

Introduction
C3 nephritic factors (C3NF) are a group of autoantibodies that permit continuous over activation of the alternative complement pathway. They are associated with a range of clinical diseases including various nephropathies, partial lipodystrophy and retinal changes as well as infections. C3NF can also be identified among patients undergoing complement studies and can be a cause of diagnostic confusion.1,2 Changes in nomenclature and treatment have prompted a new look at C3NF 3–4
C3 nephritic factors (C3NF) and alternative complement activation
The phylogenetically ancient complement system has evolved as a pivotal part of the innate immune system to help protect against infections. It is comprised of multiple proteins, which lead to an enzymatic cascade causing lysis of the target cell. The system is activated through the classical, lectin, and alternative pathways (AP). The AP pathway is continuously active at a low rate through generation of C3b molecules, the so called “tick-over” in which the internal thioester of C3, hydrolyzed by water, forms an initial convertase with factor B. Factor I and Factor H regulate this pathway and the short half-life of unstabilized C3bBb ensures the process is tightly controlled (Figure 1).
Figure 1: Alternative Complement pathway activation.
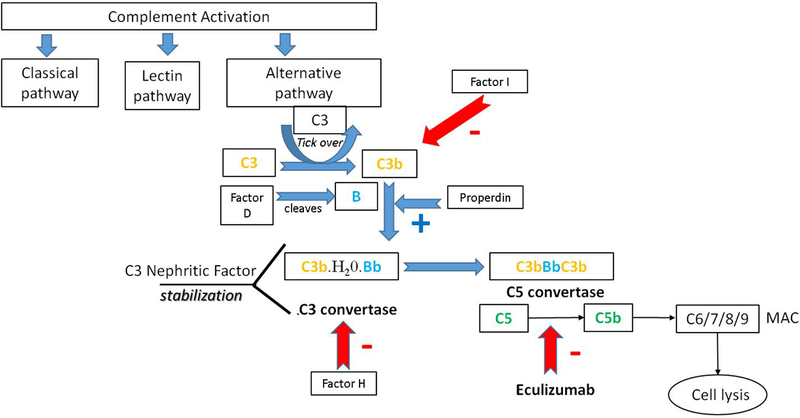
The AP pathway is activated continuously by hydrolysis of C3 (C3•H20) which binds factor B. The so called “tick-over” process in which the internal thioester of C3, hydrolyzed by water, forms an initial convertase with factor B subsequently cleaved by factor D to produce the enzyme C3 convertase (C3b(H20) Bb). This fluid phase C3 convertase can be stabilized by Properdin (P) and cleaves additional C3. Factor H binds host cell surfaces, protecting them by competing for C3b, displacing factor B. Factor I regulates the AP by cleaving C3b and inactivating it. The enzyme C3bBbC3b is termed the alternative pathway C5 convertase. Cleavage of C5 catalyzes the assembly of C5b-C9 and this is termed the membrane attack complex (MAC), a pore capable of driving cell lysis. C3NF bind to the AP C3 convertase and by doing so increase its activity duration. Eculizumab inhibits C5 cleavage.
C3NF are a group of autoantibodies that bind to the AP C3 convertase (C3bBb) increasing both convertase activity and duration of the active forms making it more stable (from minutes to hours) 4–6. These autoantibodies can be detected either by C3 convertase stabilizing activities, by semi-quantitative hemolytic assays or by measuring autoantibody levels.6 There are various types of C3 nephritic factors including both IgM and IgG which can be classified by either being heat sensitive and Properdin dependent or heat stable and Properdin independent. 7
C4 and classical complement pathway nephritic factors have been described but much less well characterized. C4NF targets the Classical pathway C3 convertase (C4bC2b) with a similar endpoint of complement pathway activation. C4NF have been described as a cause of severe infections due to C3 consumption.
C3NF are found in a small number of healthy individuals 1,2 and can be transiently induced in patients with post streptococcal glomerulonephritis(PSGN). C3NF are most common in Membranoproliferative glomerulonephritis (MPGN). Over 80% of patients with dense deposit disease(DDD) and 40–50% with C3 glomerulonephritis have C3NF 3,4.
