberg SA, Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010, 18 (4), 843–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lamers CH; Sleijfer S; van Steenbergen S; van Elzakker P; van Krimpen B; Groot C; Vulto A; den Bakker M; Oosterwijk E; Debets R; Gratama JW, Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther 2013, 21 (4), 904–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parkhurst MR; Yang JC; Langan RC; Dudley ME; Nathan DA; Feldman SA; Davis JL; Morgan RA; Merino MJ; Sherry RM; Hughes MS; Kammula US; Phan GQ; Lim RM; Wank SA; Restifo NP; Robbins PF; Laurencot CM; Rosenberg SA, T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 2011, 19 (3), 620–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fiedler WM; Wolf M; Kebenko M; Goebeler M-E; Ritter B; Quaas A; Vieser E; Hijazi Y; Patzak I; Friedrich M; Kufer P; Frankel S; Seggewiss-Bernhardt R; Kaubitzsch S, A phase I study of EpCAM/CD3-bispecific antibody (MT110) in patients with advanced solid tumors. Journal of Clinical Oncology 2012, 30 (15_suppl), 2504–2504. [Google Scholar]
- 10.Klinger M; Benjamin J; Kischel R; Stienen S; Zugmaier G, Harnessing T cells to fight cancer with BiTE(R) antibody constructs--past developments and future directions. Immunol Rev 2016, 270 (1), 193–208. [DOI] [PubMed] [Google Scholar]
- 11.Seimetz D; Lindhofer H; Bokemeyer C, Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3) as a targeted cancer immunotherapy. Cancer Treat Rev 2010, 36 (6), 458–67. [DOI] [PubMed] [Google Scholar]
- 12.Mau-Sorensen M; Dittrich C; Dienstmann R; Lassen U; Buchler W; Martinius H; Tabernero J, A phase I trial of intravenous catumaxomab: a bispecific monoclonal antibody targeting EpCAM and the T cell coreceptor CD3. Cancer Chemother Pharmacol 2015, 75 (5), 1065–73. [DOI] [PubMed] [Google Scholar]
- 13.Caruso HG; Hurton LV; Najjar A; Rushworth D; Ang S; Olivares S; Mi T; Switzer K; Singh H; Huls H; Lee DA; Heimberger AB; Champlin RE; Cooper LJ, Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res 2015, 75 (17), 3505–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu X; Jiang S; Fang C; Yang S; Olalere D; Pequignot EC; Cogdill AP; Li N; Ramones M; Granda B; Zhou L; Loew A; Young RM; June CH; Zhao Y, Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res 2015, 75 (17), 3596–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park S; Shevlin E; Vedvyas Y; Zaman M; Hsu YS; Min IM; Jin MM, Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci Rep 2017, 7 (1), 14366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drent E; Themeli M; Poels R; de Jong-Korlaar R; Yuan H; de Bruijn J; Martens ACM; Zweegman S; van de Donk N; Groen RWJ; Lokhorst HM; Mutis T, A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol Ther 2017, 25 (8), 1946–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arcangeli S; Rotiroti MC; Bardelli M; Simonelli L; Magnani CF; Biondi A; Biagi E; Tettamanti S; Varani L, Balance of Anti-CD123 Chimeric Antigen Receptor Binding Affinity and Density for the Targeting of Acute Myeloid Leukemia. Mol Ther 2017, 25 (8), 1933–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calderon AJ; Bhowmick T; Leferovich J; Burman B; Pichette B; Muzykantov V; Eckmann DM; Muro S, Optimizing endothelial targeting by modulating the antibody density and particle concentration of anti-ICAM coated carriers. J Control Release 2011, 150 (1), 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zern BJ; Chacko AM; Liu J; Greineder CF; Blankemeyer ER; Radhakrishnan R; Muzykantov V, Reduction of nanoparticle avidity enhances the selectivity of vascular targeting and PET detection of pulmonary inflammation. ACS Nano 2013, 7 (3), 2461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aires A; Cadenas JF; Guantes R; Cortajarena AL, An experimental and computational framework for engineering multifunctional nanoparticles: designing selective anticancer therapies. Nanoscale 2017, 9 (36), 13760–13771. [DOI] [PubMed] [Google Scholar]
- 21.Hong S; Leroueil PR; Majoros IJ; Orr BG; Baker JR Jr.; Banaszak Holl MM, The binding avidity of a nanoparticle-based multivalent targeted drug delivery platform. Chem Biol 2007, 14 (1), 107–15. [DOI] [PubMed] [Google Scholar]
