

Viruses usurp host cell functions to advance their replicative agenda. HSV relies on cellular proteasome activity for successful infection. Proteasome inhibitors, such as MG132, block HSV infection at multiple stages of the infectious cycle. Targeting host cell processes for antiviral intervention is an unconventional approach that might limit antiviral resistance. Here we demonstrated that the proteasome inhibitor bortezomib, which is a clinically effective cancer drug, has the in vitro features of a promising anti-HSV therapeutic. Bortezomib inhibited HSV infection during the first hours of infection at nanomolar concentrations that were minimally cytotoxic. The mechanism of bortezomib’s inhibition of early HSV infection was to halt nucleocapsid transport to the nucleus and to stabilize the ND10 cellular defense complex. Bortezomib and acyclovir acted synergistically to inhibit HSV infection. Overall, we present evidence for the repurposing of bortezomib as a novel antiherpesviral agent and describe specific mechanisms of action.
KEYWORDS: antiviral agents, bortezomib, herpes simplex virus, human herpesviruses, proteasome
ABSTRACT
Viruses commandeer host cell 26S proteasome activity to promote viral entry, gene expression, replication, assembly, and egress. Proteasomal degradation activity is critical for herpes simplex virus (HSV) infection. The proteasome inhibitor bortezomib (also known as Velcade and PS-341) is a clinically effective antineoplastic drug that is FDA approved for treatment of hematologic malignancies such as multiple myeloma and mantle cell lymphoma. Low nanomolar concentrations of bortezomib inhibited infection by HSV-1, HSV-2, and acyclovir-resistant strains. Inhibition coincided with minimal cytotoxicity. Bortezomib did not affect attachment of HSV to cells or inactivate the virus directly. Bortezomib acted early in HSV infection by perturbing two distinct proteasome-dependent steps that occur within the initial hours of infection: the transport of incoming viral nucleocapsids to the nucleus and the virus-induced disruption of host nuclear domain 10 (ND10) structures. The combination of bortezomib with acyclovir demonstrated synergistic inhibitory effects on HSV infection. Thus, bortezomib is a novel potential therapeutic for HSV with a defined mechanism of action.
INTRODUCTION
Herpes simplex viruses (HSVs) are significant causes of morbidity and mortality in humans worldwide (1, 2). HSV-1 is primarily associated with self-limiting oral mucocutaneous disease and is the leading viral cause of blindness and encephalitis (3–5). Neonatal infections occur in ∼1 in 3,200 deliveries in the United States, and the majority of these infections result in central nervous system disease (6, 7). HSV-2 infection is the most common cause of genital ulcers worldwide (8). Genital herpes increases the risk of acquisition and transmission of human immunodeficiency virus type 1 (HIV-1) (9, 10). HSV causes lifelong latent infection for which there is no cure and no clinically effective vaccine. Acyclovir, the first specific and selective antiviral drug, is a guanosine analogue that targets HSV DNA replication for termination (11). Acyclovir-resistant strains can lead to severe disease, including disseminated infection of immune-dysregulated individuals (12, 13). New, effective therapeutics with different mechanisms of action are needed.
26S proteasomes are ∼2.5-MDa, ATP-dependent multisubunit proteolytic machines present in both the nucleus and the cytoplasm of all eukaryotic cells. They are composed of a barrel-shaped, central 20S core that contains the proteolytic activities. The proteasome executes the controlled degradation of functional proteins as well as the hydrolysis of aberrantly folded polypeptides (14). Proteasome-dependent degradation plays a key role in many cellular processes, such as cell cycle control, proliferation, and apoptosis (15). Viruses can commandeer proteasome activity to promote a diversity of functions critical for their replicative cycles (16). MG132 is a peptide aldehyde that competitively inhibits the degradative activity of the proteasome. Functional proteasomes facilitate HSV entry at a postpenetration stage. MG132 impairs incoming HSV capsid transport to the nuclear periphery (17). Proteasome inhibitors also block HSV infection by preventing degradation of promyelocytic leukemia (PML) isoforms, stabilizing nuclear domain 10 (ND10), and, in turn, preventing lytic replication (18–20). Thus, the proteasome is important for early events in HSV infection. Many antiviral drugs are designed to target viral proteins to ensure specificity and avoid toxicity, but such antivirals select for drug-resistant virus mutants. In contrast, antivirals that target cellular proteins required for viral replication, such as proteasomal components, avert the development of resistance.
Bortezomib [N-(2,3-pyrazine)carbonyl-l-phenylalanine-l-leucine boronic acid] (C19H25BN4O4; Fig. 1), originally known as PS-341, is a dipeptide boronic acid inhibitor of the proteasome. Proteasome inhibitors, including bortezomib, trigger apoptosis preferentially in tumor cells and serve as novel anticancer drugs (21). Bortezomib was clinically approved by the U.S. FDA to treat multiple myeloma, an incurable white blood cell cancer, and mantle cell lymphoma, a B-cell non-Hodgkin’s lymphoma, in 2003 and 2006, respectively (22–24). The boron atom (Fig. 1) binds directly to the chymotrypsin-like active site of the proteasome, which is located on the beta-5 subunit of the 20S particle (25).
FIG 1.
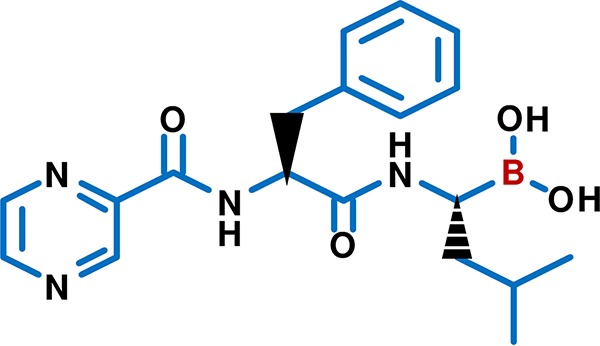
Chemical structure of the proteasome inhibitor bortezomib (C19H25BN4O4). Bortezomib [N-(2,3-pyrazine) carbonyl-l-phenylalanine-l-leucine boronic acid] inhibits the proteasome via binding of its boron atom (red) to the chymotrypsin-like active site of the proteasome. The figure was drawn with PubChem Sketcher (103).
Here, we report anti-HSV activity of the proteasome inhibitor bortezomib. Overall, we provide evidence that bortezomib inhibits infection of multiple HSV strains early in the infectious cycle, exhibits minimal cytotoxicity, mechanistically halts viral capsid transport to the nucleus and stabilizes ND10 structure, and exhibits synergy with acyclovir.
RESULTS
Bortezomib inhibits HSV infection.
To determine the effect of bortezomib on HSV infectivity, HSV and increasing concentrations of bortezomib were added to Vero cells. Bortezomib decreased the infectivity of HSV-1 strain KOS and HSV-2 strain G in a dose-dependent manner (Fig. 2A). Bortezomib also reduced the infectivity of acyclovir-resistant HSV-1 strains ACGr5, PAAr5, and dlsptk. These laboratory-constructed viruses contain mutations in the HSV thymidine kinase or DNA polymerase genes that render them resistant to acyclovir (26–28). Bortezomib was similarly effective at blocking infection by HSV-1 clinical isolate H129, derived from the brain of a herpes encephalitis patient (29). The concentrations of bortezomib that inhibited 50% of HSV infection (EC50) are depicted in Fig. 2 The EC50 values ranged from 3.7 to 50.6 nM, indicating that bortezomib, at low nanomolar concentrations, inhibits infection by wild-type, acyclovir-resistant, and clinically isolated HSV. In this assay, the EC50 value for acyclovir was 380 nM (see Fig. S1 in the supplemental material), which falls within the broad range of EC50s reported for acyclovir. Experiments with bortezomib and primary human foreskin fibroblasts (HFFs) yielded an EC50 of 6.0 nM (Fig. 2B).
FIG 2.
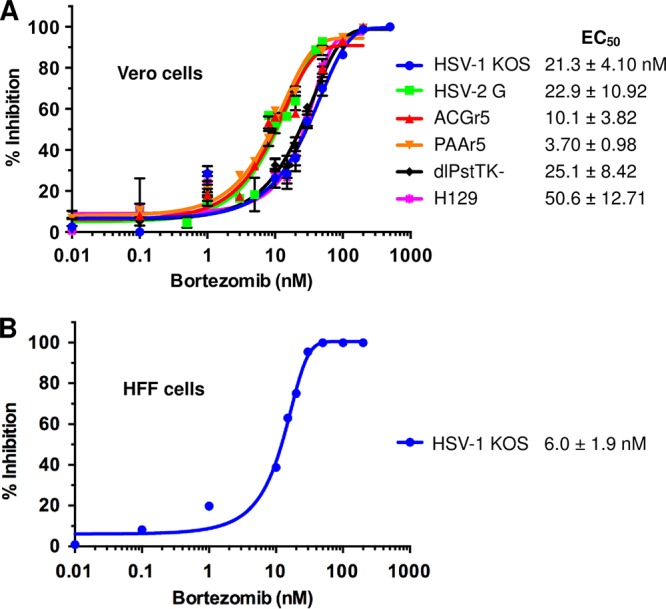
Bortezomib inhibits HSV infection. The indicated strains of HSV were added to (A) Vero cells (MOI of 0.004) or (B) HFF cells (MOI of 0.004) in the presence of increasing concentrations of bortezomib. At 18 to 24 h p.i., cells were fixed and assayed for HSV plaque formation. Plaque reduction is indicated as inhibition represented as a percentage of PFU obtained in the absence of drug. EC50 values for each virus as shown were calculated using GraphPad Prism software and range from 3.7 to 50.6 nM. Data are presented as graphed representatives of results from at least three experiments for each strain. Error bars, standard deviations (SD). EC50 data are presented as means ± standard errors of the means (SEM).
