

Abstract
Changes of intracellular Ca2+ concentration regulate many aspects of cardiac myocyte function. About 99% of the cytoplasmic calcium in cardiac myocytes is bound to buffers, and their properties will therefore have a major influence on Ca2+ signaling. This article considers the fundamental properties and identities of the buffers and how to measure them. It reviews the effects of buffering on the systolic Ca2+ transient and how this may change physiologically, and in heart failure and both atrial and ventricular arrhythmias, as well. It is concluded that the consequences of this strong buffering may be more significant than currently appreciated, and a fuller understanding is needed for proper understanding of cardiac calcium cycling and contractility.
Keywords: arrhythmias, cardiac; buffers; calcium; heart failure
The importance of changes in intracellular calcium concentration in cardiac function needs little introduction (see1,2 for reviews). The systolic rise of ionized cytoplasmic calcium concentration ([Ca2+]i) activates contraction and regulates many sarcolemmal ion currents and, thereby, the electrophysiology of the cell; abnormal Ca2+ handling is implicated in the genesis of arrhythmias. Calcium is also a major factor in the control of gene expression. Work using fluorescent Ca2+ indicators has demonstrated how alterations of Ca2+ fluxes into the cytoplasm underlie the changes of contractility in health and disease. It is often overlooked, however, that only ≈1% of cytoplasmic Ca2+ is free, with the remainder being bound to cytoplasmic buffers.3 Therefore, the properties of these buffers will potentially play as large a role as Ca2+ fluxes do in determining the size and kinetics of changes of [Ca2+]i.
Here we review recent progress in characterizing cytoplasmic buffers and their effects on the physiology of cardiac muscle, and disease mechanisms, as well. It is important to note that we also highlight the numerous areas where more work is required.
Properties of Intracellular Ca2+ Buffers
Chemistry and Kinetics of Ca2+ Binding
Calcium (Ca2+) has several chemical features that make it a ubiquitous second messenger.4,5 It forms a chemically active divalent cation in aqueous solution with an ionic radius larger than the other common divalent ion (Mg2+) resulting in higher-affinity binding. The fact that its intracellular concentration is much lower than extracellular permits large changes of concentration because of sarcolemmal fluxes. Ca2+ binding alters the tertiary structure of proteins with consequences for their catalytic activity. This is a reciprocal interaction because binding also resists changes of the free concentration of an ion by acting as a buffer.
The formation of several coordinate bonds between single Ca2+ ions and ligands is known as chelation. The electrons for these bonds typically come from nitrogen or oxygen atoms, replacing water molecules in the solvation sphere of the Ca2+ ion with a series of bonds (usually 6) in a claw-like or chelation arrangement (Figure 1A). For example, EDTA is an organic chelator designed to bind divalent cations with very high affinity. The EF hand, a helix-loop-helix configuration, is the most common Ca2+ binding motif in proteins.6 EF hand sites generally occur in pairs, and their affinity for Ca2+ and Mg2+ depends on the amino acids used to form the coordinate bonds, and the surrounding protein environment, as well. For example, calmodulin has 4 EF hand domains: (1) a high-affinity site that is normally bound at resting Ca2+ concentrations with a dissociation constant (Kd) of ≈100 nmol/L, (2) 2 further binding sites on the C terminal with Kd values of ≈300 nmol/L.7 The fourth binding site has the lowest affinity (Kd=10 µmol/L) and therefore binds negligible Ca2+ within the physiological range (0.1–1 µmol/L). It may have a role in controlling enzymes located near sarcoplasmic reticulum Ca2+ release sites in cardiac cells where [Ca2+]i rises to ≈100 µmol/L.7,8
Figure 1.
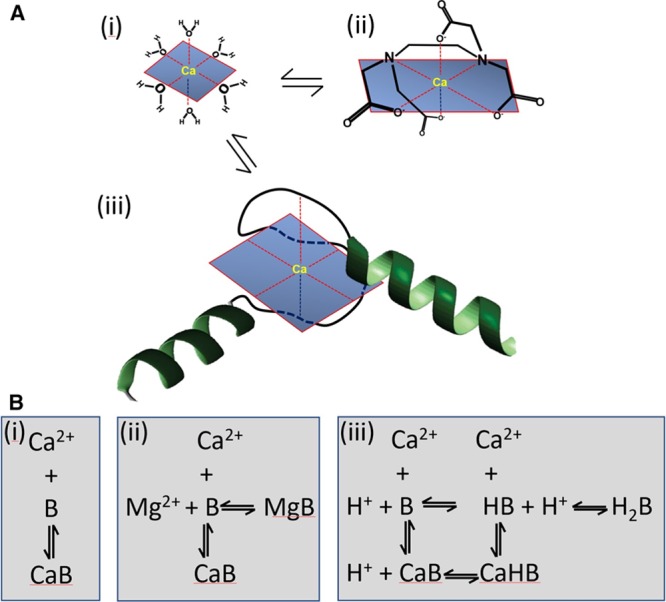
Chemistry of Ca2+ binding to buffers. A, Schematics showing Ca2+ in various environments: water (i), bound to EDTA (ii), and bound to an EF hand (iii). B, Reaction schemes for Ca2+ binding to various buffers: simple binding (i), competition with Mg2+ (ii), and competition with protons (iii).
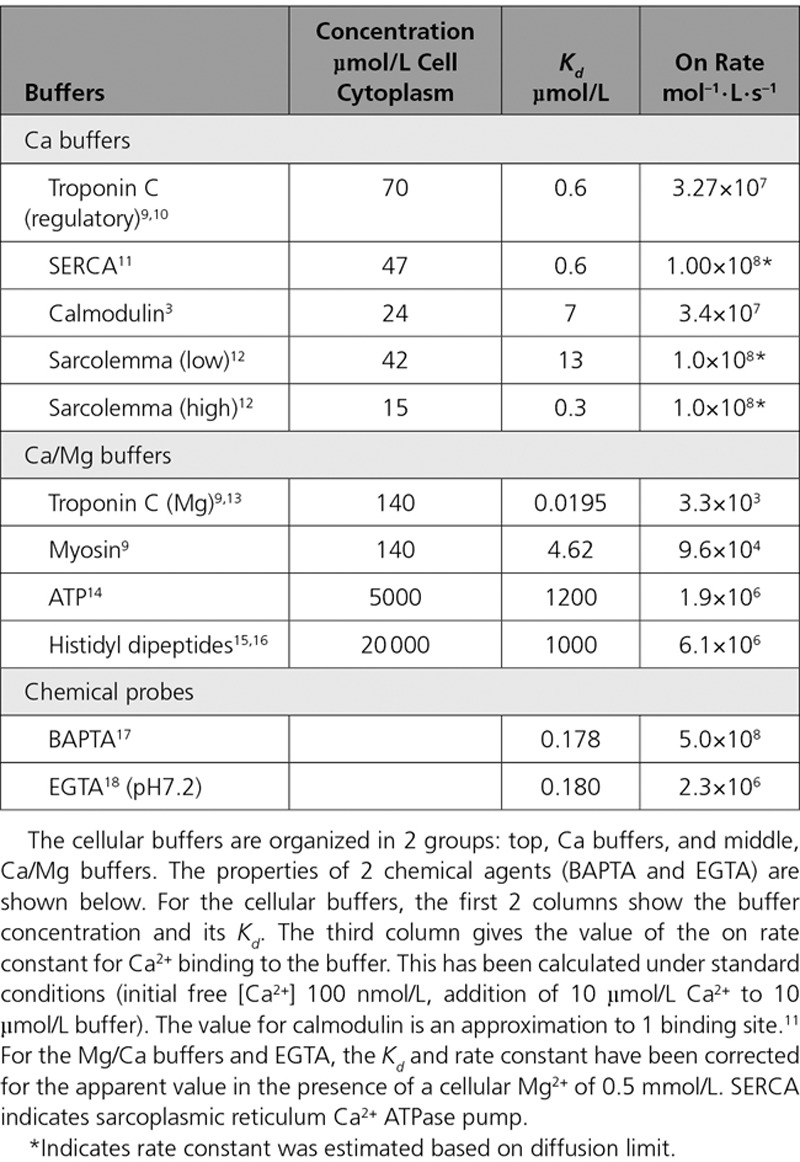
The speed of the chelation reaction depends on its complexity (Table). The fastest binding occurs with small molecules (molecular weight <1000) such as BAPTA or Ca2+ indicators (eg, Fura-2 or Fluo-3), with a forward rate constant that approaches the diffusion-controlled limit (minimally 108 mol-1·L·s-1).17 Ca2+ binding to the EF hand structure of the regulatory site of troponin C is slightly slower.9 With even more complex reaction schemes (Figure 1B) that involve displacement of ions (eg, H+ or Mg2+) before Ca2+ binding, the kinetics slows considerably. For example, Ca2+ binding to the chelator EGTA requires dissociation of protons from intermediate forms of the ligand, reducing the overall forward rate constant (Table).18 Different forms of the EF hand motif, such as the Mg2+ sites of myosin, troponin C (TnC), and parvalbumin, have high relative affinities for Mg2+ that result in significant Mg2+ bound under physiological conditions.19 The need for Mg2+ to dissociate as part of the equilibration results in a low apparent rate constant of Ca2+ binding.
Table.
Concentration and Properties of the Major Cellular Buffers in Ventricular Myocytes
Ca2+ Binding Sites in Cardiac Muscle
Based on previous work,3,11,20 the Table lists the major Ca2+ binding ligands, their estimated cytoplasmic concentrations, and dissociation constants (Kd) alongside estimates of the rate constants of Ca2+ binding. The ligands that bind appreciable amounts of Mg2+ under physiological conditions are grouped separately. The steady-state Ca2+ binding for several buffers as a function of [Ca2+] is shown in Figure 2Ai. The 2 major contributors to buffering are TnC and sarcoplasmic reticulum Ca2+ ATPase pump (SERCA).
Figure 2.
