

Abstract
Repetitive action potentials (APs) in hippocampal CA3 pyramidal cells (CA3-PCs) backpropagate to distal apical dendrites, and induce calcium and protein tyrosine kinase (PTK)-dependent downregulation of Kv1.2, resulting in long-term potentiation of direct cortical inputs and intrinsic excitability (LTP-IE). When APs were elicited by direct somatic stimulation of CA3-PCs from rodents of either sex, only a narrow window of distal dendritic [Ca2+] allowed LTP-IE because of Ca2+-dependent coactivation of PTK and protein tyrosine phosphatase (PTP), which renders non-mossy fiber (MF) inputs incompetent in LTP-IE induction. High-frequency MF inputs, however, could induce LTP-IE at high dendritic [Ca2+] of the window. We show that MF input-induced Zn2+ signaling inhibits postsynaptic PTP, and thus enables MF inputs to induce LTP-IE at a wide range of [Ca2+]i values. Extracellular chelation of Zn2+ or genetic deletion of vesicular zinc transporter abrogated the privilege of MF inputs for LTP-IE induction. Moreover, the incompetence of somatic stimulation was rescued by the inhibition of PTP or a supplement of extracellular zinc, indicating that MF input-induced increase in dendritic [Zn2+] facilitates the induction of LTP-IE by inhibiting PTP. Consistently, high-frequency MF stimulation induced immediate and delayed elevations of [Zn2+] at proximal and distal dendrites, respectively. These results indicate that MF inputs are uniquely linked to the regulation of direct cortical inputs owing to synaptic Zn2+ signaling.
SIGNIFICANCE STATEMENT Zn2+ has been mostly implicated in pathological processes, and the physiological roles of synaptically released Zn2+ in intracellular signaling are little known. We show here that Zn2+ released from hippocampal mossy fiber (MF) terminals enters postsynaptic CA3 pyramidal cells, and plays a facilitating role in MF input-induced heterosynaptic potentiation of perforant path (PP) synaptic inputs through long-term potentiation of intrinsic excitability (LTP-IE). We show that the window of cytosolic [Ca2+] that induces LTP-IE is normally very narrow because of the Ca2+-dependent coactivation of antagonistic signaling pairs, whereby non-MF inputs become ineffective in inducing excitability change. The MF-induced Zn2+ signaling, however, biases toward facilitating the induction of LTP-IE. The present study elucidates why MF inputs are more privileged for the regulation of PP synapses.
Keywords: CA3, hippocampus, intrinsic plasticity, mossy fiber, protein tyrosine phosphatase, zinc
Introduction
Mossy fibers (MFs), the axons of dentate granule cells, make sparse but strong synapses on CA3 pyramidal cells (CA3-PCs), and serve as conditional detonators (Henze et al., 2002; Bischofberger et al., 2006). MF inputs play an essential role in the initial encoding and storage of memories at direct cortical synapses on the CA3 network (Lee and Kesner, 2004). Sparse MF innervation of CA3-PCs, together with sparse firing of dentate granule cells, may help the CA3 network to reduce overlap between similar memory representations and thus contribute to pattern separation (Leutgeb et al., 2007; Neunuebel and Knierim, 2014). Temporal overlap of MF inputs with associational/commissural (A/C) or perforant path (PP) synaptic inputs facilitates long-term potentiation (LTP) of concurrent non-MF synaptic inputs by providing sufficient depolarization of postsynaptic CA3-PCs (McMahon and Barrionuevo, 2002; Kobayashi and Poo, 2004). Further studies revealed that kainate receptors or NMDA receptors of MF synapses exert the heterosynaptic potentiation on concurrent A/C synaptic inputs (Sachidhanandam et al., 2009; Hunt et al., 2013). Most postulated roles for MF input in the CA3 area are based on its heterosynaptic interactions with other synaptic inputs arriving close in time (Treves and Rolls, 1992; O'Reilly and McClelland, 1994). The long-term heterosynaptic influences of MF inputs to temporally remote non-MF synaptic inputs, however, are not well understood. Previously Tsukamoto et al. (2003) reported that high-frequency stimulation of MFs induces LTP of EPSPs specifically at PP synapses onto CA3-PCs. Subsequently, we found that this heterosynaptic potentiation of PP-EPSPs is mediated by downregulation of Kv1.2 at distal apical dendrites of CA3-PC, which is referred to as the LTP of intrinsic excitability (LTP-IE). The long-term interaction of MF and PP synapses in a CA3-PC is potentially important for understanding the role of MF inputs in the hippocampal CA3 network dynamics, but it is still obscure whether LTP-IE is uniquely linked to the MF inputs. In fact, LTP-IE could be induced not only by high-frequency stimulation of MFs but also by direct somatic stimulation that elicits a train of action potentials (APs) at 10 Hz (somatic conditioning; Hyun et al., 2013), raising a possibility that non-MF synaptic inputs may induce LTP-IE as long as they induce moderate-frequency AP firings in a postsynaptic CA3-PC.
CA3-PCs are extensively connected with each other via A/C fibers, forming an autoassociation network, which subserves pattern completion (Marr, 1971; Nakazawa et al., 2003). A/C synapses are weak but comprise the largest number of synapses on a CA3-PC (Amaral et al., 1990). Considering these features of A/C synapses along with efficient summation of EPSPs owing to slow membrane time constant (Kowalski et al., 2016), it is plausible that A/C synaptic inputs may elicit moderate-frequency AP firings to induce LTP-IE. Because MF and A/C inputs play distinct roles in the network dynamics, it is of crucial importance to address whether LTP-IE is uniquely linked to the MF inputs. It remains unanswered, however, whether AP firings evoked by A/C inputs can induce LTP-IE.
The other question related to this issue is why MF input is more robustly capable of inducing LTP-IE than somatic conditioning. Previously, we reported that the magnitude of LTP-IE by somatic AP trains at 20 or 50 Hz is less pronounced than that by AP trains at 10 Hz (Hyun et al., 2013). In contrast, LTP-IE was readily induced by MF stimulation even if the MF input-induced postsynaptic AP frequency was >10 Hz (Hyun et al., 2015). This finding implies that some mechanism unique to MF inputs may be involved in the robustness of MF inputs in the LTP-IE induction. Here, we show that the cytosolic free calcium ([Ca2+]i) window that induces LTP-IE is normally very narrow because of Ca2+-dependent coactivation of antagonistic pairs: protein tyrosine kinase (PTK) and protein tyrosine phosphatase (PTP). Although the coactivation of PTP makes non-MF inputs incompetent in inducing LTP-IE, it is evaded by intracellular Zn2+ signaling upon high-frequency MF inputs.
Materials and Methods
Animals and ethical approval.
All studies, experimental protocols, and animal manipulation protocols described in this article were conducted with the approval of the Institutional Animal Care and Use Committee. The animals were maintained in standard environmental conditions (temperature, 25 ± 2°C; humidity, 60 ± 5%; dark/light cycle, 12/12 h) and monitored under veterinary supervision by the Institute for Experimental Animals, Seoul National University College of Medicine.
Zinc transporter 3 mutant mice.
Zinc transporter 3 (ZnT3) knock-out (KO) mice (donating investigator: Richard Palmiter, University of Washington School of Medicine, Seattle; The Jackson Laboratory), hybrids of C57BL/129Sv mice, were provided by Dr. Jae-Young Koh (College of Medicine, University of Ulsan, Ulsan, Republic of Korea) and backcrossed to the C57BL/6J background for at least seven generations for homogeneity of genetic background. The targeting construct for these mice was the same as that described by Cole et al. (1999). Depletion of Zn2+ from synaptic vesicles in MF terminal is also validated by (Cole et al. (1999). The KO and wild-type (WT) mice used for the experiments were generated by breeding heterozygotes (HTs), which in turn came from the breeding of ZnT3 HT mice to C57BL/6J mice (The Jackson Laboratory). For genotyping, DNA was isolated from the tail of each mouse in the litter at the age of 6 - 8 d., as described by Cole et al. (1999). All animals were maintained on the standard environmental conditions described above.
Preparation of slices.
Acute transverse hippocampal slices were obtained from Sprague Dawley rats [postnatal day 15 (P15) to P22] or mice (P15 to P24) of either sex. Animals were anesthetized by inhalation of isoflurane. After decapitation, brain was quickly removed and chilled in ice-cold preparation solution containing the following (in mm): 75 sucrose, 87 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 25 d-glucose, 7 MgCl2, and 0.5 CaCl2, equilibrated with a carbogen mixture of 95% O2 and 5% CO2. After mounting on a slicer (model VT1200, Leica), 300-μm-thick transverse slices were prepared and incubated at 34°C for 30 min in the preparation solution and thereafter were stored at room temperature (22°C). For experiments, slices were transferred to a submersion recording chamber superfused with standard aCSF containing the following (in mm): 124 NaCl, 26 NaHCO3, 3.2 KCl, 1.25 NaH2PO4, 10 d-glucose, 2.5 CaCl2, and 1.3 MgCl2.
Electrophysiological recordings.
Whole-cell voltage-clamp or current-clamp recordings from CA3-PCs were performed at near-physiological temperature (34 ± 1°C), while the recording chamber was perfused with the standard aCSF at ∼1–1.5 ml/min. Patch pipettes were pulled from borosilicate glass tubing (outer diameter, 1.5 mm; wall thickness, 0.225 mm) with a horizontal pipette puller (P-97, Sutter Instruments) and filled with the intracellular solution containing the following (in mm): 130 K-gluconate, 7 KCl, 1 MgCl2, 2 Mg-ATP, 0.3 Na-GTP, 10 HEPES, and 0.1 EGTA, with pH adjusted to 7.20 with KOH, at 295 mOsm. Pipette resistance was ∼3–4 MΩ. Recordings were preferentially obtained from the hippocampal CA3b. After formation of whole-cell configuration on the somata of CA3-PCs, recordings were performed only from cells that had a stable resting membrane potential (RMP) between −76 and −58 mV. Cells that exhibited more positive or unstable RMP were discarded. Under this condition, input conductance (Gin) was measured from subthreshold voltage responses to −30 and +10 pA current steps of 0.5 s (Hyun et al., 2015). Gin was monitored every 10 s before and after delivery of a conditioning protocol. To avoid voltage-dependent activation of K+ channels, the membrane potential was held at −68 ± 1 mV by current injection for all recordings. Recordings were discarded when the absolute amount of current injection was >50 pA. Recordings were obtained in the presence of a GABAA receptor antagonist, picrotoxin (PTX; 100 μm) unless specified. All recordings were made using a MultiClamp 700B amplifier controlled by Clampex 10.2 software through a Digidata 1440A Data Acquisition System (Molecular Devices).
Synaptic stimulation of MF, A/C, and PP synapses.
We stimulated different types of synapses on CA3-PCs to evaluate the influence of each synaptic input on the intrinsic excitability of CA3-PCs. Afferent MFs were stimulated with a recording solution-filled glass monopolar electrode with resistance of ∼1–2 MΩ placed in stratum lucidum (SL; stimulus intensity with ∼2–20 V) using minimal stimulation techniques (Hyun et al., 2015). Afferent PP and A/C fibers were stimulated with a concentric bipolar electrode (CBAPB125, FHC) positioned at stratum radiatum (SR) of CA3 (for A/C stimulation) or stratum lacunosum moleculare (SLM) on the border of the subiculum and CA1 for PP stimulation (Perez-Rosello et al., 2011). Brief stimulation pulses (100 μs) were generated by a computer-controlled digital stimulator (DS8000, WPI) and delivered to a stimulation electrode through an isolation unit (DLS100 Stimulus Isolator, WPI). For the stimulation of A/C fibers, we made an incision through the hilus of dentate gyrus (DG) to truncate MF input (Berzhanskaya et al., 1998) and the hippocampal sulcus to truncate entorhino-hippocampal input (Tsukamoto et al., 2003). The type of synaptic inputs were identified by the 20–80% rise time of EPSCs and the sensitivity to the group II mGluR agonist (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine (DCG-IV; 2 μm). Consistent with previous reports (Tsukamoto et al., 2003), DCG-IV attenuated PP-EPSCs and MF-EPSCs by 72.8% and 72.6%, respectively, but did not attenuate A/C-EPSCs.
Dynamic clamp.
To emulate massive and distributed A/C synaptic inputs (see Figure 2), we constructed a conductance waveform mimicking A/C synaptic input by convolving a Poisson process (2 s; mean frequency between 100 and 600 Hz) with unitary conductance of A/C synaptic inputs (Perez-Rosello et al., 2011). The amplitude distribution of the unitary synaptic conductance was randomly chosen from the log normal distribution of A/C synaptic inputs that has been previously reported in (Ikegaya et al., 2013). The constructed conductance waveform [G(t)] was instantaneously converted to the current waveform [Iinj(t)] according to the equation, Iinj = G(t) (Vm − Erev), by a custom-made dynamic-clamp device that uses an analog multiplier (model AD734, Analog Devices) (Iinj, injected current; Vm, measured membrane potential; Erev, reversal potential of AMPA current). The current waveform was delivered to the patch-clamp amplifier to be injected into the cell.
Figure 2.