Acyclovir inhibition of HSV infection of Vero cells. HSV-1 KOS was added to Vero cells (MOI of 0.004) in the presence of increasing concentrations of acyclovir. Plaques were enumerated as described for Fig. 2. The EC50 was 380 nM. Download FIG S1, TIF file, 0.06 MB (58.7KB, tif) .
Copyright © 2019 Schneider et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Bortezomib exhibits low cytotoxicity at concentrations effective against HSV infection.
Proteasome inhibition ultimately leads to cell death (30). Thus, it was important to determine that bortezomib cytotoxicity did not explain the loss in HSV infectivity under our experimental conditions. We exposed Vero cells to doses of bortezomib similar to those utilized in the experiments whose results are presented Fig. 2. The cytotoxicity of bortezomib was quantitated via measurement of the levels of extracellular lactate dehydrogenase (LDH), an enzyme released upon cell death. Bortezomib-induced cytotoxicity peaked at ∼30% at concentrations of >10 µM but did not increase further, even at 1 mM (Fig. 3). Thus, the concentration of bortezomib that is cytotoxic to 50% of cells (CC50) is >1 mM. At bortezomib concentrations near the EC50 for HSV infection, cytotoxicity levels ranged from 0% to 8% (Fig. 3). Similar results were obtained with human foreskin fibroblasts (Fig. S2). Thus, bortezomib exhibits low cytotoxicity at concentrations that are effective against HSV infection.
FIG 3.
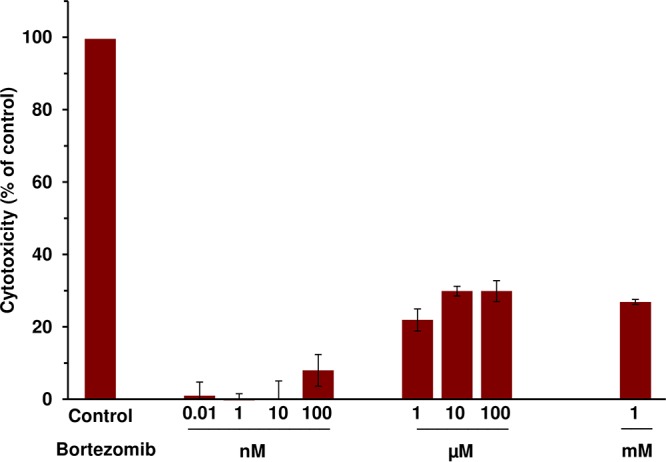
Bortezomib exhibits low cytotoxicity at concentrations effective against HSV infection. Bortezomib was added to Vero cells. At 24 h, LDH activity in the supernatant was assayed as a measure of cytotoxicity. Values are shown as percentages of detergent-lysed control values. Data presented are representative of results from at least three experiments. Error bars, SD.
Bortezomib cytotoxicity on HFF cells. Increasing concentrations of bortezomib were added to HFF cells for 24 h, and cytotoxicity was determined via LDH activity. Data presented are representative of results from three experiments. Error bars, SEM. Download FIG S2, TIF file, 0.2 MB (190.9KB, tif) .
Copyright © 2019 Schneider et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Bortezomib is effective when added prior to 3 h postinfection (p.i.).
To begin to determine the mechanism by which bortezomib blocks HSV infection, a time-of-addition study was implemented. HSV-1 KOS (multiplicity of infection [MOI] of 0.001) was added to Vero cells. At time points from 0 to 6 h p.i., 200 nM bortezomib was added to the infected cells. HSV-1 titers were determined at 24 h p.i. The later bortezomib was added, the less of an effect it had on HSV infection (Fig. 4). Bortezomib added at time zero almost completely inhibited HSV infection but consistently lost effectiveness over time, inhibiting only minimally after 3 h p.i. At least two proteasome-dependent processes occur during the first hours of HSV-1 infection. These results suggest bortezomib acts at an early step of HSV infection that requires the proteasome.
FIG 4.

Bortezomib is effective when added prior to 3 h p.i. HSV-1 KOS was added to Vero cells (MOI of 0.001). At the indicated times p.i., 200 nM bortezomib was added. At 18 to 24 h p.i., plaques were enumerated. The values representing mock-treated samples were set to 100%. Data presented are representative of results from three experiments. Error bars, SEM.
Bortezomib is not virucidal against HSV.
We determined whether the inhibitory effect of bortezomib on HSV infection was due to a direct, inactivating effect on viral particles. HSV-1 KOS was treated with 100 nM bortezomib for 1 h. The virus-drug mixture was diluted to noninhibitory concentrations of bortezomib, and titers were determined on Vero cells. The infectivity of the bortezomib-treated samples was similar to that seen with the vehicle control (Fig. 5). This result suggests that bortezomib does not have a direct, virucidal effect on the infectivity of HSV particles.
FIG 5.
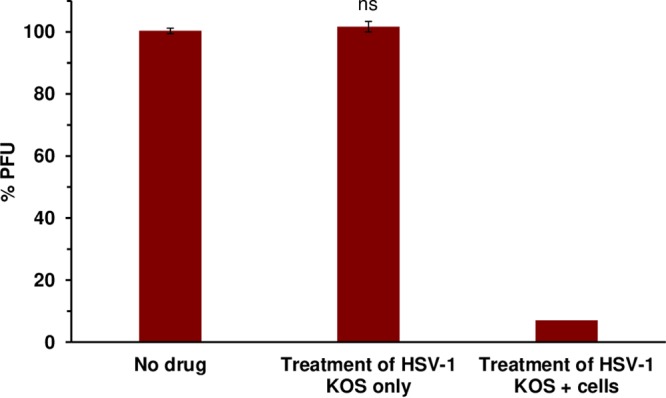
Bortezomib does not exhibit virucidal activity. HSV-1 KOS virions were treated with 100 nM bortezomib at 37°C for 1 h. Bortezomib was diluted to reach noninhibitory concentrations, and titers were determined on Vero cells. Data presented are representative of results from three experiments. Error bars, SEM. ns, not significant (compared to no-drug treatment).
HSV attachment to cells is unaltered by bortezomib.
To rule out the possibility that the bortezomib inhibition was due to an effect on viral attachment to the cell surface, HSV-1 KOS (40 genomes/cell) was added to Vero cells on ice for 1 h at 4°C in the presence of 200 nM bortezomib, a vehicle control, or a heparin treatment control. Cell-attached HSV-1 was quantitated by quantitative PCR (qPCR). Bortezomib-treated HSV-1 attached to cells in a manner similar to that seen with mock-treated HSV-1 (Fig. 6A). Control soluble heparin inhibited HSV-1 attachment to cells by >84%. These results suggest that the inhibitory activity of bortezomib is not due to a defect in HSV-1 attachment to cells.
FIG 6.

Bortezomib does not affect HSV attachment to cells but inhibits transport of the entering capsid to the nucleus. (A) HSV-1 KOS was added to Vero cells (40 genome copies/cell) in the presence of DMSO control (No drug), 500 nM bortezomib, or 2 µg/ml heparin control. Samples were subjected to spinoculation at 200 × g at 4°C for 1 h. After three washes, cell-associated HSV levels were determined by qPCR. Data presented represent means of results from three experiments. Error bars, SEM; ns, not significant; *, P value of <0.05 (compared to no drug). (B to D) HSV-1 K26GFP was added to Vero cells on coverslips in the presence of (B) DMSO control or (C) 100 nM bortezomib or (D) 500 nM bortezomib for 2.5 h. Cells were fixed and stained with DAPI nuclear stain and visualized. Data presented are representative of results from at least two experiments.
Transport of the HSV capsid to the nucleus is halted by bortezomib.
Following fusion with a cell membrane, entering HSV nucleocapsids are transported in a proteasome-dependent manner to the nucleus, the site of herpesviral genome replication (17). To determine whether this step in the viral life cycle is affected by bortezomib, HSV-1 K26GFP was added to Vero cells in the presence of 100 or 500 nM bortezomib at 37°C. Herpesviruses utilize multiple entry pathways in a cell-specific manner (31). HSV-1 entry into Vero cells proceeds via direct penetration with the host cell plasma membrane (32, 33). By 2.5 h p.i., in untreated cells, capsids were detected at the nuclear periphery (Fig. 6B). In contrast, in cells treated with bortezomib, HSV-1 capsids were halted at the cell periphery (Fig. 6C and D). Thus, bortezomib impacts HSV-1 infection at an early step, prior to capsid arrival at the nucleus.
HSV-induced ND10 disruption is prevented in the presence of bortezomib.
