

Abstract
This overview covers advances in mechanisms of chronic pain and their consequent clinical opportunities. Our research field is fractured into two separate camps: “peripheralists” and “centralists”. While the strong position of the first group is the contention that mechanisms of chronic pain can be understood within the limits of afferent inputs and spinal cord circuitry, the second group insists that the rest of the brain plays a critical role. Here we attempt to conjoin these positions, across clinical pain conditions and animal studies, and demonstrate that the effort can lead to novel translational concepts.
Introduction
Over two decades ago, our lab switched human brain imaging studies away from acute painful stimuli and their representation in healthy subjects to the study of the clinical chronic pain brain. This neuroimaging-based work, together with efforts from many other labs studying a variety of clinical pain conditions, and now being complemented with animal model studies, generally constitutes the science of “centralists”. The general interpretation of this work may be described as the notion that chronic pain is a brain state. Conversely, the more traditional studies in the field conform to the Cartesian viewpoint (pain as a labeled-line sensory system for the transduction and encoding of nociceptive inputs, “representation of the fire”), here dubbed “peripheralists”, which constitute the bulk of animal model studies: research concentrated on the peripheral afferents and spinal cord circuitry, their reorganization and underlying molecular targets in rodent models of chronic pain. This latter viewpoint can be summarized with the concept that chronic pain is determined at the interface between the periphery and the spinal cord, and then reflected, with some modifications, in nociceptive-cortical circuitry that mediate perception. This article attempts to explore convergences between these two supposedly contradicting positions.
Four components of chronic pain
A recent review summarizes chronic pain with a minimalist concept of four stages that should apply to all chronic pain conditions [2]. These consist of the presence of predisposing risk factors, which in combination with an injury, precipitate processes that transition the condition into a stable chronic state [Figure 1]. Research from Apkarian lab and complimentary studies both in humans and rodent models now provide strong evidence that brain limbic circuitry predetermine risk for chronic pain, mainly mesolimbic functional and anatomical properties, while studies of the brain in chronic pain also show that functional and anatomical properties of the neocortex change with transition to chronic pain [2].
Figure 1: Distinct stages for chronic pain.
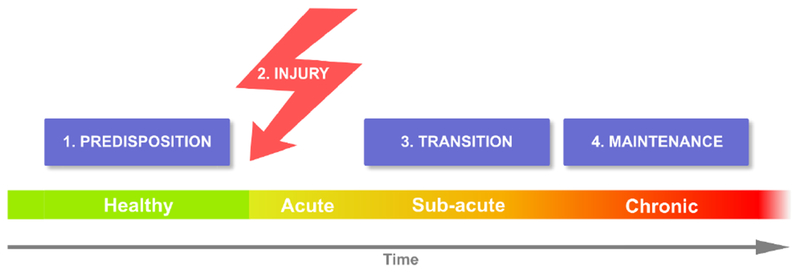
Predispositions are pre-existing factors that place some individuals at risk for developing chronic pain following an injury. Duration of the transition state may be dependent on type of injury as well as also based on predisposing factors. The interaction between injury and predispositions generate the transition state which may, or not, lead to a new brain state of maintenance of chronic pain. The “peripheralist” viewpoint posits that all four stages are determined by nociceptive properties, controlled primarily by peripheral afferents and their interaction with spinal cord circuitry. The “centralists” advance the notion that predispositions are mainly determined by limbic brain properties, and that the interaction between nociceptive circuitry and predispositions creates the transition state, where brain learning and memory circuitry interact with the cortex to give rise to the neocortical state of chronic pain.
While the extreme peripheralist position on this concept would be that all four components are controlled by spinal and peripheral circuitry, the utmost centralist viewpoint posits that only the inciting injury is dependent on peripheral nociceptors and the rest are brain mechanisms.