Optimal [Ca2+]i window at distal apical dendrites for induction of LTP-IE. Aa, Simulation of Poisson random arrivals of A/C synaptic inputs at the mean frequency of 400 Hz. The amplitude of synaptic conductance was assumed to follow a log normal distribution with a mean and SD of 0.49 and 0.5, respectively. Inset, Template waveform of uEPSG at A/C–CA3 synapses. Ab, The conductance waveform for simulated A/C inputs, which was constructed from convolution of the events of A/C inputs with the uEPSG waveform shown in Aa. Ac, The number of APs as a function of mean frequencies of simulated A/C inputs for 2 s. Ad, Simulated A/C inputs for 2 s delivered to the soma at 0 s did not reduce Gin (yellow symbols). For comparison, Gin changes caused by somatic conditioning are plotted again from Figure 1Aa (gray). Inset, Representative AP responses to the simulated A/C inputs (top) and the subthreshold voltage responses to injection of +30 and −10 pA for 0.5 s (bottom). Ae, Probability distributions for instantaneous frequencies of APs elicited by 20 Hz MF stimulation, high-K+ aCSF, and simulated A/C inputs at 300–600 Hz. The distributions for the first two conditionings are based on data from Figure 1. Note that high-frequency AP bursts exceeding 30 Hz are generated by high-K aCSF or simulated A/C inputs, but not by 20 MF stimulation. The inset is the same plot expanded in y-axis. Ba, Somatic AP responses (top) and distal dendritic CaTs (bottom) evoked by different conditioning protocols, which are categorized by capability for the induction of LTP-IE (adequate or inadequate stimulations). In each panel of CaT, an averaged trace (red) is overlapped on raw CaTs evoked by the same stimulation protocol in different cells (gray). Composite AP train, somatic 10 Hz AP train intervened by 5 APs at 50 Hz in the middle. Bb, For all individual CaTs evoked by different stimulations, their time-averaged [Ca2+] levels are plotted as a function of their peak [Ca2+] levels. Note that CaTs evoked by adequate stimulations (gray and red filled symbols) are found within a narrow window of peak [Ca2+] levels between 338 and 378 nm, and do not overlap with CaTs induced by inadequate stimuli. Ca, To elicit excessive [Ca2+]i elevation, CA3-PCs were stimulated at 0 s with a composite AP train under standard aCSF (red triangle) or a 10 Hz AP train in the presence of extracellular 4 mm Ca2+ and 10 μm BayK8644 (blue triangle). Neither reduced the Gin of CA3-PCs, in contrast to somatic conditioning (10 Hz AP train for 2 s, gray; replotted from Fig. 1A). Insets, Representative AP responses to a composite train (top) and subthreshold voltage responses to current injection for measuring Gin (middle and bottom; black, before conditioning; color, 30 min after conditioning). Cb, Summary for mean ΔGin measured at 30 min after different conditionings. The ΔGin values for the first two conditionings (10 Hz AP train and high-K+ aCSF) are repeated from Figure 1D for statistical comparison. n.s., No statistical significance. ***p < 0.005.
Elevation of network activity using high-potassium aCSF.
To enhance A/C synaptic inputs (Fig. 1), we used a modified recording solution containing the following (in mm): 124 NaCl, 26 NaHCO3, 5 KCl, 1.25 NaH2PO4, 10 d-glucose, 2.5 CaCl2, and 1.3 MgCl2 (referred to as high-K+ aCSF). To block MF inputs and PP inputs, we made two incisions through the hilus of DG and the hippocampal sulcus (Berzhanskaya et al., 1998), and added 2 μm DCG-IV to the bathing solution to attenuate MF and PP inputs. After obtaining stable membrane potential in the standard aCSF without synaptic blockers, we applied high-K aCSF to activate CA3 subregion. After high-K+ aCSF depolarized the RMP of CA3-PCs, the RMP was readjusted to that at the standard aCSF by hyperpolarizing current injection to prevent high [K+] from contributing to AP firing in the CA3-PC under observation. Because MF and PP inputs were blocked by the incision of hilus and hippocampal sulci and DCG-IV, the only excitatory inputs to the CA3-PCs are recurrent inputs from other CA3-PCs, we can assume that AP bouts caused by high-K+ aCSF originate largely from A/C inputs.
Figure 1.

A/C synaptic inputs are incapable of inducing LTP-IE of CA3-PCs. A, Induction of LTP-IE by the repetitive somatic firing at 10 Hz (gray) or 20 Hz MF stimulation (red) for 2 s delivered at 0 s. B, Neither local electrical stimulation of A/C fibers (orange) nor high-K+ aCSF (cyan) induced the LTP-IE of CA3-PCs. Electrical stimulation was delivered at 0 min. To enhance A/C fiber inputs, high-K+ aCSF containing 2 μm DCG-IV was applied as indicated by the horizontal bar. A, B, Insets, Representative traces for AP responses (left panels) to conditioning stimulations, and subthreshold voltage responses (right panels) to +30 and −10 pA recorded before (black) and 30 min after conditioning (colored). The conditioning methods are indicated by the same color code in each panel. Ca, Representative traces for membrane potential during and after applying high-K aCSF, which contained 2 μm DCG-IV to suppress MF and PP synaptic inputs (see Materials and Methods). Membrane potential (top) and current injection (bottom) are depicted on the same time axis. The gray and black boxed regions are expanded in time on the inset. To readjust the membrane potential depolarized by high-K+ aCSF to the control level, hyperpolarizing current was injected (gray box, left inset). After a sufficient number of APs were elicited, the generation of APs was stopped by the injection of additional hyperpolarizing current (black box, right inset) The current injection time points are indicated by arrowheads. Cb, Application of synaptic blockers (PTX and CNQX) completely abolished not only the firing of CA3-PCs, but also subthreshold synaptic responses caused by high-K+ aCSF. Da, Relative changes of Gin (ΔGin) as a function of somatic AP frequency elicited by conditioning stimuli (gray, 10 Hz 2 s AP train; red, 20 Hz 2 s MF stimulation; yellow, local stimulation of A/C fibers; cyan, high-K+ aCSF). Db, Summary for ΔGin measured at 30 min after indicated conditioning. The number of APs elicited by each conditioning are also shown. n.s., No statistical significance. *p < 0.05; ***p < 0.005.
Measurements of intracellular Ca2+ and Zn2+.
CA3-PCs were loaded with Ca2+ indicator dye (Fura-2 pentapotassium salt; 100 μm) or Zn2+ indicator dye (FluoZin-3 tetrapotassium salt; 50 μm) through a whole-cell patch pipette to acquire fluorescence images of them. Imaging of dendrites of CA3-PCs was performed with a 60× water-immersion object lens (numerical aperture, 0.9; LUMPlanFI, Olympus), an air-cooled scientific CMOS digital camera (model C11440–22CU, Hamamatsu Photonics), and a monochromator (xenon-lamp based, Polychrome-V, TILL-Photonics), which were operated by Digidata 1440A Data Acquisition System and custom-made software. Acquired images were analyzed with HC-Image Live 4.0 (Hamamatsu Photonics). All cellular fluorescence values in a region of interest (ROI) were used after subtracting background values measured in the cell-free area near the ROI.
Measurements of cytosolic Ca2+.
The procedures for measurement of cytosolic [Ca2+] in the slice have been described in detail previously (Lee et al., 2000; Helmchen, 2011). To increase the time resolution and minimize photobleaching, we used single-wavelength protocol in imaging of dendrites of CA3-PCs. Images were taken at 20 Hz with single wavelength excitation at 380 nm (F380) preceded and followed by excitation at an isosbestic wavelength (360 nm). The intensity of isosbestic fluorescence (Fiso) was linearly interpolated between points just before and after the period of excitation at 380 nm. Calibration parameters were calculated using an in-cell calibration protocol (Helmchen, 2011), where the minimum ratio (Rmin), maximum ratio (Rmax), and intermediate ratio (Rint) values were determined using intracellular solutions containing 10 mm EGTA, 10 mm [Ca2+], or 10 mm EGTA, and 5 mm CaCl2 ([Ca2+] ∼159 nm), respectively. Values for fluorescence ratio (Fiso/F380) in data traces were converted to [Ca2+] using the following equation: [Ca2+] = Keff (R − Rmin)/(Rmax − R). The effective dissociation constant (Keff) was calculated using the fluorescence ratio at intermediate [Ca2+].
Measurements of cytosolic Zn2+.
Because the control intracellular solution contains 0.1 mm EGTA, which chelates not only Ca2+ but also Zn2+, 0.1 mm EGTA was routinely included in the intracellular solution for Zn2+ imaging together with 50 μm FluoZin-3. Images were taken at 20 Hz with the excitation wavelength at 494 nm. To quantify the fluorescence changes in an ROI, we determined the mean value for the baseline fluorescence (F0) before electrical stimulation. Measured values for the change in fluorescence from the baseline value in the same ROI (ΔF) were divided by F0, and the ratio of ΔF/F0 was regarded as a parameter for [Zn2+]i. Because the visual field of our camera does not encompass the whole length of an apical dendrite of CA3-PC, to image MF stimulation-induced FluoZin-3 signals at multiple ROIs along an apical dendrite we repeated the same imaging protocol with the visual field shifted along the apical dendrite. But repetition of imaging in a cell did not exceed three times.
Chemicals and reagents.
Unless specifically noted, all chemicals and reagents were purchased from Sigma-Aldrich. 2-{[Bis(2-pyridinylmethyl)amino]ethylamino}benzenesulfonic acid hydrate sodium (ZX1) was purchased from Strem Chemicals. Fluorescence indicators (Fura-2 and FluoZin-3) were purchased from Thermo Fisher Scientific. For a series of antibody studies, we used a rabbit IgG antibody targeted to the whole D2 domain of receptor tyrosine phosphatase α (RPTPα; catalog #07-472, Millipore; RRID:AB_310645; Gomez et al., 2015) or rabbit IgG targeted to a synthetic peptide located between amino acids 686 and 735 of RPTPα (catalog #LS-C80716-50, LSBio; RRID:AB_1600934). Before use, antibodies were dialyzed with PBS using a dialysis kit (Thermo Fisher Scientific).
RNA interference.
To test the specificity of LS-C80716-50 antibody, HEK293T cells were transfected with the control small interfering RNA (siRNA; catalog #SN-1011; Bioneer) or two different RPTPα siRNAs (catalog #SDH-1001) by using Lipofectamine RNAiMAX Reagent (Thermo Fisher Scientific) following the manufacturer instruction. The sequences for the PTPRA-RNAi are as follows: 5786-1 (RPTPα), 5′-GUCA UGGUUACCAACCUGA-3′; and 5786-2 (RPTPα), 5′-GAUUUCUGGCGGAUGAUCU-3′. On the 48 h after transfection, HEK293T cells were lysed in an ice-cold lysis buffer containing 50 mm Tris (pH 7.4 adjusted with HCl), 150 mm NaCl, 1 mm EDTA, and 1% SDS, and the lysate was subject to immunoblotting analysis. Heat shock protein 90 (HSP90) was measured as a control with the same molecular weight. Horseradish peroxidase-conjugated goat anti-rabbit IgG was used as a secondary antibody.
Experimental design and statistical analysis.
Electrophysiological data were obtained from 96, 10, and 11 hippocampal CA3-PCs from rats, and WT and ZnT3KO mice, respectively, of either sex. Imaging data were acquired from 25 and 10 hippocampal CA3-PCs from rats and mice of either sex, respectively (WT mice, 5; ZnT3KO mice, 5). The number of slices studied per an animal were two to three and one to two for rats and mice, respectively. Only one cell was recorded from each slice. Statistical data are expressed as the mean ± SEM and the number of cells measured (denoted as n). Statistical data were evaluated for normality and variance equality with Kolmogorov–Smirnov test and Levene's test, respectively. For data that satisfy normality and equality of variances, statistical evaluations were performed with Student's t test or one-way ANOVA. For data that did not satisfy the above properties, nonparametric tests were performed for evaluation. The number of cells and statistical tests for determining statistical significance are stated in the text using following abbreviations: n.s., no statistical significance; *p < 0.05; **p < 0.01; ***p < 0.005. Statistical analyses were performed using PASW Statistics 18 (SPSS).
Results
A/C synaptic inputs are incapable of inducing LTP-IE of CA3-PCs
We confirmed our previous observations that not only does 20 Hz MF stimulation (for 2 s) cause long-term reduction of Gin in CA3-PCs, which is referred to as LTP-IE (Hyun et al., 2013, 2015), but also the somatic injection of suprathreshold current pulses (10 Hz for 2 s, somatic conditioning). Gin was measured from subthreshold voltage deflections in response to −30 and +10 pA current steps (0.5 s; Fig. 1A, right inset). Statistical values for changes in Gin are expressed as fractional changes (%) in Gin measured at 30 min after conditioning compared with each control value. The 20 Hz MF stimulation reduced Gin of the CA3-PCs (69.75 ± 4.27%; n = 5; t(4) = 4.528, p = 0.011, paired t test) to a similar extent as the somatic conditioning (70.58 ± 1.44%; n = 9; t(8) = 14.363, p < 0.005, paired t test; Fig. 1A).
