

Abstract
The conserved regulators of cell cycle progression—Cyclins, Cdc2 kinase, and String phosphatase (Cdc25)—accommodate multiple modes of regulation during Drosophila embryogenesis. During cell cycles 2–7, Cdc2/Cyclin complexes are continuously present and show little fluctuation in abundance, phosphomodification, or activity. This suggests that cycling of the mitotic apparatus does not require cytoplasmic oscillations of known regulatory activities. During cycles 8–13 a progressive increase in the degradation of Cyclins at mitosis leads to increasing oscillations of Cdc2 kinase activity. Mutants deficient in cyclin mRNAs suffer cell cycle delays during this period, suggesting that Cyclin accumulation times these cycles. During interphase 14, programmed degradation of maternal String protein leads to inhibitory phosphorylation of Cdc2 and cell cycle arrest. Subsequently, mitoses 14–16 are triggered by pulses of zygotic string transcription.
Keywords: Drosophila embryogenesis, cell cycle, Cyclin, String
In eukaryotic cells, the transition from G2 to mitosis is driven by activation of the Cdc2 protein kinase (Nurse 1990). This results in phosphorylation of numerous cellular substrates, causing structural changes in the cyto-skeleton, nuclear envelope, and chromatin. Although Cdc2 and many of its regulators are highly conserved, studies in diverse systems have revealed that different regulatory inputs can govern Cdc2 activation. For example, Cdc2 activation in Schizosaccharomyces pombe is timed by accumulation of Cdc25+ , a Cdc2-activating phosphatase (Russell and Nurse 1986; Moreno et al. 1990), whereas in Saccharomyces ceievisiae this phosphatase has no obvious role (Russell et al. 1989; Amon et al. 1992; Sorger et al. 1992). Thus, different components of the conserved machinery may play regulatory roles in different systems.
Two well-supported models for the regulation of mitotic timing have dominated the literature in recent years. Studies in marine invertebrate embryos and extracts of Xenopus eggs have emphasized the importance of accumulation of Cyclins in timing mitosis (Evans et al. 1983; Solomon et al. 1990; Hunt et al. 1992). Cdc2 requires a Cyclin subunit for activity, and in cell-free extracts of Xenopus eggs, Cyclin accumulation is rate-limiting for Cdc2 activation (Minshull et al. 1989; Murray and Kirschner 1989). It has been postulated that positive feedback involving phosphoregulation converts gradual accumulation of Cyclins into an abrupt activation of Cdc2 that actually triggers mitosis (Wasserman and Masui 1975; Gerhart et al. 1984; Solomon et al., 1990; Hoffmann et al. 1993). However, Cyclin accumulation is thought to be the dominant parameter in timing progress to mitosis, whereas the positive feedback creates a threshold Cyclin level at which abrupt Cdc2 activation occurs. In contrast, Cyclin accumulation does not time mitosis in the postblastoderm Drosophila embryo, or in the fission yeast, S. pombe (Booher and Beach 1988; Hagen et al. 1988; Lehner and O’Farrell 1989, 1990a). In these systems Cyclins are expressed in excess, and dephosphorylation of Cdc2 by newly expressed Cdc25-type phosphatases is rate-limiting for mitosis (Russell and Nurse 1986, 1987; Edgar and O’Farrell 1989, 1990; Gould and Nurse 1989; Moreno et al. 1990). In both types of cell cycle, degradation of Cyclins and consequent inactivation of Cdc2 appear to cause the exit from mitosis (Murray et al. 1989; Luca et al. 1991; Surana et al. 1993).
Although previous studies have suggested flexibility in the regulatory hardware that drives the cell cycle, differences in experimental systems allow various interpretations. In Drosophila, different types of cell cycles occur during different developmental stages, providing the opportunity to study alternate modes of cell cycle regulation in a single organism. Drosophila embryogenesis begins with 13 rapid, synchronous, syncytial mitotic cycles that are driven maternally (Rabinowitz 1941; Edgar and Schubiger 1986). Interphases lengthen gradually after the tenth mitosis, and the embryo arrests in an extended G2 phase during cycle 14 (Foe and Alberts 1983). Zygotic cell cycle control begins during interphase 14, and a series of slower, spatially patterned, G2-regulated cell cycles follow (Hartenstein and Campos-Ortega 1985; Foe 1989; Edgar and O’Farrell 1990). Here, we analyze modes of cell cycle regulation at these different stages and demonstrate how developmental changes in the relative abundance of cell cycle regulators lead to changes in the fundamental mode of operation of the engine that drives the cell cycle.
Results
Dephosphorylation of Cdc2 at Y15 limits the postblastoderm cycles
Previous studies suggested that pulses of expression of string, a Cdc2-specific tyrosine phosphatase, regulate mitoses during the postblastoderm cell cycles 14–16 (Edgar and O’Farrell 1989, 1990). We have tested and confirmed three predictions arising from this proposal.
The proposal implies that String protein, as well as string RNA, should be absent during most of interphase, and expressed in pulses prior to mitosis. Consistent with this, both immunoblots and in situ immunohistochemical staining showed that String protein declines to undetectable levels within the first 20 min of interphase 14, and only reappears just before mitosis 14 (Figs. 1 and 4).
If lack of String, a Cdc2-activating tyrosine phosphatase, limits progress to mitosis 14, Cdc2 should be inhibited by phosphorylation at Y15 during interphase 14. The different phosphoisoforms of Cdc2 can be resolved on SDS gels and detected by antibodies to either Cdc2 or phosphotyrosine (Figs. 2A and 8; see also Materials and methods). As predicted, inhibitory phosphorylation of Cdc2 on Y15 increases during cycle 14, so that by G2, Cdc2 is fully tyrosine phosphorylated (Fig. 2A). Cdc2 kinase activity also remains low during interphase 14 (Fig. 6).
Progress to mitosis is proposed to involve dephosphorylation of Cdc2. Dephosphorylated forms of Cdc2 appear as String reappears during mitosis 14 (Figs. 1 and 2A). Furthermore, abrupt, spatially uniform expression of String from a heat shock promoter resulted in a dramatic conversion of tyrosyl-phospho-Cdc2 to the active form, and induced mitosis (Fig. 2B, see also Edgar and O’Farrell 1990). Thus, String appears to dephosphorylate phospho-tyrosyl-inhibited Cdc2/Cyclin complexes in vivo, just as it does in vitro (Dunphy and Kumagai 1991; Gautier et al. 1991).
Figure 1.
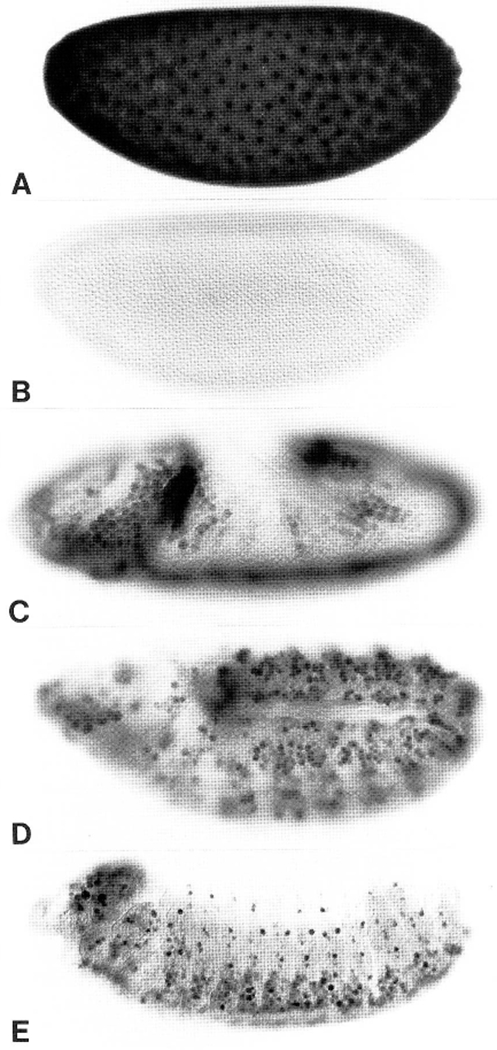
Spatial expression of String protein during embryogenesis. (A–E) Embryos at progressively more advanced stages of development, immunohistochemically stained for String protein, and photographed using DIC optics. (A) Nuclear cycle 11: Note the concentration of String in nuclei. High magnification observations revealed that String is concentrated in nuclei during the syncytial interphases and on chromosomes during all stages of mitosis (not shown). This embryo was stained severalfold less intensely than the others to highlight this nuclear staining. (B) Interphase 14, maternal String protein is completely degraded. (C) Early mitosis 14, zygotically expressed String appears in mitotic domains 1–11 (Foe 1989). (D) Patterns of mitosis 15. (E) String expression in proliferating neuroblasts of the central and peripheral nervous systems during postcycle 16 embryogenesis.
Figure 4.