The “peripheralist” viewpoint
There is substantial evidence that injuries give rise to local and extended injury-related reorganization of nociceptive, and non-nociceptive, afferents, as well as concomitant adaptation of spinal cord circuitry. The details of such reorganization seem specific to type of injury: nociceptive, inflammatory, neuropathic (and even maladaptive) [4; 5]. One of the most solid evidences involves the role of spinal cord microglia. Following a peripheral nerve injury that gives rise to tactile allodynia, microglia are activated and exhibit increased expression of P2X4 receptors (P2X4R; a subtype of ionotropic ATP receptor); blockade of such receptors suppresses tactile allodynia, whereas spinal cord administration of activated microglia with stimulated P2X4R produces tactile allodynia in naïve rats [7]. Thus, this process itself seems necessary and sufficient for neuropathic tactile allodynia.
Given such strong evidence one readily extrapolates these rodent model results to various types of clinical pain conditions as being a reflection of extent of sensitization (maladaptation) of the nociceptive circuitry from the periphery to the cortex. Here, the main tenet being that changes in peripheral and spinal cord sensitivity determine the gain of the system (amplification) which, in turn, is passively propagated to the rest of the brain. And the idea that follows from this extrapolation is the notion that probing nociceptive properties of the skin should lead to clinically meaningful concepts regarding categories of chronic pain. These include: 1) nociceptive conditions, posited as purely dependent on peripheral afferent properties, where clinical examples include osteoarthritis and angina pain; 2) inflammatory pain, which would involve periphery, dorsal root ganglion, and spinal cord sensitization, with clinical examples of tissue injury, surgery, and rheumatoid arthritis; 3) dysfunctional pain, listed as conditions where no known peripheral or central inflammation, or lesion, are observed, and thus maladaptive processes are hypothesized throughout the nociceptive system, with clinical examples including primary erythromelalgia and fibromyalgia; 4) neuropathic pains, which assume neuroimmune interactions in the periphery and the central nociceptive circuitry, where clinical examples include post-stroke, spinal cord injury and peripheral neuropathic conditions [5]. Importantly these subdivisions remain arbitrary and their combinations are the reality of most clinical pain conditions. These concepts in turn lead to quantitative sensory testing as the methodology with which personalized medicine would be rationally imposed on chronic pain. The concept is simple and many large studies have now been undertaken, unfortunately with minimal success [8].
The “centralist” viewpoint
One doubts the strong peripheralist position based on the common clinical observation that for any given common injury, only a small minority of patients with similar injuries develop chronic pain. Thus, clinically it seems that peripheral injury properties are not sufficient to explain chronic pain. As soon as the technology became available to non-invasively interrogate the human brain, it was evident that at least the neocortex of patients with chronic pain (or even only with intermittent persistent pain) presented metabolic, anatomical and functional abnormalities [1]. Yet, the bulk of such studies to date remains cross-sectional, and cannot distinguish between causes and consequences of the observed brain abnormalities, relative to the initial inciting injury. The only longitudinal study remains one done in back pain patients, and the results show that the transition to chronic pain involves anatomical and functional reorganization of the neocortex, while risk factors involve limbic brain anatomical and functional properties, with no evidence that the injury-related pain properties additionally contribute to this risk [3; 9]. Is this finding unique to back pain or does it generalize to other chronic pains? We do not know the answer; only additional studies in other pain conditions can address the question directly. Simplistically, these results have been interpreted as evidence that chronic pain is independent of afferent nociception, and that persistence of pain in time becomes a purely central state.
The exciting consequence of these human brain imaging studies is their generalization to rodent models, where brain circuitry - traditionally assumed to be irrelevant to pain/nociception - are now aggressively interrogated regarding their role in chronic pain, especially concerning the role of various components of the limbic brain [2]. In parallel to the evidence for spinal cord microglia, there is now similarly strong evidence for limbic brain neurons causally controlling tactile allodynia in neuropathic injured rodents. Chemogenetic viral vector manipulations provide the opportunity to test specific neuronal population properties in relation to behavior. In neuropathic animals, nucleus accumbens medial shell dopamine D2 receptor-containing neurons exhibit hyper-excitability, as tested in ex-vivo slice preparation. When the excitability of these same neurons is transiently upregulated, or downregulated, by a specific activator (introducing genetically tagged viruses that are only incorporated in these neurons) one observes decreased, or increased, tactile allodynia in neuropathic rodents. Thus, it is concluded that the level of excitability of these neurons are causally involved in the control of sensitivity of the behavior [6]. Such evidence is as compelling as the one observed for spinal cord microglia. In fact, there is now accumulating evidence that similar chemo- or opto-genetic manipulations in other limbic neuronal populations can similarly control persistent, inflammatory or neuropathic, pain behaviors [10; 11; 12]. The general mechanistic interpretation would be that modulating the emotional gating of nociceptive inputs, through limbic circuitry, should have a large influence on pain behavior/perception.