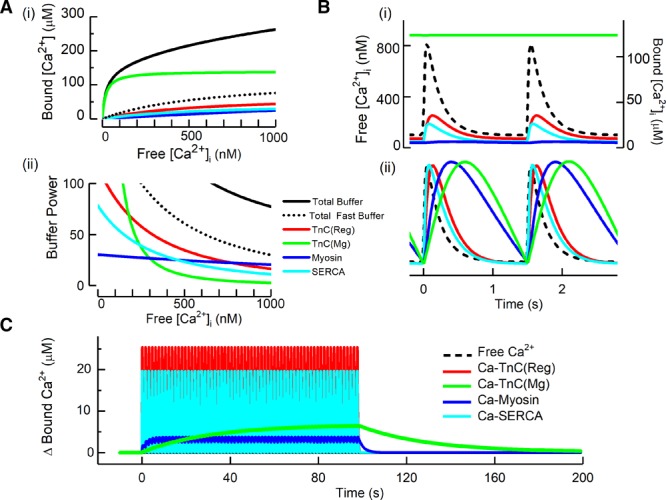
Properties of cellular cardiac Ca2+ buffers. Ai, Steady-state Ca2+ buffering showing the dependence of bound on free Ca2+. The colored curves show the contributions of the 2 Ca2+ binding sites (regulatory [Reg] and Mg) of TnC, myosin, and SERCA. The solid black line shows the total, calculated as the sum of these components and the others listed in the Table. The dotted black line represents the sum of the fast buffers (top part of Table). ii, Buffer power (calculated as in equation 2) as a function of [Ca2+]i. B, Time course of change of bound [Ca2+] in response to systolic Ca2+ transients applied at 1.5 Hz. i, Absolute levels of [Ca2+]. The dashed line shows free [Ca2+] and the colored traces show the concentration of the Ca2+-bound form of the various ligands. ii, Normalized concentrations to emphasize kinetics. C, The change of bound Ca2+ in response to a series of Ca2+ transients (not shown) applied at 1.5 Hz. SERCA indicates sarcoplasmic reticulum Ca2+ ATPase pump; and TnC, troponin C.
Troponin C
TnC has 2 classes of Ca2+ binding sites (Figure 2A): (1) a single, lower-affinity, regulatory site that modulates myofibril activation and thence force; and (2) 2 high-affinity sites that can also bind Mg2+, the Mg2+ sites. There is ample evidence that the affinity for Ca2+ of the regulatory site changes in various situations. For example, acidification decreases the binding of Ca2+.21 Work using a fluorescent TnC showed that phosphorylation of troponin I, as occurs during β-adrenergic stimulation, shifts the relationship between fluorescence and [Ca2+]i to higher [Ca2+]i, indicating decreased Ca2+ affinity. A similar approach has shown that troponin and tropomyosin mutations affect Ca2+ affinity,22 and such mutations have been directly shown to affect Ca2+ buffering.23,24 It should, however, be noted that there are many circumstances (see below for discussion of heart failure) where the only available data are of a shift in the relationship between [Ca2+]i and force. It is often not certain whether this shift results from a direct effect on Ca2+ binding or a subsequent step in the contraction mechanism.25 For example, caffeine shifts the relationship to lower [Ca2+]i but this effect is not accompanied by increased Ca2+ binding.26 Finally, much less is known about the properties of the Mg2+ site on troponin than the regulatory one. One issue, which also applies to other cellular buffers, is that studies of Ca2+ binding are generally performed in vitro using artificial solutions as opposed to cytoplasm. Given that many cellular constituents may affect the properties of this important buffer, it is important to characterize Ca2+ binding to the Mg2+ sites under more physiological conditions. These sites can be mutated, and normal contraction requires only 1 of the 2 Mg2+ sites.27 It would be interesting to know the effects on cardiac function and Ca2+ cycling of the expected large decrease of Ca2+ buffering.
Sarcoplasmic Reticulum Ca2+ ATPase Pump
The inclusion of SERCA as a buffer emphasizes that it has 2 roles in decreasing cytoplasmic [Ca2+]i.28 Initial buffering by binding is followed by active sequestration. In rabbit ventricle, systole involves an increase of ≈60 µmol/L total Ca2+ resulting in a rise of free [Ca2+]i of ≈0.6 µmol/L. Because of the affinity of SERCA binding sites, ≈30 µmol/L binds immediately and, with a peak uptake rate of ≈200 µmol·L–1·s–1, only ≈2 pump cycles are required to sequester the Ca2+ associated with a Ca2+ transient. This emphasizes the importance of the initial binding/buffering by SERCA in addition to its turnover in determining the rate of decay of the cytoplasmic Ca2+ transient.29
Other Ligands
One important distinction is whether the buffers are immobile, or can diffuse. The Table gives values for the fixed sarcolemmal binding sites. The highly diffusible ATP binds Mg2+ and Ca2+ with moderately fast kinetics, but, although present at 5 mmol/L, its low affinity results in only a modest contribution to buffering. Other diffusible ligands include creatine phosphate and histidyl dipeptides that also bind Ca2+ and Mg2+. In heart, the predominant forms of this latter group of compounds include homocarnosine and anserine with a total concentration of ≈20 mmol/L.15 The affinities of Ca2+ and Mg2+ for these histidyl dipeptides are lower than that of ATP, and together they constitute the bulk of the diffusible Ca2+ buffers. One feature of diffusible Ca2+ buffers is their ability to increase the apparent diffusion coefficient of Ca2+ through diffusion of the Ca-bound form.30 The histidyl dipeptides are also weak acids and contribute to the pH buffer power of the cytosol, thereby linking intracellular pH and Ca2+ buffering (see below).
Buffer Kinetics
The importance of the different kinetics of the major buffers is illustrated in Figure 2Bi. The amount of Ca2+ bound to the regulatory site of TnC lags slightly behind free [Ca2+]. The lag is much greater for the slower buffers (here the Mg2+ sites of myosin and TnC) and this is emphasized in the normalized data of Figure 2Bii. During a train of stimuli (Figure 2C), the slow kinetics of these buffers results in a beat-to-beat increase of bound Ca2+. Even at 1.5 Hz, these 2 slow sites together accumulate a total of ≈10 µmol/L Ca2+ and, at higher rates, when diastolic [Ca2+]i increases, greater binding is to be expected.
Measurement of Ca2+ Buffering
As discussed above, buffering depends on the summed effects of a variety of Ca2+ binding molecules. It is often convenient to approximate this with a composite buffer value described by a single dissociation constant and ligand concentration. The simplest method is by titration. Solaro et al31 studied isolated cardiac myofilaments and calculated that about 22 µmol of Ca2+ per kg heart is required to produce 50% maximum contraction. This was accompanied by a rise of free Ca2+ of ≈1.4 µmol/L, indicating that the myofilaments alone can bind >90% of the total Ca2+. A subsequent approach, using cardiac homogenates, found that to raise free [Ca2+] to 1 µmol/L required 72 µmol/kg total Ca2+.32 Hove-Madsen and Bers33 performed similar experiments using permeabilized cells. This removed complications of extracellular components and allowed study of mitochondrial and sarcoplasmic reticulum (SR) buffering separately from cytoplasmic. They found that cytoplasmic buffering could be described by a Kd of 0.42 µmol/L, plus a much lower-affinity component (Kd=79 µmol/L).
The methods described above involve destruction of the cell membrane. It is also important to be able to measure buffering under physiological conditions. This was first done by depolarizing ventricular myocytes and measuring the total entry of calcium through the L-type Ca2+ current20,34 under conditions in which Ca2+ removal mechanisms were inhibited (see Figure 3A and 3B). Berlin et al34 compared Ca2+ entry with the rise of [Ca2+]i, giving a Kd of 0.96 µmol/L and a maximum buffer capacity of 123 µmol/L. A related method compared the entry of Ca2+ through sodium calcium exchange (NCX) with [Ca2+]i as estimated indirectly from changes of cell length.37 A limitation of the method of Berlin et al is that it requires the irreversible SERCA inhibitor thapsigargin, precluding repeated measurements before and after other interventions. An alternative approach uses rapid application of caffeine to release calcium from the SR resulting in an abrupt increase of [Ca2+]i which then decays as NCX removes Ca2+ from the cell. Integrating the NCX current gives a measure of the change of total Ca2+ concentration that is compared continuously with the change of free [Ca2+]i to characterize buffers36 (Figure 3C through 3E). The caffeine response typically decays with a time constant of 1 to 2 s38 so this cannot detect slower buffers. In ferret ventricular myocytes, this method gave a Kd of 0.59 µmol/L with a maximum capacity of 114 µmol/L cell equivalent to 175 µmol/L cytoplasm36 and, in rat ventricular myocytes, a Kd of 0.49 µmol/L and a maximum capacity of 149 µmol/L cytoplasm.39 This is stronger Ca2+ buffering than that found by Berlin et al.34 This may be because, in part, the caffeine method includes buffering by SERCA because thapsigargin is not present. Consistent with this, addition of thapsigargin has been shown to decrease buffer power.40
Figure 3.
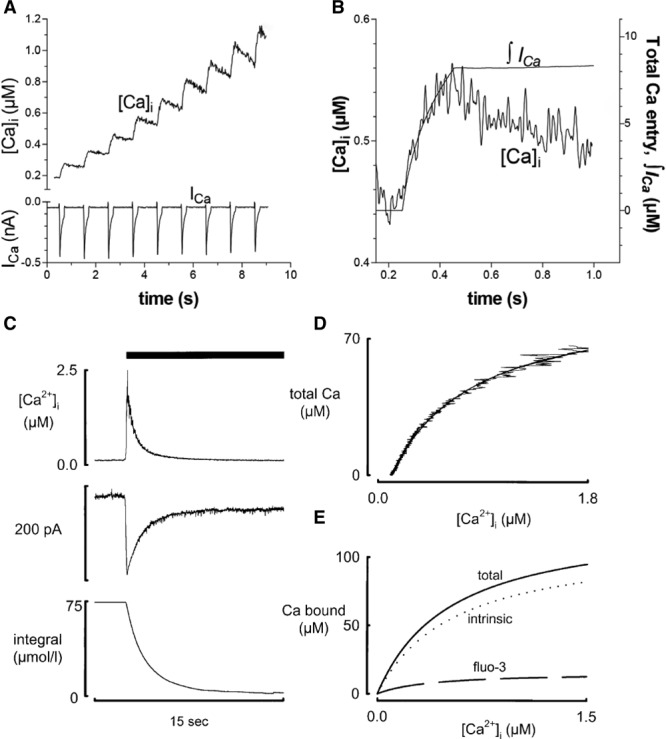
Measurement of buffering in intact cells. A, Comparison of the effects of Ca2+ influx through the L-type Ca2+ channel (bottom) with the resulting increase of [Ca2+]i. B, Relationship between the integral of the L-type Ca2+ current and the change of [Ca2+]i. Ca2+ removal by SR, mitochondria, NCX, and PMCA were inhibited with thapsigargin, a mitochondrial uncoupler, Na-free solution and elevated external Ca2+ concentration, respectively. Figure reproduced from Bers35 from an original article34 with permission. Copyright © 2001, Kluwer Academic. C, Determination of Ca2+ buffering from the caffeine-evoked release of Ca2+ from the SR. Traces show (from top to bottom): [Ca2+]i, NCX current, integral of current. Caffeine (10 mmol/L) was applied as shown by the bar. D, Relationship between total Ca2+ (estimated from the integral of NCX current) and [Ca2+]i. E, Separation of buffering into total, the contribution from the Ca-sensitive indicator (fluo-3) and the calculated intrinsic buffering of the cytoplasm. Reproduced from Trafford et al.36 NCX indicates sodium calcium exchange; PMCA, plasma membrane Ca2+ ATPase; and SR, sarcoplasmic reticulum.