Host cell ND10 nuclear bodies contain many proteins responsible for normal functions, including cell cycle regulation, apoptosis, gene transcription, and antiviral defense mechanisms (34–36). A hallmark of infection by viruses, including HSV, is disruption of ND10 (35). HSV-induced disruption of ND10 is proteasome mediated and dependent on viral immediate early protein infected cell protein 0 (ICP0) (20). MG132 halts virus-mediated ND10 dispersal (20). To determine if bortezomib blocks the HSV-triggered dissolution of host ND10, we infected Vero cells with HSV-1 KOS and visualized PML, a major protein component of ND10, via immunofluorescence microscopy. In uninfected, untreated control samples, PML was present in punctate nuclear dots (Fig. S3). Upon infection, confirmed by the presence of HSV-1 ICP4, vehicle control-treated samples lacked punctate dots. Instead, there was diffuse PML throughout the nucleus (Fig. 7A to C), indicating ND10 disruption triggered by HSV infection. The staining in Fig. 7 is consistent with antibody detection of ICP4 expressed by the infected cell (37). Tegument ICP4 is present at ∼150 copies per virion (38) and has not been detected in infected cells. When 200 or 500 nM bortezomib was added prior to infection, the punctate PML staining was sustained during infection (Fig. 7G and K, open triangles). Uninfected (ICP4-negative) cells were also detected when bortezomib was present (Fig. 7G and K, closed triangles), which might have been due to effects of bortezomib on incoming capsid transport. Interestingly, ICP4 staining was still detected in a subset of nuclei, suggesting that viral entry and ICP4 gene expression had occurred while the ND10 structure remained intact (open triangles). Transport of incoming HSV-1 capsids to the nucleus is largely proteasome dependent. However, proteasome-independent transport also occurs and is particularly notable when tegument ICP0 is missing from incoming particles (17, 39). The results represented in Fig. 6 and 7 are consistent with the notion that bortezomib shuts down HSV-1 infection at two steps. If the drug is ineffective at the earlier capsid transport step, it has a second opportunity to inhibit infection by preserving ND10. Overall, these results suggest that bortezomib prevents the disruption of the ND10 host cell defense complex, which is coincident with successful lytic replication of HSV.
FIG 7.
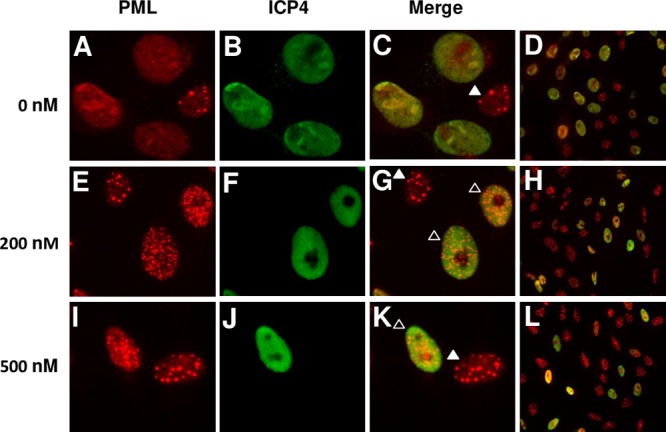
Bortezomib prevents virus-induced ND10 disruption. Vero cells were pretreated with (A to D) DMSO control or (E to H) 200 nM bortezomib or (I to L) 500 nM bortezomib for 15 to 18 min at 37°C. HSV-1 KOS was added to Vero cells (MOI of ∼0.8) for 6 h at 37°C in the continued presence of agent. Cells were fixed, permeabilized, and stained for PML and ICP4. Panels D, H, and L represent zoomed-out views to show more of the surrounding cells. The ICP4 staining results were consistent with ICP4 expressed by the infected cell (37). Data presented are representative of results from three experiments.
ND10 phenotype in uninfected or HSV-1-infected Vero cells. Mock inoculum (left) or HSV-1 (MOI of ∼0.8; right) was added to Vero cells. At 6 h p.i., cells were fixed, and PML was detected by immunofluorescence to demarcate ND10 structures. ND10 structures appear as nuclear dots in uninfected cells (left). Virus infection-induced ND10 disruption is marked by diffuse nuclear staining of PML in HSV-1-infected cells (right). Download FIG S3, TIF file, 0.5 MB (529.7KB, tif) .
Copyright © 2019 Schneider et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Acyclovir and bortezomib work synergistically to inhibit HSV infection.
Combination therapies with two or more drugs have the potential to successfully inhibit viral infection more effectively than either drug alone (40). To determine if this is the case for acyclovir and bortezomib, HSV-1 KOS (MOI of 0.1) was added to Vero cells in the presence of acyclovir and bortezomib simultaneously at various concentrations. At 24 h p.i., the cells and supernatant were collected, and the titers of each drug combination sample were determined. Increasing concentrations of either bortezomib alone (topmost bars) or acyclovir alone (blue bars) reduced HSV infectivity (Fig. 8A). Selected combinations of the two drugs also inhibited infectivity. These combinations resulted in minimal cytotoxicity (Fig. S4). Results were analyzed with CompuSyn software, which provided a combination index (CI) value that suggested whether the effect of two drugs was additive, antagonistic, or synergistic. At software-determined concentrations that resulted in 50%, 75%, or 90% inhibition of HSV infection, the CI values were all less than 1, indicating synergy (Fig. 8B and C). CI values for most of the individual concentrations tested were also indicative of synergy, with a minority indicating additivity or antagonism (see Table S1 in the supplemental material). These results suggest that the combination of bortezomib and acyclovir inhibits HSV infection more effectively than treatment with either drug alone, indicative of a synergistic relationship.
FIG 8.

Bortezomib and acyclovir act synergistically to inhibit HSV infection. HSV-1 KOS was added to Vero cells (MOI of 0.1) in the presence of various combinations of acyclovir and bortezomib. At 24 h p.i., cells were fixed, and titers were determined on Vero cells. (A) 3D graph depicting viral titers at the various drug combinations. (B) Isobologram depicting synergistic profiles of bortezomib and acyclovir. “Fa” (fraction affected) refers to fraction inhibition. Each colored line depicts a certain level of fraction inhibition, with endpoints signifying the amount of each drug alone needed to achieve that amount of inhibition. Colored symbols signify how much of each of the two drugs working together is needed to achieve the same inhibition. Symbols below the respective colored lines indicate synergism, those on or near the respective lines indicate additivity, and those above the respective lines indicate antagonism. Data presented are representative of results from three experiments. (C) Software-determined CI values at the specified fractions affected (Fa). Here, “Fa” refers to inhibition of HSV-1 plaque formation (fraction of control). Data are presented as means of results from three experiments ± SEM.
Cytotoxicity profiles of HSV-infected cells treated with combinations of bortezomib and acyclovir. HSV-1 KOS was added to Vero cells (MOI of 0.1) in the presence of various combinations of acyclovir and bortezomib as described for Fig. 8. At 24 h p.i., a sample of supernatant from each condition was assayed for LDH activity as a measure of cytotoxicity. Data represent means of results from three experiments. Download FIG S4, TIF file, 0.3 MB (327.2KB, tif) .
Copyright © 2019 Schneider et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
CI values for a representative acyclovir-bortezomib synergy assay. The superscript 1 in the column 1 and 2 headings indicates that the data represent doses tested on the basis of previously determined EC50 values for the two drugs. The superscript 2 in the column 3 heading indicates that the data represent fractions of inhibition of plaque formation. The superscript 3 in the column 4 heading indicates that the data represent combination index (CI) values for the drug combinations as determined via CompuSyn software. CI values of <1 indicate synergy, those of ∼1 indicate additivity, and those of >1 indicate antagonism. Download Table S1, TIF file, 0.3 MB (273.9KB, tif) .
Copyright © 2019 Schneider et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
The proteasome is a host cell component required for successful infection by HSV. It is needed at multiple steps in the viral life cycle and thus is an attractive target for therapeutic intervention. Targeting a host cell component is expected to decrease the development of viral resistance. Here, we demonstrate that proteasome inhibition by bortezomib effectively inhibits HSV infection in cell culture and identify a mechanism of action. Bortezomib inhibits wild-type, acyclovir-resistant, and clinically isolated HSV infection. It has low cytotoxicity at effective concentrations. Mechanistically, bortezomib acts during the capsid transport and ND10 disruption steps of the HSV life cycle. Bortezomib did not affect HSV attachment to cells and was not virucidal. Bortezomib exhibited synergy with acyclovir. Overall, we present evidence that proteasome inhibition represents an attractive intervention for HSV infection and that the anticancer drug bortezomib might be able to be repurposed as a novel anti-HSV therapeutic.
Incoming HSV-1 nucleocapsid transport to the nucleus is blocked by the proteasome inhibitors lactacystin, epoxomicin, MG132, and bortezomib (17) (Fig. 6C and D). Nuclear delivery of HSV capsids is facilitated by ICP0 present in the virion tegument but is independent of E1 activation of ubiquitin (17, 39, 41). The substrate targeted by the proteasome for HSV capsid transport is not currently known. Nuclear transport of capsids from incoming virions that lack tegument ICP0 is not blocked by proteasome inhibitors; these virions remain infectious in a proteasome-independent manner (39). Our results indicate that while bortezomib blocks the majority of capsid transport to the nucleus, a subset of virions might still enter. HSV-1 ICP4 is detected in nuclei even after bortezomib treatment (Fig. 7). This is consistent with the notion that HSV capsids, while largely dependent on proteasomal activity, can also reach the nucleus via a proteasome-independent mechanism.