Hence the general “centralist” position can be summarized as the concept that past experiences, related motivations, and accumulated learned memory traces all influence everyday pain experience, and that such influences are a dominant factor in many, if not most, chronic pain conditions. The strong clinical implication of this perspective then becomes the notion that chronic pain requires manipulation of the central nervous system’s emotional circuitry, rather than nociceptive circuitry.
Reconciling “peripheralists” and “centralists”
A minimal definition of pain would be a somatically embodied negative emotional state, associated with strong urges to modify the current state of beingness. A corollary is that negative emotions and moods by themselves are not pain; and injury per se also does not imply pain. Instead, conscious subjective evaluation of the injury and related nociceptive inputs are necessary and enable identification of associated body parts (even if it is ambiguous, as in visceral pain; requiring nociceptive representation in the neocortex, i.e., body site-specific activations in parts of primary/secondary somatosensory and/or insular cortices). Within this construct chronic pain may be viewed as a spectrum of persistent conditions, where the gain of the nociceptive system may be amplified through either or both nociceptive (peripheral and spinal cord) and non-nociceptive (central limbic) routes, eventually creating and maintaining the chronic pain brain state that seems to be a self-referential emotional state. It should be emphasized that modulation of nociceptive inputs at spinal, mesolimbic and prefrontal cortical levels must be considered fundamental to endowing pain (both acute and chronic) and its multi-dimensionality, as well as to defining its motivational-emotional-attentional subjectivity.
Even though chronic back pain seems to be dominantly a consequence of limbic brain predispositions, it still responds at least transiently to non-steroidal anti-inflammatories and such patients clearly embody their pain to their back. Thus, there is little doubt that nociceptive afferents are still engaged in the condition. On the other hand, if we are ever to cure chronic back pain, one may assume that it will require therapies directed at the limbic brain and not the nociceptive circuitry. It is quite likely that even in chronic back pain, some individuals’ pain is more nociceptively driven than in others. We have long ways to go in our ability to categorize the extent of differential involvement of central limbic circuits versus nociceptive circuits in the variety of chronic pain conditions but, again, simple generalizations are possible. For example, as knee osteoarthritis pain can be relieved by a peripheral knee replacement surgery in about 80% of such patients, we have to accept that the peripheral injury remains the main nociceptive drive, although this chronic state most likely is still controlled by amplification of limbic circuitry given that knee injury itself is poorly correlated with pain [13].
If we recognize the opposing peripheralist and centralist camps actually complementing each other, novel opportunities for personalized medicine become obvious. For example, a strong peripheralist position is espoused by evidence that lidocaine applied to intrathecally or to the dorsal root ganglion level can abolish phantom pains [14]. The alternative centralist response is the evidence that purely visual illusions also eliminate phantom pains [15]. A more complex, conjoined viewpoint would be that peripheral inputs are the marker of somatic/visceral marker but its subjectivity necessarily involves continuous central interpretation of nociceptive inputs, which would translate between nociception and pain.
Similarly, as a treatment option one assumes that decreasing spinal microglia activation and simultaneously diminishing excitability of nucleus accumbens shell dopamine D2 neurons would be more effective than controlling either alone, at least in some chronic pain conditions. Clinically for example, we should be able to identify chronic pain conditions where peripheral interventions are more effective, others where central limbic manipulations would have a higher chance for pain relief (even identifying cases where cognitive interventions may be the better therapy option), and others where interventions need to incorporate multiple therapies targeting both nociception and the emotional brain.