Buffering and the Systolic Ca2+ Transient
Alterations of buffering power affect the systolic Ca2+ transient and thence contraction. Incorporation of Ca-sensitive indicators has the side effect of increasing Ca2+ buffering, and this decreases systolic and increases diastolic force, and slows the rate of mechanical relaxation, as well.41,42 Subsequent work found a decrease of both the amplitude and rate constant of decay of the Ca2+ transient because, the higher the buffer power, the smaller the change of free [Ca2+] resulting from a given rate of Ca2+ pumping.43 Adding exogenous buffer also decreases the rate of spontaneous beating of sinoatrial node cells, presumably by decreasing the changes of [Ca2+]i that contribute to pacemaker activity.44 In recent years, much work has been done using transgenic animals that express calcium indicators. In principle, the additional buffering could be a concern, but it has been demonstrated that, at the concentrations expressed, this is not an issue.45
The effect of increased buffering also depends on the kinetics of the added buffer. Although fast buffers simply slow the Ca2+ transient, slower buffers produce a biphasic decay. The initial, fast phase reflects the time taken for cytoplasmic Ca2+ to bind to the buffer with the slower phase depending on the kinetics of Ca2+ removal from the cytoplasm.43
It should be noted that, in the steady state, averaged over the cardiac cycle, Ca2+ efflux must equal influx. This efflux is determined by [Ca2+]i. If one assumes that Ca2+ efflux is proportional to [Ca2+]i, then, in the steady state, the decrease of amplitude of the Ca2+ transient resulting from increased buffering must exactly balance the slowing of decay of the transient and increased diastolic level such that the average level of [Ca2+]i is unaffected.46,47 Increasing stimulation rate will load cytoplasmic Ca2+ buffers (Figure 2C). An interruption of beating will result in this extra Ca2+ being taken up by the SR and then being available for release.23 This may affect contractility and (see below) contribute to Ca-dependent arrhythmias. A more complicated question is what is the effect of increased buffering on SR Ca2+ content in the steady state? Experimental studies have found that adding exogenous cytoplasmic buffers decreases SR Ca2+.43,48 One explanation is that SERCA activity depends in a cooperative manner on [Ca2+]i,49 whereas NCX has a linear dependence.50 The decreased amplitude of the systolic Ca2+ transient may therefore decrease SERCA activity more than NCX, leading to a net loss of SR Ca2+. Further studies are required to see if the decrease of SR content with increased buffering is a general phenomenon.
Factors that Alter Ca2+ Buffering
Diastolic [Ca2+]i
For a simple buffer, total ([CaT]) and free ([Ca2+]) are related by:
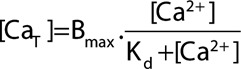 |
(1) |
where Bmax is the total buffer concentration and Kd is the concentration of Ca2+ at which 50% of the buffer has Ca2+ bound. The upper graph of Figure 4A shows such relationships for 3 values of Kd.
Figure 4.
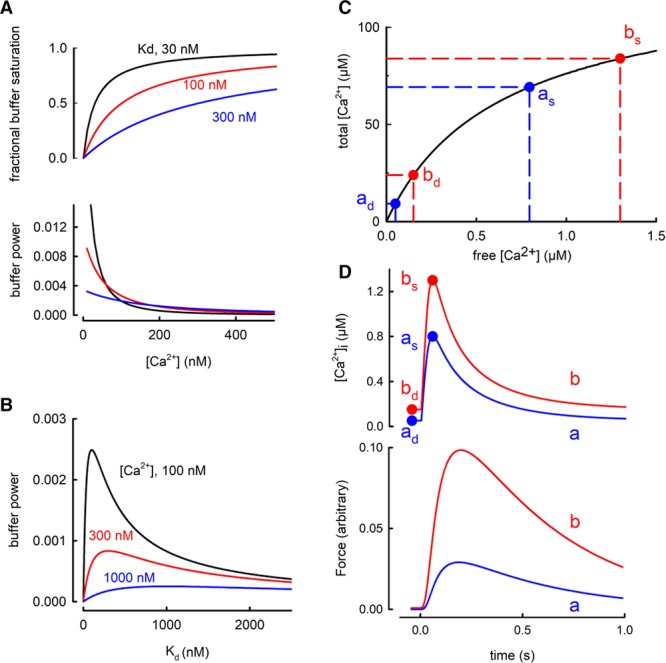
Effects of [Ca2+]i and buffer Kd on buffer power. A, Effects of [Ca2+]. The top graph shows fractional saturation of a single buffer as a function of [Ca2+]. Curves are shown for 3 different values of Kd. The lower graph shows calculated buffer power (change of total Ca2+/change of [Ca2+]i) as a function of [Ca2+] for these values of Kd. Colors correspond to those above. B, Dependence of buffer power on Kd at the 3 values of [Ca2+] indicated. C, The effects of a small increase of diastolic [Ca2+]i on the change of systolic [Ca2+]i produced by the addition of a fixed amount of total [Ca2+]. The buffer curve (fast buffers only) shows 2 levels of diastolic [Ca2+]i indicated as ad (50 nmol/L) and bd (150 nmol/L). The systolic levels (respectively, as and bs) are obtained by adding 60 µmol/L total [Ca2+] resulting in a larger increase of systolic [Ca2+]i in b compared with a. D, Simulation of the effect of adding and removing 60 µmol/L total Ca2+ with kinetics designed to represent a Ca2+ transient. Labels correspond to points in C. Upper, simulated Ca2+ transients; Lower, predicted force responses calculated using a published model.53
Buffer power (β) is defined as the change of total Ca2+ divided by that of free Ca.
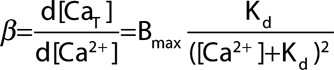 |
(2) |
The individual contributions of the major individual buffers to the total buffer power are shown in Figure 2Aii. At [Ca2+]i ≈100 nmol/L, the Mg2+ sites on TnC make the largest contribution, whereas, at >200 nmol/L, these are tending to saturation, and the regulatory site and SERCA contribute most. The lower graph of Figure 4A shows that buffer power has its highest value (equal to Bmax/Kd) at low [Ca2+] and decreases as [Ca2+] increases. When [Ca2+]=Kd the buffer power is 30% of the value at 0.1 Kd and, at 2 Kd, it is only 13% of this level. Consequently, the greater the diastolic level of [Ca2+]i, the larger will be the increase of [Ca2+]i produced by a given release of total Ca from the SR.51,52 Some appreciation of the importance of this effect is provided by the buffer curve of Figure 4C. Because of the flattening of the buffer curve at elevated [Ca2+]i, an increase of 60 µmol/L total [Ca2+] produces a larger increase of free [Ca2+] when applied from a higher diastolic [Ca2+]i than from a lower. This is clear in the simulated transients of the upper graph of Figure 4D. An increase of diastolic [Ca2+]i of only 100 nmol/L (from 50 to 150) increases systolic [Ca2+]i by 500 nmol/L. Therefore, an increase of diastolic [Ca2+]i alone can lead to an increase of systolic, which is predicted (see lower graph Figure 4D) to result in a large increase of developed force with little change of resting force. Finally, the decrease of buffer power at elevated [Ca2+]i has also been suggested to account for a rapid initial rate of decay of the Ca2+ transient.54
This consequence of changes of diastolic [Ca2+]i will add to the inotropic effects of manoeuvers such as the addition of cardiac glycosides55 or β-adrenergic stimulation56 that can increase diastolic [Ca2+]i. It is also a possible explanation for changes of the amplitude of the Ca2+ transient and force under the many conditions where there are no measurements of diastolic [Ca2+]i. Testing this will require obtaining and comparing absolute measurements of [Ca2+]i between cells or tissues from different animals or patients. There is a dearth of such measurements in the literature57 because it is much easier to measure changes of fluorescence of a Ca2+-sensitive indicator than absolute levels of [Ca2+]i. Properly calibrated measurements, ideally using ratiometric indicators, are required.
Stimulation Rate
Repetitive stimulation will load slower Ca buffers (Figure 2C). This will decrease buffer power and might therefore increase the rise of [Ca2+]i produced by a given increase of total cytoplasmic Ca2+, contributing to the inotropic effects of increased rate.52 This effect is analogous to that discussed above for elevated diastolic [Ca2+]i, but the slow kinetics result in a memory so that, following a change of rate, the effects on systolic [Ca2+]i and thence on the action potential duration may outlast those of diastolic [Ca2+]i. Such effects may also contribute to the slow effects of changes of rate on parameters such as action potential duration.58
Buffer Kd
Equation 2 (Figure 4A) shows that, at lower values of [Ca2+]i, buffer power is greater the lower the value of Kd because this results in stronger Ca2+ binding. In contrast, at higher [Ca2+]i, the lower the Kd, the less the buffer power as the buffers become saturated (see23 for experimental demonstration). Figure 4B shows the biphasic dependence of buffer power on Kd with the maximum being reached when Kd=[Ca2+]i. Therefore, increasing buffer affinity will increase buffering at diastolic levels of [Ca2+]i but decrease it at peak systolic ones.