Following deposition of the incoming HSV-1 genome into the nucleus, virion subassemblies appear in the vicinity of ND10 structures (42). The mechanism of ND10 antagonism of the viral replicative cycle is not completely understood. However, it likely involves the coordinated action of the major ND10 protein components PML, Sp100, and Daxx, as well as chromatin repression of the viral genome (35, 43). The antiviral activity of host ND10 has been documented for many DNA viruses, including HSV, human cytomegalovirus (HCMV), varicella zoster virus (VZV), Epstein-Barr virus (EBV), Kaposi’s sarcoma-associated herpesvirus (KSHV), adenovirus, human papillomavirus (HPV), and simian virus 40 (SV40) (44). ND10 might also have protective roles during RNA virus infection (44). Dispersal of ND10 structures by herpesvirus infection is thought to facilitate the lytic replication cycle. ND10 disruption typically depends on the proteasome for virus-induced degradation of major ND10 protein components. This is mediated by HSV-1 ICP0 (19, 20), HCMV immediate early 1 (IE1) (45), and EBV BZLF1 (46). Bortezomib blocks ND10 disruption (Fig. 7), as do lactacystin and MG132 (20), supporting the notion that bortezomib acts on another early proteasome-dependent step of HSV infection in addition to capsid transport. Bortezomib blocks infection by vesicular stomatitis virus, influenza virus, hepatitis B virus, Venezuelan equine encephalitis virus, dengue virus, Rift Valley fever virus, Zika virus, African swine fever virus, and Nipah virus (47–56). MG132 inhibits cell infection by the veterinary alphaherpesviruses bovine herpesvirus 1 and pseudorabies virus (57, 58). It is tempting to speculate that bortezomib inhibits other herpesviruses and might, in fact, represent a broad-spectrum antiviral agent.
HSV infection is also facilitated by the proteasome’s degradative activity at later steps in the viral life cycle. Following HSV genome entry into the nucleus, the viral DNA relies partly on nuclear factor kappa-β (NFκ-β) for transcriptional activation (59). Induction of NFκ-β depends on proteasomal degradation of the inhibitor of kappa-β kinase (IκK). Proteasome inhibitors downregulate NFκ-β and, consequently, the HSV transcripts that depend on NFκ-β induction (60). Future efforts will test directly the effect of bortezomib on the expression of HSV-1 immediate early, early, and late genes and the localization of their gene products. HSV ICP0-mediated proteasomal degradation of DNA-dependent protein kinase (61) and centromeric proteins CENP-A (62) and CENP-C (63) has also been described. Proteasome activity is also required during HSV reactivation from latency (64). Here we demonstrate that bortezomib blocks infection by inhibiting two key, proteasome-dependent processes that occur during the first hours of infection: HSV capsid transport to the nucleus (Fig. 6B to D) and ND10 disruption (Fig. 7).
The primary treatment for HSV infection uses the acyclovir family of drugs. Acyclovir is a guanosine analogue that interferes with viral DNA replication. Direct pressure on the viral kinase results in the development of HSV strains resistant to acyclovir (26). Immunocompromised patients are particularly susceptible to infection by acyclovir-resistant strains, which can result in disseminated disease and death (12, 13). Second-line treatments for these severe infections include the use of foscarnet, a pyrophosphate analogue, and of cidofovir, a cytosine analogue, both of which target viral DNA replication (65). Thus, treating acyclovir-resistant strains with these second-line agents might result in the development of multiresistant strains, as has been reported previously for foscarnet (66).
The CC50 values for foscarnet and cidofovir on Vero cells are 50 mM and 560 µM, respectively (67, 68). We were unable to reach 50% Vero cell cytotoxicity with concentrations as high as 1 mM. The foscarnet and cidofovir EC50 values for HSV-1 on Vero cells are 32.6 µM and 6.4 µM, respectively (69, 70). Bortezomib yields ∼1,000-times-lower EC50 values. Moreover, foscarnet and cidofovir both exhibit toxicity in vivo, particularly in the form of nephrotoxicity (71, 72). Other side effects of cidofovir include neutropenia and metabolic acidosis (73). The therapeutic dose of foscarnet is 40 to 90 mg/kg of body weight, and its level of toxicity in mice is 500 mg/kg (74). The therapeutic dose of cidofovir is 5 mg/kg, but its level of toxicity in animals is 0.25 to 1 mg/kg (75). The therapeutic dose of bortezomib in cancer treatment is 1.3 mg/m2. The level of bortezomib toxicity in animals is 0.6 to 0.9 µg/m2 (76). Of course, toxicity in animal models is not always predictive of toxicity in humans. Bortezomib, unlike acyclovir, foscarnet, and cidofovir, targets a host process; thus, antiviral resistance might be less likely.
Host cell processes have been targeted to combat viral infections. Cyclin-dependent kinase (CDK) inhibitors, such as roscovitine, have anti-HSV properties (77, 78), as do cyclooxygenase 2 (COX-2) inhibitors (79). Strategies to inhibit influenza viruses and hepatitis B viruses have also targeted cellular processes (80, 81). Maraviroc is an FDA-approved, clinically effective anti-HIV drug that binds to host chemokine receptors (82–84).
The EC50 values for bortezomib against HSV infection ranged from 3.7 to 50.6 nM (Fig. 2). Similar concentrations inhibited proteasome activity and kill cancer cells in culture models of B cell lymphoma and mantle cell lymphoma (85–87). Clinical side effects of bortezomib treatment include peripheral neuropathy (PN) and thrombocytopenia (88, 89). PN might be due to serine protease inhibition by bortezomib in mitochondria, which has a role in the survivability of neurons (90). At a dose of 1.45 to 2.0 mg/m2, bortezomib has a half-life of 23 days or 8.7 to 14.8 h in whole blood or plasma, respectively (91, 92). Bortezomib is given twice weekly, and the mean maximum plasma concentration is 200 to 300 nM (92, 93). Thus, the EC50s that we obtained were well below the clinically achievable plasma concentration. Therefore, it is possible that plasma concentrations of bortezomib lower than those used for multiple myeloma might be effective against HSV, potentially ameliorating side effects. Bortezomib is clinically effective in patients with immune deficiency resulting from plasma cancers. Thus, bortezomib might be appropriate for administration to the immunocompromised, an important target population for new anti-HSV therapeutics. Bortezomib delivery by subcutaneous administration might be safer than delivery by intravenous administration as suggested by a previous report from a clinical trial in multiple myeloma patients (94). Herpes zoster (shingles), caused by varicella zoster virus, is an adverse event associated with bortezomib use (95). For this reason, acyclovir prophylaxis is recommended for multiple myeloma patients under treatment (96). For the HSV-infected immunocompromised population, acyclovir and low-concentration bortezomib therapy could be imagined and would also reduce the risk of shingles.
Repurposing of existing drugs has been successful in treating other medical conditions. For example, eflornithine is an ornithine decarboxylase inhibitor used to treat African sleeping sickness that was repurposed to treat female facial hirsutism (97). Thalidomide, an anti-inflammatory medication for treatment of leprosy, has recently been approved for treatment of multiple myeloma (98). Bortezomib, like these examples, has already been FDA approved for treatment, which would aid in streamlining the redevelopment process. Altogether, we provide evidence that bortezomib is a novel potential therapeutic for HSV with a defined mechanism of action. The results warrant preclinical testing of bortezomib efficacy in an animal model of HSV infection.
MATERIALS AND METHODS
Cells and viruses.
African green monkey kidney (Vero) cells (American Type Culture Collection, Manassas, VA) and human foreskin fibroblasts (HFFs; American Type Culture Collection) were maintained in Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific, Waltham, MA) containing 10% fetal bovine serum (FBS; Atlanta Biologicals, Atlanta, GA). HSV-1 strain KOS (Priscilla Schaffer, Harvard University); HSV-2 strain G (ATCC); acyclovir-resistant HSV-1 KOS derivatives (Don Coen, Harvard University) ACGr5, containing a mutation in the thymidine kinase gene (99), PAAr5, containing an Arg-to-Ser mutation at residue 842 of the viral DNA polymerase gene (26, 100), and dlsptk, containing a 360-bp deletion in the thymidine kinase gene (27); and HSV-1 strain H129 (Richard Dix, Georgia State University), a clinical isolate from the brain of an encephalitis patient (29), were all propagated and their titers determined on Vero cells.
Chemicals.
Stocks of 50 mM bortezomib (Selleckchem, Houston, TX, or Sigma, St. Louis, MO) and 20 mM acyclovir (Sigma) were prepared in dimethyl sulfoxide (DMSO; Fisher Scientific, Fair Lawn, NJ) and stored at −80°C and −20°C, respectively. Stocks of 0.5 mg/ml heparin (Sigma) were prepared in water and stored at −20°C.
HSV plaque assay.
HSV-infected Vero cells were incubated at 37°C and 5% CO2 for 18 to 24 h. Cells were fixed with methanol-acetone (2:1), dried, and stained with rabbit polyclonal antibody HR50 to HSV (Fitzgerald Industries, Acton, MA) and with horseradish peroxidase-conjugated protein A (Thermo Fisher Scientific). 4-Choloro-1-naphthol substrate (Sigma) and H2O2 catalyst (VWR International, Inc., Radnor, PA) were added to visualize plaques.