It is important to note that by design here we have forced concepts into extreme dichotomous views, to simplify and clarify the discussion. Moreover, our interpretation of individual studies and related authors into one camp or the other is our own interpretation and may importantly diverge from the actual authors’ opinions. Most likely the reality is that most scientists in our field hold positions closer to the “reconciled” position that we advance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Apkarian AV, Bushnell MC, Treede RD, Zubieta JK. Human brain mechanisms of pain perception and regulation in health and disease. Eur J Pain 2005;9(4):463–484. [DOI] [PubMed] [Google Scholar]
- [2].Baliki MN, Apkarian AV. Nociception, Pain, Negative Moods, and Behavior Selection. Neuron 2015;87(3):474–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Baliki MN, Petre B, Torbey S, Herrmann KM, Huang L, Schnitzer TJ, Fields HL, Apkarian AV. Corticostriatal functional connectivity predicts transition to chronic back pain. Nature neuroscience 2012;15(8):1117–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell 2009;139(2):267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annual review of neuroscience 2009;32:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ren W, Centeno MV, Berger S, Wu Y, Na X, Liu X, Kondapalli J, Apkarian AV, Martina M, Surmeier DJ. The indirect pathway of the nucleus accumbens shell amplifies neuropathic pain. Nature neuroscience 2016;19(2):220–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 2003;424(6950):778–783. [DOI] [PubMed] [Google Scholar]
- [8].Smith SM, Dworkin RH, Turk DC, Baron R, Polydefkis M, Tracey I, Borsook D, Edwards RR, Harris RE, Wager TD, Arendt-Nielsen L, Burke LB, Carr DB, Chappell A, Farrar JT, Freeman R, Gilron I, Goli V, Haeussler J, Jensen T, Katz NP, Kent J, Kopecky EA, Lee DA, Maixner W, Markman JD, McArthur JC, McDermott MP, Parvathenani l, Raja SN, Rappaport BA, Rice ASC, Rowbotham MC, Tobias JK, Wasan AD, Witter J. The potential role of sensory testing, skin biopsy, and functional brain imaging as biomarkers in chronic pain clinical trials: IMMPACT considerations. The Journal of Pain. 2017. July 1;18(7):757–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Vachon-Presseau E, Tetreault P, Petre B, Huang L, Berger SE, Torbey S, Baria AT, Mansour AR, Hashmi JA, Griffith JW, Comasco E, Schnitzer TJ, Baliki MN, Apkarian AV. Corticolimbic anatomical characteristics predetermine risk for chronic pain. Brain 2016; 139(7), 1958–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Apkarian AV, Wei X, Ren W, Centeno M, Procissi D, Xu T, Jabakhanji R, Martina M, Radulovic J, Surmeier D, Liu X. Dorsal Hippocampal Activation Suppresses Neuropathic Pain Behaviors: Chronic pain as extinction-resistant pain-related memory traces. bioRxiv. 2018. January 1:292094. [Google Scholar]
- [11].Schwartz N, Miller C, Fields HL. Cortico-Accumbens Regulation of Approach-Avoidance Behavior Is Modified by Experience and Chronic Pain. Cell reports. 2017. May 23;19(8):1522–31. [DOI] [PubMed] [Google Scholar]
- [12].Watanabe M, Narita M, Hamada Y, Yamashita A, Tamura H, Ikegami D, Kondo T, Shinzato T, Shimizu T, Fukuchi Y, Muto A, et al. Activation of ventral tegmental area dopaminergic neurons reverses pathological allodynia resulting from nerve injury or bone cancer. Molecular pain. 2018. January;14:1744806918756406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Eberly L, Richter D, Comerci G, Ocksrider J, Mercer D, Mlady G, Wascher D, Schenck R. Psychosocial and demographic factors influencing pain scores of patients with knee osteoarthritis. PloS one 2018, 13(4), e0195075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vaso A. et al. Peripheral nervous system origin of phantom limb pain. Pain 155, 1384–1391, doi: 10.1016/j.pain.2014.04.018 (2014). [DOI] [PubMed] [Google Scholar]
- [15].Ramachandran VS & Rogers-Ramachandran D Synaesthesia in phantom limbs induced with mirrors. Proc R.Soc Lond B Biol Sci 263, 377–386 (1996). [DOI] [PubMed] [Google Scholar]