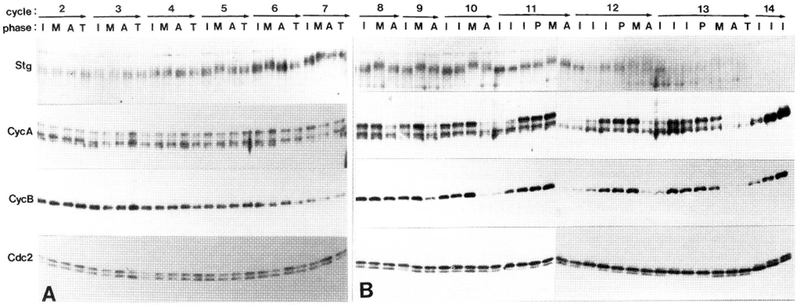
Changes in cell cycle regulators during cycles 2–7 (A) and 8–14 (B). We show time-course immunoblots probed with antibodies to String (Stg|, Cyclin A (CycA), Cyclin B (CycB), and Cdc2 (Cdc2). Cycle numbers and phases are denoted (top). The embryos were precisely staged as interphase (I), prophase (P), metaphase (M), anaphase (A) or telophase (T). In later cycles, several interphase time points were ordered using nuclear volume, which increases with time (Fig. 3). Phosphatase treatment indicated that mobility shifts of String are due to relative hyperphosphorylation during mitosis. Lower mobility bands of Cyclin A are likewise due to phosphorylation (see Materials and methods). Phosphorylation of Cdc2 at T161 causes a downshift, whereas Y15 and T14 phosphorylation cause upshifts (in cycle 14 only; see Materials and methods and Fig. 8). Both unphosphorylated and T161-phospho-Cdc2 are continuously present during cycles 2–9. One embryo was loaded per lane in A, and three embryos of identical stage were loaded in B. The same blots were serially probed with each antibody, except for String in A, which was from a separate experiment. These experiments were repeated four times (A) or six times (B), with similar results.
Figure 2.
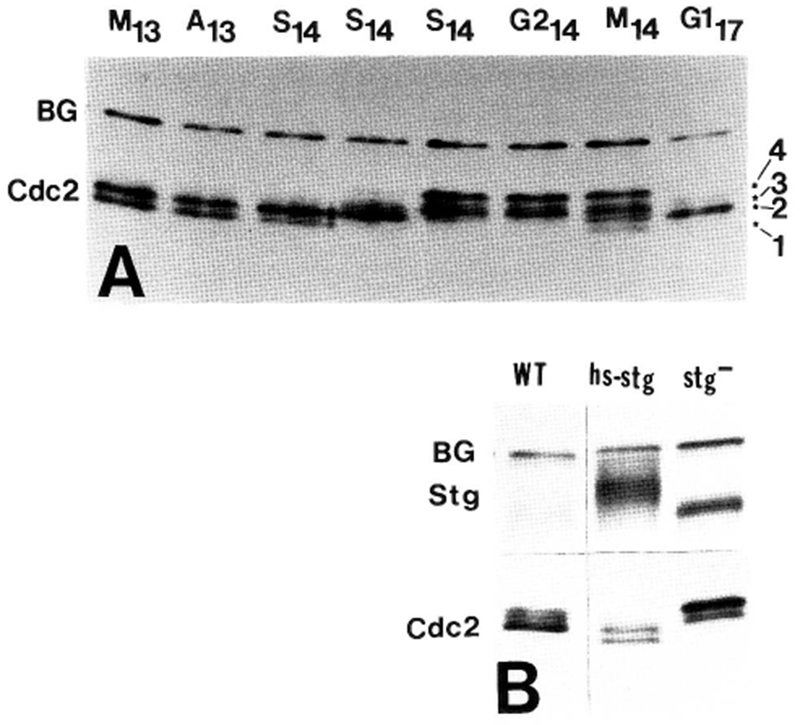
Regulation of Cdc2 by phosphorylation during cycles 13 and 14. (A) Time-course immunoblot of Cdc2 from metaphase and anaphase of cycle 13 (M13, A13) through S phase, G2, and mitosis of cycle 14. Methanol-fixed embryos were staged as described in Fig. 3 and Materials and methods. S phase and G2 time points were ordered by the degree of cellularization (McKnight and Miller 1977; Edgar and Schubiger 1986.) The four phosphoisoforms of Cdc2 can be distinguished in the M14 lane, and are labeled (right, 1–4). Active T161-phospho-Cdc2 (form 1) is the fastest migrating, unphosphorylated Cdc2 is above this (form 2), and the two tyrosine-phosphorylated forms (Y15- and Y15, T14-phospho) are slowest migrating (forms 3 and 4; Fig. 8). Active Cdc2 disappears during early S phase 14, and is replaced by tyrosine-phosphorylated forms. During mitosis 14, active T161-phospho-Cdc2 reappears, and during the first G1 period (G117), Cdc2 is unphosphorylated. (B) Immunoblots of wild-type embryos in G214 (WT), similarly aged embryos (genotype stg7B hs–stg/stg7B hs–stg), which were driven into mitosis 14 by expression of String from the heat shock promotor (hs–stg), and embryos of the genotype stg7B/stg7B arrested in G214 by the string mutation (stg−). Note that induction of String causes tyrosine dephosphorylation Cdc2, leading to increased mobility. (BG) Background bands.
Figure 8.
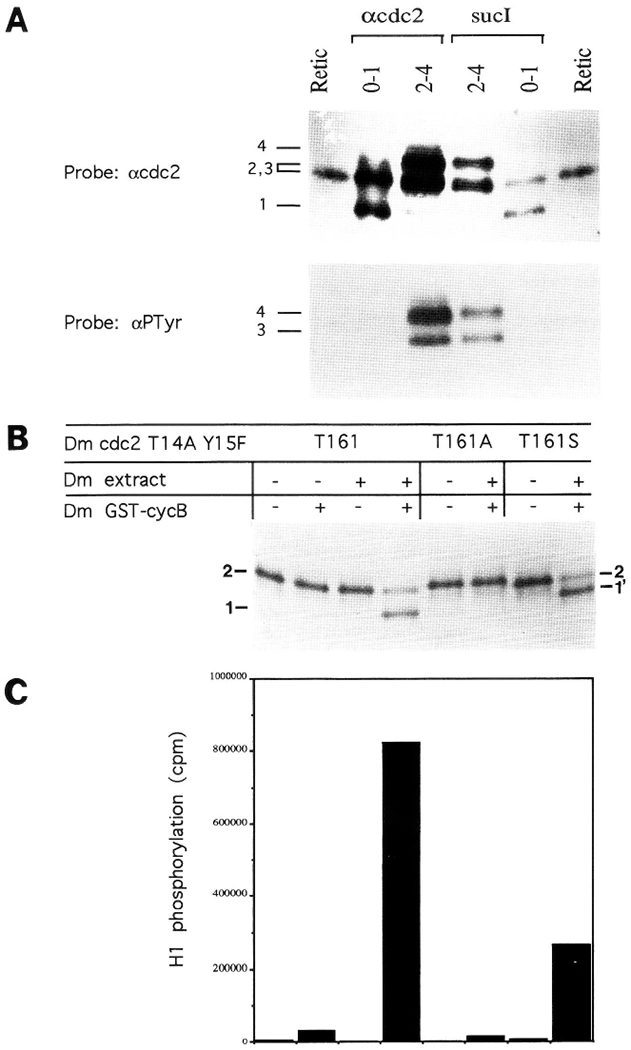
Identification of Cdc2 phosphoisoforms. (A) Tyrosine phosphorylation of Cdc2 results in reduced electrophoretic mobility. Cdc2 was precipitated from 0- to 1-hr (cycles 1–6) or 2- to 4-hr (cycles 13–15) embryos using either anti-Cdc2 antisera or sucl beads (top). The precipitated proteins, along with in vitro-translated Cdc2 (Retie) were immunoblotted with either anti-Cdc2 antibodies (top) or anti-phosphotyrosine antibodies (lower). Note that the two upper bands (forms 3 and 4) in the 2- to 4-hr samples contain phosphotyrosine, and that no phosphotyrosine is found in the two faster migrating Cdc2 isoforms (forms 1 and 2) found in 0- to 1-hr embryos. In this gel, forms 2 and 3 comigrated (see Fig. 2A for separation). Numbers referring to the different Cdc2 forms (1–4) are to the left of the gels. (B) Phosphorylation at T161 increases Cdc2 mobility. HA-tagged, 35S-labeled, in vitro-translated Drosophila Cdc2 containing nonphosphory-latable amino acid substitutions at T14 (A) and Y15 (F) was incubated in the presence (+) or absence (−) of extracts of early Drosophila embryos, and GST–CyclinB (above). The labeled protein was then analyzed by SDS-PAGE. Both Cyclin B and the embryo extract, which contains a T161 kinase activity, are required to generate the downshifted isoform (form 1). When T161 is mutated to the non-phosphorylatable residue alanine (T161A), this downshift does not occur; but when T161 is replaced by phosphorylatable serine (T161S) a somewhat less extreme downshift occurs (1′). (C) Phospho-T161-Cdc2 (form 1) is the active H1 kinase. A fraction of each of the samples shown in B was immunoprecipitated using anti-HA antibodies, and its kinase activity was measured using histone H1 as a substrate. The order of samples in B and C is the same. Only samples containing the down-shifted (form 1) Cdc2 have high kinase activity. See Fig. 6 for the analogous experiment in vivo.
Figure 6.