Cytotoxicity of bortezomib.
Bortezomib or a DMSO vehicle control was added to confluent cell monolayers in 96-well plates. At 24 h, a sample of supernatant was assayed for lactate dehydrogenase (LDH) activity using a Pierce LDH cytotoxicity assay kit (Thermo Scientific, Rockford, IL) according to the manufacturer’s instructions. All concentrations were tested in triplicate. As a positive-control sample, cells were lysed with 1% SDS for 30 min. Cytotoxicity is reported as LDH activity as a percentage of the detergent-lysed sample.
Time of addition of bortezomib.
HSV-1 strain KOS (MOI of 0.001) was added to Vero cells. Bortezomib or vehicle control was added at a concentration of 200 nM at various times postinfection (p.i.) from 0 to 6 h. Cells were incubated at 37°C and 5% CO2 for 18 to 24 h in total and were subjected to HSV plaque assay. Plaques were quantitated, and data are presented as percent inhibition of vehicle control infectivity.
HSV attachment to cells.
HSV-1 KOS was treated with 2 μg/ml DNase (Turbo DNAFree; Thermo Fisher Scientific) according to the manufacturer's instructions. This treatment removes any free HSV-1 DNA that is not protected inside viral capsids. Virus was diluted in ice-cold binding medium (carbonate-free, serum-free DMEM supplemented with 20 mM HEPES and 0.2% bovine serum albumin [BSA]). Prechilled Vero cells were simultaneously exposed to 200 or 500 nM bortezomib or 2 µg/ml heparin control and virus (40 genome copies/cell). Virus was subjected to spinoculation onto the cells at 200 × g for 1 h at 4°C. Cells were washed twice with ice-cold phosphate-buffered saline (PBS; Thermo Fisher Scientific), and cell-associated HSV-1 DNA was isolated with a QIAamp DNA blood minikit (Qiagen, Germany). The HSV-1 ICP22 copy number was determined via qPCR. HSV-1 was quantitated using a CFX96 real-time PCR detection system (Bio-Rad). Primers (Integrated DNA Technologies [IDT], Coralville, IA) were based on KOS ICP22 sequences, both forward (5′ GAG TTT GGG GAG TTT G 3′) and reverse (5′ GGC AGG CGG TGG AGA A 3′) (101, 102). A standard curve was generated for the assay using known quantities of a plasmid containing the HSV-1 ICP22 coding region diluted in glycogen.
Direct effect of bortezomib on viral particles.
HSV-1 KOS virions (∼3 × 107 PFU) were directly treated with 100 nM bortezomib–culture medium for 1 h at 37°C. Control samples were treated with DMSO vehicle. Samples were diluted 10-fold in culture medium, and HSV-1 titers were determined on Vero cells. The concentration of residual bortezomib in diluted virus preparations (0.01 nM) does not inhibit HSV-1 infection.
Capsid transport of HSV.
HSV-1 K26GFP (Prashant Desai, Johns Hopkins University) (MOI of ∼30) was added to Vero cells grown on coverslips in the presence of 100 or 500 nM bortezomib and 0.5 mM cycloheximide. At 2.5 h p.i., cultures were washed thrice with PBS and fixed with 3% paraformaldehyde–PBS. Nuclei were counterstained with 5 ng/ml of 4,6–diamidino-2-phenylindole dihydrochloride (DAPI; Roche). Coverslips were mounted on slides with Fluoromount G (Electron Microscopy Sciences, Hatfield, PA) and visualized with a Leica D4000 epifluorescence microscope at ×63 magnification. Images were processed with ImageJ (https://imagej.nih.gov/ij/) and Adobe Photoshop CS5.1.
Disruption of ND10 nuclear bodies.
Vero cells were seeded on glass coverslips and infected with HSV-1 KOS (MOI of ∼0.8) in the presence of 200 or 500 nM bortezomib or vehicle control. At 6 h p.i., cultures were fixed with 3% paraformaldehyde, quenched with 50 mM ammonium chloride, and permeabilized with 0.1% Triton X-100. Coverslips were stained for ND10 with 1:500 rabbit polyclonal primary antibody against PML (Santa Cruz Biotechnology, Dallas, TX) and 1:1,000 goat-anti-rabbit Cy3 secondary antibody (Thermo Fisher Scientific) and were stained for HSV-1 infection with 1:1,000 mouse monoclonal primary antibody against ICP4 (H1A021; Santa Cruz Biotechnology) and 1:1,000 goat-anti-mouse Alexa Fluor 488 secondary antibody. Nuclei were counterstained with DAPI. Coverslips were mounted onto slides with Fluoromount G and visualized with a Leica D4000 microscope (magnification, ×40). Images were processed with ImageJ (https://imagej.nih.gov/ij/) and Adobe Photoshop CS5.1.
Synergy of bortezomib and acyclovir.
Vero cells were infected with HSV-1 KOS (MOI of 0.1) and exposed simultaneously to various combinations of acyclovir and bortezomib. Cells were incubated at 37°C and 5% CO2 for 24 h. A sample of supernatant was taken for LDH cytotoxicity analysis as described above. Cells were collected and the titers determined via HSV plaque assay as described above. Data are presented as percent inhibition compared to an untreated control. The three-dimensional (3D) graph was constructed using Microsoft Excel. Combination indices and isobolograms were generated with CompuSyn software.
Ethics statement.
Low-passage-number HSV-1 isolate H129 was part of an already-existing, publicly available collection (26). Institutional review board (IRB) approval was not sought. Samples were anonymized in the previous study.
ACKNOWLEDGMENTS
We thank Darin Weed for early contributions to this project and Jean Celli for use of the fluorescence microscope.
This work was supported by Public Health Service grants AI119159 and AI113619 to A.V.N. from the National Institute of Allergy and Infectious Diseases.
Footnotes
Citation Schneider SM, Pritchard SM, Wudiri GA, Trammell CE, Nicola AV. 2019. Early steps in herpes simplex virus infection blocked by a proteasome inhibitor. mBio 10:e00732-19. https://doi.org/10.1128/mBio.00732-19.
REFERENCES
- 1.Looker KJ, Magaret AS, May MT, Turner KME, Vickerman P, Gottlieb SL, Newman LM. 2015. Global and regional estimates of prevalent and incident herpes simplex virus type 1 infections in 2012. PLoS One 10:e0140765. doi: 10.1371/journal.pone.0140765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Looker KJ, Magaret AS, Turner KM, Vickerman P, Gottlieb SL, Newman LM. 2015. Global estimates of prevalent and incident herpes simplex virus type 2 infections in 2012. PLoS One 10:e114989. doi: 10.1371/journal.pone.0114989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levitz RE. 1998. Herpes simplex encephalitis: a review. Heart Lung 27:209–212. doi: 10.1016/S0147-9563(98)90009-7. [DOI] [PubMed] [Google Scholar]
- 4.Dawson CR, Togni B. 1976. Herpes simplex eye infections: clinical manifestations, pathogenesis and management. Surv Ophthalmol 21:121–135. doi: 10.1016/0039-6257(76)90090-4. [DOI] [PubMed] [Google Scholar]
- 5.Roizman B, Knipe DM, Whitley R. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 6.Brown ZA, Wald A, Morrow RA, Selke S, Zeh J, Corey L. 2003. Effect of serologic status and cesarean delivery on transmission rates of herpes simplex virus from mother to infant. JAMA 289:203–209. doi: 10.1001/jama.289.2.203. [DOI] [PubMed] [Google Scholar]
- 7.Whitley RJ. 2012. The use of antiviral drugs during the neonatal period. Clin Perinatol 39:69–81. doi: 10.1016/j.clp.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schiffer JT, Corey L. 2009. New concepts in understanding genital herpes. Curr Infect Dis Rep 11:457–464. doi: 10.1007/s11908-009-0066-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freeman EE, Weiss HA, Glynn JR, Cross PL, Whitworth JA, Hayes RJ. 2006. Herpes simplex virus 2 infection increases HIV acquisition in men and women: systematic review and meta-analysis of longitudinal studies. AIDS 20:73–83. doi: 10.1097/01.aids.0000198081.09337.a7. [DOI] [PubMed] [Google Scholar]
- 10.Wald A, Link K. 2002. Risk of human immunodeficiency virus infection in herpes simplex virus type 2-seropositive persons: a meta-analysis. J Infect Dis 185:45–52. doi: 10.1086/338231. [DOI] [PubMed] [Google Scholar]
- 11.Furman PA, St Clair MH, Spector T. 1984. Acyclovir triphosphate is a suicide inactivator of the herpes simplex virus DNA polymerase. J Biol Chem 259:9575–9579. [PubMed] [Google Scholar]
- 12.Stranska R, Schuurman R, Nienhuis E, Goedegebuure IW, Polman M, Weel JF. 2005. Survey of acyclovir-resistant herpes simplex virus in the Netherlands: prevalence and characterization. J Clin Virol 32:7–18. doi: 10.1016/j.jcv.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 13.Piret J, Boivin G. 2014. Antiviral drug resistance in herpesviruses other than cytomegalovirus. Rev Med Virol 24:186–218. doi: 10.1002/rmv.1787. [DOI] [PubMed] [Google Scholar]