One issue that has received no attention is whether Ca2+ buffering is the same in all cells in the ventricle. Given the regional differences of expression of other proteins including pumps59 and channels,60 heterogeneity of buffering would not be surprising. Likewise, possible variations of Ca2+ buffering between individuals, because of mutations and polymorphisms, do not appear to have been considered.
Physiological Modulation of Buffering
β-Adrenergic Stimulation
The 2 major Ca2+ buffers, TnC and SERCA, are regulated by the phosphorylation of troponin I and phospholamban, respectively, resulting in increased affinity of Ca2+ for SERCA61 and decreased affinity of Ca2+ for TnC.62 One might therefore expect that β-adrenergic stimulation would alter the buffer power. Experimental measurements, however, found no such effect,40 possibly because of 2 opposing factors: phosphorylation increases the affinity of Ca2+ binding to SERCA, but lowers it for troponin. If the Kd values are above the range of [Ca2+]i considered, these effects will respectively increase and decrease buffer power (Figure 4A). Subsequent experiments, performed on transgenic mice in which either troponin could not be phosphorylated or lacking phospholamban, found the expected increase and decrease, respectively, of buffer power on phosphorylation. Further work is required to investigate the possibility that, in other species, the balance is less exact, and, therefore, phosphorylation may have a net effect on buffer power. As mentioned above, it should also be noted that the effects of a change of Ca2+ affinity on buffer power will depend on the range of [Ca2+]i under investigation.
Effects of Changes of pH on Buffering
Many Ca2+ buffers can bind protons as an alternative to Ca2+ ions. Direct measurements have shown that acidification decreases Ca2+ binding to troponin.21 Therefore, acidification will decrease the affinity for Ca2+ with a decrease of Ca2+ buffering power predicted at values of [Ca2+]i below the Kd (Figure 4B). It is surprising that intracellular acidification had no effect on Ca2+ buffering.63 We suggest that this may occur because, although a decrease of Ca2+ affinity of low-affinity buffers will decrease buffer power, decreased affinity of very-high-affinity buffers will increase their contribution to buffering. Acidification has been shown to increase resting [Ca2+]i in rat ventricular myocytes, an effect attributed to displacement of Ca2+ from buffers.15 It is not clear, however, why such displacement should produce the observed maintained increase of [Ca2+]i; one would expect a transient increase that decays back to baseline as Ca2+ is pumped out of the cell. It may result from the inhibition of Ca2+ efflux on NCX by acidification.64 If this is the case then the maintained effect on [Ca2+]i is presumably a consequence of the NCX effect and not of altered buffering. This question could be resolved by directly measuring the effects of pH on NCX activity.
Cardiac Dysfunction and Ca2+ Buffering
Atrial Buffering, Fibrillation, and Failure
The total concentration of Ca2+ buffers in rat atrial myocytes has been reported to be about 3 times greater than in ventricular myocytes with no difference in apparent Kd,65 possibly because of higher SERCA expression in the atrium than in the ventricle. Changes in Ca2+ buffering have been suggested to be important in both normal and abnormal atrial function. For example, in sheep atria, buffer power increases with age because of an increase of Ca2+ affinity of the buffers.66 This decreased both the amplitude and rate of decay of the systolic Ca2+ transient. Atrial myocytes from many species, including rabbits and cats, have few or no t-tubules (67 for review) and the systolic Ca2+ transient begins at the periphery of the cell and then propagates toward the center.68,69 Increasing Ca2+ buffering by incorporation of EGTA can prevent this propagation.70 A modeling study also predicted this inhibitory effect of high buffer concentrations but pointed out that lower concentrations of mobile buffers such as ATP facilitate propagation.30 Greiser et al71 investigated the effects of rapid atrial pacing in rabbits to mimic the effects of atrial fibrillation. This resulted in a 2- to 3-fold increase of buffering power, at least in part, because of decreased phosphorylation of troponin I which was accompanied by (Figure 5A) decreased centripetal propagation. Evidence for a causal link between increased buffering and decreased propagation was provided by showing that incorporation of BAPTA to increase buffering mimicked the effect on propagation. The effects of rapid pacing to induce heart failure have also been studied on sheep atrial myocytes where a decrease of buffer power was observed.74 This was accompanied by a decrease of the amplitude of the central calcium transient attributed to the loss of transverse tubules rather than a change of buffering.75 A similar decrease of calcium buffering in the sheep has been observed during atrial fibrillation where it was suggested to lead to arrhythmogenic Ca2+ waves, thereby contributing to atrial fibrillation.76 More work is required on changes of atrial Ca2+ buffering and their importance in atrial function. Finally, it is worth noting that the atrial studies reviewed above could not exclude the effects of small changes of end-diastolic [Ca2+]i on the measured buffer power.
Figure 5.
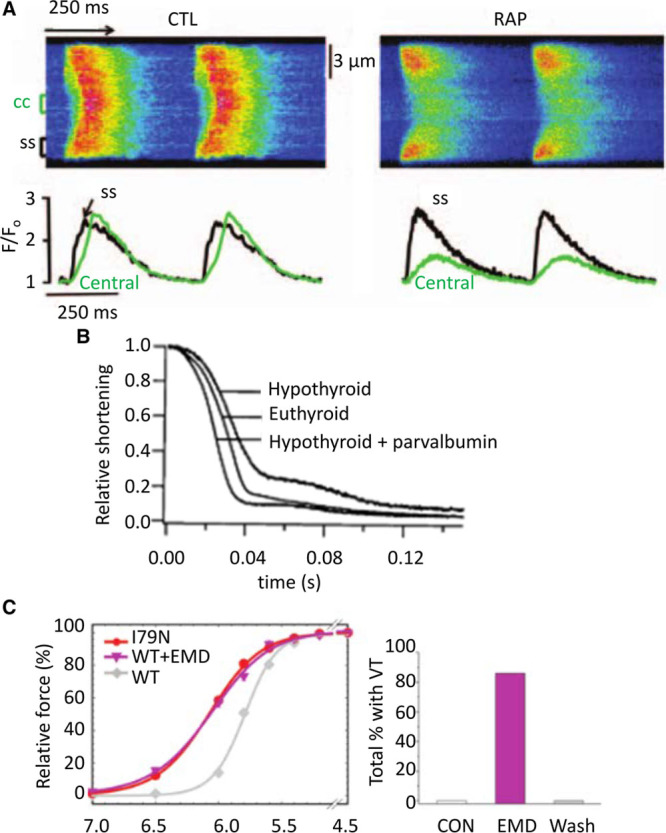
Effects of altered buffer power. A, Abolition of centripetal propagation of the Ca2+ transient by rapid atrial pacing (RAP). Upper traces are linescan images. Lower records show [Ca2+]i measured at surface of cell (black) and in center (green). Left, Records from control. Right, Records after rapid stimulation. Reproduced from.71 B, Relaxation of contraction in isolated rat myocytes. Records show control (euthyroid), hypothyroid, and hypothyroid with parvalbumin expressed by gene transfer. Reproduced from.72 C, Left, comparison of the effects of the I79N mutation of troponin T with that of the Ca2+ sensitizing agent EMD 57033 on the relationship between pCa and force. Right, Effects of EMD 57033 on the occurrence of ventricular tachycardia (VT). Reproduced from Greiser et al.73
Ca2+ Buffering and Heart Failure
Ca2+ buffering is unaffected by pacing-induced heart failure in both dogs77 and sheep.78 In contrast, in samples from human ventricle, the Ca2+ sensitivity of contraction was increased in dilated cardiomyopathy, possibly because of the decreased phosphorylation of troponin I.79 Increased Ca2+ sensitivity was also found in canine dilated cardiomyopathy80 and mouse infarct models.81,82 As mentioned earlier, changes of Ca2+ sensitivity of contraction do not necessarily indicate altered Ca2+ binding and buffering; direct measurements of Ca2+ binding are therefore required. Increased myofilament Ca sensitivity by itself will decrease cardiac relaxation and thereby contribute to diastolic heart failure. In addition, any consequential increase of Ca2+ buffering will slow the decay of [Ca2+]i, worsening relaxation. In contrast to the data discussed above, either the induction in rats of pressure overload–induced left ventricular hypertrophy or heart failure following myocardial ischemia resulted in a decreased Ca2+ sensitivity for activation of contraction, an effect attributed to alterations in troponin.83 Some of the controversies in this area have been reviewed.84 As far as myocardial ischemia is concerned, it is well known that troponin is lost from the heart and, indeed, the appearance of troponin I and troponin T in plasma is diagnostic of cardiac damage. Troponin release has also been detected in myocardium in conditions not associated with obvious cellular degeneration, but this only represents a small fraction (≈3%) of the total troponin85 and will not therefore significantly affect cellular buffering.
Finally, it should be noted that many studies of heart failure find a decrease of SERCA expression.86 We speculate that the consequent decrease of Ca2+ buffering would compensate in those situations where an increase of myofilament buffering is expected and worsen where there is a decrease. Again, it will be important to repeat these studies of heart failure while measuring buffering directly.
It is also important to reemphasize the potential effects (Figure 4C) of changes of diastolic [Ca2+]i and, thence, of buffering power on the amplitude of the calcium transient. A major problem here is the paucity of measurements of diastolic [Ca2+]i. It is essential that studies on heart failure ask the simple question: how accurately has diastolic [Ca2+]i been measured and can it be excluded that changes (for example, between animals or in disease) account for the observed changes of systolic [Ca2+]i?
Work from Metzger and colleagues has demonstrated that changes of Ca2+ buffering may not simply be involved in the development and consequences of heart failure, but may also be used to treat it. They suggested that impaired relaxation in heart failure could be ameliorated by adding intracellular buffers. They noted that fast Ca2+ buffers slow both the rise and fall of [Ca2+]i and decrease the amplitude of the Ca2+ transient, and, instead, they advocated the use of parvalbumin, a skeletal muscle Ca2+ buffer. This has the important property that it binds Ca2+ slowly because Mg2+ has to dissociate first and, therefore, there is little attenuation of the peak Ca2+ transient. It will bind Ca2+ during diastole thereby improving diastolic performance. Incorporation of α-parvalbumin was shown to accelerate the decay of [Ca2+]i with no effect on peak [Ca2+]i and (see Figure 5B) also reversed the slowing of relaxation produced by experimental hypothyroidism72 and in the Dahl salt-sensitive rat model of diastolic dysfunction.87 Subsequent work has turned to altering the structure and thence the relative Ca2+ and Mg2+ affinities of parvalbumin analogs to improve the effects.88,89 In general, these effects of parvalbumin highlight the potential importance of endogenous slow buffers such as the Mg2+ site of TnC and myosin.