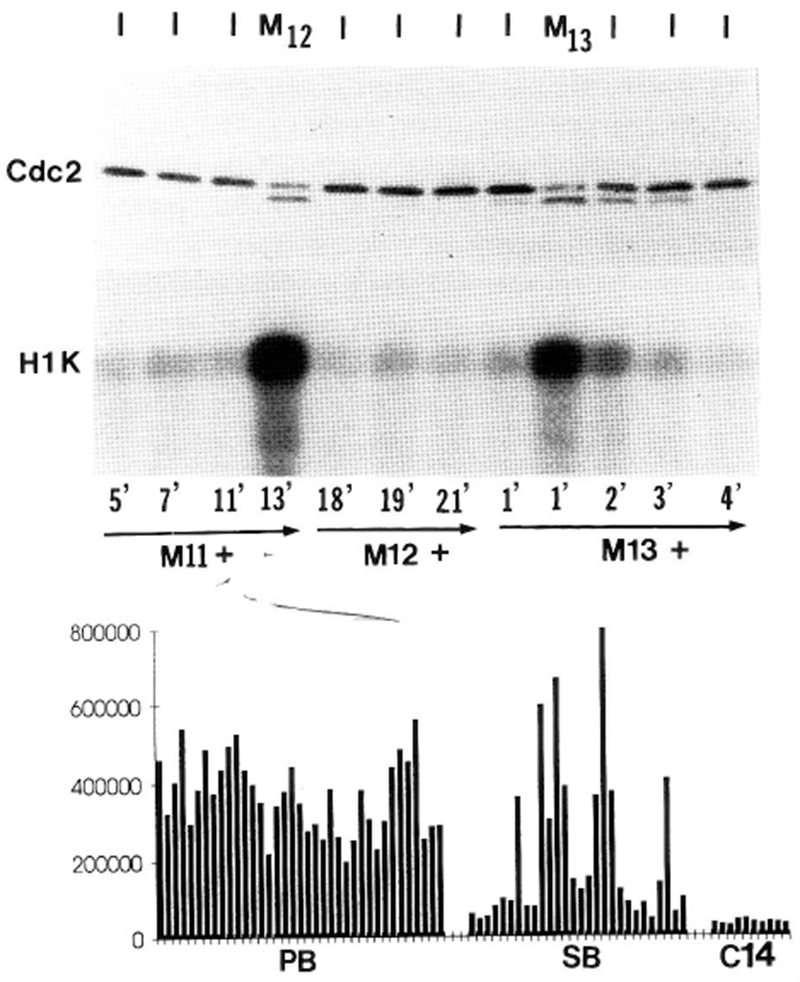
Cdc2 kinase activity oscillates in synchrony with T161-phospho-Cdc2 during the syncytial blastoderm cycles, but does not oscillate during the preblastoderm cycles. (Top) Both the state of Cdc2 and its kinase activity in single embryos are shown from cycles 12, 13, and early 14. We staged individual living embryos visually, keeping track of the minutes elapsed between mitosis and the time of lysis (noted below lower gel). Mitoses were scored by the disappearance of the nuclear envelope using DIC optics, and cycle numbers were determined using nuclear density (Foe and Alberts 1983). The cycle phase of each embryo at the time of lysis is noted along the top (I = interphase, M = mitosis). Half of each embryo lysate was immunoblotted for Cdc2 (upper gel; Cdc2), and the other half was tested for kinase activity after immunoprecipitation with anti-Cdc2 antisera (lower; H1K). Note that high levels of Cdc2-associated H1 kinase activity are confined to those embryos with the faster-migrating form of Cdc2, which is T161 phosphorylated (Fig. 8). This experiment was repeated four times with similar results. (Below) We show the H1 kinase activity of Cdc2 immunoprecipitates of 38 random single preblastoderm embryos (PB; cycles 2–8), 28 random syncytial blastoderm embryos (SB; cycles 10–13), and 10 interphase 14 (C14) embryos. H1 kinase activity was quantified by Phosphorimaging, after SDS–gel electrophoresis and electroblotting. Note that Cdc2 kinase activity fluctuates little in the preblastoderm embryos, fluctuates dramatically in the syncytial blastoderm embryos, and is uniformly low in interphase 14 embryos.
Dephosphorylation of Cdc2 at Y15 does not regulate the early cycles
Unlike the postblastoderm divisions, the first 13 mitoses require only maternal proteins and RNA, and thus cannot be regulated by periodic transcription of string. Although these early divisions are synchronous, they are so rapid (8.6 min each at 25°C) they have been intractable to biochemical analysis. To study these cycles, we developed a method for measuring protein levels and phosphorylation states in individual, precisely staged embryos. Embryos were rapidly fixed in methanol, stained with a DNA dye, selected visually (Fig. 3) and subjected to SDS-PAGE and immunoblotting. Using this method, we found that levels of String protein, which are very low at fertilization, rise during the first 8 cycles and then decline gradually (Figs. 4, 7). The accumulation of String and its gradual disappearance occur without changes in the level of maternal string RNA, which is stable until interphase 14 (Delia Lakich, unpubl.). This suggests translational or post-translational control of String. As anticipated by work in other systems (Gautier et al. 1991; Izumi et al. 1992; Hoffmann et al. 1993), String is phosphorylated, and the degree of phosphorylation, which begins to fluctuate in cycle 5 or 6, peaks at mitosis (Fig. 4). If these fluctuations in String phosphorylation control its activity, and thus influence Cdc2 activity, the level of Y15-phospho-Cdc2 should fluctuate during these early cycles. However, except for trace amounts in interphase 13, we detected no Y15-phospho-Cdc2 until the complete disappearance of String in cycle 14 (Figs. 2, 4, 6, and 8). This suggests that inhibitory phosphorylation of Cdc2 does not regulate the early cycles. We cannot exclude, however, the possibility that significant inhibitory phosphorylation of Cdc2 might occur in an extremely small subcellular compartment within the early embryo.
Figure 3.
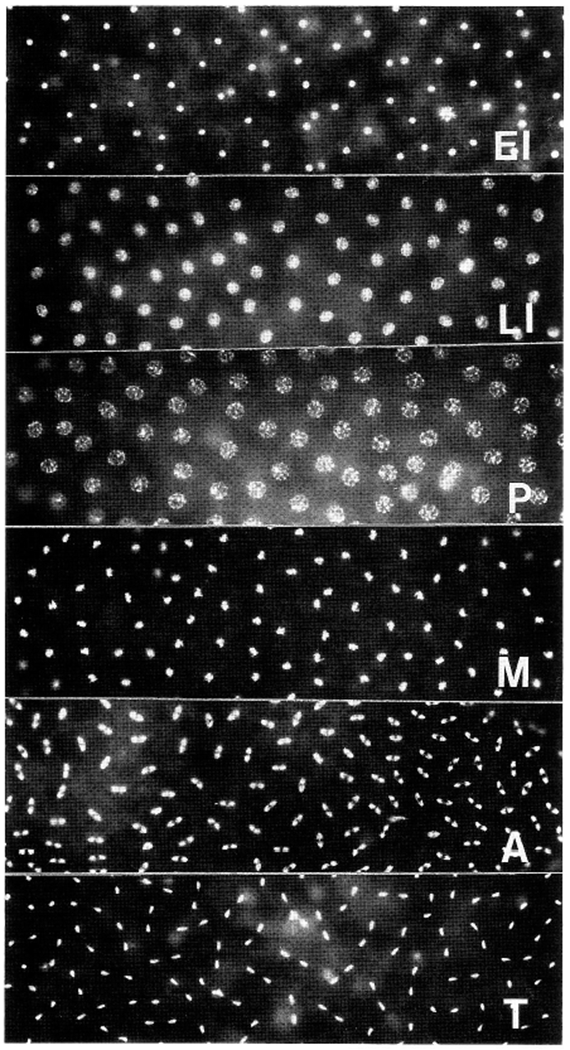
Mitotic synchrony and staging of methanol-fixed embryos. Each panel shows a field that covers ~80% of the surface of a methanol-fixed, Hoecht 33258-stained cycle 10 embryo, such as those used in our immunoblotting experiments. Only the poles of the embryos (which would be out of the focal plane) are not shown; however, nuclei in the poles exhibit the same synchrony as those shown. Between early interphase (EI) and late interphase (LI), nuclei increase their volume several fold. At prophase (P), chromatin condenses onto the nuclear envelope; at metaphase (M), condensed chromosomes congregate to form the metaphase plate. Anaphases (A) and telophases (T) also occur synchronously.
Figure 7.
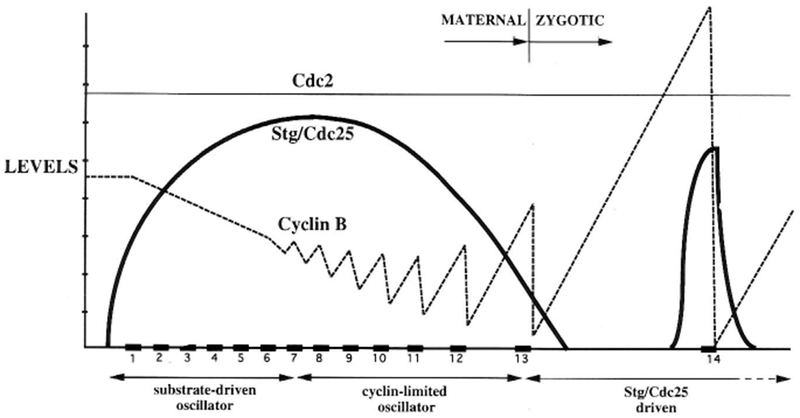
Summary, showing fluctuations in levels of String (bold line), Cyclin B (dashed line), and Cdc2 (fine line) during the first 14 cell cycles. Protein levels are depicted along the y-axis on a linear scale, with arbitrary units. Time is indicated along the x-axis, with periods of mitosis boxed in black and numbered. During cycles 1–7, there is little fluctuation in Cyclin levels or Cdc2 kinase activity. Beginning at cycle 8, Cyclin degradation at metaphase/anaphase transitions becomes apparent, and the cycles begin to slow. At this point Cdc2 kinase activity also begins to fluctuate, due to gain and loss of the activating phosphate at T161. It is note-worthy that greater amounts of Cyclin are degraded with progressively later cycles. Because nuclei and mitotic apparatuses (presumed Cdc2/Cyclin substrates) increase exponentially in number, this suggests a connection between Cyclin utilization and its degradation. Accordingly, we propose that cycle slowdown following mitosis 8 is effected by titration and depletion of maternal Cyclins (Table 1). Mitosis 14 is timed by a distinct mechanism, in which inactive Cdc2/Cyclin complexes are activated by String translated from new zygotic transcripts.
The onset of Cyclin oscillation is gradual
Because the early cycles lack inhibitory phosphorylation of Cdc2, it seemed that Cyclin levels might limit their progress. Immunoblots of precisely staged embryos from cell cycles 2–14 showed a gradual onset of oscillation in Cyclin levels (Fig. 4). Quantitative measurements indicated that <10% of Cyclin A and B was degraded at mitosis 5, and that ~30%, 60%, and 80% was degraded at mitoses 8, 11, and 13, respectively (Fig. 5A).