- 22.Silpe JE; Sumit M; Thomas TP; Huang B; Kotlyar A; van Dongen MA; Banaszak Holl MM; Orr BG; Choi SK, Avidity modulation of folate-targeted multivalent dendrimers for evaluating biophysical models of cancer targeting nanoparticles. ACS Chem Biol 2013, 8 (9), 2063–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Dongen MA; Silpe JE; Dougherty CA; Kanduluru AK; Choi SK; Orr BG; Low PS; Banaszak Holl MM, Avidity mechanism of dendrimer-folic acid conjugates. Mol Pharm 2014, 11 (5), 1696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huskens J; Mulder A; Auletta T; Nijhuis CA; Ludden MJ; Reinhoudt DN, A model for describing the thermodynamics of multivalent host-guest interactions at interfaces. J Am Chem Soc 2004, 126 (21), 6784–97. [DOI] [PubMed] [Google Scholar]
- 25.Martinez-Veracoechea FJ; Frenkel D, Designing super selectivity in multivalent nano-particle binding. Proc Natl Acad Sci U S A 2011, 108 (27), 10963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rhoden JJ; Dyas GL; Wroblewski VJ, A Modeling and Experimental Investigation of the Effects of Antigen Density, Binding Affinity, and Antigen Expression Ratio on Bispecific Antibody Binding to Cell Surface Targets. J Biol Chem 2016, 291 (21), 11337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Michele C; De Los Rios P; Foffi G; Piazza F, Simulation and Theory of Antibody Binding to Crowded Antigen-Covered Surfaces. PLoS Comput Biol 2016, 12 (3), e1004752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Curk T; Dobnikar J; Frenkel D, Optimal multivalent targeting of membranes with many distinct receptors. Proc Natl Acad Sci U S A 2017, 114 (28), 7210–7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKenzie M; Ha SM; Rammohan A; Radhakrishnan R; Ramakrishnan N, Multivalent Binding of a Ligand-Coated Particle: Role of Shape, Size, and Ligand Heterogeneity. Biophys J 2018, 114 (8), 1830–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kane RS, Thermodynamics of multivalent interactions: influence of the linker. Langmuir 2010, 26 (11), 8636–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoon JH; Kim DK; Na M; Lee SY, Multi-ligand functionalized particle design for cell targeting and drug delivery. Biophys Chem 2016, 213, 25–31. [DOI] [PubMed] [Google Scholar]
- 32.Dubacheva GV; Curk T; Mognetti BM; Auzely-Velty R; Frenkel D; Richter RP, Superselective targeting using multivalent polymers. J Am Chem Soc 2014, 136 (5), 1722–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dubacheva GV; Curk T; Auzely-Velty R; Frenkel D; Richter RP, Designing multivalent probes for tunable superselective targeting. Proc Natl Acad Sci U S A 2015, 112 (18), 5579–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hadzhieva M; Pashov AD; Kaveri S; Lacroix-Desmazes S; Mouquet H; Dimitrov JD, Impact of Antigen Density on the Binding Mechanism of IgG Antibodies. Sci Rep 2017, 7 (1), 3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cairo CW; Gestwicki JE; Kanai M; Kiessling LL, Control of multivalent interactions by binding epitope density. J Am Chem Soc 2002, 124 (8), 1615–9. [DOI] [PubMed] [Google Scholar]
- 36.Mazor Y; Oganesyan V; Yang C; Hansen A; Wang J; Liu H; Sachsenmeier K; Carlson M; Gadre DV; Borrok MJ; Yu XQ; Dall’Acqua W; Wu H; Chowdhury PS, Improving target cell specificity using a novel monovalent bispecific IgG design. MAbs 2015, 7 (2), 377–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mazor Y; Hansen A; Yang C; Chowdhury PS; Wang J; Stephens G; Wu H; Dall’Acqua WF, Insights into the molecular basis of a bispecific antibody’s target selectivity. MAbs 2015, 7 (3), 461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carlson CB; Mowery P; Owen RM; Dykhuizen EC; Kiessling LL, Selective tumor cell targeting using low-affinity, multivalent interactions. ACS Chem Biol 2007, 2 (2), 119–27. [DOI] [PubMed] [Google Scholar]
- 39.Mazor Y; Sachsenmeier KF; Yang C; Hansen A; Filderman J; Mulgrew K; Wu H; Dall’Acqua WF, Enhanced tumor-targeting selectivity by modulating bispecific antibody binding affinity and format valence. Sci Rep 2017, 7, 40098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramakrishnan N; Tourdot RW; Eckmann DM; Ayyaswamy PS; Muzykantov VR; Radhakrishnan R, Biophysically inspired model for functionalized nanocarrier adhesion to cell surface: roles of protein expression and mechanical factors. R Soc Open Sci 2016, 3 (6), 160260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carlson JCT; Jena SS; Flenniken M; Chou T.-f.; Siegel RA; Wagner CR, Chemically Controlled Self-Assembly of Protein Nanorings. Journal of the American Chemical Society 2006, 128 (23), 7630–7638. [DOI] [PubMed] [Google Scholar]