Clinical manifestations
C3 glomerulopathy
MPGN is now classified based on immunofluorescent findings rather than on light and electron microscopic findings. This classification groups MPGN into those with immune complex deposition (previously MPGN type 1 and 3) versus complement staining with no or almost no immune complex formation (formerly MPGN Type 2 (DDD)). The formation of immune complexes is usually secondary to chronic infections, autoimmune disease or paraproteins, while the usually isolated deposition of C3 is secondary to chronic AP activation. Thus DDD with intramembranous isolated C3 deposition by EM and MPGN is now classified as C3 glomerulopathy (C3GN). 3–4.
C3GN is a rare cause of GN and can present with proteinuria, hematuria, nephrotic syndrome, nephritic syndrome or nephrotic/nephritic syndrome. It most often occurs in children between the ages of 5–15 years. Complement studies (such as factor levels as well as function) demonstrate a low serum C3 concentration and positive C3NF. Historically, over 50% of patients with DDD develop end stage kidney disease (ESRD) within 10 years of diagnosis. After kidney transplant, recurrence of the disease occurs in over 80% of patients with a rate of over 50% graft failure. However, patients with C3GN may have a slightly better long-term prognosis than previously described.3–4. Of note, there have been isolated reports of C3NF in membranous nephropathy and PSGN with both having better clinical outcomes than C3GN.
Partial lipodystrophy
Acquired partial lipodystrophy (APL) is a disease that presents in early childhood (mean 7 years of age) and is more common in girls. It is characterized by loss of upper extremity and facial fat with sparing of the lower extremities. It may precede kidney disease (in 22%) by 8 years or more. When present, kidney disease manifests as C3GN with over 80% having low serum C3 concentration in the presence of C3NF. The mechanism of APL is due to AP complement destruction of adipocytes via factor D. Prognosis depends on the renal disease and additional systemic manifestations can include pancreatitis, liver dysfunction, diabetes mellitus and retinitis pigmentosa 8
Ocular manifestations
Macular degeneration may be present in patients with C3GN. Common findings on fluorescein angiography include macular hyperpigmentation, drusen and neovascularization. Genetic polymorphisms in the AP proteins are also associated with macular degeneration highlighting the importance of complement in endothelial protection9. The pathophysiological mechanism of macular degeneration with C3NF is not yet clear.
Diagnostic approach
C3NF is associated with a low CH50 and a low C3 with preserved C4 levels. The presence of C3NF is sought by direct autoantibody analysis and/or by using a semi quantitative hemolytic method to analyze the stability of C3 convertase 5. The tests for C3NF can be requested through reference labs as part of a complement pathway workup or through available complement study panels. (Table 1)
Table 1:
Laboratory findings among various phenotypes presenting with C3NF
| Clinical Phenotype | Prevalence of C3NF in disease state |
Laboratory findings |
|
|---|---|---|---|
| C3 Glomerulopathy | Dense deposit Disease (DDD) |
80% | Low C3, Low CH50, Normal C4 |
| C3 glomerulonephritis |
50% | Low C3, Low CH50, Normal C4 |
|
| Partial Lipodystrophy | 80% | Low C3, Low CH50, Normal C4 |
|
| Macular Degeneration | unknown | Low C3, Low CH50, Normal C4 |
|
| Healthy General Population | Rare/unknown | unclear | |
| C4NF | unknown | Low C3, Low CH50, Low C4 |
|
| C3 Classical NF | Unknown | Low C3, Low CH50, Normal C4 |
|
Therapeutic modalities
Asymptomatic patients should have monitoring of renal function and periodic eye exams. Treatment of MPGN depends on the clinical presentation with an early decline in kidney function requiring more aggressive therapy. First line therapy includes glucocorticoids, cyclophosphamide, mycophenolate mofetil and tacrolimus although success has been limited with these approaches. 3–4. Plasmapheresis with use of rituximab to remove these autoantibodies in patients with C3GN has shown conflicting results 4.
The recent emergence of a specific, humanized monoclonal antibody C5 inhibitor (Eculizumab) holds therapeutic promise. Multiple case reports have shown benefits either prior to or after recurrence in the renal allograft 10. Despite promise, further studies are required to determine long term outcomes.