Figure 5.
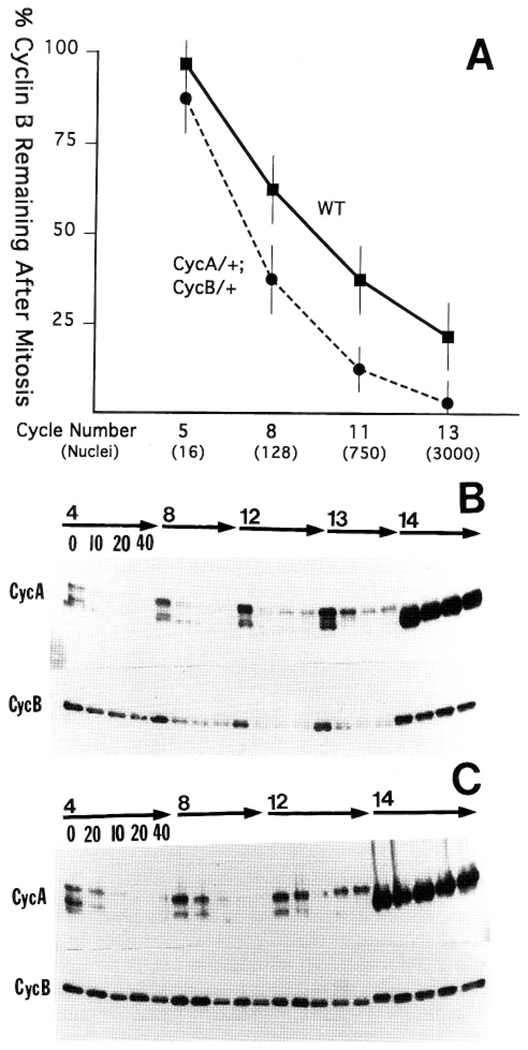
(A) Mitotic degradation of Cyclin B increases as the nuclei proliferate. Cyclin B levels were measured by scanning densitometry of dilution-series immunoblots. Metaphase and anaphase levels for each of cycles 5, 8, 11, and 13 were measured, using samples containing 10 embryos of identical stage. (WT) Wild-type embryos; (CycA/+; CycB/+ ) embryos from mothers heterozygous for a cyclin A mutation (neo114) and a deficiency covering the cyclin B gene (Df(2R)59AD). These embryos receive half the normal dose of cyclin mRNA. Standard deviations were derived from multiple loadings and exposures of a sample, and levels were normalized to Cdc2, a stable protein. (B) Cyclin turnover in embryos arrested in interphase by cycloheximide. Permeabilized embryos were treated with 20 μg/ml of cycloheximide for 0, 10, 20, and 40 min and then methanol-fixed. Three embryos arrested in interphase of each of cycles 4, 8, 12, 13, and 14 were selected, pooled, and subjected to immunoblot analysis. Numbers above the arrows indicate the cycle of arrest, and numbers below the arrows indicate minutes of treatment with cycloheximide (these follow the same order for each cycle displayed). Note that Cyclin A (CycA) is degraded rapidly and completely after arrest during the earlier cycles and that degradation becomes less complete in later cycles. In contrast, a greater proportion of Cyclin B (CycB) is degraded with progressively later cycles. Both Cyclins are stable during interphase 14. The 0 min time points represent late interphase embryos that were not treated with cycloheximide. (C) Cyclin stability in embryos arrested in metaphase by colcemid. Permeabilized embryos were first treated with 20 μg/ml of colcemid for 20 min. This depolymerizes microtubules and results in metaphase arrest. Following the 20-min colcemid treatment, embryos were treated with 20 μg/ml of cycloheximide and 20 μg/ml of colcemid for 10, 20, or 40 min to assess the stability of the Cyclins during metaphase arrest. The embryos were then methanol-fixed, and three embryos arrested in metaphases 4, 8, 12, and interphase 14 from each time point were selected, pooled, and subjected to immunoblot analysis. Note that Cyclin A is unstable during metaphase arrest, whereas Cyclin B is essentially stable. 0-Min time points represent untreated metaphase embryos (except for the cycle 14 time points, which are all interphase embryos).
To assess Cyclin turnover, embryos were treated with cycloheximide and then processed for immunoblotting with Cyclin antibodies (Fig. 5B). Cycloheximide treatment of rapidly cycling embryos is usually followed by one mitosis prior to interphase arrest. Cyclin A appeared to be unstable throughout the early cycles (4, 8, 12, and 13), because it dropped to low levels within 10 min of cycloheximide treatment. This may explain the protein synthesis requirement in the preblastoderm embryo. In contrast, Cyclin B was largely stable after cycloheximide arrest in cycle 4, but progressively greater fractions were degraded after arrest in cycles 8, 12, and 13. The bulk of Cyclin degradation occurred within the first 10 min after cycloheximide treatment, and thus probably occurred in the final metaphase/anaphase transition before arrest. Both Cyclins A and B were stable during interphase 14, consistent with mitotic instability.
Coupling of Cyclin degradation to the metaphase/anaphase transition was tested by blocking embryos in metaphase with the microtubule inhibitor colcemid and then treating them with cycloheximide. Cyclin B levels remained virtually constant after addition of cycloheximide to metaphase-arrested embryos (Fig. 5C). Apparently, the mitotic block stabilizes Cyclin B. In contrast, Cyclin A was not stabilized: Cyclin A levels fell rapidly after addition of cycloheximide to metaphase-arrested embryos (Fig. 5C). Similar selective stabilization of Cyclin B during metaphase arrest has been noted in a variety cells, including Drosophila neuroblasts (Whitfield et al. 1990). In summary, Cyclin B degradation appears to be coupled to mitosis, and progressively more Cyclin is destroyed in successive cycles. We suggest (see Discussion) that coupling of Cyclin B degradation to the exponential increase in nuclei is responsible for this progressive change. In contrast, the instability of Cyclin A correlates with the presence of active Cdc2 kinase.
Cyclin synthesis is limiting for mitoses 10–13
Cycles 2–9 average 8.6 min each, whereas cycles 11, 12, and 13 last 10, 13, and 18 min, respectively (Foe and Alberts 1983). This lengthening of the cell cycle might result from increasing Cyclin degradation, which would cause each successive cycle to require more time to reaccumulate Cyclins to a level adequate to trigger mitosis. To test this possibility, we timed the mitotic cycles in embryos from mothers heterozygous for a mutation in the cyclin A gene and/or a small deletion covering the cyclin B gene (Table 1). Such embryos receive half the normal dose of cyclin mRNA during oogenesis, and have a reduced capacity to synthesize Cyclin proteins. Western analysis confirmed that these embryos had reduced Cyclin levels (not shown). Furthermore, the proportion of Cyclin degraded was higher in Cyclin-deficient embryos than in comparably staged normal embryos (Fig. 5A). Importantly, Cyclin deficient embryos also had altered cell cycle times. Although careful timing of embryos between egg deposition and cycle 10 failed to detect delays, progress from cycle 10 to mitosis 13 was slightly delayed in Cyclin B-deficient embryos, and severely delayed in Cyclin A/B doubly deficient embryos, which took 30% longer to complete these cycles. Interestingly, 10% of these embryos did not even initiate mitosis 13; this might result from reducing Cyclin synthetic capacity below the level required to complete 13 divisions before maternal String is degraded. These experiments indicate that accumulation of Cyclins is rate-limiting for progress of cycles 10–13, and suggest that the gradually increasing degradation of Cyclins causes the gradual lengthening of these cycles.
Table 1.
Division timing in cyclin-deficient embryos
| Maternal genotype | Egg deposition to cycle 10 (min) |
Cycle 10 to mitosis 13 (min) |
Number |
|---|---|---|---|
| +/+ | 103 ± 11 | 47 ± 3 | 58 |
| stg−/+ | 112 ± 6 | 43 ± 3 | 14 |
| cycA−/+ | 107 ± 7 | 45 ± 3 | 19 |
| cycB−/+ | 105 ± 7 | 52 ± 4 | 16 |
| cycA−/+; cycB−/+ | 102 ± 10 | 62 ± 5 | 60/54a |
6/60 embryos derived from cycA−/+; cycB−/+ mothers failed to initiate mitosis 13 altogether. These embryos cellularized and gastrulated during an extended interphase 13.
The first seven cycles occur without Cyclin or Cdc2 oscillation
Although we cannot be sure exactly when Cyclins begin to limit cell cycle progression, Cyclin accumulation does not appear to time the earliest cycles. We detected no oscillation in Cyclin levels during cycles 2–7 (Fig. 4A), and reductions in Cyclin levels did not detectably prolong these cycles (Table 1). In an effort to define what might regulate these cycles, we examined other cell cycle components, and found that all oscillatory behaviors paralleled those of the Cyclins. For example, whereas phosphorylation of String and Cyclin A oscillates in cycles 11–13, both proteins retain high levels of phosphorylation through all phases of cycles 2–7 (Fig. 4A). Of particular importance, phosphorylation of Cdc2 at T161, an essential activating modification, is constitutive during cycles 2–7 (Fig. 4A). Reproducibly detectable decreases in T161 phosphorylation begin in interphase 10 or 11, and progressively greater oscillations occur in successive interphases, such that by cycle 13 the active (T161-phospho) form of Cdc2 is undetectable during much of interphase (Fig. 4B).