- 14.Goldberg AL. 2007. Functions of the proteasome: from protein degradation and immune surveillance to cancer therapy. Biochem Soc Trans 35:12–17. doi: 10.1042/BST0350012. [DOI] [PubMed] [Google Scholar]
- 15.Tanaka K. 2009. The proteasome: overview of structure and functions. Proc Jpn Acad Ser B Phys Biol Sci 85:12–36. doi: 10.2183/pjab.85.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao G, Luo H. 2006. The ubiquitin-proteasome pathway in viral infections. Can J Physiol Pharmacol 84:5–14. doi: 10.1139/y05-144. [DOI] [PubMed] [Google Scholar]
- 17.Delboy MG, Roller DG, Nicola AV. 2008. Cellular proteasome activity facilitates herpes simplex virus entry at a postpenetration step. J Virol 82:3381–3390. doi: 10.1128/JVI.02296-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maul GG, Guldner HH, Spivack JG. 1993. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). J Gen Virol 74:2679–2690. doi: 10.1099/0022-1317-74-12-2679. [DOI] [PubMed] [Google Scholar]
- 19.Everett RD, Maul GG. 1994. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J 13:5062–5069. doi: 10.1002/j.1460-2075.1994.tb06835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Everett RD, Freemont P, Saitoh H, Dasso M, Orr A, Kathoria M, Parkinson J. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J Virol 72:6581–6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ. 1999. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res 59:2615–2622. [PubMed] [Google Scholar]
- 22.Kane RC, Farrell AT, Sridhara R, Pazdur R. 2006. United States Food and Drug Administration approval summary: bortezomib for the treatment of progressive multiple myeloma after one prior therapy. Clin Cancer Res 12:2955–2960. doi: 10.1158/1078-0432.CCR-06-0170. [DOI] [PubMed] [Google Scholar]
- 23.Kane RC, Dagher R, Farrell A, Ko C-W, Sridhara R, Justice R, Pazdur R. 2007. Bortezomib for the treatment of mantle cell lymphoma. Clin Cancer Res 13:5291–5294. doi: 10.1158/1078-0432.CCR-07-0871. [DOI] [PubMed] [Google Scholar]
- 24.Utecht KN, Kolesar J. 2008. Bortezomib: a novel chemotherapeutic agent for hematologic malignancies. Am J Health Syst Pharm 65:1221–1231. doi: 10.2146/ajhp070272. [DOI] [PubMed] [Google Scholar]
- 25.Groll M, Berkers CR, Ploegh HL, Ovaa H. 2006. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure 14:451–456. doi: 10.1016/j.str.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 26.Coen DM, Schaffer PA. 1980. Two distinct loci confer resistance to acycloguanosine in herpes simplex virus type 1. Proc Natl Acad Sci U S A 77:2265–2269. doi: 10.1073/pnas.77.4.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coen DM, Kosz-Vnenchak M, Jacobson JG, Leib DA, Bogard CL, Schaffer PA, Tyler KL, Knipe DM. 1989. Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc Natl Acad Sci U S A 86:4736–4740. doi: 10.1073/pnas.86.12.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schang LM, Bantly A, Knockaert M, Shaheen F, Meijer L, Malim MH, Gray NS, Schaffer PA. 2002. Pharmacological cyclin-dependent kinase inhibitors inhibit replication of wild-type and drug-resistant strains of herpes simplex virus and human immunodeficiency virus type 1 by targeting cellular, not viral, proteins. J Virol 76:7874–7882. doi: 10.1128/JVI.76.15.7874-7882.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dix RD, McKendall RR, Baringer JR. 1983. Comparative neurovirulence of herpes simplex virus type 1 strains after peripheral or intracerebral inoculation of BALB/c mice. Infect Immun 40:103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nunes AT, Annunziata CM. 2017. Proteasome inhibitors: structure and function. Semin Oncol 44:377–380. doi: 10.1053/j.seminoncol.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nicola AV. 2016. Herpesvirus entry into host cells mediated by endosomal low pH. Traffic 17:965–975. doi: 10.1111/tra.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nicola AV, McEvoy AM, Straus SE. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J Virol 77:5324–5332. doi: 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koyama AH, Uchida T. 1987. The mode of entry of herpes simplex virus type 1 into Vero cells. Microbiol Immunol 31:123–130. doi: 10.1111/j.1348-0421.1987.tb03075.x. [DOI] [PubMed] [Google Scholar]
- 34.Zhong S, Salomoni P, Pandolfi PP. 2000. The transcriptional role of PML and the nuclear body. Nat Cell Biol 2:E85–E90. doi: 10.1038/35010583. [DOI] [PubMed] [Google Scholar]
- 35.Gu H, Zheng Y. 2016. Role of ND10 nuclear bodies in the chromatin repression of HSV-1. Virol J 13:62. doi: 10.1186/s12985-016-0516-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geoffroy MC, Chelbi-Alix MK. 2011. Role of promyelocytic leukemia protein in host antiviral defense. J Interferon Cytokine Res 31:145–158. doi: 10.1089/jir.2010.0111. [DOI] [PubMed] [Google Scholar]
- 37.Pritchard SM, Cunha CW, Nicola AV. 2013. Analysis of herpes simplex virion tegument ICP4 derived from infected cells and ICP4-expressing cells. PLoS One 8:e70889. doi: 10.1371/journal.pone.0070889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao F, Courtney RJ. 1989. A major transcriptional regulatory protein (ICP4) of herpes simplex virus type 1 is associated with purified virions. J Virol 63:3338–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delboy MG, Nicola AV. 2011. A pre-immediate-early role for tegument ICP0 in the proteasome-dependent entry of herpes simplex virus. J Virol 85:5910–5918. doi: 10.1128/JVI.00267-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pirrone V, Thakkar N, Jacobson JM, Wigdahl B, Krebs FC. 2011. Combinatorial approaches to the prevention and treatment of HIV-1 infection. Antimicrob Agents Chemother 55:1831–1842. doi: 10.1128/AAC.00976-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delboy MG, Siekavizza-Robles CR, Nicola AV. 2010. Herpes simplex virus tegument ICP0 is capsid associated, and its E3 ubiquitin ligase domain is important for incorporation into virions. J Virol 84:1637–1640. doi: 10.1128/JVI.02041-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maul GG, Ishov AM, Everett RD. 1996. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology 217:67–75. doi: 10.1006/viro.1996.0094. [DOI] [PubMed] [Google Scholar]
- 43.Glass M, Everett RD. 2013. Components of promyelocytic leukemia nuclear bodies (ND10) act cooperatively to repress herpesvirus infection. J Virol 87:2174–2185. doi: 10.1128/JVI.02950-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rivera-Molina YA, Martínez FP, Tang Q. 2013. Nuclear domain 10 of the viral aspect. World J Virol 2:110–122. doi: 10.5501/wjv.v2.i3.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Korioth F, Maul GG, Plachter B, Stamminger T, Frey J. 1996. The nuclear domain 10 (ND10) is disrupted by the human cytomegalovirus gene product IE1. Exp Cell Res 229:155–158. doi: 10.1006/excr.1996.0353. [DOI] [PubMed] [Google Scholar]
- 46.Adamson AL, Kenney S. 2001. Epstein-barr virus immediate-early protein BZLF1 is SUMO-1 modified and disrupts promyelocytic leukemia bodies. J Virol 75:2388–2399. doi: 10.1128/JVI.75.5.2388-2399.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neznanov N, Dragunsky EM, Chumakov KM, Neznanova L, Wek RC, Gudkov AV, Banerjee AK. 2008. Different effect of proteasome inhibition on vesicular stomatitis virus and poliovirus replication. PLoS One 3:e1887. doi: 10.1371/journal.pone.0001887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dudek SE, Luig C, Pauli EK, Schubert U, Ludwig S. 2010. The clinically approved proteasome inhibitor PS-341 efficiently blocks influenza A virus and vesicular stomatitis virus propagation by establishing an antiviral state. J Virol 84:9439–9451. doi: 10.1128/JVI.00533-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bandi P, Garcia ML, Booth CJ, Chisari FV, Robek MD. 2010. Bortezomib inhibits hepatitis B virus replication in transgenic mice. Antimicrob Agents Chemother 54:749–756. doi: 10.1128/AAC.01101-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Widjaja I, de Vries E, Tscherne DM, Garcia-Sastre A, Rottier PJ, de Haan CA. 2010. Inhibition of the ubiquitin-proteasome system affects influenza A virus infection at a postfusion step. J Virol 84:9625–9631. doi: 10.1128/JVI.01048-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Amaya M, Keck F, Lindquist M, Voss K, Scavone L, Kehn-Hall K, Roberts B, Bailey C, Schmaljohn C, Narayanan A. 2015. The ubiquitin proteasome system plays a role in Venezuelan equine encephalitis virus infection. PLoS One 10:e0124792. doi: 10.1371/journal.pone.0124792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choy MM, Zhang SL, Costa VV, Tan HC, Horrevorts S, Ooi EE. 2015. Proteasome inhibition suppresses Dengue virus egress in antibody dependent infection. PLoS Negl Trop Dis 9:e0004058. doi: 10.1371/journal.pntd.0004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keck F, Amaya M, Kehn-Hall K, Roberts B, Bailey C, Narayanan A. 2015. Characterizing the effect of bortezomib on Rift Valley fever virus multiplication. Antiviral Res 120:48–56. doi: 10.1016/j.antiviral.