Ca2+ Buffering and Hypertrophic Cardiomyopathy
Several studies have examined the molecular basis of familial hypertrophic cardiomyopathy (FHC). Much of this work involves the effects of mutations in thin filament proteins such as troponin and tropomyosin, which are among the causes of FHC. Robinson et al22 showed that mutations causing hypertrophic cardiomyopathy increased the binding affinity of Ca2+ to myofilaments (as assessed with a fluorescent troponin) and presumably therefore Ca2+ buffering. They proposed that alterations of buffering might lead to pathological changes of the Ca2+ transient. Troponin mutations were subsequently investigated in a mouse model of the related condition of restrictive cardiomyopathy and the predicted decreased amplitude and slowed decay of the Ca2+ transient observed.23,90 In addition, myofilament Ca2+ sensitization with EMD 57033 mimicked the effects of troponin T mutations on Ca2+ buffering and the Ca2+ transient.23 A recent study used adenovirus to infect isolated myocytes with troponin or tropomyosin mutations and, again, found an increase of diastolic [Ca2+]i.24 Although the above results would be expected from an increase of buffering power, it has been reported that there is a decrease of SERCA expression that may also contribute.91 This study also found that the late Na+ current inhibitor ranolazine abolished the slowing of decay of the Ca2+ current. Although no data are available, it seems unlikely that ranolazine would affect Ca2+ buffering. It may therefore be that some of the effects of thin filament mutations are directly attributable to Ca2+ buffering, and others are a secondary consequence of the resulting heart failure, possibly attributable to decreased SERCA.
As mentioned in an earlier section, the Mg2+ sites on troponin are important contributors to buffering at low [Ca2+]i. It is therefore interesting that one of the mutations associated with FHC (D145E) greatly decreases the affinity of Ca2+ binding to these sites.92 At first sight, this might appear to contrast with the association between FHC and the increased affinity reviewed above. These observations may be reconciled by noting that a decrease in affinity of the very-high-affinity Mg2+ TnC sites will actually increase Ca2+ buffering power in the systolic range of [Ca2+]i.
Ca2+ Buffering and Arrhythmias
Ventricular arrhythmias constitute a major cause of death in FHC.93 The Knollmann group has investigated the underlying mechanisms in transgenic mice. Incorporation of mutations in troponin T or tropomyosin led to ventricular tachycardia. These mutations also sensitized the contractile machinery to activation by Ca2+ (Figure 5C) with those that produced the greatest incidence of ventricular tachycardias and arrhythmias having the greatest Ca-sensitizing effect.73 A causal link between Ca sensitization and arrhythmogenesis was provided by showing both that EMD 57033 caused arrhythmias and the contractile uncoupler blebbistatin decreased both Ca sensitivity of the contractile machinery and arrhythmia susceptibility. These arrhythmias were accompanied by a shortening and triangulation of the action potential, and electric repolarization alternans, as well (see below). Subsequent work using myocytes derived from human-induced pluripotent stem cells reproduced these effects of increased Ca2+ buffering by myofilaments on action potential shape and suggested that the shortened, triangulated action potential could be attributable to increased buffering decreasing the amplitude of the systolic Ca2+ transient and thereby the inward (depolarizing) NCX current.94 Although this is an attractive explanation, it is also worth noting that (see above), as well as decreasing the amplitude of the Ca2+ transient, increased buffering slows decay, making it harder to predict the net effect of increased buffering on NCX current. Another article showed that, when regular pacing was terminated by a pause, the next Ca2+ transient was larger than control and this effect was more prominent in troponin T mutations that sensitize to activation by Ca2+.23 This effect was attributed to a higher cell Ca2+ content in the mutant during stimulation, with the excess Ca2+ being taken up by the SR such that release after a pause results in a prolonged action potential, increasing the probability of an arrhythmogenic early afterdepolarization. Any increase of diastolic [Ca2+]i and consequent decrease of buffer power may also increase the rise of [Ca2+]i caused by release from the SR. In the work reviewed above, the increase of buffering was a consequence of genetic changes. Similar increases of myofilament Ca2+ sensitivity have been reported following myocardial infarction81 where manoeuvers that decrease Ca2+ sensitivity were found to abolish ventricular tachycardia following pauses of stimulation. As reviewed above, an increase of Ca2+ buffering is proarrhythmogenic. It has also been shown, however, that addition of the buffer EGTA can prevent the propagation of arrhythmogenic Ca2+ waves,95 and therefore the net effect may be more complicated.
The induction of alternans of the action potential duration by increased Ca2+ buffering attributable to thin filament mutations73 may be a consequence of the slowed decay of the Ca2+ transient, resulting in incomplete recovery at the time of the next stimulus at increased rates. This would be analogous to the idea that the increased propensity of endocardium in comparison with epicardium to alternans is associated with a more slowly decaying Ca2+ transient because of lower SERCA expression.59 In contrast, a modeling study has predicted that increasing cytoplasmic Ca2+ buffering should decrease the probability of alternans occurring by decreasing the probability that Ca2+ released from the SR induces further Ca2+ release from neighboring release sites.96 This is consistent with the experimental demonstration, in whole mouse hearts, that addition of the buffer EGTA decreased the occurrence of alternans48 and may be related to the experimental observation that increasing cytoplasmic Ca2+ buffering decreases the frequency of propagating Ca2+ waves97 and makes Ca2+ sparks terminate earlier.98 Further work is clearly required in understanding the relationship between Ca2+ buffering and alternans.
Why Do Cells Have Ca2+ Buffers?
A high level of Ca2+ buffering is not unique to cardiac myocytes. For example, ≈99% of the Ca2+ entering chromaffin cells binds to cytoplasmic buffers.99 The presence of Ca2+ buffers means that much larger movements of total Ca2+ are required to produce a given change of [Ca2+]i. Given that calcium movements account for up to 30% of the total energy consumption of the heart,100 one might wonder why evolution has resulted in such strong buffering. There are several explanations. (1) It may be an inescapable consequence of the fact that using Ca2+ as a second messenger requires high concentrations of Ca2+ binding proteins, for example, to activate contraction. (2) A high Ca2+ buffering may stabilize Ca2+ signaling by stopping an abnormal increase of [Ca2+]i in one part of a cell propagating throughout the cell. In this context, it is worth noting that (Figure 2Aii) the dependence of buffer power on [Ca2+]i means that the buffer power is much lower in systole than diastole. This may help Ca2+ release during systole, but protect against it in diastole. (3) The need for buffering may relate to the low intracellular concentration of calcium. A diastolic concentration of 100 nmol/L equates to 6×1016 ions per liter corresponding to a mean distance between ions of 0.25 µm. Soeller and Cannell8 have modeled Ca2+ fluxes into the space between the transverse tubule and SR (dyad). They calculated that, at a concentration of 100 nmol/L, each dyad would contain between 0.007 and 0.028 Ca2+ ions. At 10 µmol/L, there will be between 0.7 and 2.8 ions. This would make it impossible to control [Ca2+]i in a stable manner because a single Ca2+ transported into or out of the space would result in an enormous fractional change of [Ca2+]i. As pointed out previously by Bers,35 at such low concentrations, chance will determine whether a transporter interacts with an ion. In contrast, if total Ca2+ is 100 times the free then there will be between 70 and 280 ions per cleft. (4) If troponin was the only buffer, then virtually all the total Ca2+ would be bound to troponin irrespective of its Kd. This would make it impossible to change force by altering Kd because this requires other buffers to take up a fraction of the total Ca.
Conclusions
The concentration of buffered calcium in cytoplasm is 2 orders of magnitude greater than that of the free concentration, and, therefore, the buffers have an enormous effect on calcium signaling. There is a need for more work investigating whether changes of buffer properties, either directly or secondary to changes of diastolic [Ca2+]i, contribute to alterations of calcium handling and contractility. The limited human data reviewed above and extrapolated from animal models argue that changes of Ca2+ buffering are important in determining both inotropy and proarrhythmic status in conditions such as cardiomyopathies (dilated cardiomyopathy and hypertrophic cardiomyopathy) and ischemic heart failure in both health and disease. Clarification will require more work on human tissue.
Acknowledgments
We are indebted to F. Burton and S. Wray for comments on an earlier version of the manuscript and to Q. Lachaud for the design of Figure 1.
Sources of Funding
This work from is supported by grants from the British Heart Foundation (PG/17/12/32847 to Dr Smith and CH/2000004/12801 to Dr Eisner).
Disclosures
None.