To directly assess the activity of Cdc2, we immunoprecipitated Cdc2 from single embryos and measured its kinase activity using histone H1 as a substrate. During cycles 11–14, Cdc2 kinase activity peaked at mitoses and decreased at least 10-fold during interphases, following oscillations in Cyclins and T161-phospho-Cdc2 (Figure 6; see also Fenton and Glover 1993). Because of the difficulty in staging live embryos prior to cycle 9, we used a statistical approach for the earliest cycles. We assayed 108 individual preblastoderm embryos (cycles 1–9) for Cdc2 kinase activity, and found that 103/108 of these embryos had kinase activity at least four times greater than the average interphase level in cycles 11–14 (see Fig. 6). Immunoblots done in parallel indicated that the few embryos that had reduced kinase activity also had reduced levels of Cyclins A and B. Based on our immunoblot analysis of staged embryos (Fig. 4), these particular embryos arc likely to be cycle 8 or 9 embryos. In a tabulation of mitotic stages from a population of methanol-fixed, cycle 2–7 embryos, we found 45% in various stages of interphase, 10% in prophase or prometaphase, 28% in metaphase, and 17% in anaphase and telophase (n = 146). According to accepted models ~60% of these embryos (those in interphase, anaphase, and telophase) should exhibit low Cdc2 kinase activity. Because we find high kinase activity in 95% of embryos during these cycles, our results imply that Cdc2 remains active through all phases of these cycles. We suggest that cycles 2–7 are timed by a mechanism unlike any of those described previously.
Discussion
During Drosophila development, changing levels of cell cycle regulators alter the rate-limiting step and, thus, the mechanism, governing progress of the cell cycle. Here we define three phases in the developmental progression of cell cycle regulation (Fig. 7). In contrast to other known cycles, the first seven cycles in Drosophila run in the presence of constitutively active Cdc2 kinase. Later, during cycles 8–13, increasing mitotic destruction of Cyclins drives oscillations in Cdc2 activity and makes Cyclin accumulation rate-limiting for mitosis. Degradation of maternally supplied String (Cdc25), a Cdc2-activating phosphatase, causes tyrosine dephosphorylation of Cdc2 to become rate-limiting for mitosis beginning in cycle 14.
String-limited cycles (14–16)
Our understanding of the postblastoderm divisions is the most thorough. These cycles are regulated at the G2/M transition by new transcription of a single gene, string, whose product is both required and sufficient to trigger mitosis (Edgar and O’Farrell 1989, 1990). An intricate spatiotemporal pattern of string transcription produces pulses of RNA that anticipate mitosis in every embryonic cell (Edgar and O’Farrell 1989, and unpubl.). Immunoblots (Fig. 2) and immunohistochemical staining of embryos (Fig. 1) show that these episodes of string mRNA production are associated with transient accumulation of String protein. String, a functional homolog of Cdc25 of S. pombe, acts as a specific Cdc2-activating phosphatase in vitro (Dunphy and Kumagai 1991; Gautier et al. 1991). We find that tyrosine phosphorylated Cdc2 accumulates during interphase 14, after degradation of maternal String, and that Cdc2 is dephosphorylated upon zygotic production of String (Fig. 2). Thus, String appears to act in vivo just as it does in vitro.
Cyclin-limited cycles (~8–13)
Unlike the postblastoderm cycles, cycles 1–13 do not require zygotic transcription (O’Farrell et al. 1989) and therefore cannot be controlled as the later cycles are. The early cycles do require translation, and we initially suspected that they might operate as proposed for the translation-dependent divisions in early Xenopus and marine invertebrate embryos. The key features of this proposal are that mitosis is driven by accumulation of Cyclins to a threshold required to activate Cdc2, and that mitotic degradation of Cyclins inactivates Cdc2 and allows mitotic exit (Murray and Kirschner 1989; Murray et al. 1989; Hunt et al. 1992). These features of the regulatory mechanism are relevant to Drosophila, but apply during only a subset of the early divisions. We find that progressively increasing Cyclin oscillations begin to drive detectable oscillations in Cdc2 activity at about cycle 9 or 10. To test whether, and when, reaccumulation of Cyclins limits mitosis, we timed cycle progression in mutant embryos with reduced levels of maternal Cyclin RNAs and found that transit through cycles 10–14 was prolonged when both Cyclins A and B were reduced (Table 1). This is the first in vivo demonstration that Cyclin accumulation times an embryonic cell cycle. We emphasize, however, that Cyclin accumulation does not time later cycles (14–16) or earlier ones (~1–9).
The Cyclin-dependent cycles we describe differ from those of Xenopus egg extracts in one significant respect. In both systems, Cyclin accumulation is gradual, whereas Cdc2 kinase activation is abrupt. In Xenopus extracts, Cdc2 activation is delayed by inhibitory phosphorylation at Y15, and positive feedback that stimulates dephosphorylation of Cdc2 is thought to produce the abrupt activation (Solomon et al. 1990; Izumi et al. 1992; Smythe and Newport 1992; Hoffmann et al. 1993). In Drosophila embryos however, we detected no inhibitory phosphorylation of Cdc2 during the early cycles, and thus there must a different explanation for this phenomenon. A single report of observations made in vivo in Xenopus suggests that early cycles in the frog may also lack inhibitory phosphorylation of Cdc2 and thus may actually be similar to the early cycles in Drosophila (Ferrell et al. 1991).
Cycles lacking oscillation in Cdc2 activity (~2–1)
Perhaps our most striking observation is that the earliest cell cycles occur without pronounced oscillations in Cyclins, Cdc2 phosphorylation, or Cdc2 activity. Although we cannot eliminate the possibility of slight oscillations in Cdc2 activity, these would have to be within the variation of our measurements (~2×). The persistence of Cyclins and the active T161 phosphorylated form of Cdc2 through interphases gives us considerable confidence that there is negligible cycling of Cdc2 kinase activity during the early cycles. Consistent with this, we found that antigens recognized by the MPM-2 antibody begin oscillations only after cycle 10 (data not shown). MPM-2 recognizes a variety of phosphopeptide antigens that appear during mitosis, and are correlated with high Cdc2 or MPF activity (Davis et al. 1983). Moreover, because maternal Cdc2 is required for the early division cycles (Stem et al. 1993), it is unlikely that these cycles are controlled by oscillations of an unknown regulator that bypasses the Cdc2 kinase.
How do the early cycles ran with constitutively active Cdc2? The observation that Cyclin degradation at mitosis increases gradually suggests that a special pool of Cyclin, which expands with the proliferation of nuclei, is subject to degradation (Figs. 4 and 5A). We infer that during the earliest cycles mitotic Cyclin degradation probably occurs, but at undetectable levels. What is the nature of the degradable Cyclin pool? In situ immunostaming has failed to visualize local Cyclin degradation in early embryos, suggesting that degradable Cyclin may be confined to a very small subcellular compartment (Maldonado-Codina and Glover 1992; B.A. Edgar, unpubl). We propose that only Cyclin associated with nuclear and/or spindle substrates is targeted for degradation. In support of this idea, Cyclin B associates with spindles in many cell types, including the early Drosophila embryo (Alfa et al. 1990; Leiss et al. 1992; Maldonado-Codina and Glover 1992; Verde et al. 1992; Kellogg 1993). Moreover, spindle disruption by microtubule depolymerizing agents blocks Cyclin B degradation in most systems, indicating that spindles are required for degradation (Whitfield et al. 1990; Schimke et al. 1991; Hunt et al. 1992; Fig. 5C). Because spindles accumulate with successive cycles, spindle-dependent degradation could explain the progressive increase in Cyclin B degradation that we observe. The fact that this increase is not exponential may reflect reductions in spindle size with later cycles (Karr and Alberts 1986). Unlike Cyclin B, Cyclin A turns over rapidly in even the earliest embryos (Fig. 5B,C), and thus its behavior is inconsistent with substrate-dependent degradation. Because Cyclin A appears to be unstable whenever active Cdc2 is present, oscillations in Cyclin A may result from its periodic stabilization during the later interphases, when Cdc2 is tran-siently inactivated. In this case, substrate-driven oscillations in Cyclin B would effect Cyclin A oscillations.
A model in which Cyclin B degradation is substrate-dependent has interesting implications. It assumes that even when there is no detectable degradation (cycles 2–7), the functionally relevant Cyclin (that which is substrate-associated) is in fact degraded at mitosis. This might allow transient dephosphorylation of substrates in the spindle and nucleus, causing return to the interphase configuration despite high Cdc2 activity in the cytoplasm. In this instance, mitotic timing during cycles 2–7 would depend not upon oscillating Cdc2 activity, but upon the kinetics of substrate response; how long it takes, for instance, to assemble and disassemble the mitotic spindle and the nuclear envelope. Such a mechanism would allow each individual mitotic apparatus (spindle and nucleus) to cycle autonomously during the earliest cycles, when Cdc2 is continuously active. Interestingly, nuclei injected into preblastoderm embryos do cycle out of phase with surrounding host nuclei until about cycle 9, when they fall into synchrony with host nuclei (Okada et al. 1980). This is exactly what we would expect if mitoses are timed by mechanisms intrinsic to each mitotic apparatus during the preblastoderm cycles, but become entrained with each other by oscillations in cytoplasmic Cyclin/Cdc2 at about cycle 9.
Finally, we note that in the mechanism we propose, oscillations of the enzymatic regulators of the cycle (Cyclins and Cdc2) are driven by, rather than driving, oscillations of the mitotic apparatus. This makes the mechanism inherently self-limiting, since increases in Cyclin B substrates would cause ever greater depletion of the Cyclins at mitosis, and this would cause the cycle to slow. Titration and depletion of Cyclins by the proliferating mitotic apparati provides an attractive explanation of results showing that slowing of the maternally controlled divisions is caused by increases in the nucleo: cytoplasmic ratio not only in Drosophila, but in embryos of diverse species (Koboyakawa and Kubota 1981; Newport and Kirschner 1982; Mita and Obata 1984; Edgar et al. 1986; Yasuda and Schubiger 1992). The transition to a Cyclin-limited cell cycle, followed by destruction of maternal String and transition to a String-limited cycle, are major developmental events that are coupled with the onset of transcription, and initiation of zygotically programmed morphogenesis, respectively.