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 54.Barrows NJ, Campos RK, Powell ST, Prasanth KR, Schott-Lerner G, Soto-Acosta R, Galarza-Muñoz G, McGrath EL, Urrabaz-Garza R, Gao J, Wu P, Menon R, Saade G, Fernandez-Salas I, Rossi SL, Vasilakis N, Routh A, Bradrick SS, Garcia-Blanco MA. 2016. A screen of FDA-approved drugs for inhibitors of Zika virus infection. Cell Host Microbe 20:259–270. doi: 10.1016/j.chom.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barrado-Gil L, Galindo I, Martinez-Alonso D, Viedma S, Alonso C. 2017. The ubiquitin-proteasome system is required for African swine fever replication. PLoS One 12:e0189741. doi: 10.1371/journal.pone.0189741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang YE, Park A, Lake M, Pentecost M, Torres B, Yun TE, Wolf MC, Holbrook MR, Freiberg AN, Lee B. 2010. Ubiquitin-regulated nuclear-cytoplasmic trafficking of the Nipah virus matrix protein is important for viral budding. PLoS Pathog 6:e1001186. doi: 10.1371/journal.ppat.1001186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pastenkos G, Lee B, Pritchard SM, Nicola AV. 15 October 2018, posting date. Bovine herpesvirus 1 entry by a low-pH endosomal pathway. J Virol doi: 10.1128/JVI.00839-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miller JL, Weed DJ, Lee BH, Pritchard SM, Nicola AV. 2018. Low pH endocytic entry of the porcine alphaherpesvirus pseudorabies virus. J Virol 93:e01849-18. doi: 10.1128/JVI.01849-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Amici C, Rossi A, Costanzo A, Ciafrè S, Marinari B, Balsamo M, Levrero M, Santoro MG. 2006. Herpes simplex virus disrupts NF-kappaB regulation by blocking its recruitment on the IkappaBalpha promoter and directing the factor on viral genes. J Biol Chem 281:7110–7117. doi: 10.1074/jbc.M512366200. [DOI] [PubMed] [Google Scholar]
- 60.La Frazia S, Amici C, Santoro MG. 2006. Antiviral activity of proteasome inhibitors in herpes simplex virus-1 infection: role of nuclear factor-kappaB. Antivir Ther 11:995–1004. [PubMed] [Google Scholar]
- 61.Parkinson J, Lees-Miller SP, Everett RD. 1999. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J Virol 73:650–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lomonte P, Sullivan KF, Everett RD. 2001. Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J Biol Chem 276:5829–5835. doi: 10.1074/jbc.M008547200. [DOI] [PubMed] [Google Scholar]
- 63.Everett RD, Earnshaw WC, Findlay J, Lomonte P. 1999. Specific destruction of kinetochore protein CENP-C and disruption of cell division by herpes simplex virus immediate-early protein Vmw110. EMBO J 18:1526–1538. doi: 10.1093/emboj/18.6.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Everett RD, Orr A, Preston CM. 1998. A viral activator of gene expression functions via the ubiquitin-proteasome pathway. EMBO J 17:7161–7169. doi: 10.1093/emboj/17.24.7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Piret J, Boivin G. 2011. Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob Agents Chemother 55:459–472. doi: 10.1128/AAC.00615-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schmit I, Boivin G. 1999. Characterization of the DNA polymerase and thymidine kinase genes of herpes simplex virus isolates from AIDS patients in whom acyclovir and foscarnet therapy sequentially failed. J Infect Dis 180:487–490. doi: 10.1086/314900. [DOI] [PubMed] [Google Scholar]
- 67.Smee DF, Bray M, Huggins JW. 2001. Antiviral activity and mode of action studies of ribavirin and mycophenolic acid against orthopoxviruses in vitro. Antivir Chem Chemother 12:327–335. doi: 10.1177/095632020101200602. [DOI] [PubMed] [Google Scholar]
- 68.Ledbetter EC, Spertus CB, Pennington MR, Van de Walle GR, Judd BE, Mohammed HO. 2015. In vitro and in vivo evaluation of cidofovir as a topical ophthalmic antiviral for ocular canine herpesvirus-1 infections in dogs. J Ocul Pharmacol Ther 31:642–649. doi: 10.1089/jop.2015.0024. [DOI] [PubMed] [Google Scholar]
- 69.Piret J, Desormeaux A, Cormier H, Lamontagne J, Gourde P, Juhasz J, Bergeron MG. 2000. Sodium lauryl sulfate increases the efficacy of a topical formulation of foscarnet against herpes simplex virus type 1 cutaneous lesions in mice. Antimicrob Agents Chemother 44:2263–2270. doi: 10.1128/AAC.44.9.2263-2270.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Piret J, Goyette N, Boivin G. 2016. Novel method based on real-time cell analysis for drug susceptibility testing of herpes simplex virus and human cytomegalovirus. J Clin Microbiol 54:2120–2127. doi: 10.1128/JCM.03274-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bacon TH, Levin MJ, Leary JJ, Sarisky RT, Sutton D. 2003. Herpes simplex virus resistance to acyclovir and penciclovir after two decades of antiviral therapy. Clin Microbiol Rev 16:114–128. doi: 10.1128/CMR.16.1.114-128.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Deray G, Martinez F, Katlama C, Levaltier B, Beaufils H, Danis M, Rozenheim M, Baumelou A, Dohin E, Gentilini M, Jacobs C. 1989. Foscarnet nephrotoxicity: mechanism, incidence and prevention. Am J Nephrol 9:316–321. doi: 10.1159/000167987. [DOI] [PubMed] [Google Scholar]
- 73.Zabawski EJ Jr, Cockerell CJ. 1998. Topical and intralesional cidofovir: a review of pharmacology and therapeutic effects. J Am Acad Dermatol 39:741–745. doi: 10.1016/S0190-9622(98)70046-5. [DOI] [PubMed] [Google Scholar]
- 74.Omar RF, Gourde P, Desormeaux A, Tremblay M, Beauchamp D, Bergeron MG. 1996. In vivo toxicity of foscarnet and zidovudine given alone or in combination. Toxicol Appl Pharmacol 139:324–332. doi: 10.1006/taap.1996.0172. [DOI] [PubMed] [Google Scholar]
- 75.Hitchcock MJM, Lacy SA, Lindsey JR, Kern ER. 1995. The cyclic congener of cidofovir has reduced nephrotoxicity in three species. Antiviral Res 26:A358. doi: 10.1016/0166-3542(95)94952-X. [DOI] [Google Scholar]
- 76.Bross PF, Kane R, Farrell AT, Abraham S, Benson K, Brower ME, Bradley S, Gobburu JV, Goheer A, Lee S-L, Leighton J, Liang CY, Lostritto RT, McGuinn WD, Morse DE, Rahman A, Rosario LA, Verbois SL, Williams G, Wang Y-C, Pazdur R. 2004. Approval summary for bortezomib for injection in the treatment of multiple myeloma. Clin Cancer Res 10:3954–3964. doi: 10.1158/1078-0432.CCR-03-0781. [DOI] [PubMed] [Google Scholar]
- 77.Schang LM, Rosenberg A, Schaffer PA. 2000. Roscovitine, a specific inhibitor of cellular cyclin-dependent kinases, inhibits herpes simplex virus DNA synthesis in the presence of viral early proteins. J Virol 74:2107–2120. doi: 10.1128/JVI.74.5.2107-2120.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schang LM. 2002. Cyclin-dependent kinases as cellular targets for antiviral drugs. J Antimicrob Chemother 50:779–792. doi: 10.1093/jac/dkf227. [DOI] [PubMed] [Google Scholar]
- 79.Gebhardt BM, Varnell ED, Kaufman HE. 2005. Inhibition of cyclooxygenase 2 synthesis suppresses herpes simplex virus type 1 reactivation. J Ocul Pharmacol Ther 21:114–120. doi: 10.1089/jop.2005.21.114. [DOI] [PubMed] [Google Scholar]
- 80.Smallwood HS, Duan S, Morfouace M, Rezinciuc S, Shulkin BL, Shelat A, Zink EE, Milasta S, Bajracharya R, Oluwaseum AJ, Roussel MF, Green DR, Pasa-Tolic L, Thomas PG. 2017. Targeting metabolic reprogramming by influenza infection for therapeutic intervention. Cell Rep 19:1640–1653. doi: 10.1016/j.celrep.2017.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zheng Z, Li J, Sun J, Song T, Wei C, Zhang Y, Rao G, Chen G, Li D, Yang G, Han B, Wei S, Cao C, Zhong H. 2011. Inhibition of HBV replication by theophylline. Antiviral Res 89:149–155. doi: 10.1016/j.antiviral.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 82.Fätkenheuer G, Pozniak AL, Johnson MA, Plettenberg A, Staszewski S, Hoepelman AIM, Saag MS, Goebel FD, Rockstroh JK, Dezube BJ, Jenkins TM, Medhurst C, Sullivan JF, Ridgway C, Abel S, James IT, Youle M, van der Ryst E. 2005. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med 11:1170–1172. doi: 10.1038/nm1319. [DOI] [PubMed] [Google Scholar]