To examine whether LTP-IE can be induced by synaptic inputs other than MFs, Gin was monitored before and after 20 Hz stimulation delivered to A/C fibers for 2 s using a bipolar electrode positioned at SR (see Materials and Methods). Stimulation intensity was adjusted such that the amplitudes of EPSCs evoked by A/C fiber stimulation (A/C-EPSCs) were similar to those of minimally evoked MF-EPSCs. Because of the strong short-term depression of A/C synaptic inputs, the 2 s 20 Hz stimulation of A/C fibers elicited only a few APs in the early phase of the train and did not reduce Gin (99.81 ± 4.31%, n = 8). To attain the effective somatic AP frequency (10–20 Hz) required for the induction of LTP-IE (Hyun et al., 2013), we tested a theta burst stimulation (10 bursts at 5 Hz, 10 pulses at 100 Hz for each burst). Although theta burst stimulation of A/C fibers increased the number of elicited APs (range, ∼0.5–13 Hz; mean, 7.2 ± 2.1 Hz), it still failed to induce LTP-IE (97.99 ± 3.77%; n = 5; t(4) = 0.805, p = 0.466, paired t test; Fig. 1B, orange).
Because local electrical stimulation cannot elicit A/C synaptic inputs that are widely distributed over a dendritic arbor, we tested whether the enhancement of CA3 network activity can induce LTP-IE. To this end, we applied an aCSF bathing solution containing 5 mm KCl (high-K+ aCSF) in the presence of DCG-IV (for procedure details, see Materials and Methods), after measuring the baseline Gin of a CA3-PC under the normal aCSF conditions. High-K+ aCSF depolarized RMP of the CA3-PC typically by 5 mV before spontaneous EPSPs began. In order for synaptic input-induced AP generation not to be facilitated by depolarized RMP, the RMP was readjusted to the value in normal aCSF by the injection of hyperpolarizing current (Fig. 1Ca, right inset, boxed by gray broken line). Under these conditions, spontaneous EPSPs and AP bouts were elicited in most slices examined (9 of 10), and these APs were abolished by synaptic blockers 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and PTX (Fig. 1Cb), indicating that A/C synaptic inputs are responsible for the spontaneous APs. Once spontaneous APs had lasted >2 s and their averaged frequency reached ∼10 Hz, the spontaneous APs were stopped by the injection of additional hyperpolarizing current (Fig. 1Ca); then, we resumed the monitoring of Gin after a complete change of bathing solution to normal aCSF (Fig. 1Ca, inset, boxed by black broken line). As shown in Figure 1B, the enhancement of CA3 network activity using high-K+ aCSF did not induce LTP-IE (99.10 ± 2.27%; n = 6; t(5) = 0.345, p = 0.821, paired t test). Figure 1D summarizes the change of Gin (denoted by ΔGin) caused by different stimulation protocols. The number of APs elicited by local A/C stimulation, which occurred for last 2 s in high-K+ aCSF, were not significantly different from that elicited by 20 Hz MF stimulation (local A/C stimulation, 14.4 ± 4.2; high-K+ aCSF, 12.2 ± 2.2; MF stimulation, 19.2 ± 3.5; one-way ANOVA, F(3,22) = 2.75, p = 0.067). Nevertheless, neither the local simulation of A/C fibers nor the high-K+ aCSF induced LTP-IE in CA3-PCs, indicating that the optimal number of APs is necessary but is not the only factor needed for the induction of LTP-IE.
Only a narrow window of distal dendritic [Ca2+]i allows the induction of LTP-IE
Given that LTP-IE depends on backpropagating AP (bAP)-induced Ca2+ signaling at distal dendrites, we hypothesized that the induction of LTP-IE is regulated not only by the mean frequency of APs but also by their instantaneous frequencies. We investigated distal dendritic [Ca2+] evoked by different patterns of somatic APs. To this end, we have to elicit APs in more controlled way, because it is not technically easy to study Ca2+ signaling evoked by spontaneous APs, as in Figure 1C. Most of the A/C synaptic inputs are weak in synaptic strength (Ikegaya et al., 2013; Guzman et al., 2016). Such weak but high-frequency A/C synaptic inputs may elevate the membrane potential to a subthreshold depolarization level, as is evident in Figure 1C. To emulate the high-frequency A/C inputs, we injected a conductance waveform mimicking randomly arriving A/C synaptic inputs (GA/C waveform) for 2 s under the dynamic clamp mode (see Materials and Methods). To make the GA/C waveform, we made a waveform of excitatory postsynaptic conductance of unitary amplitude (uEPSG) from the averaged trace of minimally evoked A/C-EPSCs (Fig. 2Aa, inset). The amplitude distribution of uEPSG was assumed to follow the log normal distribution reported by Ikegaya et al. (2013). Assuming that A/C synaptic inputs of variable amplitudes arrive in a Poisson process (Fig. 2Aa), we constructed a GA/C waveform (referred to as “simulated A/C noise”; Fig. 2Ab) by convolving the uEPSG with the event trace. The stimulation intensity was set by the mean frequency of GA/C events (100–600 Hz for 2 s). The mean number of somatic APs was proportional to the mean frequency of GA/C events (Fig. 2Ac). To test whether the simulated A/C noise can induce LTP-IE in CA3-PCs, the frequency of simulated A/C noise was adjusted such that it evokes 10–30 somatic APs for 2 s, which has been noted as the optimal number of APs for the LTP-IE induction (Hyun et al., 2013, 2015). Similar to high-K+ aCSF, the simulated A/C noise did not induce LTP-IE (115.39 ± 8.78%; n = 7; t(6) = −1.959, p = 0.090, paired t test; Fig. 2Ad), indicating that the optimal AP frequency (10–20 Hz) does not ensure LTP-IE induction. The distribution of instantaneous AP frequencies upon 20 Hz MF stimulation is compared with those upon high-K+ aCSF and upon simulated A/C noise at different frequencies (Fig. 2Ae). The 20 Hz MF stimulation was distinguished from other stimulation in that it lacked instantaneous AP frequencies of >30 Hz (referred to as “AP bursts”), implying that excessively high [Ca2+]i elevation resulting from AP bursts may inhibit the induction of LTP-IE.
Figure 2Ba shows distal dendritic Ca2+ transients (CaTs) evoked by different stimulation protocols. We categorized stimulation protocols as “adequate stimulation” (10 Hz AP train, 20 Hz MF stimulation; Fig. 2Ba) and “inadequate stimulation” (other stimulation protocols; Fig. 2Ba, boxed by purple broken line) based on their capability of LTP-IE induction that has been observed from this and our previous studies (Hyun et al., 2013, 2015). The elevated [Ca2+]i plateau was a characteristic feature of CaTs evoked by adequate stimulation (Fig. 2Ba). We compared individual CaTs evoked by different stimulation on the plane of peak versus mean [Ca2+]i (Fig. 2Bb). Remarkably, CaTs evoked by adequate stimulation well overlapped with each other on this plane, and the peak [Ca2+]i levels of adequate stimulation were found within a narrow range (338–378 nm), with relatively high mean [Ca2+]i levels (168–243 nm) for their peaks. These results imply that [Ca2+]i elevation within a narrow window is required for the induction of LTP-IE, and an excessive [Ca2+]i peak may prevent the induction of LTP-IE. This view is consistent with our previous observation that LTP-IE induced by 20 and 50 Hz somatic AP trains are significantly less than that induced by a 10 Hz train (Hyun et al., 2013).
To test the hypothesis that AP bursts induced excessive [Ca2+]i elevation prevents the induction of LTP-IE, we examined Gin changes caused by two different stimulation conditions that induce high [Ca2+]i elevation in CA3-PCs. First, we inserted a bout of AP bursts (5 APs at 50 Hz) in the middle of the somatic conditioning protocol (referred to as “composite train”). The composite train under standard aCSF conditions induced the CaTs with high [Ca2+]i peaks out of the optimal range at distal apical dendrites (Fig. 2Ba, rightmost trace). Second, because the activation of L-type voltage-dependent calcium channels (VDCCs) is required for induction of LTP-IE (Hyun et al., 2013), a 10 Hz AP train for 2 s was delivered to the soma in the presence of 10 μm Bay K8644, an L-type VDCC agonist, and 4 mm extracellular Ca2+ (denoted as “high-Ca2+ aCSF”). Consistent with our hypothesis, neither the composite train nor the 10 Hz AP train under high-Ca2+ aCSF induced LTP-IE of CA3-PCs. (composite AP train: 98.83 ± 9.58%, n = 5, t(4) = 0.341, p = 0.751, paired t test; high-Ca2+ aCSF: 107.6 ± 10.7%, n = 5, t(4) = −0.964, p = 0.39, paired t test; Fig. 2Ca). In Figure 2Cb, the mean values for ΔGin caused by high-K+ aCSF, simulated A/C noise, the composite train, and 10 Hz AP under high-Ca2+ aCSF conditions are compared with those obtained by standard somatic conditioning (10 Hz AP train for 2 s).
Inhibition of PTP enables inadequate stimulation to induce LTP-IE
LTP-IE is mediated by Ca2+-dependent activation of PTKs (Hyun et al., 2013). Given that CaTs with excessively high peaks prevent the LTP-IE induction, such a high [Ca2+] may activate not only PTKs, but also a signaling process antagonizing PTKs, probably PTPs. Previous studies have suggested that Kv1.2 subunits undergo reciprocal regulation by PTKs and RPTPα (Lev et al., 1995; Tsai et al., 1999). Under the hypothesis that excessive [Ca2+]i elevation may activate RPTPα and antagonize the action of PTKs, we tested whether phosphatase inhibitors can enable inadequate stimulation to induce LTP-IE. As an inadequate stimulation, we used a 50 Hz somatic AP train (2 s), which normally does not induce LTP-IE (Hyun et al., 2013). Indeed, when 100 μm sodium orthovanadate, a general protein phosphatase inhibitor, was applied through the whole-cell patch pipette, the 50 Hz AP train was able to induce LTP-IE (68.99 ± 2.24%; n = 6; t(5) = 12.106, p < 0.001, paired t test; Fig. 3Aa). The peak values for distal dendritic [Ca2+]i evoked by the 50 Hz AP train were not significantly altered by intracellular application of Na3VO4 (control: 1167.2 ± 146.25 nm, n = 5; Na3VO4: 1225.6 ± 228.87 nm, n = 5; t(8) = 0.395, p = 0.835; independent t test; Fig. 3Ab). To narrow down the type of phosphatase involved in the regulation of Kv1.2, we intracellularly perfused the CA3-PCs with a rabbit IgG antibody targeted to the whole D2 domain of RPTPα (denoted as “anti-RPTPα AbM”; catalog #07-472, Millipore) or that targeted to the synthetic peptide located between amino acids 686 and 735 of the intracellular catalytic domain of RPTPα (denoted as “anti-RPTPα AbL”; 1 μg/ml; catalog #LS-C80716M-50, LSBio; see Materials and Methods) through the whole-cell patch pipette. Because anti-RPTPα AbM was previously validated (Gomez et al., 2015), we tested only for the specificity of anti-RPTPα AbL by immunoblotting of HEK293T cell lysates that were transfected with control or RPTPα-targeting siRNA (Fig. 3Ba). RNA interference of RPTPα significantly reduced the immunoblot stained by anti-RPTPα AbL at the 90 kDa band that corresponds to the expected molecular weight of RPTPα. Similar to Na3VO4, the incompetence of 50 Hz AP trains in LTP-IE induction was readily rescued by intracellular perfusion with one of two different anti-RPTPα antibodies (anti-RPTPα AbL: 69.19 ± 4.96%, n = 4, t(3) = 4.445, p = 0.004; anti-RPTPα AbM: 70.61 ± 2.67%, n = 5, t(4) = 7.920, p = 0.001), but not by intracellular perfusion of rabbit IgG isotype control antibody as a negative control (1 μg/ml; catalog #LS-C351731, LSBio; 105.31 ± 12.35%, n = 5, t(4) = −0.615, p = 0.572, paired t test; Fig. 3Bb,Bc). In the presence of phosphatase inhibitors, the changes in Gin caused by the 50 Hz AP train were comparable to those caused by somatic conditioning (t(8) = −1.207, p = 0.262, independent t test; Fig. 3Bc). Next, given that protein kinase C (PKC) is a positive regulator of PTPRα (Tsai et al., 1999; Brandt et al., 2003), we examined whether LTP-IE induction by somatic conditioning (10 Hz AP train, 2 s) is inhibited by phorbol ester. In the presence of 100 nm phorbol 12-myristate 13-acetate (PMA) in aCSF, somatic conditioning did not reduce Gin (96.78 ± 3.24%; n = 5; t(4) = 1.173, p = 0.306, paired t test; Fig. 3Bb,Bc). These findings suggest that the activation of RPTPα is responsible for the incompetence of inadequate stimulation in LTP-IE induction. Because inadequate stimulation excessively elevates distal dendritic [Ca2+]i, RPTPα may be activated by such a high [Ca2+]i level and antagonize the action of PTKs that induce LTP-IE. The mean values for ΔGin 30 min after conditioning shown in Figure 3 are summarized in Figure 3Bc. The baseline intrinsic properties under different experimental conditions are summarized in Table 1.
Figure 3.