Materials and Methods
Antibodies
Rabbit anti-Cdc2 antisera was raised against a gel-purified, full-length T7–DmCdc2 fusion-protein produced in Escherichia coli, by Cocalico, Inc. The sera were column affinity purified using gel-purified β-galactosidase–Dm Cdc2 fusion protein cross-linked to Affigel matrix. Immunoblots to in vitro-produced DmCdc2 and DmCdc2c showed that our anti-Cdc2 antibodies did not cross-react substantially with DmCdc2c, a highly homologous protein that is a stmctural but not a functional homolog of S. pombe Cdc2+ and S. cerevisiae CDC28 (Lehner and O’Farrell 1990b). Anti-String antibodies (AP4), were produced and affinity purified in a similar fashion, using the services of the Berkeley Antibody Company. Tests using transcription-null string mutant embryos (stgAR2/stgAR2) confirmed the identity of String protein on immunoblots and indicated that the other prominent band recognized by the antibody was not String-related. Affinity purified, rabbit anti-Cyclin A antibodies were the gift of Christian Lehner (Lehner and O’Farrell 1989). We used hybridoma supernatants of a mouse monoclonal antibody (F2F4) for detection of Cyclin B (Knoblich and Lehner 1993).
Precipitation of Cdc2 from Drosophila embryos
Preparation of Drosophila embryo extracts and immunoprecipitation was performed as described (Sprenger et al. 1992). p13suc1 beads were prepared as described (Solomon et al. 1990).
In vitro activation of Cdc2
Drosophila extract was prepared by resuspending 0.1 gram of embryos (0–1 hr at 25°C) in 1 ml of EB (extraction buffer). EB contained 10 mm Tris at pH 7.5, 80 mm Naβ-glycerophosphate at pH 7.5, 20 mm EGTA, 15 mm MgCl2, 2 mm Na3VO4, 1 mm benzamidine, 1 mm sodium meta-bisulfite, and 0.2 mm PMSF. Embryos were then subjected to six strokes in a ground-glass Dounce homogenizer (Wheaton) and incubated on ice for 15 min. Insoluble material was removed by centrifugation at 4°C for 10 min at 10,000g. Drosophila Cyclin B was kindly provided by Doug Kellogg (University of California, San Francisco) as a bacterially expressed fusion protein (GST–CycB) containing glutathione-S-transferase at the amino terminus. The following Cdc2 containing plasmids were used for the expression of Cdc2 proteins in vitro: pSF259 (HA cdc2 T14A Y15F), pSF292 (HA cdc2 T14A Y15F T161A), and pSF287 (HA cdc2 T14A Y15F T161S). For the construction of these plasmids, the coding region of the Dmcdc2 gene (Lehner and O’Farrell 1990b) was inserted into the vector pSPB4, a pSP64 derivative containing the Xenopus β-globin leader fragment (Siegel and Walter 1988). In vitro mutagenesis using oligonucleotides and the polymerase chain reaction were performed to introduce the indicated mutations at the codons for threonine 14 (T14A), tyrosine 15 (Y15F) and threonine 161 (T161A and T161S). All of these plasmids also code for Cdc2 proteins that contain at the carboxy terminus the influenza hemagglutinin (HA) epitope GRPPYPYDVPDYASLG. This peptide epitope is recognized by a monoclonal antibody 12CA5 (Field et al. 1988). These plasmids were linearized with ApaL1, extracted with phenol, precipitated and dissolved in TE and RNA was transcribed in vitro as described by Driever et al. (1990). For in vitro translation, 100 μl of reticulocyte lysate (Promega) containing 0.05 mm amino acids (minus methionine), 40 units RNase inhibitor (Boehringer Mannheim), 5 μl [35S]methionine (Expre35s35s protein labeling mix, NEN), and 5 μg of in vitro-synthesized RNA were incubated for 1 hr at 37°C. To obtain activation of Cdc2 in vitro, 20 μl of Cdc2 expressing reticulocyte lysate was mixed with 20 μl of Drosophila extract and incubated in the presence of 0.3 mg/ml of GST–CycB, 2 mm ATP and 15 mm MgCl2 for 30 min at room temperature. Immunoprecipitation of the reticulocyte expressed HA-tagged Cdc2 proteins were performed by diluting with 1 ml of EB buffer and incubating the lysate with mAb 12CA5 for 2 hr at 4°C. Protein A–Sepharose beads were used to collect the antigen–antibody complexes. Immunoprecipitates were analyzed by SDS-PAGE and autoradiography of the dried gel. To measure Cdc2 kinase activity, an H1–kinase assay was performed on a fraction of the immunoprecipitate.
Phosphoisoforms of Cdc2
Four phosphoisoforms of Cdc2 were identified as described below and in Figure 8. These forms are denoted 1–4, in order of decreasing electrophoretic mobility on SDS gels.
Form 1
Cdc2 had a greater electrophoretic mobility than unmodified, in vitro-produced Cdc2. It was not recognized by anti-phosphotyrosine antibodies (Fig. 8A). In coimmunoprecipitation assays from Drosophila embryo extracts using anti-Cyclin antibodies, it was associated with Cyclins A and B (not shown). Form 1 Cdc2 was positively correlated with high levels of Cdc2-associated H1 kinase activity in vitro and in vivo (Figs. 6 and 8). Work on Xenopus, starfish, goldfish, and clam Cdc2 (Luca et al. 1991; Yamashita et al. 1991; Clarke et al. 1992; Lorca et al. 1992) and human Cdk2 (Gu et al. 1992) has described a similar fast-migrating isoform that is phosphorylated on a highly conserved threonine that is at position 161 in the Drosophila DmCdc2 protein. Phosphorylation of this threonine is required for Cdc2 kinase activation at mitosis in a number of species (Gould et al. 1991; Norbury et al. 1991; Solomon et al. 1990). We found that in vitro-produced Cdc2, which comigrated with form 2, could be converted to form 1 by the addition of bacterially produced Cyclin B and extracts of Drosophila embryos (Fig. 8B). This conversion did not occur on Cdc2 in which threonine 161 was replaced by alanine (T161A), which cannot be phosphorylated. The T161A mutation abolished the H1 kinase activity of Cdc2 (Fig. 8C). When T161 was replaced by serine (T161S), Cdc2 could be phosphorylated and a form with greater electrophoretic mobility than unmodified Cdc2 was observed (Fig. 8B). However, this mobility shift was distinct from that of normal form 1 Cdc2, and the kinase activity of the mutant T161S–Cdc2 was diminished. All of these data indicate that the increase in electrophoretic mobility of form 1 Cdc2 is due to phosphorylation at T161, a modification required for Cdc2 kinase activity.
Form 2
Cdc2 had the same electrophoretic mobility as in vitro-translated DmCdc2. Its mobility was not affected by added phosphatases, and it was not recognized by anti-phosphotyrosine antibodies (Fig. 8A). Unlike forms 1, 3, and 4, form 2 Cdc2 was not associated with Cyclin A or B in coimmunoprecipitation assays. Form 2 is thus most likely unmodified Cdc2.
Forms 3 and 4
The two slowest migrating isoforms, forms 3 and 4, were both recognized by anti-phosphotyrosine antibodies (Fig. 8A). Both were also associated with Cyclins A and B in coimmunoprecipitation assays. Forms 3 and 4 Cdc2 from embryos could be converted into forms 1 and 2 by incubation with either alkaline or acid phosphatase, or bacterially produced String protein (not shown). In vivo, forms 3 and 4 accumulated in embryos arrested in interphase by mutations in string (Fig. 2B). When String was expressed in these interphase-arrested embryos, they rapidly entered mitosis (Edgar and O’Farrell 1990), and the accumulated form 3 and 4 Cdc2 was rapidly replaced by forms 1 and 2 (Fig. 2B). In other systems Cdc2 kinase is subject to inhibitory phosphorylation on tyrosine 15 (Y15), which can be counteracted by String/Cdc25 phosphatases. Our results indicate that forms 3 and 4 must correspond to phospho-Y15 Cdc2. By analogy with other systems, form 4 Cdc2 probably differs from form 3 in having an additional inhibitory phosphate at T14 .
Embryo staging and immunoblots
Embryos were dechorionated in 50% bleach for 2 min, rinsed well, and rapidly devitellinized and fixed with a two-phase mixture of 99% methanol, 1 mm Na3VO4, and heptane for 5 min with gentle mixing. They were then rinsed three times in ice-cold 99% methanol, 1 mm Na3VO4, and twice in EB containing 0.05% Tween 20. In some experiments a similar buffer containing 10 mm EDTA instead of 15 mm MgCl2 was used, with similar results. The embryos were then stained for DNA with 4 μg/ml of bisbenzamid in EB for 5 min, rinsed twice in EB, transferred to 40% EB/60% glycerol, and allowed to sit on ice for 1 hr to clear. Fixed embryos could be stored in this solution at −80°C without deterioration. Embryos were staged and selected visually using UV illumination on a compound microscope with 6.3 and 20× objectives, and a tungsten needle and pipetman for manipulation. The various mitotic phases (prophase, metaphase, anaphase, telophase) could be easily distinguished by nuclear morphology (Fig. 3), and cycle numbers were determined unambiguously by the number of nuclei present (2 = cycle 2, 4 = cycle 3, 8 = cycle 4, etc.) and their position in the egg (Zalokar and Erk 1976; Foe and Alberts 1983). Embryos with mitotic asynchronies or pronounced mitotic waves (Foe and Alberts 1983) were not selected. Handpicked embryos (generally 1–10 per sample) were dissolved in 50% EB, 50% 4× SDS sample buffer, mixed well, boiled for 5 min, and subjected to SDS-PAGE followed by immunoblotting. We used 22-cm, 10.5% polyacrylamide gels (29:1) containing 1 mm Na3VO4. Cdc2 protein was run ~20 cm. For transfers we used an LKB semidry electroblotter, and Immobilon P membranes (Millipore). Blots were probed according to standard protocols using 3% milk as a blocking agent, HRP-conjugated secondary antibodies, and ECL chemiluminescent detection (Amersham). Filters were stripped for reprobing by incubation in 50 mm Tris at pH 7.5, 2% SDS, 100 mm β-mercaptoethanol at 50°C for 30 min.