- 42.Li Q; So CR; Fegan A; Cody V; Sarikaya M; Vallera DA; Wagner CR, Chemically Self-Assembled Antibody Nanorings (CSANs): Design and Characterization of an Anti-CD3 IgM Biomimetic. Journal of the American Chemical Society 2010, 132 (48), 17247–17257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Csizmar CM; Petersburg JR; Hendricks A; Stern LA; Hackel BJ; Wagner CR, Engineering Reversible Cell-Cell Interactions with Lipid Anchored Prosthetic Receptors. Bioconjug Chem 2018, 29 (4), 1291–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petersburg J; Shen J; Csizmar CM; Murphy KA; Spanier J; Gabrielse K; Griffith TS; Fife B; Wagner CR, Eradication of Established Tumors by Chemically Self-Assembled Nanoring (CSAN) Targeted T-cells. ACS Nano 2018, 12 (7), 6563–6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen J; Vallera DA; Wagner CR, Prosthetic Antigen Receptors. J Am Chem Soc 2015, 137 (32), 10108–11. [DOI] [PubMed] [Google Scholar]
- 46.Stern LA; Csizmar CM; Woldring DR; Wagner CR; Hackel BJ, Titratable Avidity Reduction Enhances Affinity Discrimination in Mammalian Cellular Selections of Yeast-Displayed Ligands. ACS Combinatorial Science 2017, 19 (5), 315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Went PT; Lugli A; Meier S; Bundi M; Mirlacher M; Sauter G; Dirnhofer S, Frequent EpCam protein expression in human carcinomas. Hum Pathol 2004, 35 (1), 122–8. [DOI] [PubMed] [Google Scholar]
- 48.Baeuerle PA; Gires O, EpCAM (CD326) finding its role in cancer. Br J Cancer 2007, 96 (3), 417–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gires O; Klein CA; Baeuerle PA, On the abundance of EpCAM on cancer stem cells. In Nat Rev Cancer, England, 2009; Vol. 9, p 143; author reply 143. [DOI] [PubMed] [Google Scholar]
- 50.Patriarca C; Macchi RM; Marschner AK; Mellstedt H, Epithelial cell adhesion molecule expression (CD326) in cancer: a short review. Cancer Treat Rev 2012, 38 (1), 68–75. [DOI] [PubMed] [Google Scholar]
- 51.Shah R; Petersburg J; Gangar AC; Fegan A; Wagner CR; Kumarapperuma SC, In Vivo Evaluation of Site-Specifically PEGylated Chemically Self-Assembled Protein Nanostructures. Mol Pharm 2016, 13 (7), 2193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jencks WP, On the attribution and additivity of binding energies. Proc Natl Acad Sci U S A 1981, 78 (7), 4046–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaufman EN; Jain RK, Effect of bivalent interaction upon apparent antibody affinity: experimental confirmation of theory using fluorescence photobleaching and implications for antibody binding assays. Cancer Res 1992, 52 (15), 4157–67. [PubMed] [Google Scholar]
- 54.Fegan A; Kumarapperuma SC; Wagner CR, Chemically Self-Assembled Antibody Nanostructures as Potential Drug Carriers. Molecular Pharmaceutics 2012, 9 (11), 3218–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zahnd C; Kawe M; Stumpp MT; de Pasquale C; Tamaskovic R; Nagy-Davidescu G; Dreier B; Schibli R; Binz HK; Waibel R; Pluckthun A, Efficient tumor targeting with high-affinity designed ankyrin repeat proteins: effects of affinity and molecular size. Cancer Res 2010, 70 (4), 1595–605. [DOI] [PubMed] [Google Scholar]
- 56.Okarvi SM; Jammaz IA, Preparation and evaluation of bombesin peptide derivatives as potential tumor imaging agents: effects of structure and composition of amino acid sequence on in vitro and in vivo characteristics. Nucl Med Biol 2012, 39 (6), 795–804. [DOI] [PubMed] [Google Scholar]
- 57.Adams GP; Schier R; McCall AM; Simmons HH; Horak EM; Alpaugh RK; Marks JD; Weiner LM, High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res 2001, 61 (12), 4750–5. [PubMed] [Google Scholar]
- 58.Schmidt MM; Wittrup KD, A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Mol Cancer Ther 2009, 8 (10), 2861–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tolmachev V; Tran TA; Rosik D; Sjoberg A; Abrahmsen L; Orlova A, Tumor targeting using affibody molecules: interplay of affinity, target expression level, and binding site composition. J Nucl Med 2012, 53 (6), 953–60. [DOI] [PubMed] [Google Scholar]
- 60.Landmark KJ; Dimaggio S; Ward J; Kelly C; Vogt S; Hong S; Kotlyar A; Myc A; Thomas TP; Penner-Hahn JE; Baker JR; Holl MM; Orr BG, Synthesis, characterization, and in vitro testing of superparamagnetic iron oxide nanoparticles targeted using folic Acid-conjugated dendrimers. ACS Nano 2008, 2 (4), 773–83. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.