Footnotes
References
- 1.Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- 2.Eisner DA, Caldwell JL, Kistamás K, Trafford AW. Calcium and excitation-contraction coupling in the heart. Circ Res. 2017;121:181–195. doi: 10.1161/CIRCRESAHA.117.310230. doi: 10.1161/CIRCRESAHA.117.310230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol. 1983;245:C1–14. doi: 10.1152/ajpcell.1983.245.1.C1. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- 4.Williams RJ. The evolution of calcium biochemistry. Biochim Biophys Acta. 2006;1763:1139–1146. doi: 10.1016/j.bbamcr.2006.08.042. doi: 10.1016/j.bbamcr.2006.08.042. [DOI] [PubMed] [Google Scholar]
- 5.Carafoli E, Krebs J. Why calcium? How calcium became the best communicator. J Biol Chem. 2016;291:20849–20857. doi: 10.1074/jbc.R116.735894. doi: 10.1074/jbc.R116.735894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kretsinger RH, Nockolds CE. Carp muscle calcium-binding protein. II. Structure determination and general description. J Biol Chem. 1973;248:3313–3326. [PubMed] [Google Scholar]
- 7.Saucerman JJ, Bers DM. Calmodulin mediates differential sensitivity of CaMKII and calcineurin to local Ca2+ in cardiac myocytes. Biophys J. 2008;95:4597–4612. doi: 10.1529/biophysj.108.128728. doi: 10.1529/biophysj.108.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soeller C, Cannell MB. Numerical simulation of local calcium movements during L-type calcium channel gating in the cardiac diad. Biophys J. 1997;73:97–111. doi: 10.1016/S0006-3495(97)78051-2. doi: 10.1016/S0006-3495(97)78051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robertson SP, Johnson JD, Potter JD. The time-course of Ca2+ exchange with calmodulin, troponin, parvalbumin, and myosin in response to transient increases in Ca2+. Biophys J. 1981;34:559–569. doi: 10.1016/S0006-3495(81)84868-0. doi: 10.1016/S0006-3495(81)84868-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao WD, Backx PH, Azan-Backx M, Marban E. Myofilament Ca2+ sensitivity in intact versus skinned rat ventricular muscle. Circ Res. 1994;74:408–415. doi: 10.1161/01.res.74.3.408. [DOI] [PubMed] [Google Scholar]
- 11.Shannon TR, Ginsburg KS, Bers DM. Reverse mode of the sarcoplasmic reticulum calcium pump and load-dependent cytosolic calcium decline in voltage-clamped cardiac ventricular myocytes. Biophys J. 2000;78:322–333. doi: 10.1016/S0006-3495(00)76595-7. doi: 10.1016/S0006-3495(00)76595–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Post JA, Langer GA. Sarcolemmal calcium binding sites in heart: I. Molecular origin in “gas-dissected” sarcolemma. J Membr Biol. 1992;129:49–57. doi: 10.1007/BF00232054. [DOI] [PubMed] [Google Scholar]
- 13.Pan BS, Solaro RJ. Calcium-binding properties of troponin C in detergent-skinned heart muscle fibers. J Biol Chem. 1987;262:7839–7849. [PubMed] [Google Scholar]
- 14.Picht E, Zima AV, Shannon TR, Duncan AM, Blatter LA, Bers DM. Dynamic calcium movement inside cardiac sarcoplasmic reticulum during release. Circ Res. 2011;108:847–856. doi: 10.1161/CIRCRESAHA.111.240234. doi: 10.1161/CIRCRESAHA.111.240234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swietach P, Youm JB, Saegusa N, Leem CH, Spitzer KW, Vaughan-Jones RD. Coupled Ca2+/H+ transport by cytoplasmic buffers regulates local Ca2+ and H+ ion signaling. Proc Natl Acad Sci USA. 2013;110:E2064–E2073. doi: 10.1073/pnas.1222433110. doi: 10.1073/pnas.1222433110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baran EJ. Metal complexes of carnosine. Biochemistry (Mosc) 2000;65:789–797. [PubMed] [Google Scholar]
- 17.Naraghi M. T-jump study of calcium binding kinetics of calcium chelators. Cell Calcium. 1997;22:255–268. doi: 10.1016/s0143-4160(97)90064-6. doi: 10.1016/S0143-4160(97)90064–6. [DOI] [PubMed] [Google Scholar]
- 18.Smith PD, Liesegang GW, Berger RL, Czerlinski G, Podolsky RJ. A stopped-flow investigation of calcium ion binding by ethylene glycol bis(beta-aminoethyl ether)-N,N’-tetraacetic acid. Anal Biochem. 1984;143:188–195. doi: 10.1016/0003-2697(84)90575-x. [DOI] [PubMed] [Google Scholar]
- 19.Kawasaki H, Kretsinger RH. Calcium-binding proteins. 1: EF-hands. Protein Profile. 1994;1:343–517. [PubMed] [Google Scholar]
- 20.Sipido KR, Wier WG. Flux of Ca2+ across the sarcoplasmic reticulum of guinea-pig cardiac cells during excitation-contraction coupling. J Physiol. 1991;435:605–630. doi: 10.1113/jphysiol.1991.sp018528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blanchard EM, Solaro RJ. Inhibition of the activation and troponin calcium binding of dog cardiac myofibrils by acidic pH. Circ Res. 1984;55:382–391. doi: 10.1161/01.res.55.3.382. [DOI] [PubMed] [Google Scholar]
- 22.Robinson P, Griffiths PJ, Watkins H, Redwood CS. Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ Res. 2007;101:1266–1273. doi: 10.1161/CIRCRESAHA.107.156380. doi: 10.1161/CIRCRESAHA.107.156380. [DOI] [PubMed] [Google Scholar]
- 23.Schober T, Huke S, Venkataraman R, Gryshchenko O, Kryshtal D, Hwang HS, Baudenbacher FJ, Knollmann BC. Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause-dependent Ca-triggered arrhythmia. Circ Res. 2012;111:170–179. doi: 10.1161/CIRCRESAHA.112.270041. doi: 10.1161/CIRCRESAHA.112.270041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson P, Liu X, Sparrow A, Patel S, Zhang YH, Casadei B, Watkins H, Redwood C. Hypertrophic cardiomyopathy mutations increase myofilament Ca2+ buffering, alter intracellular Ca2+ handling, and stimulate Ca2+-dependent signaling. J Biol Chem. 2018;293:10487–10499. doi: 10.1074/jbc.RA118.002081. doi: 10.1074/jbc.RA118.002081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siddiqui JK, Tikunova SB, Walton SD, Liu B, Meyer M, de Tombe PP, Neilson N, Kekenes-Huskey PM, Salhi HE, Janssen PM, Biesiadecki BJ, Davis JP. Myofilament calcium sensitivity: consequences of the effective concentration of troponin I. Front Physiol. 2016;7:632. doi: 10.3389/fphys.2016.00632. doi: 10.3389/fphys.2016.00632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Powers FM, Solaro RJ. Caffeine alters cardiac myofilament activity and regulation independently of Ca2+ binding to troponin C. Am J Physiol. 1995;268(6 Pt 1):C1348–C1353. doi: 10.1152/ajpcell.1995.268.6.C1348. doi: 10.1152/ajpcell.1995.268.6.C1348. [DOI] [PubMed] [Google Scholar]
- 27.Negele JC, Dotson DG, Liu W, Sweeney HL, Putkey JA. Mutation of the high affinity calcium binding sites in cardiac troponin C. J Biol Chem. 1992;267:825–831. [PubMed] [Google Scholar]
- 28.Higgins ER, Cannell MB, Sneyd J. A buffering SERCA pump in models of calcium dynamics. Biophys J. 2006;91:151–163. doi: 10.1529/biophysj.105.075747. doi: 10.1529/biophysj.105.075747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shannon TR, Wang F, Puglisi J, Weber C, Bers DM. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J. 2004;87:3351–3371. doi: 10.1529/biophysj.104.047449. doi: 10.1529/biophysj.104.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michailova A, DelPrincipe F, Egger M, Niggli E. Spatiotemporal features of Ca2+ buffering and diffusion in atrial cardiac myocytes with inhibited sarcoplasmic reticulum. Biophys J. 2002;83:3134–3151. doi: 10.1016/S0006-3495(02)75317-4. doi: 10.1016/S0006-3495(02)75317-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Solaro RJ, Wise RM, Shiner JS, Briggs FN. Calcium requirements for cardiac myofibrillar activation. Circ Res. 1974;34:525–530. doi: 10.1161/01.res.34.4.525. [DOI] [PubMed] [Google Scholar]
- 32.Pierce GN, Philipson KD, Langer GA. Passive calcium-buffering capacity of a rabbit ventricular homogenate preparation. Am J Physiol. 1985;249(3 pt 1):C248–C255. doi: 10.1152/ajpcell.1985.249.3.C248. doi: 10.1152/ajpcell.1985.249.3.C248. [DOI] [PubMed] [Google Scholar]
- 33.Hove-Madsen L, Bers DM. Passive Ca buffering and SR Ca uptake in permeabilized rabbit ventricular myocytes. Am J Physiol. 1993;264(3 pt 1):C677–C686. doi: 10.1152/ajpcell.1993.264.3.C677. doi: 10.1152/ajpcell.1993.264.3.C677. [DOI] [PubMed] [Google Scholar]
- 34.Berlin JR, Bassani JW, Bers DM. Intrinsic cytosolic calcium buffering properties of single rat cardiac myocytes. Biophys J. 1994;67:1775–1787. doi: 10.1016/S0006-3495(94)80652-6. doi: 10.1016/S0006-3495(94)80652-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. 2nd ed. Dordrecht, The Netherlands: Kluwer Academic; 2001. [Google Scholar]
- 36.Trafford AW, Díaz ME, Eisner DA. A novel, rapid and reversible method to measure Ca buffering and time-course of total sarcoplasmic reticulum Ca content in cardiac ventricular myocytes. Pflugers Arch. 1999;437:501–503. doi: 10.1007/s004240050808. doi: 10.1007/s004240050808. [DOI] [PubMed] [Google Scholar]
- 37.Kuratomi S, Matsuoka S, Sarai N, Powell T, Noma A. Involvement of Ca2+ buffering and Na+/Ca2+ exchange in the positive staircase of contraction in guinea-pig ventricular myocytes. Pflugers Arch. 2003;446:347–355. doi: 10.1007/s00424-003-1023-1. doi: 10.1007/s00424-003-1023-1. [DOI] [PubMed] [Google Scholar]
- 38.Negretti N, O’Neill SC, Eisner DA. The relative contributions of different intracellular and sarcolemmal systems to relaxation in rat ventricular myocytes. Cardiovasc Res. 1993;27:1826–1830. doi: 10.1093/cvr/27.10.1826. doi: 10.1093/cvr/27.10.1826. [DOI] [PubMed] [Google Scholar]