Inhibition of PTP enables inadequate stimulation to induce LTP-IE. Somatic current pulse injection for 2 s, which elicits a 50 Hz AP train, was used as an inadequate conditioning stimulation. For inhibition of PTP, 100 μm Na3VO4 or anti-RPTPα antibody (1 μg/ml) was added to the whole-cell patch pipette. Aa, Relative changes of Gin caused by a 50 Hz AP train was delivered at 0 s with (cyan) or without (purple) Na3VO4 in the patch pipette. The LTP-IE caused by the 50 Hz AP train in the presence of intracellular Na3VO4 was similar to the somatic conditioning (10 Hz for 2 s, gray), which was reproduced from Figure 1A for comparison. Insets, AP responses to 50 Hz 2 s somatic AP train (top) and subthreshold voltage responses for measuring Gin with the same color code as the main panel (bottom; black, control). Ab, The peak values for distal dendritic [Ca2+]i evoked by a 50 Hz AP train are compared between conditions with or without intracellular Na3VO4. Ba, Test for specificity of the anti-RPTPα antibody. Ctrl siRNA, Nontargeting siRNA; RPTPα siRNA, RPTPα-targeting siRNA. Bb, Relative changes of Gin caused by a 50 Hz AP train with intracellular application of anti-RPTPα antibody (green filled symbols) or isotype antibody (green open symbols). In addition, the effects of PMA on Gin changes after a 10 Hz AP train are superimposed (orange). For comparison, somatic conditioning-induced ΔGin (gray) was reproduced from Figure 1A. Insets, Subthreshold voltage responses for measuring Gin with the same color codes as the main panel. Bc, Summary for ΔGin measured at 30 min after different conditionings: 10 Hz AP train (control), 50 Hz AP train (50 Hz), 50 Hz AP train with intracellular Na3VO4 (50 Hz + VO4), anti-RPTPα AbL (50 Hz + AbL), anti-RPTPα AbM (50 Hz + AbM), non-immunized Ab (50 Hz + ctrl Ab) and 10 Hz AP in the presence of PMA (10 Hz + PMA). n.s., No statistical significance. ***p < 0.005.
Table 1.
Parameters for intrinsic excitability
| Input conductance (nS) | AP onset time (ms) | First spike latency (ms) | |
|---|---|---|---|
| Control (CA3-PC in SD rats; n = 9) | 3.74 ± 0.17 | 10.0 ± 0.16 | 469.1 ± 13.12 |
| TPEN (n = 16) | 4.73 ± 0.30 | 10.6 ± 0.4 | 640.2 ± 34.9 |
| 100 μm ZX1 (n = 16) | 4.83 ± 0.36 | 9.4 ± 0.2 | 599.6 ± 112.7* |
| 50 μm ZX1 (n = 5) | 5.27 ± 0.67 | 8.82 ± 0.2 | 591.7 ± 40.7* |
| Na3VO4 (n = 11) | 4.50 ± 0.25 | 11.03 ± 0.3 | 538.2 ± 35.0 |
| Anti-RPTPα antibody (LS-C80176–50) (n = 8) | 5.70 ± 0.33* | 10.2 ± 0.2 | 684.7 ± 55.8* |
| Anti-RPTPα antibody (07–472) (n = 5) | 6.35 ± 0.67* | 9.6 ± 0.2 | 740.6 ± 50.9*** |
| Nonimmunized antibody (n = 5) | 4.34 ± 0.4 | 8.1 ± 0.46 | 578.17 ± 58.46 |
| ZnCl2 supplement (n = 10) | 5.31 ± 0.32 | 11.0 ± 0.3 | 620.5 ± 32.85 |
| 8 ∼ 14 week-old mice (n = 10) | 5.25 ± 0.52 | 9.6 ± 0.8 | 554.1 ± 40.2 |
| ∼8- to 14-week-old mice + ZnCl2 (n = 5) | 6.46 ± 1.49 | 8.48 ± 0.8 | 566.2 ± 99.25 |
| ZnT3+/+ (n = 5)a | 3.52 ± 0.34 | 8.8 ± 0.8 | 493.8 ± 22.5 |
| ZnT3−/− (n = 10) | 3.62 ± 0.23 | 9.4 ± 0.5 | 502.3 ± 39.9 |
| ZnT3−/− and ZnCl2 supplement (n = 6) | 3.45 ± 0.09 | 8.3 ± 0.3 | 471.2 ± 37.9 |
Statistical values and significance for the effects of conditioning on Gin, AP onset time, and the first spike latency were determined at 30 min after conditioning; AP onset time and the first spike latency are defined in Hyun et al. (2013). All values are shown as the mean ± SEM. Statistical significance was tested in comparison with control values using one-way ANOVA and Bonferroni correction.
aData from ZnT3+/+ CA3-PCs were used for control of data from ZnT3−/− mice. Otherwise, data from Sprague Dawley rats were regarded as control.
*p < 0.05;
***p < 0.005.
Zn2+ released from MF terminals enables MF inputs to induce LTP-IE even at high postsynaptic firing rates
Our results suggest that excessively high peak CaTs evoked by AP bursts prevent the induction of LTP-IE. However, this prediction has a caveat. The 50 Hz MF stimulation (1 s) reduced Gin to the same extent as the 20 Hz MF stimulation (2 s; Fig. 4Aa; Hyun et al., 2015), despite the fact that the distal dendritic peak [Ca2+]i evoked by 50 Hz MF stimulation was significantly higher than that by 20 Hz MF stimulation (50 Hz: 452.00 ± 18.24 nm, n = 5; 20 Hz: 352.42 ± 6.1 nm, n = 6; t(9) = 2.687, p = 0.021, independent t test; Fig. 4Ab). Not all of CaTs evoked by 50 Hz MF stimulation fell into the optimal [Ca2+] window (Fig. 4Ab, broken line box) on the plane of mean versus peak [Ca2+]i. This may be ascribed to postsynaptic AP bursts that occurred during the 50 Hz MF stimulation (Fig. 4Ac). How is the 50 Hz MF stimulation capable of inducing LTP-IE despite the presence of an excessive distal dendritic [Ca2+]i level?
Figure 4.

Zn2+ released from MF terminals is responsible for the robustness of MF inputs in LTP-IE induction. Aa, The 50 Hz MF stimulation (for 1 s) reduced the Gin of postsynaptic CA3-PCs, similar to the 20 Hz MF stimulation (for 2 s; reproduced from Fig. 1A). Ab, The 50 Hz MF stimulation-induced CaTs at distal apical dendrites are plotted on the mean vs peak [Ca2+]i plane. Data for CaTs evoked by somatic conditioning or 20 Hz MF train are reproduced from Figure 2Bb. The broken line box indicates the optimal Ca2+ window into which adequate CaTs fell. Insets, Representative somatic voltage response (top) and distal dendritic CaTs (bottom) to 50 Hz MF stimulation. Raw (gray) and averaged (red) CaTs are overlapped in the bottom. Ac, Probability distribution for instantaneous AP frequency evoked by MF stimulation. B, C, Relative Gin changes caused by MF stimulation at 20 Hz (2 s; Ba, Ca), 50 Hz (1 s; Bb, Cb), and in the presence of 1 μm TPEN (B) or ZX1 (C) in bathing solution. As a control, we observed the Gin time profiles upon somatic conditioning (10 Hz AP; gray) in the presence of 1 μm TPEN (B) or ZX1 (C) in bathing solution. Conditioning stimuli were delivered at 0 min (arrowheads). In the presence of TPEN, anti-RPTPa AbM or anti-RPTPa AbL was intracellularly perfused to block RPTPα (Bb). Insets, Representative subthreshold voltage responses before (black) and 30 min after MF stimulation (colored) or somatic conditioning (gray). Calibration: 5 mV, 0.2 s. D, Relative changes of Gin as a function of postsynaptic AP frequencies. Color codes are the same as in A–C. The data for ΔGin caused by MF and somatic stimulations under control conditions (gray symbols) are reproduced from our previous reports (Hyun et al., 2013, 2015). Inset, Magnified view for the area of postsynaptic APs between 5 and 15 Hz of the plot in D, showing that zinc chelators did not suppress the induction of LTP-IE upon the 10 Hz AP train or 20 Hz MF stimulation. Note that Zn2+ chelators render the 50 Hz MF stimulation incompetent in the induction of LTP-IE (colored symbol).
Given that PTP inhibitors rescued inadequate stimulation for LTP-IE induction (Fig. 3), these results raise a possibility that PTP may be suppressed by one of the neurotransmitters released from MF terminals. Previously, it has been shown that Zn2+ inhibits PTPs at picomolar concentrations (Brautigan et al., 1981; Wilson et al., 2012). Zinc ions, coreleased with glutamate from MF terminals (Qian and Noebels, 2005), enter postsynaptic pyramidal cells (Li et al., 2001a,b; Takeda et al., 2007).
We hypothesized that Zn2+ released from MF terminals may widen the calcium window for the induction of LTP-IE by the inhibition of PTPs. To test this hypothesis, we first examined the effect of bath-applied 1 μm TPEN, a membrane-permeable Zn2+ chelator, on the MF-induced LTP-IE. TPEN did not suppress the LTP-IE induced by 20 Hz MF stimulation (74.66 ± 4.76%; n = 5; t(4) = 3.742, p = 0.005, paired t test) or somatic conditioning (71.93 ± 3.79%; n = 5; t(4) = 5.583, p = 0.00, paired t test; Fig. 4Ba), which elicits adequate CaTs (Fig. 2). In contrast, 50 Hz MF stimulation for 1 s did not induce LTP-IE in the presence of TPEN (105.54 ± 3.95%; n = 6; t(5) = −1.249, p = 0.267, paired t test; Fig. 4Bb). Moreover, intracellular perfusion with either of two different anti-RPTPα antibodies rescued the competence of 50 Hz MF stimulation for the induction of LTP-IE despite the presence of TPEN (anti-RPTPα AbL: 60.81 ± 2.88%, n = 5, t(4) = −8.652, p = 0.001; anti-RPTPα AbM: 64.61 ± 0.92%, n = 5, t(4) = −14.304, p < 0.001, paired t test; Fig. 4Bb), suggesting that the TPEN effect is mediated by the inhibition of RPTPα. Ca2+ influx associated with glutamatergic synaptic activation can depolarize mitochondria (Bindokas et al., 1998) and evoke the release of mitochondrial free Zn2+ to the cytosol (Sensi et al., 2003). TPEN, a membrane-permeable chelator, may deplete Zn2+ in intracellular organelles. Because we cannot address which organellar or synaptically released Zn2+ is responsible for the TPEN effect, we examined the effect of bath-applied 100 μm ZX1, a fast membrane-impermeable zinc chelator, on the induction of LTP-IE. Although 100 μM ZX1 did not suppress the induction of LTP-IE caused by 20 Hz MF stimulation (79.00 ± 2.94%; n = 6; t(5) = 5.603, p = 0.003, paired t-test) or somatic conditioning (70.32 ± 3.20%; n = 6; t(5) = 5.355, p = 0.006, paired t-test; Fig. 4Ca), it significantly supressed the LTP-IE following 50 Hz MF stimulation (97.09 ± 3.65%; n = 5; t(4) = 0.890, p = 0.414, paired t test; Fig. 4Cb). Similar to the effect of ZX1 on LTP at MF–CA3 synapses (Pan et al., 2011), 50 μm ZX1 did not suppress the LTP-IE induced by 50 Hz MF stimulation (60.81 ± 2.89%; n = 5; t(4) = 3.819, p = 0.019, paired t test; Fig. 4Cb), suggesting that Zn2+ influx channels may be located in a spatial range close to MF terminals. Because the effects of membrane-impermeable ZX1 on LTP-IE were similar to those of TPEN, extracellular Zn2+, rather than Zn2+ released from the intracellular organelle, may mediate the facilitation of MF-induced LTP-IE induction. In Figure 4D, the changes of Gin are plotted as a function of MF-induced postsynaptic firing rates with or without Zn2+ chelators. Whereas MF stimulation induced LTP-IE at a wide range of postsynaptic AP frequencies under the control conditions (Fig. 4D, gray circles; adapted from Hyun et al., 2015), in the presence of Zn2+ chelator, it induced LTP-IE only at a narrow range of AP frequencies (10–15 Hz; Fig. 4D) similar to direct somatic AP stimulation (Fig. 4D, filled squares). These results suggest that Zn2+ signaling is responsible for the robustness of MF inputs to postsynaptic firing rates in the LTP-IE induction.
Deletion of vesicular zinc transporter (ZnT3) abolishes the privilege of MF inputs in the LTP-IE induction
To test whether synaptic vesicular Zn2+ plays an essential role in facilitating the MF input-induced LTP-IE, we studied LTP-IE in CA3-PCs from ZnT3 KO mice, in which synaptic vesicular Zn2+ is depleted (Cole et al., 1999; Vergnano et al., 2014). The EPSC amplitudes evoked by minimal stimulation of MFs in CA3-PCs were not different between WT and ZnT3KO mice (WT mice: 71.9 ± 7.4 pA, n = 10; ZnT3KO mice: 66.1 ± 7.5 pA, n = 10; t(18) = 0.549, p = 0.59, independent t test). The reduction of Gin induced by MF stimulation at 50 Hz was not different from that at 20 Hz in WT littermates of ZnT3KO mice (20 Hz: 74.24 ± 2.4%, n = 5; 50 Hz: 74.54 ± 2.1%, n = 5; t(8) = −0.095, p = 0.926, independent t test; Fig. 5Aa). These values were similar to those from Sprague Dawley rats shown in Figure 1 (F(2,16) = 3.067, p = 0.075). In ZnT3 KO mice, however, the extent of MF-induced ΔGin was heavily dependent on the postsynaptic AP frequencies (Fig. 5Ab,B). Whereas the 20 Hz MF stimulation that elicited postsynaptic AP frequency between 10 and 15 Hz reduced Gin to 75.22 ± 2.39% of the baseline (n = 5; t(4) = 7.965, p = 0.001, paired t test), 50 Hz MF stimulation that elicited higher AP frequencies did not reduce Gin (107.7 ± 3.20%; n = 5; t(4) = −1.976, p = 0.119, paired t test; Fig. 5B). These results are consistent with the effects of Zn2+ chelators on the ΔGin induced by MF stimulation [Fig. 5B, pale-colored symbols (adapted from Fig. 4D)]. The intrinsic excitability of CA3-PCs from ZnT3 KO mice was not different from that of WT littermates (Table 1). These results suggest that vesicular zinc ions confer robustness in the LTP-IE induction on the MF inputs such that MF inputs can induce LTP-IE even at very high frequencies.