- 83.Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, Mori J, Rickett G, Smith-Burchnell C, Napier C, Webster R, Armour D, Price D, Stammen B, Wood A, Perros M. 2005. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother 49:4721–4732. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gulick RM, Lalezari J, Goodrich J, Clumeck N, DeJesus E, Horban A, Nadler J, Clotet B, Karlsson A, Wohlfeiler M, Montana JB, McHale M, Sullivan J, Ridgway C, Felstead S, Dunne MW, van der Ryst E, Mayer H. 2008. Maraviroc for previously treated patients with R5 HIV-1 infection. N Engl J Med 359:1429–1441. doi: 10.1056/NEJMoa0803152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vigneron N, Abi Habib J, Van den Eynde BJ. 2015. The capture proteasome assay: a method to measure proteasome activity in vitro. Anal Biochem 482:7–15. doi: 10.1016/j.ab.2015.04.019. [DOI] [PubMed] [Google Scholar]
- 86.Pham LV, Tamayo AT, Yoshimura LC, Lo P, Ford RJ. 2003. Inhibition of constitutive NF-kappa B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis. J Immunol 171:88–95. doi: 10.4049/jimmunol.171.1.88. [DOI] [PubMed] [Google Scholar]
- 87.Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, Anderson KC. 2001. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res 61:3071–3076. [PubMed] [Google Scholar]
- 88.Yi Y-S, Chung J-S, Song M-K, Shin H-J, Seol Y-M, Choi Y-J, Cho G-J, Lee G-W, Moon J-H, Hwang I-H, Ahn K-H, Lee H-S, Shin K-H, Hwang J-M. 2010. The risk factors for herpes zoster in bortezomib treatment in patients with multiple myeloma. Korean J Hematol 45:188–192. doi: 10.5045/kjh.2010.45.3.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Field-Smith A, Morgan GJ, Davies FE. 2006. Bortezomib (Velcade trade mark) in the treatment of multiple myeloma. Ther Clin Risk Manag 2:271–279. doi: 10.2147/tcrm.2006.2.3.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Arastu-Kapur S, Anderl JL, Kraus M, Parlati F, Shenk KD, Lee SJ. 2011. Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: a link to clinical adverse events. Clin Cancer Res 17:2734–2743. doi: 10.1158/1078-0432.CCR-10-1950. [DOI] [PubMed] [Google Scholar]
- 91.Osawa T, Naito T, Kaneko T, Mino Y, Ohnishi K, Yamada H, Kawakami J. 2014. Blood distribution of bortezomib and its kinetics in multiple myeloma patients. Clin Biochem 47:54–59. doi: 10.1016/j.clinbiochem.2014.06.077. [DOI] [PubMed] [Google Scholar]
- 92.Papandreou CN, Daliani DD, Nix D, Yang H, Madden T, Wang X, Pien CS, Millikan RE, Tu S-M, Pagliaro L, Kim J, Adams J, Elliott P, Esseltine D, Petrusich A, Dieringer P, Perez C, Logothetis CJ. 2004. Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen-independent prostate cancer. J Clin Oncol 22:2108–2121. doi: 10.1200/JCO.2004.02.106. [DOI] [PubMed] [Google Scholar]
- 93.Ogawa Y, Tobinai K, Ogura M, Ando K, Tsuchiya T, Kobayashi Y, Watanabe T, Maruyama D, Morishima Y, Kagami Y, Taji H, Minami H, Itoh K, Nakata M, Hotta T. 2008. Phase I and II pharmacokinetic and pharmacodynamic study of the proteasome inhibitor bortezomib in Japanese patients with relapsed or refractory multiple myeloma. Cancer Sci 99:140–144. doi: 10.1111/j.1349-7006.2007.00638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Moreau P, Pylypenko H, Grosicki S, Karamanesht I, Leleu X, Grishunina M, Rekhtman G, Masliak Z, Robak T, Shubina A, Arnulf B, Kropff M, Cavet J, Esseltine D-L, Feng H, Girgis S, van de Velde H, Deraedt W, Harousseau J-L. 2011. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, phase 3, non-inferiority study. Lancet Oncol 12:431–440. doi: 10.1016/S1470-2045(11)70081-X. [DOI] [PubMed] [Google Scholar]
- 95.Chanan-Khan A, Sonneveld P, Schuster MW, Stadtmauer EA, Facon T, Harousseau J-L, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, Neuwirth R, Anderson KC, Richardson PG. 2008. Analysis of herpes zoster events among bortezomib-treated patients in the phase III APEX study. J Clin Oncol 26:4784–4790. doi: 10.1200/JCO.2007.14.9641. [DOI] [PubMed] [Google Scholar]
- 96.Pour L, Adam Z, Buresova L, Krejci M, Krivanova A, Sandecka V, Zahradova L, Buchler T, Vorlicek J, Hajek R. 2009. Varicella-zoster virus prophylaxis with low-dose acyclovir in patients with multiple myeloma treated with bortezomib. Clin Lymphoma Myeloma 9:151–153. doi: 10.3816/CLM.2009.n.036. [DOI] [PubMed] [Google Scholar]
- 97.Coyne PE., Jr. 2001. The eflornithine story. J Am Acad Dermatol 45:784–786. doi: 10.1067/mjd.2001.117853. [DOI] [PubMed] [Google Scholar]
- 98.Bartlett JB, Dredge K, Dalgleish AG. 2004. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer 4:314–322. doi: 10.1038/nrc1323. [DOI] [PubMed] [Google Scholar]
- 99.Coen DM, Dixon RAF, Ruby SW, Schaffer PA. 1980. Genetics of acycloguanosine resistance and the thymidine kinase gene in HSV-1, p 581–590. In Fields B, Jenisch R, Fox CF (ed), Animal virus genetics. Academic Press, New York, NY. [Google Scholar]
- 100.Gibbs JS, Chiou HC, Bastow KF, Cheng YC, Coen DM. 1988. Identification of amino acids in herpes simplex virus DNA polymerase involved in substrate and drug recognition. Proc Natl Acad Sci U S A 85:6672–6676. doi: 10.1073/pnas.85.18.6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Komala Sari T, Pritchard SM, Cunha CW, Wudiri GA, Laws EI, Aguilar HC, Taus NS, Nicola AV. 2013. Contributions of herpes simplex virus 1 envelope proteins to entry by endocytosis. J Virol 87:13922–13926. doi: 10.1128/JVI.02500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Walker EB, Pritchard SM, Cunha CW, Aguilar HC, Nicola AV. 2015. Polyethylene glycol-mediated fusion of herpes simplex type 1 virions with the plasma membrane of cells that support endocytic entry. Virol J 12:190. doi: 10.1186/s12985-015-0423-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ihlenfeldt WD, Bolton EE, Bryant SH. 2009. The PubChem chemical structure sketcher. J Cheminform 1:20. doi: 10.1186/1758-2946-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Acyclovir inhibition of HSV infection of Vero cells. HSV-1 KOS was added to Vero cells (MOI of 0.004) in the presence of increasing concentrations of acyclovir. Plaques were enumerated as described for Fig. 2. The EC50 was 380 nM. Download FIG S1, TIF file, 0.06 MB (58.7KB, tif) .
Copyright © 2019 Schneider et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Bortezomib cytotoxicity on HFF cells. Increasing concentrations of bortezomib were added to HFF cells for 24 h, and cytotoxicity was determined via LDH activity. Data presented are representative of results from three experiments. Error bars, SEM. Download FIG S2, TIF file, 0.2 MB (190.9KB, tif) .
Copyright © 2019 Schneider et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
ND10 phenotype in uninfected or HSV-1-infected Vero cells. Mock inoculum (left) or HSV-1 (MOI of ∼0.8; right) was added to Vero cells. At 6 h p.i., cells were fixed, and PML was detected by immunofluorescence to demarcate ND10 structures. ND10 structures appear as nuclear dots in uninfected cells (left). Virus infection-induced ND10 disruption is marked by diffuse nuclear staining of PML in HSV-1-infected cells (right). Download FIG S3, TIF file, 0.5 MB (529.7KB, tif) .
Copyright © 2019 Schneider et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Cytotoxicity profiles of HSV-infected cells treated with combinations of bortezomib and acyclovir. HSV-1 KOS was added to Vero cells (MOI of 0.1) in the presence of various combinations of acyclovir and bortezomib as described for Fig. 8. At 24 h p.i., a sample of supernatant from each condition was assayed for LDH activity as a measure of cytotoxicity. Data represent means of results from three experiments. Download FIG S4, TIF file, 0.3 MB (327.2KB, tif) .
Copyright © 2019 Schneider et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
CI values for a representative acyclovir-bortezomib synergy assay. The superscript 1 in the column 1 and 2 headings indicates that the data represent doses tested on the basis of previously determined EC50 values for the two drugs. The superscript 2 in the column 3 heading indicates that the data represent fractions of inhibition of plaque formation. The superscript 3 in the column 4 heading indicates that the data represent combination index (CI) values for the drug combinations as determined via CompuSyn software. CI values of <1 indicate synergy, those of ∼1 indicate additivity, and those of >1 indicate antagonism. Download Table S1, TIF file, 0.3 MB (273.9KB, tif) .
Copyright © 2019 Schneider et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.