- 39.Trafford AW, Díaz ME, Sibbring GC, Eisner DA. Modulation of CICR has no maintained effect on systolic Ca2+: simultaneous measurements of sarcoplasmic reticulum and sarcolemmal Ca2+ fluxes in rat ventricular myocytes. J Physiol. 2000;522(pt 2):259–270. doi: 10.1111/j.1469-7793.2000.t01-2-00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Briston SJ, Dibb KM, Solaro RJ, Eisner DA, Trafford AW. Balanced changes in Ca buffering by SERCA and troponin contribute to Ca handling during β-adrenergic stimulation in cardiac myocytes. Cardiovasc Res. 2014;104:347–354. doi: 10.1093/cvr/cvu201. doi: 10.1093/cvr/cvu201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steenbergen C, Murphy E, Levy L, London RE. Elevation in cytosolic free calcium concentration early in myocardial ischemia in perfused rat heart. Circ Res. 1987;60:700–707. doi: 10.1161/01.res.60.5.700. [DOI] [PubMed] [Google Scholar]
- 42.Harding DP, Smith GA, Metcalfe JC, Morris PG, Kirschenlohr HL. Resting and end-diastolic [Ca2+]i measurements in the Langendorff-perfused ferret heart loaded with a 19F NMR indicator. Magn Reson Med. 1993;29:605–615. doi: 10.1002/mrm.1910290505. [DOI] [PubMed] [Google Scholar]
- 43.Díaz ME, Trafford AW, Eisner DA. The effects of exogenous calcium buffers on the systolic calcium transient in rat ventricular myocytes. Biophys J. 2001;80:1915–1925. doi: 10.1016/S0006-3495(01)76161-9. doi: 10.1016/S0006-3495(01)76161-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yaniv Y, Stern MD, Lakatta EG, Maltsev VA. Mechanisms of beat-to-beat regulation of cardiac pacemaker cell function by Ca2+ cycling dynamics. Biophys J. 2013;105:1551–1561. doi: 10.1016/j.bpj.2013.08.024. doi: 10.1016/j.bpj.2013.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaestner L, Scholz A, Tian Q, Ruppenthal S, Tabellion W, Wiesen K, Katus HA, Müller OJ, Kotlikoff MI, Lipp P. Genetically encoded Ca2+ indicators in cardiac myocytes. Circ Res. 2014;114:1623–1639. doi: 10.1161/CIRCRESAHA.114.303475. doi: 10.1161/CIRCRESAHA.114.303475. [DOI] [PubMed] [Google Scholar]
- 46.Neher E. Usefulness and limitations of linear approximations to the understanding of Ca++ signals. Cell Calcium. 1998;24:345–357. doi: 10.1016/s0143-4160(98)90058-6. doi: 10.1016/S0143-4160(98)90058-6. [DOI] [PubMed] [Google Scholar]
- 47.Sankaranarayanan R, Kistamás K, Greensmith DJ, Venetucci LA, Eisner DA. Systolic [Ca2+ ]i regulates diastolic levels in rat ventricular myocytes. J Physiol. 2017;595:5545–5555. doi: 10.1113/JP274366. doi: 10.1113/JP274366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kornyeyev D, Reyes M, Escobar AL. Luminal Ca(2+) content regulates intracellular Ca(2+) release in subepicardial myocytes of intact beating mouse hearts: effect of exogenous buffers. Am J Physiol Heart Circ Physiol. 2010;298:H2138–H2153. doi: 10.1152/ajpheart.00885.2009. doi: 10.1152/ajpheart.00885.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lytton J, Westlin M, Burk SE, Shull GE, MacLennan DH. Functional comparisons between isoforms of the sarcoplasmic or endoplasmic reticulum family of calcium pumps. J Biol Chem. 1992;267:14483–14489. [PubMed] [Google Scholar]
- 50.Barcenas-Ruiz L, Beuckelmann DJ, Wier WG. Sodium-calcium exchange in heart: membrane currents and changes in [Ca2+]i. Science. 1987;238:1720–1722. doi: 10.1126/science.3686010. [DOI] [PubMed] [Google Scholar]
- 51.MacQuaide N, Dempster J, Smith GL. Measurement and modeling of Ca2+ waves in isolated rabbit ventricular cardiomyocytes. Biophys J. 2007;93:2581–2595. doi: 10.1529/biophysj.106.102293. doi: 10.1529/biophysj.106.102293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gattoni S, Røe ÅT, Frisk M, Louch WE, Niederer SA, Smith NP. The calcium-frequency response in the rat ventricular myocyte: an experimental and modelling study. J Physiol. 2016;594:4193–4224. doi: 10.1113/JP272011. doi: 10.1113/JP272011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hunter PJ, McCulloch AD, ter Keurs HE. Modelling the mechanical properties of cardiac muscle. Prog Biophys Mol Biol. 1998;69:289–331. doi: 10.1016/s0079-6107(98)00013-3. [DOI] [PubMed] [Google Scholar]
- 54.Díaz ME, Trafford AW, Eisner DA. The role of intracellular Ca buffers in determining the shape of the systolic Ca transient in cardiac ventricular myocytes. Pflugers Arch. 2001;442:96–100. doi: 10.1007/s004240000509. doi: 10.1007/s004240000509. [DOI] [PubMed] [Google Scholar]
- 55.Wier WG, Hess P. Excitation-contraction coupling in cardiac Purkinje fibers. Effects of cardiotonic steroids on the intracellular [Ca2+] transient, membrane potential, and contraction. J Gen Physiol. 1984;83:395–415. doi: 10.1085/jgp.83.3.395. doi: 10.1016/S0079-6107(98)00013-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sankaranarayanan R, Li Y, Greensmith DJ, Eisner DA, Venetucci L. Biphasic decay of the Ca transient results from increased sarcoplasmic reticulum Ca leak. J Physiol. 2016;594:611–623. doi: 10.1113/JP271473. doi: 10.1113/JP271473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eisner DA. Ups and downs of calcium in the heart. J Physiol. 2018;596:19–30. doi: 10.1113/JP275130. doi: 10.1113/JP275130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eisner DA, Dibb KM, Trafford AW. The mechanism and significance of the slow changes of ventricular action potential duration following a change of heart rate. Exp Physiol. 2009;94:520–528. doi: 10.1113/expphysiol.2008.044008. doi: 10.1113/expphysiol.2008.044008. [DOI] [PubMed] [Google Scholar]
- 59.Laurita KR, Katra R, Wible B, Wan X, Koo MH. Transmural heterogeneity of calcium handling in canine. Circ Res. 2003;92:668–675. doi: 10.1161/01.RES.0000062468.25308.27. doi: 10.1161/01.RES.0000062468.25308.27. [DOI] [PubMed] [Google Scholar]
- 60.Soltysinska E, Olesen SP, Christ T, Wettwer E, Varró A, Grunnet M, Jespersen T. Transmural expression of ion channels and transporters in human nondiseased and end-stage failing hearts. Pflugers Arch. 2009;459:11–23. doi: 10.1007/s00424-009-0718-3. doi: 10.1007/s00424-009-0718-3. [DOI] [PubMed] [Google Scholar]
- 61.Kirchberber MA, Tada M, Katz AM. Phospholamban: a regulatory protein of the cardiac sarcoplasmic reticulum. Recent Adv Stud Cardiac Struct Metab. 1975;5:103–115. [PubMed] [Google Scholar]
- 62.Robertson SP, Johnson JD, Holroyde MJ, Kranias EG, Potter JD, Solaro RJ. The effect of troponin I phosphorylation on the Ca2+-binding properties of the Ca2+-regulatory site of bovine cardiac troponin. J Biol Chem. 1982;257:260–263. [PubMed] [Google Scholar]
- 63.Choi HS, Trafford AW, Orchard CH, Eisner DA. The effect of acidosis on systolic Ca2+ and sarcoplasmic reticulum calcium content in isolated rat ventricular myocytes. J Physiol. 2000;529(pt 3):661–668. doi: 10.1111/j.1469-7793.2000.00661.x. doi: 10.1111/j.1469-7793.2000.00661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boyman L, Hagen BM, Giladi M, Hiller R, Lederer WJ, Khananshvili D. Proton-sensing Ca2+ binding domains regulate the cardiac Na+/Ca2+ exchanger. J Biol Chem. 2011;286:28811–28820. doi: 10.1074/jbc.M110.214106. doi: 10.1074/jbc.M110.214106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walden AP, Dibb KM, Trafford AW. Differences in intracellular calcium homeostasis between atrial and ventricular myocytes. J Mol Cell Cardiol. 2009;46:463–473. doi: 10.1016/j.yjmcc.2008.11.003. doi: 10.1016/j.yjmcc.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 66.Clarke JD, Caldwell JL, Pearman CM, Eisner DA, Trafford AW, Dibb KM. Increased Ca buffering underpins remodelling of Ca2+ handling in old sheep atrial myocytes. J Physiol. 2017;595:6263–6279. doi: 10.1113/JP274053. doi: 10.1113/JP274053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Trafford AW, Clarke JD, Richards MA, Eisner DA, Dibb KM. Calcium signalling microdomains and the t-tubular system in atrial myocytes: potential roles in cardiac disease and arrhythmias. Cardiovasc Res. 2013;98:192–203. doi: 10.1093/cvr/cvt018. doi: 10.1093/cvr/cvt018. [DOI] [PubMed] [Google Scholar]
- 68.Blatter LA, Kockskämper J, Sheehan KA, Zima AV, Hüser J, Lipsius SL. Local calcium gradients during excitation-contraction coupling and alternans in atrial myocytes. J Physiol. 2003;546(pt 1):19–31. doi: 10.1113/jphysiol.2002.025239. doi: 10.1113/jphysiol.2002.025239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bootman MD, Higazi DR, Coombes S, Roderick HL. Calcium signalling during excitation-contraction coupling in mammalian atrial myocytes. J Cell Sci. 2006;119(Pt 19):3915–3925. doi: 10.1242/jcs.03223. doi: 10.1242/jcs.03223. [DOI] [PubMed] [Google Scholar]