Figure 5.
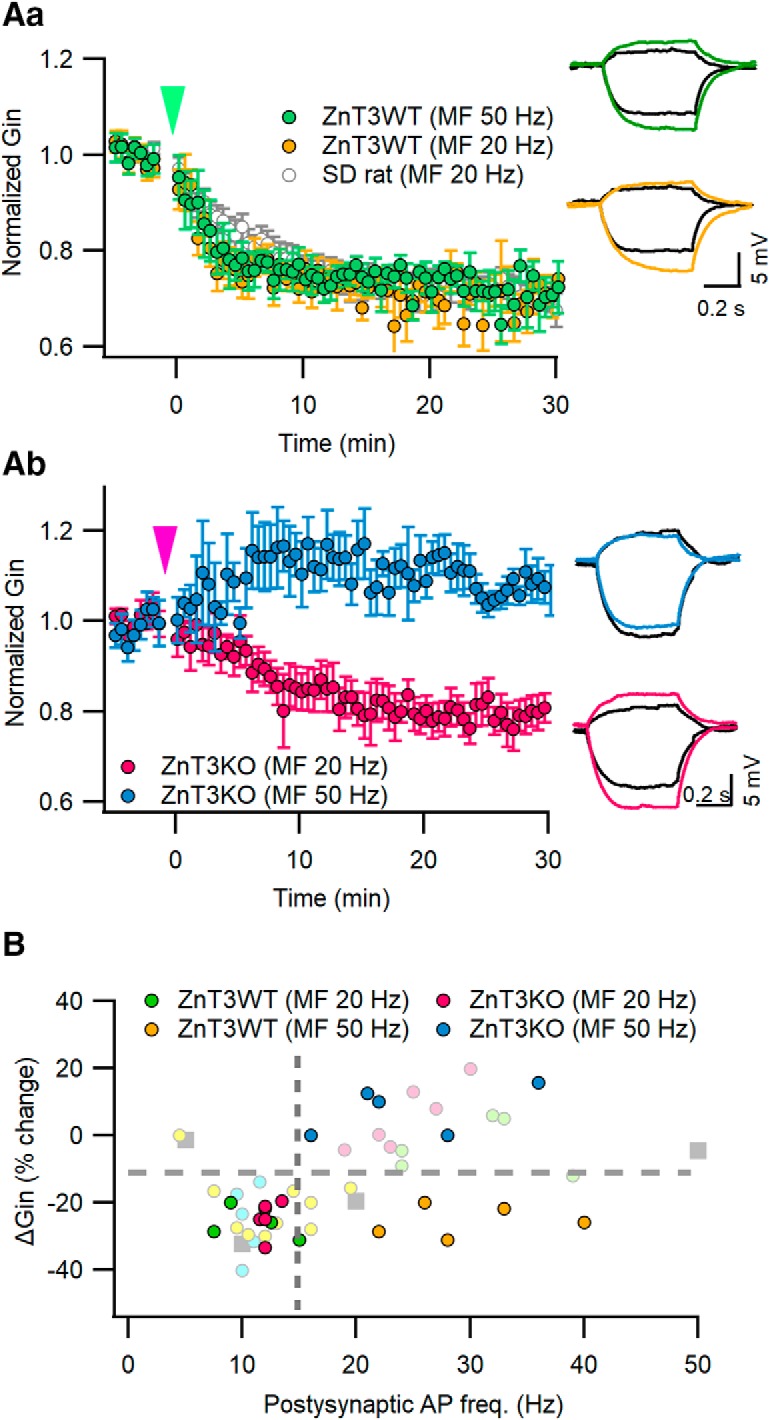
Deletion of vesicular zinc transporter (ZnT3) abolishes the privilege of MF inputs in LTP-IE induction. A, MF stimulation induced relative Gin changes in postsynaptic CA3-PCs from ZnT3 KO mice (Ab) or their WT littermates (Aa). MF stimulations at 20 or 50 Hz were given at t = 0 s (arrowheads). For comparison, Gin changes upon 20 MF stimulation in CA3-PCs from Sprague Dawley rats are reproduced from Figure 1A (gray). Inset, Subthreshold voltage responses before (black) and 25 min after MF stimulation (same color codes as the main panel). B, MF stimulation-induced ΔGin as a function of postsynaptic AP frequencies in WT and ZnT3KO CA3-PCs. The same color codes as in A are used for ΔGin data for WT or ZnT3KO mice (filled symbols). ΔGin data for Zn2+ chelators are reproduced from Figure 4 (pale colored symbols) for comparison.
Supplement of Zn2+ to extracellular solution disinhibits the induction of LTP-IE upon inadequate stimulation
Given that the activation of PTP is responsible for the incompetence of inadequate stimulation in the LTP-IE induction, the role of Zn2+ in MF-induced LTP-IE may be mediated by the inhibition of PTPs. Because [Zn2+] in the cleft of MF synapses induced by a few stimuli is between 20 nm and 1 μm (Vergnano et al., 2014), we tested whether a supplement of 100 nm ZnCl2 to bathing solution allows inadequate stimulation to induce LTP-IE similar to PTP inhibitors. Because inorganic phosphate forms metal precipitation with Zn2+, to dissolve ZnCl2 in aCSF we added 200 nm histidine to standard aCSF, which forms both mono-histidine and bis-histidine complexes with zinc (Rumschik et al., 2009). When 100 nm ZnCl2 was added to aCSF, LTP-IE was able to be induced by two different types of inadequate stimulation, which were the composite train (78.20 ± 3.86%; n = 5; t(4) = 4.322, p = 0.012, paired t test) and the 50 Hz somatic AP train (73.06 ± 4.43%; n = 5; t(4) = 7.003, p = 0.002, paired t test; Fig. 6A). Next, we tested whether supplement of Zn2+ rescues LTP-IE in ZnT3KO CA3-PCs. In the presence of 100 nm ZnCl2, 50 Hz MF stimulation (1 s) reduced Gin in CA3-PCs from ZnT3KO mice to a similar extent as in CA3-PCs from WT mice (KO: 71.68 ± 2.41%, n = 5; WT: 74.54 ± 2.1%, n = 5; t(8) = 0.406, p = 0.695, independent t test; Fig. 6B). Histidine alone, however, did not help the 50 Hz MF stimulation to induce LTP-IE in ZnT3-KO CA3-PCs (106.96 ± 3.49%; n = 5; t(4) = −1.951, p = 0.123, paired t test; Fig. 6B). Whereas the dependence of MF-induced LTP-IE on postsynaptic firing rates was biphasic in ZnT3KO CA3-PCs, similar to that induced by direct somatic stimulation, the supplement of Zn2+ made the dependence rather similar to that in WT CA3-PCs (Fig. 6C). The mean values for ΔGin under the conditions shown in Figure 6, A and B, are summarized in Figure 6D. Although the majority of results were obtained in young 2- to 3-week-old mice or rats [postnatal week 2 (PW2) to PW3], we have previously shown that LTP-IE can be induced in CA3-PCs of 6-week-old rats (Hyun et al., 2015). To test whether the facilitation of LTP-IE by Zn2+ occurs in an adult rodent brain, we replicated experiments shown in Figure 6A in 8- to 14-week-old mice (PW8 to PW14) (Rollenhagen et al., 2007). In CA3-PCs of PW8 to PW14 mice, LTP-IE was readily induced by the 10 Hz AP train (PW8 to PW14: 70.58 ± 3.34%, n = 5; PW2–3: 69.61 ± 1.16%, n = 9; t(4.981) = −0.304, p = 0.716), but not by the 50 Hz AP train (PW8 to PW14: 110.34 ± 9.12%, n = 5; PW2 to PW3: 94.71 ± 4.10%, n = 9; t(12) = −1.809, p = 0.096; Fig. 6E). Similar to young rats, the supplement of 100 nm ZnCl2 allowed the 50 Hz AP train to induce LTP-IE (PW8 to PW14: 70.65 ± 4.99%, n = 5, t(8) = 0.361, p = 0.727; PW2 to PW3: 73.06 ± 4.44%, n = 5; Fig. 6E), indicating that the facilitation of LTP-IE by Zn2+ is not restricted to the developing brain.
Figure 6.
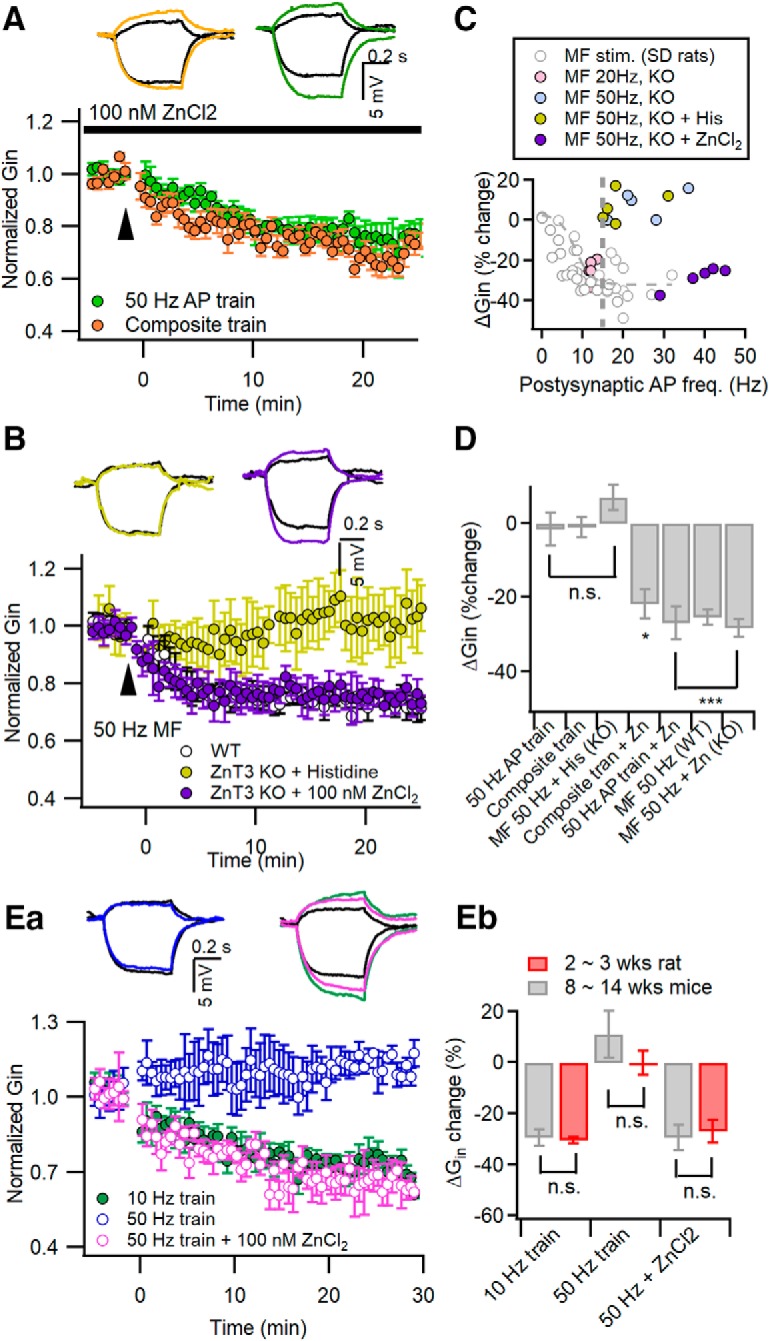
Supplement of Zn2+ to extracellular solution disinhibits the induction of LTP-IE upon inadequate stimulation. A, Supplement of 100 nm ZnCl2 in the extracellular solution enabled the composite train (orange) and 50 Hz AP train (green) to induce LTP-IE. Insets, Representative traces for Gin before (black) and 25 min after conditioning (the same color codes as the main panel). B, MF stimulation at 50 Hz for 1 s readily induced LTP-IE in CA3-PCs of ZnT3KO mice in the presence of 100 nm ZnCl2 in bathing solution (purple). Histidine alone did not allow 50 Hz MF stimulation to induce LTP-IE in CA3-PCs of ZnT3KO mice (yellow). The extent of ΔGin in ZnT3KO was not different from that in WT as control (open circles, reproduced from Fig. 4Aa). Insets, Representative subthreshold voltage responses before (black) and 25 min after conditioning. C, Plot of 50 Hz MF stimulation-induced ΔGin (purple) as a function of the number of postsynaptic APs in ZnT3-KO CA3-PCs with 100 nm ZnCl2 added to the bathing solution. For comparison, data for ΔGin induced by 20 or 50 Hz MF stimulation in KO CA3-PCs and in Sprague Dawley rats are reproduced from Figures 5B and 4D (gray symbols), respectively. Note that the nadir on the plot occurred around the postsynaptic AP frequency at 10 Hz for MF-induced ΔGin in KO CA3-PCs, but supplement of ZnCl2 converted their relationship similar manner as those of Sprague Dawley rats. D, Summary for the mean values of ΔGin induced by conditioning, as indicated on the abscissa. The control values (ΔGin values under the conditions without Zn2+ supplement) are reproduced from Hyun et al. (2013) (for 50 Hz AP train), Figure 2C (for composite train), and Figure 5A (for 50 MF stimulation in WT). Ea, Experiments similar to those in Aa were replicated in CA3-PCs from 8- to 14-week-old mice. LTP-IE was readily induced by 10 Hz somatic AP trains (green) but not by the 50 Hz AP train (blue). With 100 nm Zn2+ supplemented to aCSF, LTP-IE was induced by the 50 Hz AP train (purple). Eb, Mean values for the extent of ΔGin were not different between 2- to 3-week-old rats (red) and 8- to 14-week-old mice (gray) for each condition. n.s., No statistical significance. *p < 0.05; ***p < 0.005.