Conclusions
C3NF are a heterogeneous group of autoantibodies that results in a secondary AP hypocomplementemia and are associated with a variety of clinical conditions (Box 1). New treatments show promise.
Text Box 1.
-
○
C3NF are autoantibodies that activate the AP and may be found as part of a glomerulonephritis work-up as well as among asymptomatic individuals.
-
○
MPGN has been newly classified leading to a grouped syndrome of C3 glomerulopathy, which encompasses DDD and C3GN, both with predominant C3 deposition in the glomerulus.
-
○
The phenotype can include lipodystrophy, pancreatitis, and macular degeneration.
-
○
New treatment modalities such as Eculizumab may alter the course of C3 glomerulopathy.
Supplementary Material
Abbreviations/Acronyms:
- (C3NF)
C3 nephritic factors
- (AP)
Alternative pathways
- (C3bBb)
C3b-Complement factor 3b, AP C3 convertase
- (C4bC2b)
Classical pathway C3 convertase
- (PSGN)
post streptococcal glomerulonephritis
- (MPGN)
Membranoproliferative glomerulonephritis
- (DDD)
dense deposit disease
- (C3GN)
C3 Glomerulonephritis
- (MAC)
membrane attack complex
- (P)
Properdin
Footnotes
Financial Disclosure: This research did not receive any specific grant from funding agencies in the public, commercial, or not-for profit sectors.
Conflict of Interest: The authors have no conflict of interest to disclose.
References
- 1.Egan M, Sullivan K, Frazer-Abel A, Cunningham-Rundles C. A healthy female with C3 hypocomplementemia and C3 Nephritic Factor. Clin.Immunol.2016;169:14–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spitzer RE, Stitzel AE, Tsokos GC. Autoantibody to the alternative pathway C3/C5 convertase and its anti-idiotypic response. A study in affinity. J Immunol. 1992. January 1;148(1):137–41 [PubMed] [Google Scholar]
- 3.Riedl M, Thorner P, Licht C. C3 Glomerulopathy. Pediatr Nephrol. 2017. January;32(1):43–57. [DOI] [PubMed] [Google Scholar]
- 4.Bomback AS, Appel GB. Pathogenesis of the C3 glomerulopathies and reclassification of MPGN. Nat Rev Nephrol. 2012. November;8(11):634–42. [DOI] [PubMed] [Google Scholar]
- 5.Wen L, Atkinson JP, Giclas PC. Clinical and laboratory evaluation of complement deficiency. J Allergy Clin Immunol. 2004. April;113(4):585–93; quiz 594. [DOI] [PubMed] [Google Scholar]
- 6.Paixão-Cavalcante D, Ló Pez-Trascasa M, Skattum L, Giclas PC, Goodship TH, Rodríguez De Có Rdoba S, et al. Sensitive and specific assays for C3 nephritic factors clarify mechanisms underlying complement dysregulation. Kidney Int 2012;82:1084–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tanuma Y, Ohi H, Hatano M. Two types of C3 nephritic factor: properdin-dependent C3NeF and properdin-independent C3NeF. Clin Immunol Immunopathol. 1990. August;56(2):226–38. [DOI] [PubMed] [Google Scholar]
- 8.Misra A, Peethambaram A, Garg A. Clinical features and metabolic and autoimmune derangements in acquired partial lipodystrophy: report of 35 cases and review of the literature. Medicine (Baltimore). 2004. January;83(1):18–34. [DOI] [PubMed] [Google Scholar]
- 9.Zipfel PF, Heinen S, Józsi M, Skerka C. Complement and diseases: Defective alternative pathway control results in kidney and eye diseases. Mol Immunol 2006;43:97–106. [DOI] [PubMed] [Google Scholar]
- 10.Sánchez-Moreno A, De la Cerda F, Cabrera R, Fijo J, López-Trascasa M, Bedoya R, Rodríguez de Córdoba S, Ybot-González P. Eculizumab in dense-deposit disease after renal transplantation. Pediatr Nephrol. 2014. October;29(10):2055–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.