- 70.Sheehan KA, Blatter LA. Regulation of junctional and non-junctional sarcoplasmic reticulum calcium release in excitation-contraction coupling in cat atrial myocytes. J Physiol. 2003;546(pt 1):119–135. doi: 10.1113/jphysiol.2002.026963. doi: 10.1113/jphysiol.2002.026963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Greiser M, Kerfant BG, Williams GS, Voigt N, Harks E, Dibb KM, Giese A, Meszaros J, Verheule S, Ravens U, Allessie MA, Gammie JS, van der Velden J, Lederer WJ, Dobrev D, Schotten U. Tachycardia-induced silencing of subcellular Ca2+ signaling in atrial myocytes. J Clin Invest. 2014;124:4759–4772. doi: 10.1172/JCI70102. doi: 10.1172/JCI70102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wahr PA, Michele DE, Metzger JM. Parvalbumin gene transfer corrects diastolic dysfunction in diseased cardiac myocytes. Proc Natl Acad Sci USA. 1999;96:11982–11985. doi: 10.1073/pnas.96.21.11982. doi: 10.1073/pnas.96.21.11982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, Potter JD, Knollmann BC. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest. 2008;118:3893–3903. doi: 10.1172/JCI36642. doi: 10.1172/JCI36642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Clarke JD, Caldwell JL, Horn MA, Bode EF, Richards MA, Hall MC, Graham HK, Briston SJ, Greensmith DJ, Eisner DA, Dibb KM, Trafford AW. Perturbed atrial calcium handling in an ovine model of heart failure: potential roles for reductions in the L-type calcium current. J Mol Cell Cardiol. 2015;79:169–179. doi: 10.1016/j.yjmcc.2014.11.017. doi: 10.1016/j.yjmcc.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dibb KM, Clarke JD, Horn MA, Richards MA, Graham HK, Eisner DA, Trafford AW. Characterization of an extensive transverse tubular network in sheep atrial myocytes and its depletion in heart failure. Circ Heart Fail. 2009;2:482–489. doi: 10.1161/CIRCHEARTFAILURE.109.852228. doi: 10.1161/CIRCHEARTFAILURE.109.852228. [DOI] [PubMed] [Google Scholar]
- 76.Macquaide N, Tuan HT, Hotta J, Sempels W, Lenaerts I, Holemans P, Hofkens J, Jafri MS, Willems R, Sipido KR. Ryanodine receptor cluster fragmentation and redistribution in persistent atrial fibrillation enhance calcium release. Cardiovasc Res. 2015;108:387–398. doi: 10.1093/cvr/cvv231. doi: 10.1093/cvr/cvv231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hobai IA, O’Rourke B. Enhanced Ca(2+)-activated Na(+)-Ca(2+) exchange activity in canine pacing-induced heart failure. Circ Res. 2000;87:690–698. doi: 10.1161/01.res.87.8.690. [DOI] [PubMed] [Google Scholar]
- 78.Briston SJ, Caldwell JL, Horn MA, Clarke JD, Richards MA, Greensmith DJ, Graham HK, Hall MC, Eisner DA, Dibb KM, Trafford AW. Impaired β-adrenergic responsiveness accentuates dysfunctional excitation-contraction coupling in an ovine model of tachypacing-induced heart failure. J Physiol. 2011;589(pt 6):1367–1382. doi: 10.1113/jphysiol.2010.203984. doi: 10.1113/jphysiol.2010.203984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wolff MR, Buck SH, Stoker SW, Greaser ML, Mentzer RM. Myofibrillar calcium sensitivity of isometric tension is increased in human dilated cardiomyopathies: role of altered beta-adrenergically mediated protein phosphorylation. J Clin Invest. 1996;98:167–176. doi: 10.1172/JCI118762. doi: 10.1172/JCI118762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wolff MR, Whitesell LF, Moss RL. Calcium sensitivity of isometric tension is increased in canine experimental heart failure. Circ Res. 1995;76:781–789. doi: 10.1161/01.res.76.5.781. [DOI] [PubMed] [Google Scholar]
- 81.Venkataraman R, Baldo MP, Hwang HS, Veltri T, Pinto JR, Baudenbacher FJ, Knollmann BC. Myofilament calcium de-sensitization and contractile uncoupling prevent pause-triggered ventricular tachycardia in mouse hearts with chronic myocardial infarction. J Mol Cell Cardiol. 2013;60:8–15. doi: 10.1016/j.yjmcc.2013.03.022. doi: 10.1016/j.yjmcc.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Waard MC, van der Velden J, Bito V, Ozdemir S, Biesmans L, Boontje NM, Dekkers DH, Schoonderwoerd K, Schuurbiers HC, de Crom R, Stienen GJ, Sipido KR, Lamers JM, Duncker DJ. Early exercise training normalizes myofilament function and attenuates left ventricular pump dysfunction in mice with a large myocardial infarction. Circ Res. 2007;100:1079–1088. doi: 10.1161/01.RES.0000262655.16373.37. doi: 10.1161/01.RES.0000262655.16373.37. [DOI] [PubMed] [Google Scholar]
- 83.Belin RJ, Sumandea MP, Kobayashi T, Walker LA, Rundell VL, Urboniene D, Yuzhakova M, Ruch SH, Geenen DL, Solaro RJ, de Tombe PP. Left ventricular myofilament dysfunction in rat experimental hypertrophy and congestive heart failure. Am J Physiol Heart Circ Physiol. 2006;291:H2344–H2353. doi: 10.1152/ajpheart.00541.2006. doi: 10.1152/ajpheart.00541.2006. [DOI] [PubMed] [Google Scholar]
- 84.Solaro RJ, van der Velden J. Why does troponin I have so many phosphorylation sites? Fact and fancy. J Mol Cell Cardiol. 2010;48:810–816. doi: 10.1016/j.yjmcc.2010.02.014. doi: 10.1016/j.yjmcc.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Marjot J, Kaier TE, Martin ED, Reji SS, Copeland ON, Iqbal M, Goodson B, Hamren S, Harding SE, Marber MS. Quantifying the release of biomarkers of myocardial necrosis from cardiac myocytes and intact myocardium. Clinical Chem. 2017;63:990–996. doi: 10.1373/clinchem.2016.264648. doi: 10.1373/clinchem.2016.264648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Meyer M, Schillinger W, Pieske B, Holubarsch C, Heilmann C, Posival H, Kuwajima G, Mikoshiba K, Just H, Hasenfuss G. Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation. 1995;92:778–784. doi: 10.1161/01.cir.92.4.778. doi: 10.1161/01.CIR.92.4.778. [DOI] [PubMed] [Google Scholar]
- 87.Rodenbaugh DW, Wang W, Davis J, Edwards T, Potter JD, Metzger JM. Parvalbumin isoforms differentially accelerate cardiac myocyte relaxation kinetics in an animal model of diastolic dysfunction. Am J Physiol Heart Circ Physiol. 2007;293:H1705–H1713. doi: 10.1152/ajpheart.00232.2007. doi: 10.1152/ajpheart.00232.2007. [DOI] [PubMed] [Google Scholar]
- 88.Zhang J, Shettigar V, Kindell D, Liu X, Lopez J, Yerrimuni V, Davis G, Davis J. Engineering parvalbumin for the heart: optimizing the Mg2+ binding properties of rat β-parvalbumin. Front Physiol. 2011;2:77. doi: 10.3389/fphys.2011.00077. doi: 10.3389/fphys.2011.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Asp ML, Sjaastad FV, Siddiqui JK, Davis JP, Metzger JM. Effects of modified parvalbumin EF-hand motifs on cardiac myocyte contractile function. Biophys J. 2016;110:2094–2105. doi: 10.1016/j.bpj.2016.03.037. doi: 10.1016/j.bpj.2016.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li Y, Zhang L, Jean-Charles PY, Nan C, Chen G, Tian J, Jin JP, Gelb IJ, Huang X. Dose-dependent diastolic dysfunction and early death in a mouse model with cardiac troponin mutations. J Mol Cell Cardiol. 2013;62:227–236. doi: 10.1016/j.yjmcc.2013.06.007. doi: 10.1016/j.yjmcc.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Coppini R, Mazzoni L, Ferrantini C, Gentile F, Pioner JM, Laurino A, Santini L, Bargelli V, Rotellini M, Bartolucci G, Crocini C, Sacconi L, Tesi C, Belardinelli L, Tardiff J, Mugelli A, Olivotto I, Cerbai E, Poggesi C. Ranolazine prevents phenotype development in a mouse model of hypertrophic cardiomyopathy. Circ Heart Fail. 2017;10:e003565. doi: 10.1161/CIRCHEARTFAILURE.116.003565. doi: 10.10.1161/CIRCHEARTFAILURE.116.003565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Swindle N, Tikunova SB. Hypertrophic cardiomyopathy-linked mutation D145E drastically alters calcium binding by the C-domain of cardiac troponin C. Biochemistry. 2010;49:4813–4820. doi: 10.1021/bi100400h. doi: 10.1021/bi100400h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Maron BJ. Sudden death in young athletes. N Engl J Med. 2003;349:1064–1075. doi: 10.1056/NEJMra022783. doi: 10.1056/NEJMra022783. [DOI] [PubMed] [Google Scholar]
- 94.Wang L, Kryshtal DO, Kim K, Parikh S, Cadar AG, Bersell KR, He H, Pinto JR, Knollmann BC. myofilament calcium-buffering dependent action potential triangulation in human-induced pluripotent stem cell model of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2017;70:2600–2602. doi: 10.1016/j.jacc.2017.09.033. doi: 10.1016/j.jacc.2017.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.MacQuaide N, Ramay HR, Sobie EA, Smith GL. Differential sensitivity of Ca2+ wave and Ca2+ spark events to ruthenium red in isolated permeabilised rabbit cardiomyocytes. J Physiol. 2010;588(pt 23):4731–4742. doi: 10.1113/jphysiol.2010.193375. doi: 10.1113/jphysiol.2010.193375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nivala M, Qu Z. Calcium alternans in a couplon network model of ventricular myocytes: role of sarcoplasmic reticulum load. Am J Physiol Heart Circ Physiol. 2012;303:H341–H352. doi: 10.1152/ajpheart.00302.2012. doi: 10.1152/ajpheart.00302.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nivala M, Ko CY, Nivala M, Weiss JN, Qu Z. Criticality in intracellular calcium signaling in cardiac myocytes. Biophys J. 2012;102:2433–2442. doi: 10.1016/j.bpj.2012.05.001. doi: 10.1016/j.bpj.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bovo E, Mazurek SR, Fill M, Zima AV. Cytosolic Ca2+ buffering determines the intra-SR Ca2+ concentration at which cardiac Ca2+ sparks terminate. Cell Calcium. 2015;58:246–253. doi: 10.1016/j.ceca.2015.06.002. doi: 10.1016/j.ceca.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Neher E, Augustine GJ. Calcium gradients and buffers in bovine chromaffin cells. J Physiol. 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gibbs CL, Loiselle DS, Wendt IR. Activation heat in rabbit cardiac muscle. J Physiol. 1988;395:115–130. doi: 10.1113/jphysiol.1988.sp016911. [DOI] [PMC free article] [PubMed] [Google Scholar]