Zn2+ supplement allows somatic stimulation at an inadequate frequency to induce reduction of D-type K current and EPSP-spike (E-S) potentiation of PP-EPSPs
Previously, we reported that somatic conditioning-induced LTP-IE accompanies the reduction of D-type K+ current (IKD) and enhancement of PP-EPSP-to-spike coupling (i.e., E-S potentiation; Hyun et al., 2013, 2015). We tested whether the supplement of Zn2+ allows somatic stimulation at an inadequate frequency (50 Hz) not only to reduce Gin, but also to induce the reduction of IKD and E-S potentiation of PP-EPSPs. IKD was isolated as a low-concentration 4-aminopyridine (4-AP)-sensitive K+ current (IK) using digital subtraction as described previously (Hyun et al., 2013). Figure 7A shows representative outward IK traces without and with 30 μm 4-AP at the test potential of −40 mV (black), −30 mV (blue), −20 mV (red). IKD traces depicted in Figure 7Ab show that IKD after 50 Hz somatic stimulation is significantly larger under control conditions than in the presence of 100 nm ZnCl2 in aCSF or anti-RTPTα antibody in the patch pipette. Peak values of IKD for different conditions are summarized in Figure 7Ac and Table 2. Next, we tested whether the rescue of LTP-IE by the Zn2+ supplement accompanies E-S potentiation. E-S coupling was monitored from the number of APs elicited by temporal summation of PP-EPSPs, which were evoked by 20 Hz five-pulse bursts every 1 min. The PP stimulation intensity was adjusted such that postsynaptic voltage responses to PP stimulation were just below the AP threshold before somatic conditioning. To avoid erroneous multisynaptic MF inputs, double incisions were made in hippocampal sulcus and hilus. The number of APs was not increased after the 50 Hz somatic AP train alone. With 100 nm Zn2+ bath applied, however, the number of APs elicited by PP-EPSPs was increased after the 50 Hz somatic AP train (Fig. 7B). These results indicate that LTP-IE induced by 50 Hz somatic stimulation under Zn2+-supplemented conditions leads to the same consequence as the LTP-IE by somatic conditioning under control conditions, in that both induce not only a reduction of Gin, but also a reduction of IKD and E-S potentiation of PP synaptic inputs.
Figure 7.

Supplement of Zn2+ to aCSF allows 50 Hz somatic stimulation to induce reduction of D-type K current and E-S potentiation of PP inputs. Aa, Ab, Outward K+ IK elicited by a depolarizing step to −20 mV (red), −30 mV (blue), and −40 mV (black) from −70 mV before (Aa, top row) and after bath application of 30 μm 4-AP (Aa, bottom row) in the naive and conditioned CA3-PCs. For each condition, the arithmetical subtraction of IK under the 4-AP (bottom row) from the total IK (top row) at the same depolarizing step was regarded as IKD. Representative traces for IKD are shown in Ab. Ac, Mean values for peak amplitudes of IKD induced by a step depolarization to −20, −30, and −40 mV under different conditions. Ctrl, Without conditioning; 50 Hz, after 50 Hz somatic AP trains; 50 Hz + AbL and 50 Hz + AbM, intracellular perfusion of anti-RPTPα AbL and anti-RPTPα AbM, respectively; 50 Hz + Zn, 100 nm Zn2+ supplemented to aCSF. Ba, Number of APs elicited by five PP-EPSP trains at 20 Hz before and after 50 Hz somatic AP trains (arrowhead at 0 min). The 50 Hz AP train alone did not enhance the number of APs induced by PP synaptic inputs (black symbols). With 100 nm ZnCl2 supplemented to aCSF, however, the 50 Hz AP train enhanced the number of APs (red symbols. For comparison, AP numbers elicited by PP-EPSP bursts at 20 Hz after a 10 Hz somatic AP train are reproduced from Hyun et al. (2015) (gray symbols). Bb, Exemplar traces for temporal summation of 20 Hz PP-EPSPs before (black) and after (blue) the 50 Hz AP train without (left) and with (right) ZnCl2 supplement. n.s., No statistical significance. *p < 0.05; ***p < 0.005.
Table 2.
Summary for the peak of IKD
| −40 mV (pA) | −30 mV (pA) | −20 mV (pA) | |
|---|---|---|---|
| Control (n = 5) | 129.18 ± 8.35 | 269.91 ± 31.75 | 459.99 ± 45.39 |
| 50 Hz AP train (n = 5) | 129.46 ± 20.73 | 292.20 ± 35.18 | 420.04 ± 43.49 |
| (p = 1.00) | (p = 1.00) | (p = 1.00) | |
| 50 Hz AP train + intracellular anti-RPTPα Ab (LS-C08176-50) (n = 5) | 65.04 ± 17.22 | 139.76 ± 18.21 | 218.74 ± 16.10 |
| (p = 0.03) | (p = 0.019) | (p < 0.001) | |
| 50 Hz AP train + intracellular anti-RPTPα Ab (07-472) (n = 5) | 47.22 ± 8.00 | 145.58 ± 7.94 | 224.92 ± 12.63 |
| (p = 0.003) | (p = 0.028) | (p < 0.001) | |
| 50 Hz AP train + extracellular 100 nm ZnCl2 (n = 5) | 24.83 ± 6.43 | 115.18 ± 26.18 | 203.28 ± 11.57 |
| (p < 0.001) | (p = 0.004) | (p < 0.001) | |
| One-way ANOVA | F(4,20) = 12.763 | F(4,20) = 10.107, | F(4,20) = 17.055, |
| (p < 0.001) | (p = 0.001) | (p < 0.001) |
Values are the mean SEM, unless otherwise indicated. The 30 μm 4-AP-sensitive D-type K+ current at the test potentials of −40, −30, and −20 mV. Statistical values and significance for the effects of various conditions described above on peak IKD current. Statistical significance was tested in comparison with control values using one-way ANOVA and Bonferroni correction.
Zn2+ facilitates MF-induced heterosynaptic potentiation of PP synaptic inputs
We have shown that LTP-IE involves the downregulation of Kv1.2 at distal apical dendrites, which results in not only a reduction of Gin, but also specific potentiation of PP-EPSPs (Hyun et al., 2013, 2015). Our previous studies predict that Zn2+ signaling facilitates the MF input-induced potentiation of PP-EPSPs too. To test this prediction, we examined the effects of ZX1 and ZnT3-KO on MF-induced potentiation of PP-EPSPs. Neither extracellular ZX1 (50 or 100 μm) nor ZnT3-KO altered the amplitude of EPSCs, EPSPs, and EPSP/EPSC ratio (one-way ANOVA, F(5,41) = 1.210, p = 0.324; Fig. 8A). In the presence of 100 μm ZX1, heterosynaptic potentiation of PP-EPSPs was induced by 20 Hz MF stimulation (1.69 ± 0.11; n = 6; t(5) = −4.791, p = 0.005, paired t test) or somatic conditioning (1.78 ± 0.18; n = 5; t(4) = −6.807, p = 0.002, paired t test; Fig. 8Ba), but not by 50 Hz MF stimulation (0.93 ± 0.05; n = 5; t(4) = 1.347, p = 0.249, paired t test; Fig. 8Bb). Consistent with the concentration dependence of ZX1 effects on ΔGin (Fig. 4Cb), 50 μm ZX1 had little effect on heterosynaptic potentiation of PP-EPSPs following 50 Hz MF stimulation (1.60 ± 0.05; n = 5; t(4) = −8.412, p = 0.001, paired t test). MF stimulation at 20 Hz, but not at 50 Hz, induced heterosynaptic potentiation of PP-EPSPs in the KO CA3-PCs (20 Hz: 1.78 ± 0.07, n = 5, t(4) = −6.492, p = 0.003; 50 Hz: 0.88 ± 0.07, n = 5, t(4) = 1.669, p = 0.170, paired t test; Fig. 8Ca), whereas it was induced by MF stimulation at both frequencies in WT CA3-PCs (20 Hz: 1.86 ± 0.09, n = 4, t(3) = −5.972, p = 0.009 for WT; 50 Hz: 1.80 ± 0.06, n = 5, t(4) = −9.129, p = 0.001; paired t test; Fig. 8Cb). The 100 nm Zn2+ supplement in the bathing solution, however, rescued the heterosynaptic potentiation of PP-EPSPs upon 50 Hz MF stimulation in the KO CA3-PCs (1.81 ± 0.06; n = 6; t(5) = −6.165, p = 0.002, paired t test; Fig. 8Ca). The extent of MF-induced heterosynaptic potentiation of PP-EPSPs as a function of postsynaptic firing rates under different conditions is plotted in Figure 8Da, and the mean values are summarized in Figure 8Db. These plots demonstrate that, similar to the MF-induced reduction of ΔGin, heterosynaptic potentiation of PP-EPSPs becomes dependent on the postsynaptic firing rates without the help of Zn2+ signaling.
Figure 8.

Zn2+ facilitates MF-induced heterosynaptic potentiation of PP synaptic inputs. Aa, Ab, The relationship between PP-EPSP and PP-EPSC amplitudes measured at the same CA3-PCs (Aa), and the mean values for the EPSP/EPSC ratio (Ab) at PP-CA3 synapses under the conditions tested in Ba to Cb (control, control conditions in CA3-PCs of Sprague Dawley rats; ZX1, in the presence of 100 or 50 μm ZX1 in Sprague Dawley rats; ZnT3KO and ZnT3WT, ZnT3 KO mice and their littermates; Zn suppl., ZnT3 KO with supplement of Zn2+). Ba, Bb, Relative amplitudes of PP-EPSPs before and after MF stimulation at 20 Hz (Ba) or at 50 Hz (Bb) in the presence of 100 or 50 μm ZX1. Data for PP-CA3 EPSPs after somatic conditioning (10 Hz AP train for 2 s) are repeated in Ba and Bb as a control (open circles). Amplitudes are normalized to their mean baseline values. Conditioning MF stimuli were delivered at t = 0 (arrowhead). Ca, Cb, Relative amplitudes of PP-EPSPs before and after MF stimulation at 20 or 50 Hz in CA3-PCs from ZnT3-KO mice (Ca) or their WT littermates (Cb). MF stimulation at 20 Hz (green), but not at 50 Hz (gray), induced LTP of PP-EPSPs in KO CA3-PCs (Ca). Supplement of ZnCl2 rescued the incompetence of 50 Hz MF stimulation in KO (blue). In contrast, MF stimulation induced LTP of PP-EPSPs both at 20 and 50 Hz in WT CA3-PCs (Cb). Ba–Cb, Insets, PP-EPSP traces before (gray) and 25 min after a conditioning (same color codes as in main panels). Da, Plot of PP-EPSP potentiation as a function of the number of postsynaptic APs elicited by conditioning stimuli, as indicated in the boxed graph legend. MF-induced potentiation of PP-EPSPs under control conditions (gray triangles) are reproduced from Hyun et al. (2015) for comparison. Db, Summary for potentiation of PP-EPSPs induced by different conditionings. n.s., No statistical significance. ***p < 0.005.
High-frequency MF stimulation induces influx of Zn2+ into apical dendrites of CA3-PCs
Our results indicate that the role of Zn2+ in MF-induced LTP-IE is similar to that of PTP inhibitors. Translocation of synaptically released Zn2+ into postsynaptic CA3-PCs is a prerequisite for Zn2+-mediated inhibition of PTP in postsynaptic PTP. We examined postsynaptic intracellular [Zn2+] changes (Δ[Zn2+]i) induced by MF stimulation using a membrane-impermeable fluorescent indicator, FluoZin-3 (50 μm), included in the patch pipette (Kay and Tóth, 2006).