H1 kinase assays
For single embryo H1 kinase assays, live embryos were dechorionated with 50% bleach, visually staged (Foe and Alberts 1983), lysed by crushing with a glass rod in a 0.75 ml Eppendorf tube, and homogenized in 15 μl of EB containing 2.5 mm β-mercaptoethanol and 0.1% NP-40 instead of Tween 20. Lysate (9 μl) was then removed for analysis by immunoblotting. EB (50 μl) and crude anti-Cdc2 antisera (2 μ1) were added to the 6 μl of lysate remaining, and the mixture was incubated on ice for 1 hr. EB (250 μl) and protein A–Sepharose beads (10 μl) were then added and the mixture was rocked at 4°C for 1 hr. The beads were washed four times with 0.5 ml of EB, drained briefly, and assayed for H1 kinase activity by the addition of 10 μl of H1 kinase buffer (Solomon et al. 1990) containing 250 μg/ml of H1, 100 μm ATP, and 0.5 mCi/ml of [γ-32P]ATP. After 20 min of incubation at room temperature, 8 μl of 4× SDS sample buffer was added, and the samples were boiled for 5 min and run on 12.5% SDS-PAGE. The amount of antisera used was the minimum required for maximum recovery of H1 kinase activity. The Cdc2-associated H1 kinase activity recovered by this assay constituted ~5–10% of the total embryonic H1K kinase. H1 kinase activity from total embryo lysates showed no cell cycle-specific oscillation.
Phosphatase assays
Visually staged, living embryos (5–10) were crushed with a glass rod, homogenized in phosphatase buffer, and incubated for 40 min at 37°C. The samples were then analyzed by SDS-PAGE as described above. Potato Acid phosphatase (Boehringer Mannheim) was used at 0.2 mg/ml in 10 mm PIPES at pH 6.1, 2.5 mm MgCl2, 0.05% NP-40, 100 mm NaCl, 1 mm PMSF, 2 μg/ml of aprotinin, and 1 μg/ml of leupeptin. Calf alkaline phosphatase was used in the buffer supplied (Boehringer Mannheim). Control incubations contained 3 mm Na3VO4 in addition to phosphatases.
Inhibitor experiments
Colcemid and/or cycloheximide (Sigma) were delivered by permeabilization with octane, essentially as described (Edgar and O’Farrell 1990). Both were used at 20 μg/ml in Schneider’s medium. Permeabilized embryos were incubated for various periods, and then methanol-fixed and sorted as described above.
Timing of mutant embryos
To time the early divisions in embryos from mutant mothers, we collected eggs for 15 min, dechorionated them with 50% bleach, and observed them under halocarbon oil using 10× and 20× objectives and DIC optics. Typically, 20 wild-type and 20 mutant embryos were followed simultaneously, and the stage of each embryo was noted every 4–6 min. Temperatures ranged from 23.5° to 24.5°C. +/+ flies were of the Sevelin strain. Heterozygous mutant females of the genotypes cycAneo114/TM3 Sb, Df(2R)59AD/CyO, and stg7B/TM3 Sb were crossed to males of the same genotypes. Females of the genotype cycAneo114/+; Df(2R)59AD/+ were crossed to +/+ Sevelin males. We also did several experiments with the cyclin B deficiency Df(2R)59AB and found cell-cycle delays similar to those associated with the cyclin B deficiency Df(2R)59AD. We noted, interestingly, that Cyclin A,B-deficient embryos completed nuclear migration one cycle early. Posterior pole buds formed during cycle 8 in these mutants, and somatic nuclei reached the cortex at cycle 9. Our observations also confirm the synergistic action of Cyclins A and B, noted previously in studies of the zygotic phenotypes of cyclin mutants (Knoblich and Lehner 1993).
Acknowledgments
We thank Christian Lehner for Cdc2 protein, anti-Cyclin A antibodies, and the cyclin B mutant flies, and Deborah Morrison for the anti-phosphotyrosine antibody. Doug Kellogg suggested a key technique: Western analysis of methanol-fixed embryos, and also provided us with a gift of GST–DmCycB. Garry Odell and Tabitha Doniach provided stimulating ideas concerning mitotic oscillators, and Shelagh Campbell helped with a critical experiment. We thank Jim Roberts, Linda Breeden, and Christian Lehner for comments on the manuscript. B.A.E. is a Lucille P. Markey Scholar, and this work was supported in part by a grant from the Lucille P. Markey Charitable Trust. This work was also supported by the European Molecular Biology Laboratory and the Human Frontiers in Science Program (to F.S.), the Cancer Research Fund of the Damon Runyon–Walter Winchell Foundation (DRG-1161) to R.J.D., and the National Institutes of Health (R01-GM57193) to P.O’F.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
References
- Alfa CE, Ducommun B, Beach D, and Hyams JS. 1990. Distinct nuclear and spindle pole body population of cyclin-cdc2 in fission yeast. Nature 347: 680–682. [DOI] [PubMed] [Google Scholar]
- Amon A, Surana U, Muroff I, and Nasmyth K. 1992. Regulation of p34CDC28 tyrosine phosphorylation is not required for entry into mitosis in S. cerevisiae. Nature 355: 368–371. [DOI] [PubMed] [Google Scholar]
- Booher R and Beach D. 1988. Involvement of cdc13+ in mitotic control in Schizosaccharomyces pombe; Possible interaction of the gene product with microtubules. EMBO J. 6: 3441–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PR, Leiss D, Pagano M, and Karsenti E. 1992. Cyclin A- and Cyclin B-dependent kinases are regulated by different mechanisms in Xenopus egg extracts. EMBO J. 11: 1751–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis FM, Tsao TY, Fowler SK, and Roa PN. 1983. Monoclonal antibodies to mitotic cells. Proc. Nat. Acad. Sci 80: 2926–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driever W, Siegel V, and Nusslein-Volhard C. 1990. Autonomous determination of anterior structures in the early Drosophila embryo by the bicoid morphogen. Development 109:811–820. [DOI] [PubMed] [Google Scholar]
- Dunphy WG and Kumagai A. 1991. The Cdc25 protein contains an intrinsic phosphatase activity. Cell 67: 189–196. [DOI] [PubMed] [Google Scholar]
- Edgar BA and O’Farrell PH. 1989. Genetic control of cell division patterns in the Drosophila embryo. Cell 57: 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar BA and O’Farrell PH. 1990. The three postblastoderm cell cycles of Drosophila embryogenesis are regulated in G2 by String. Cell 62: 469–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar BA and Schubiger G. 1986. Parameters controlling transcriptional activation during early Drosophila development. Cell 44: 871–877. [DOI] [PubMed] [Google Scholar]
- Edgar BA, Kiehle CP, and Schubiger G 1986. Cell cycle control by the nucleo-cytoplasmic ratio in early Drosophila development. Cell 44: 365–372. [DOI] [PubMed] [Google Scholar]
- Evans T, Rosenthal ET, Youngblom J, Distel D, and Hunt T. 1983. Cyclin: A protein specified by maternal mRNA in sea urchin eggs that is destroyed with each cleavage division. Cell 33: 389–396. [DOI] [PubMed] [Google Scholar]
- Fenton B and Glover DM. 1993. A conserved mitotic kinase active at late anaphase-telophase in syncytial Drosophila embryos. Nature 363: 637–640. [DOI] [PubMed] [Google Scholar]
- Ferrell JE, Wu M, Gerhart JC, and Martin GS. 1991. Cell cycle tyrosine phosphorylation of p34 Cdc2 and a microtubule associated protein kinase homolog in Xenopus oocytes and eggs. Mol. Cell. Biol 11: 1965–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field J, Nikawa J, Broek D, MacDonald B, Rodgers L, Wilson IA, Lerner RA, and Wigler M. 1988. Purification of a RAS-responsive adenyl cyclase complex from Saccharomyces cerevisiae by use of an epitope addition method. Mol. Cell. Biol 8: 2159–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foe VE 1989. Mitotic domains reveal early commitment of cells in Drosophila embryos. Development 107: 1–22. [PubMed] [Google Scholar]
- Foe VA and Alberts BM 1983. Studies of nuclear and cytoplasmic behaviour during the five mitotic cycles that precede gastrulation in Drosophila embryogenesis. J. Cell Sci 61:31–70. [DOI] [PubMed] [Google Scholar]
- Gautier J, Solomon MJ, Booher RN, Bazan JF, and Kirschner M 1991. Cdc25 is a specific tyrosine phosphatase that directly activates p34Cdc2. Cell 67: 197–211. [DOI] [PubMed] [Google Scholar]
- Gerhart J, Wu M, and Kirschner M. 1984. Cell cycle dynamics of an M-phase-specific cytoplasmic factor in Xenopus leavis oocytes and eggs. J. Cell Biol 98: 1247–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould KL and Nurse P. 1989. Tyrosine phosphorylation of the fission yeast Cdc2+ protein kinase regulates entry into mitosis. Nature 342: 39–45. [DOI] [PubMed] [Google Scholar]
- Gould KF, Moreno S, Owen DJ, Sazer S, and Nurse P. 1991. Phosphorylation at Thrl67 is required for Schizosaccharomyces pombe p34Cdc2 function. EMBO J. 10: 3297–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Rosenblatt J, and Morgan DO. 1992. Cell cycle regulation of CDK2 activity by phosphorylation of Thrl60 and Tyrl5. EMBO J. 11: 3995–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan I, Hayles J, and Nurse P. 1988. Cloning and sequencing of the cyclin related cdc13+ gene and a cytological study of its role in fission yeast mitosis. J. Cell Sci. 91: 587–595. [DOI] [PubMed] [Google Scholar]