Previous in vitro studies implied that the fluorescence change of FluoZin-3 to Δ[Zn2+] is higher at least by five orders of magnitude than for the same Δ[Ca2+] (Devinney et al., 2005; Zhao et al., 2008). An in-cell study showed that the FluoZin-3 signal was little affected by ionomycin-induced [Ca2+]i elevation (Kiedrowski, 2011). Nevertheless, the contribution of Δ[Ca2+]i to a FluoZin-3 signal can be comparable to that of Δ[Zn2+]i when the Δ[Ca2+]i/Δ[Zn2+]i ratio is high enough to cancel out the affinity ratio of FluoZin-3 for Zn2+ over Ca2+.
To test whether Δ[Zn2+]i contributes to the MF-induced FluoZin-3 signal, we compared relative fluorescence changes of FluoZin-3 (ΔF/F0) elicited by 20 Hz MF stimulation for 2 s at the proximal apical dendrite under control conditions and in the presence of TPEN. The 20 Hz MF stimulation elicited 20.6 ± 1.2 (n = 5) APs in the postsynaptic cells. The peak fluorescence changes (ΔF/F0) at proximal dendrites under control conditions were significantly higher than those in the presence of TPEN (control: 0.38 ± 0.10, n = 5; TPEN: 0.11 ± 0.01, n = 5; t(8) = 3.199, p = 0.027, independent t test; Fig. 9A). Moreover, the MF-induced ΔF/F0 in the presence of TPEN was rather similar to ΔF/F0 induced by somatic conditioning (10 Hz APs for 2 s: 0.09 ± 0.01, n = 5; t(8) = 1.319, p = 0.202, independent t test; Fig. 9A). Assuming that the Δ[Ca2+]i induced by somatic conditioning is comparable to that induced by 20 Hz MF stimulation, as shown in Figure 2B, the contribution of Δ[Ca2+] to the MF-induced FluoZin-3 signal (ΔF/F0) may be <30%, and thus the rest may be caused by intracellular [Zn2+] increase.
Figure 9.

High-frequency MF stimulation induces influx of Zn2+ to apical dendrites of CA3-PCs. A, FluoZin3 fluorescence changes (ΔF/F0) at proximal apical dendrites evoked by somatic conditioning (10 Hz APs, 2 s; broken line) and by 20 Hz minimal stimulation of MFs (2 s) under control conditions (black) and in the presence of 1 μm TPEN in aCSF (cyan). Bottom bar graph, peak ΔF/F0 of FluoZin-3 fluorescence. Ba, Representative FluoZin-3 fluorescence image of a CA3-PC. Colored boxes are ROIs where fluorescence was measured. For each ROI, its distance from the soma is indicated. Bb, Averaged FluoZin-3 fluorescence changes (ΔF/F0) were measured at corresponding ROIs (same color code as in Ba), while afferent MFs were minimally stimulated at 20 Hz for 2 s (horizontal bar), which typically induced an AP response, as shown in the topmost panel. In each panel, ΔF/F0 traces measured in ZnT3 WT (solid line) and KO (broken line) CA3-PCs were superimposed. Bc, Bd, Mean values for FluoZin-3 fluorescence changes measured at 2 s (at the end of MF stimulation; Bc) and at 5 s (Bd) from ROIs at different distances from the soma in WT and ZnT3KO CA3-PCs. Be, The number of MF-induced postsynaptic APs as a function of baseline MF-EPSC amplitudes for WT (open circles) and ZnT3KO (filled circles) CA3-PCs. Bf, Left, The FluoZin-3 signals at distal apical dendrites (200 μm from the soma) of WT (blue) and KO (purple; reproduced from Bb) mice. For comparison, distal dendritic FluoZin-3 signal evoked by somatic conditioning in a CA3-PC of Sprague Dawley rats is overlapped. Right, The same FluoZin-3 signals are normalized to their peak values to compare decay kinetics. The distal dendritic Fura-2 trace is superimposed (green trace, reproduced from Fig. 2Ba). Blue horizontal bars, 2 s period when conditioning stimuli were given. Ca, The number of MF-induced postsynaptic APs as a function of baseline MF-EPSC amplitudes in the control conditions (black) and after bath application of 10 nm TTX (red). Insets, Representative traces for MF-EPSCs (top) and MF-induced AP responses (bottom) under control conditions (black) and in the presence of 10 nm TTX (red) in the same CA3-PC. Cb, Averaged traces for ΔF/F0 of FluoZin-3 measured at proximal (left) and distal (right) apical dendrites in control (black) and 10 nm TTX (red) conditions. The difference trace (gray) was obtained by subtraction of the latter trace from the former one. Cc, Summary for normalized ΔF/F0 values measured from proximal and distal dendrites at 2 and 5 s from the start of MF stimulation in control conditions (open bars) and after TTX application (gray bars). n.s., No statistical significance. *p < 0.05; **p < 0.01; ***p < 0.005.
Next, we examined the MF-induced fluorescence changes (ΔF/F0) in WT and ZnT3-KO CA3-PCs along apical dendrites. We set multiple ROIs along an apical dendrite, and acquired MF stimulation-induced FluoZin-3 signals at SL, SR, and SLM. Figure 9Ba shows representative ROIs along the apical dendritic FluoZin-3 image. The averaged traces for MF-induced ΔF/F0 at each ROI are compared between WT and KO CA3-PCs in Figure 9Bb. The fluorescence changes at the end of MF stimulation (denoted as 2 s assuming that t = 0 at the start of stimulation) were significantly higher in WT CA3-PCs than in KO CA3-PCs at four proximal ROIs (25, 50, 100, and 150 μm), but was not different between WT and KO mice at distal ROIs (≥200 μm; Fig. 9Bb,Bc). The fluorescence changes at 5 s, however, were significantly higher in WT CA3-PCs than in KO CA3-PCs not only at proximal ROIs but also at distal ROIs (Fig. 9Bd). The fluorescence changes at 2 and 5 s for WT and KO CA3-PCs are summarized in Table 3. Neither the MF-EPSC amplitudes nor the number of elicited APs were significantly different between the two genotypes, arguing against the contribution of different MF synaptic inputs (Fig. 9Be). The averaged traces for MF-induced ΔF/F0 at distal apical dendrites (200 μm from the soma) are compared between WT and KO CA3-PCs on an expanded scale (Fig. 9Bf, left). The averaged ΔF/F0 traces show that the FluoZin-3 signal in WT CA3-PCs, but not in KO CA3-PCs, persisted even 4 s after the cessation of MF stimulation. In contrast, The MF-induced FluoZin-3 signal in KO CA3-PCs and the somatic conditioning-induced FluoZin-3 signal decayed in 2 s after the end of stimulation. When the averaged traces are normalized to the peak, the decay phases of MF-induced FluoZin-3 signals in KO and somatic conditioning-induced signals overlapped to a large degree with the decay phase of the MF-induced Fura-2 signal (Fig. 9Bf, right). These results suggest that Δ[Ca2+] may contribute to the early phase of FluoZin-3 signal, which decayed in 2 s after the end of stimulations, but the later phase of the FluoZin-3 signal represents Δ[Zn2+]i at distal apical dendrites.
Table 3.
Summary for MF stimulation-induced fluorescence changes (ΔF/F0) of FluoZin-3
| Distance from soma | ZnT3 WT (2 s) | ZnT3 KO (2 s) | Comparisona (2 s) | ZnT3 WT (5 s) | ZnT3 KO (5 s) | Comparisona (5 s) |
|---|---|---|---|---|---|---|
| 25 μm | 0.46 ± 0.02 | 0.08 ± 0.02 | t(8) = 12.794 | 0.27 ± 0.02 | 0.03 ± 0.01 | t(8) = 11.718 |
| p < 0.001 | p < 0.001 | |||||
| 50 μm | 0.36 ± 0.03 | 0.10 ± 0.02 | t(8) = 7.402 | 0.20 ± 0.02 | 0.05 ± 0.03 | t(8) = 4.635 |
| p < 0.001 | p = 0.002 | |||||
| 100 μm | 0.34 ± 0.03 | 0.08 ± 0.01 | t(7) = 7.143 | 0.03 ± 0.02 | 0.01 ± 0.02 | t(6) = 7.592 |
| p < 0.001 | p < 0.001 | |||||
| 150 μm | 0.25 ± 0.02 | 0.09 ± 0.04 | t(6) = 3.843 | 0.18 ± 0.02 | 0.01 ± 0.02 | t(6) = 5.982 |
| p = 0.009 | p = 0.001 | |||||
| 200 μm | 0.18 ± 0.04 | 0.1 ± 0.02 | t(6) = 1.729 | 0.16 ± 0.04 | 0.00 ± 0.03 | t(6) = 4.684 |
| p = 0.135 | p = 0.003 | |||||
| 250 μm | 0.13 ± 0.01 | 0.09 ± 0.03 | t(6) = 1.317 | 0.12 ± 0.03 | 0.00 ± 0.02 | t(6) = 3.441 |
| p = 0.236 | p = 0.014 |
All values are shown as the mean ± SEM. Statistical significance was tested between ZnT3WT and ZnT3KO for the same distance using an independent t test.
aFluorescence intensities for apical dendrites of CA3-PCs were compared between WT and KO mice, at the same distance and time.
To further examine the time extent of Ca2+ contribution to MF-induced FluoZin-3 signals at distal dendrites, we studied the effects of 10 nm tetrodotoxin (TTX) in CA3-PCs of Sprague Dawley rats. TTX at low concentration (10–30 nm) selectively blocks the propagation of bAPs with little effect on neurotransmitter release (Mackenzie and Murphy, 1998; Haase and Maret, 2003; Tsay et al., 2007; Hyun et al., 2013). When the same number of somatic APs were elicited, 10 nm TTX reduced Δ[Ca2+]i to 47% of control at distal apical dendrites, whereas somatic Δ[Ca2+]i was little affected (Hyun et al., 2013). In light of the results of Figure 9B, we expected that 10 nm TTX would reduce the distal dendritic FluoZin-3 signal at the end of stimulations (2 s). Moreover, the effects of TTX on the distal dendritic FluoZin-3 signal at 5 s, which is dominated by Δ[Zn2+], will provide us a clue whether influx of Zn2+ to distal apical dendrites depends on the propagation of bAPs. If Zn2+ influx into distal dendrites depends on bAPs, 10 nm TTX would reduce the FluoZin3 signal at 5 s, and vice versa.
In the presence of 10 nm TTX, stronger MF stimulation (MF-EPSCs: in control, 106 ± 11.3 pA, n = 6; in TTX, 197 ± 19.6 pA, n = 5; t(9) = −4.199, p = 0.002, independent t test; Fig. 9Ca) was needed to get a similar number of APs in postsynaptic CA3-PCs as a control (AP numbers: in control, 25.1 ± 4.1, n = 6; in TTX, 21.8 ± 5.4, n = 5; t(9) = 0.483, p = 0.641, independent t test), probably because 10 nm TTX elevated the threshold potential of postsynaptic cells. When the similar numbers of APs were elicited, the peak values and the time course of FluoZin-3 signals (ΔF/F0) at proximal dendrites were not significantly different between control and TTX conditions both at 2 s (control: 0.38 ± 0.1, n = 5; TTX: 0.29 ± 0.05, n = 5; t(8) = 0.776, p = 0.460, independent t test) and at 5 s (0.17 ± 0.05, n = 5 in control; 0.15 ± 0.03, n = 5 in 10 nm TTX; t(8) = 0.455, p = 0.661, independent t test; Fig. 9Cb, top). A small effect of TTX on proximal dendritic FluoZin-3 signal may result from the electrical compactness of CA3-PCs (Johnston, 1981). At the distal dendrites, however, 10 nm TTX significantly lowered the FluoZin-3 signal at t = 2 s (control, 0.19 ± 0.03, n = 5; TTX, 0.10 ± 0.02, n = 5; t(8) = 2.465, p = 0.039, independent t test). Notably, 10 nm TTX did not reduce the MF-induced FluoZin3 signal at 4 s and later (control at 5 s: 0.09 ± 0.03, n = 5; TTX: 0.07 ± 0.01, n = 5; t(8) = 0.800, p = 0.447,independent t test; Fig. 9Cb, bottom). The difference trace between the FluoZin-3 signals in control and TTX conditions decayed in 4 s (Fig. 9Cb, gray trace), suggesting that TTX may specifically block Ca2+ influx at distal dendrites, and that Zn2+ influx to distal apical dendrites does not depend on bAP. The effects of 10 nm TTX on the FluoZin-3 fluorescence changes at 2 s (the end of MF stimulations) and 3 s later (denoted as 5 s) are summarized in Figure 9Cc for proximal and distal apical dendrites. Only the early-phase distal dendritic FluoZin3 signal at 2 s was reduced by 10 nm TTX. Altogether, these results suggest that 20 Hz MF stimulation elicits the increases of cytosolic [Zn2+] both at proximal and distal apical dendrites of CA3-PCs, and that the distal dendritic [Zn2+] elevation is slower and independent of bAP.