- Hartenstein V and Campos-Ortega FA 1985. Fate-mapping in wild-type Drosophila melanogaster I. The spatio-temporal pattern of embryonic cell divisions. Wilhelm Roux’s Arch. Dev. Biol 194: 181–195. [Google Scholar]
- Hoffmann I, Clarke PA, Marcote MJ, Karsenti E, and Draetta G 1993. Phosphorylation and activation of human Cdc25-C by Cdc2-CyclinB and its involvement in the self amplfication of MPF at mitosis. EMBO J. 12: 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt T, Luca FC, and Ruderman JV 1992. The requirements for protein synthesis and degradation, and the control of destruction of Cyclins A and B in the meiotic and mitotic cell cycles of the clam embryo. J. Cell Biol 116: 707–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi T, Walker DH, and Maller JL. 1992. Periodic changes in phosphorylation of the Xenopus Cdc25 phosphatase regulates its activity. Mol. Biol. Cell 3: 927–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karr TL and Alberts BM. 1986. Organization of the cytoskeleton in early Drosophila embryos. J. Cell. Biol 102: 1494–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg D 1993. The Drosophila mitotic cyclins interact with microtubule-associated proteins. J. Cell Biol, (in press). [Google Scholar]
- Knoblich JA and Lehner CF. 1993. Synergistic action of Drosophila cyclins A and B during the G2-M transition. EMBO J. 12: 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayakawa Y and Kubota H. 1981. Temporal pattern of cleavage and the onset of gastrulation in amphibian embryos developed from eggs with the reduced cytoplasm. J. Embryol Exp. Morphol 62: 83–94. [PubMed] [Google Scholar]
- Lehner CF and O’Farrell PH. 1989. Expression and Function of Drosophila cyclin A during embryonic cell cycle progression. Cell 56: 957–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner CF and O’Farrell PH. 1990a. The Roles of Drosophila cyclins A and B in mitotic control. Cell 61: 535–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner CF and O’Farrell PH. 1990b. Drosophila Cdc2 homologs: A functional homolog is coexpressed with a cognate variant. EMBO J. 9: 3573–3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiss D, Felix MA, and Karsenti E. 1992. Association of cyclin-bound p34cdc2 with subcellular structures in extracts of Xenopus eggs. J. Cell Sci 102: 285–297. [DOI] [PubMed] [Google Scholar]
- Lorca T, Labbe JC, Devault A, Fesquet D, Capony JP, Cavadore JC, Le Bouffant F, and Doree M 1992. Dephosphorylation of Cdc2 on threonine 161 is required for Cdc2 kinase inactivation and normal anaphase. EMBO J. 11: 2381–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luca FC, Shibuya EK, Dohrmann CE, and Ruderman JV. 1991. Both Cyclin AD60 and BD97 are stable and arrest cells in M phase, but only BD97 turns on Cyclin destruction. EMBO /. 10: 4311–4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado-Codina G and Glover DM. 1992. Cyclins A and B associate with chromatin and the polar regions of spindles, respectively, and do not undergo complete degradation at anaphase in syncycial Drosophila embryos. J. Cell Biol 116: 967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight SL and Miller OL. 1977. Electron microscopic analysis of chromatin replication in the cellular blastoderm Drosophila melanogaster embryo. Cell 12: 795–804. [DOI] [PubMed] [Google Scholar]
- Minshull J, Blow JJ, and Hunt T. 1989. Translation of Cyclin mRNA is necessary for extracts of activated Xenopus eggs to enter mitosis. Cell 56: 947–956. [DOI] [PubMed] [Google Scholar]
- Mita I and Obata C 1984. Timing of early morphogenetic events in tetraploid starfish embryos. J. Exp. Zool 229: 215–222. [Google Scholar]
- Moreno S, Nurse P, and Russell P. 1990. Regulation of mitosis by cyclic accumulation of p80Cdc25 mitotic inducer in fission yeast. Nature 344: 549–552. [DOI] [PubMed] [Google Scholar]
- Murray AW and Kirschner M. 1989. Cyclin synthesis drives the early embryonic cell cycle. Nature 339: 275–280. [DOI] [PubMed] [Google Scholar]
- Murray AW, Solomon MJ, and Kirschner M 1989. The role of Cyclin synthesis and degradation in the control of maturation promoting factor activity. Nature 339: 280–286. [DOI] [PubMed] [Google Scholar]
- Newport J and Kirschner M. 1982. A major developmental transition in Xenopus embryos: I. Characterization of timing of cellular changes at the midblastula stage. Cell 30: 675–686. [DOI] [PubMed] [Google Scholar]
- Norbury C, Blow J, and Nurse P. 1991. Regulatory phosphorylation of the p34Cdc2 protein kinase in vertebrates. EMBO J. 10: 3321–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurse P 1990. Universal control mechanism regulating onset of M-phase. Nature 344: 503–508. [DOI] [PubMed] [Google Scholar]
- O’Farrell PH, Edgar BA, Lakich D, and Lehner CF. 1989. Directing cell division during development. Science 246: 635–640. [DOI] [PubMed] [Google Scholar]
- Okada g., Komatsu H, and Okumura M. 1980. Behavior of interphase embryonic nuclei transplanted in nuclear multiplication stage embryos of Drosophila. Dev. Growth Differ 22: 599–610. [DOI] [PubMed] [Google Scholar]
- Rabinowitz M 1941. Studies on the cytology and early embryology of the egg in Drosophila melanogaster. J. Morphol 69: 1–49. [Google Scholar]
- Russell P and Nurse P. 1986. Cdc25+ functions as an inducer in mitotic control of fission yeast. Cell 45: 145–153. [DOI] [PubMed] [Google Scholar]
- Russell P and Nurse P. 1987. Negative regulation of mitosis by weel+, a gene encoding a protein kinase homologue. Cell 49: 559–567. [DOI] [PubMed] [Google Scholar]
- Russell P, Moreno S, and Reed SI. 1989. Conservation of mitotic controls in fission and budding yeasts. Cell 57: 295–303. [DOI] [PubMed] [Google Scholar]
- Schimke RT, Kung AL, Rush DF, and Sherwood SW. 1991. Differences in mitotic control among mammalian cells. Cold Spring Harbor Symp. Quant. Biol 56: 417–425. [DOI] [PubMed] [Google Scholar]
- Siegel V and Walter P. 1988. Each of the activities of signal recognition particle (SRP) is contained within a distinct domain: Analysis of biochemical mutants of SRP. Cell 52: 39–49. [DOI] [PubMed] [Google Scholar]
- Smythe C and Newport JW. 1992. Coupling of mitosis to the completion of S phase in Xenopus occurs via modulation of the tyrosine kinase that phosphorylates p34cdc2. Cell 68: 787–797. [DOI] [PubMed] [Google Scholar]
- Solomon MJ, Glotzer M, Lee TH, Philippe M, and Kirschner M. 1990. Cyclin activation of p34Cdc2. Cell 63: 1013–102,4. [DOI] [PubMed] [Google Scholar]
- Sorger PK and Murray AW. 1992. S-phase feedback control in budding yeast independent of tyrosine phosphorylation of p34cdc28. Nature 355: 365–368. [DOI] [PubMed] [Google Scholar]
- Sprenger F, Trosclair MM, and Morrison DK. 1992. Biochemical analysis of torso and D-Raf during Drosophila embryogenesis: Implications for terminal signal transduction. Mol. Cell. Biol 13: 1163–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern B, Ried G, Clegg NJ, Grigliatti TA, and Lehner CF 1993. Genetic analysis of the Drosophila cdc2 homolog. Development 117: 219–232. [DOI] [PubMed] [Google Scholar]
- Surana U, Amon A, Dowzer C, McGrew J, Byers B, and Nasmyth K. 1993. Destruction of the CDC28/CLB mitotic kinase is not required for the metaphase to anaphase transition in budding yeast. EMBO J. 12: 1969–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verde F, Dogterom M, Stelzer E, Karsenti E, and Leibler S. 1992. Control of microtuble dynamics and length by Cyclin A- and B-dependent kinases in Xenopus egg extracts. J. Cell Biol 118: 1097–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman WJ and Masui Y. 1975. Effects of cycloheximide on a cytoplasmic factor for meiotic maturation in Xenopus oocytes. Exp. Cell Res 91: 381–388. [DOI] [PubMed] [Google Scholar]
- Whitfield WGF, Gonzalez C, Maldonado-Codina G, and Glover DM. 1990. The A- and B-type Cyclins of Drosophila are accumulated and destroyed in temporally distinct events that define separable phases of the G2-M transition. EMBO J. 9: 2563–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Yoshikuni M, Hirai T, Fukada S, and Nagahama Y. 1991. A monclonal antibody against the PSTAIR sequence of p34cdc2, catalytic subunit of maturation promoting factor and key regulator of the cell cycle, Dev. Growth Differ 33: 617–624. [DOI] [PubMed] [Google Scholar]
- Yasuda GK and Schubiger G. 1992. Temporal regulation in the early embryo: Is MBT too good to be true? Trends Genet. 8: 124–127. [DOI] [PubMed] [Google Scholar]
- Zalokar M and Erk I. 1976. Division and migration of nuclei during early embryogenesis of Drosophila melanogaster. J. Microbiol. Cell 25: 97–106. [Google Scholar]