MF input-induced proximal dendritic [Zn2+] elevation linearly depends on the number of postsynaptic APs
We examined the dependence of FluoZin-3 signals evoked by 20 Hz MF stimulation on the baseline MF-EPSC amplitudes and the number of elicited APs. Each fluorescence change of FluoZin3 (ΔF/F0) was plotted as a function of corresponding baseline EPSC amplitude or the number of postsynaptic APs for CA3-PCs of Sprague Dawley rats under control and TTX conditions measured at 2 and 5 s (Fig. 10A). The same relationships were plotted for the WT (Fig. 10B, left) and ZnT3KO (Fig. 10B, right) mice. The FluoZin-3 signals at proximal dendrites depended on the baseline MF-EPSC amplitudes and the number of postsynaptic APs in CA3-PCs from both rats and mice (Fig. 10Aa,Ba), whereas the dependence of FluoZin3 signal at distal apical dendrites was not as clear as that at proximal dendrites (Fig. 10Ab,Bb). As already mentioned, larger MF-EPSCs were elicited in the presence of TTX to meet the control postsynaptic AP numbers, and thus [Zn2+] at the synaptic cleft may be higher in the TTX condition than that in the control condition (Qian and Noebels, 2005). Nevertheless, the FluoZin3 signals at proximal dendrites were not different between control and TTX conditions. Moreover, the proximal FluoZin-3 signal under the two conditions overlapped to a large degree on the plot of ΔF/F0 versus AP numbers, but not on the plot of ΔF/F0 versus MF-EPSCs (Fig. 10Aa). To further test whether FluoZin3 signals are more dependent on AP numbers than on MF-EPSCs in CA3-PCs of mice, we compared FluoZin3 signals evoked by MF stimulation at 20 and 50 Hz with the pulse number fixed at 40 (Fig. 10Ba,C). Because of more pronounced short-term facilitation of MF-EPSCs at 50 Hz than at 20 Hz (Fig. 10Ca), the cumulative amplitudes of MF-EPSCs were higher at 50 Hz (Fig. 10Cb). Despite higher quantal release at 50 Hz, the proximal FluoZin3 signals evoked by 50 Hz MF stimulations were not higher than those by 20 Hz when AP numbers were comparable (Fig. 10Ba,Cc). Given that extracellular Zn2+ permeates the plasma membrane through VDCCs or Ca2+-permeable AMPA receptors (Sensi et al., 1997; Kerchner et al., 2000; Jia et al., 2002), these results imply that the Zn2+ influx into CA3-PCs is limited by AP-dependent activation of VDCCs located at the proximal apical dendrites.
Figure 10.

MF input-induced proximal dendritic [Zn2+] elevation linearly depends on the number of postsynaptic APs. A, MF stimulation-induced ΔF/F0 values measured at 2 s (open symbols) and 5 s (filled symbols) as a function of the number of postsynaptic APs and baseline MF-EPSC amplitudes. In each panel, ΔF/F0 values are compared for the control (black), 10 nm TTX (red), and TPEN (purple) conditions. All data are obtained from the same cells of Figure 9C. B, MF stimulation-induced ΔF/F0 values at proximal (25 μm from soma; Ba) and distal (>200 μm; Bb) dendrites of CA3-PCs from WT (open symbols) and ZnT3KO (filled symbols) mice as a function of the number of postsynaptic APs and baseline MF-EPSC amplitudes. All data were obtained from the same cells seen in Figure 9B, except for red symbols, which represent peak ΔF/F0 at proximal dendrites evoked by 50 Hz MF stimulation with the same pulse number in WT CA3-PCs. For proximal and distal dendrites, ΔF/F0 values were measured at 2 and 5 s, respectively. Ca, Top, Representative EPSC traces in mouse CA3-PCs evoked by MF stimulations at 20 Hz (black) and 50 Hz (red). Bottom, Mean values for MF-EPSC amplitudes normalized to the baseline as a function of pulse number. Cb, Sum of normalized EPSC amplitudes evoked by 40 pulses at 20 and 50 Hz delivered to MFs. Cc, Voltage responses evoked by MF stimulation with 40 pulses at 20 Hz (black) and 50 Hz (red). Bottom, Averaged traces for FluoZin3 fluorescence changes evoked by 20 and 50 Hz (40 pulses). Horizontal bars indicate the time for MF stimulations. *p < 0.05.
Discussion
The present study demonstrated that there is an optimal [Ca2+] window at distal apical dendrites for the induction of LTP-IE unless PTP is inhibited, which is consistent with our previous finding that the extent of ΔGin is not proportional to the somatic firing frequency but displays the biphasic relationship with an optimal frequency of 10 Hz (Hyun et al., 2013). The bidirectional role of Ca2+, however, is not compatible with the monophasic dependence of ΔGin on MF stimulation frequency (Fig. 4D, gray symbols). Moreover, Tsukamoto et al. (2003) showed that MF stimulation at 100 Hz can induce heterosynaptic potentiation of PP-EPSPs, implying that MF can induce LTP-IE at postsynaptic firing frequencies of wider range. Our study presented evidence in two ways for MF input-induced postsynaptic Zn2+ signaling as a key mechanism that resolves the discrepancy in the LTP-IE induction between somatic and MF stimulations. First, when Zn2+ signaling was inhibited by Zn2+ chelators or by the deletion of ZnT3, MF input-induced LTP-IE exhibited biphasic dependence on the postsynaptic firing rates similar to somatic conditioning (Figs. 4, 5). Second, in the presence of 100 nm Zn2+ in aCSF, direct somatic stimulation could induce LTP-IE even at a very high somatic firing rate, similar to MF stimulations (Fig. 6). The effects of Zn2+ on the somatic AP train-induced LTP-IE was essentially the same as those of PTP inhibitors, in that it abolished the biphasic dependence of LTP-IE induction on somatic firing rates (Figs. 3, 6), suggesting that the role of Zn2+ is mediated by the inhibition of PTP.
What mediates the calcium-dependent activation of RPTPα?
Previous studies have shown that the surface expression of Kv1.2 is bidirectionally regulated by PTK and RPTPα in a heterologous expression system (Huang et al., 1993; Tsai et al., 1999). The present study showed that the inhibition of phosphatase or RPTPα can unmask the capability of inadequate stimulation protocols to induce LTP-IE, suggesting that excessively high peak [Ca2+] may activate PTPs as well as PTK. Similar to our findings, NMDA receptors in CA3-PCs have been reported to be bidirectionally regulated by PTK and striatal enriched PTP (STEP) pairs in Ca2+-dependent manner (Salter and Kalia, 2004). Both of PTK and STEP are activated by Ca2+ via PKC and calcineurin, respectively (Paul et al., 2003). The direction of Ca2+-dependent modulation of NMDA receptors is determined by the Ca2+ sensitivity of these two opposing pathways. Consistently, Grishin et al. (2005) showed that strong Ca2+ buffering or moderate Ca2+ signaling favored the kinase action to NMDA receptor, whereas strong Ca2+ signaling under low buffer conditions favored PTP. Activity-dependent activation of PTPs is not well understood. Calmodulin-dependent activation of PTPs has been suggested as a mechanism underlying the depression of NMDA receptor current caused by muscarinic agonist (Grishin et al., 2005). For PTPRα, however, PKCδ and CaMKII have been suggested as positive regulators (Brandt et al., 2003; Bodrikov et al., 2008). The regulation and biological effects of the PTPRα may depend on the signaling microdomain where PTPRα resides. Thus, it requires more investigation to address what signaling mechanisms are involved in the PTP activation caused by inadequate stimuli in the CA3-PCs.
Possible mechanisms underlying the permissive role of Zn2+ in induction of LTP-IE
Activation of PTKs is essential for the induction of LTP-IE (Hyun et al., 2015), suggesting that the facilitating role of Zn2+ is closely related to its positive regulation of PTK activity. Because the chelation of Zn2+ or the deletion of vesicular Zn2+ did not prevent adequate stimulation from inducing LTP-IE (Figs. 4, 5), Zn2+ signaling may not be essential for the activation of PTK. Instead, the zinc supplement, like the phosphatase inhibitors, had permissive action on the induction of LTP-IE by inadequate stimulation (Fig. 6). Given that the induction of LTP-IE is determined by the balance between PTK and PTP activity, it is likely that the action of Zn2+ is mediated by the inhibition of PTP, especially RPTPα. Consistent with this view, different kinds of PTPs have been shown to be inhibited by picomolar [Zn2+] (Haase and Maret, 2003; Wilson et al., 2012). Although Zn2+ inhibition of RPTPα has not been demonstrated, the binding sites in PTP1B and SHP-1 (Src homology region 2 domain-containing phosphatase-1) for inhibitory Zn2+ are located in their catalytic site (VVHCSAG), which is also highly conserved in PTPRα, suggesting a high probability for the zinc inhibition of PTPRα. Moreover, recent studies at MF–CA3 synapses showed that Zn2+ inhibits PTP to enhance the activities of pErk at presynaptic compartments (Sindreu et al., 2011).
MF input-induced intracellular Zn2+ signaling at apical dendrites
It has been shown that zinc ions are coreleased with glutamate upon stimulations of MFs (Qian and Noebels, 2005), and are taken up by postsynaptic CA3-PCs in rat hippocampal slices (Li et al., 2001a,b; Takeda et al., 2007). The present study confirmed the previous notion at proximal apical dendrites located in SL. We showed that the MF-induced FluoZin-3 signals at proximal dendrites are unequivocally higher in control conditions than those in the conditions that do not elevate [Zn2+]i such as in the presence of TPEN or in the ZnT3-KO CA3-PCs or upon somatic conditioning (collectively referred to as “no zinc conditions”; Fig. 9A,B), indicating that high-frequency MF stimulation induces an increase in [Zn2+]i at proximal apical dendrites.
For distal dendrites, it was not easy to evaluate the FluoZin-3 signal because of low signal-to-noise ratio, small ΔF/F0, and a relatively high Ca2+ component of FluoZin-3 signal compared with proximal dendrites. The FluoZin-3 signals at distal dendrites were distinct from the corresponding Fura-2 signals, in that the FluoZin-3 signal persisted even 4 s after the end of MF stimulation (Fig. 9Bf). The slow decay of the FluoZin-3 signal is consistent with previous reports on the time constant for clearance of intracellular Zn2+ in neurons (Ohana et al., 2009; Kiedrowski, 2011), which is slower than somatic Ca2+ clearance by two orders of magnitude (Kim et al., 2003). In contrast, the FluoZin-3 signals under no zinc conditions decayed within 2 s after the end of MF stimulation (referred to as “early phase”), similar to the decay time course of corresponding Ca2+ transients (Fig. 9Bf). Moreover, the effect of bAP inhibition by 10 nm TTX on the FluoZin-3 signal was confined to the early phase with no effect on the later phase (Fig. 9Cb), indicating that the Ca2+ component of the distal dendritic FluoZin-3 comes to an end within the early phase, and thus the later FluoZin-3 signal represents elevation of [Zn2+]i. Because 10 nm TTX had little effect on the later-phase FluoZin-3 signal, the distal dendritic [Zn2+]i elevation may not depend on the propagation of bAPs. Given that 10 nm TTX had an effect only on Δ[Ca2+]i, the time course of the distal Δ[Zn2+]i is better represented by the FluoZin-3 signal in the presence of TTX than in control conditions. Considering that 10 nm TTX partially inhibits the distal Δ[Ca2+]i, and that remaining Δ[Ca2+]i may still contribute to the early phase of FluoZin-3 signal, the distal [Zn2+]i elevation could be rather slower than the FluoZin-3 signal in the presence of TTX. The slow increase of distal dendritic [Zn2+]i together with its small dependence on bAPs implies intracellular diffusion of Zn2+ from proximal dendrites rather than the direct influx of Zn2+ through distal dendritic plasma membrane.
Physiological implications
Our study showed that high-frequency MF inputs are privileged for the induction of LTP-IE in postsynaptic CA3-PCs by the help of intracellular Zn2+ signaling, which probably suppresses Ca2+-dependent activation of PTP. In contrast, high-frequency A/C inputs can hardly induce LTP-IE because they elicit bursting APs in the soma and thus transient and excessively high [Ca2+]i elevation at distal dendrites, resulting in the activation of PTP. In vivo studies on firing patterns of hippocampal granule cells in awake animals revealed that a high proportion of their firings are in bursts (Mistry et al., 2011; Pernía-Andrade and Jonas, 2014). Since the intraburst firing rates are well above 100 Hz (Henze et al., 2002; Gundlfinger et al., 2010), the MF input-induced postsynaptic Ca2+ signaling in vivo may exhibit high fluctuation, which is different from the plateau-like CaTs evoked by MF stimulation at uniform frequency in this study (Fig. 2B). If MF inputs are composed of many bursts, as observed in vivo, Zn2+ signaling will be essential for the induction of LTP-IE.
The increase in dendritic excitability can lower the threshold of synaptic inputs required for the induction of synaptic LTP. Therefore, it is expected that high-frequency MF input can induce metaplastic changes at PP synapses, and thus prime a postsynaptic CA3-PC for the subsequent PP synaptic inputs. It remains to be tested whether LTP-IE accompanies such metaplastic changes at PP synapses. Furthermore, given that the dentate gyrus plays an important role in pattern separation, we imagine that the lack of MF-induced metaplasticity can impair the performance for this behavioral task if it is an essential part of the role of MF inputs to CA3-PCs. It has been shown that KO of ZnT3 causes impairment in Erk signaling at MF terminals and in the behavioral discrimination task (Sindreu et al., 2011). Our study raises a possibility that the behavioral impairment in ZnT3-KO mice may be caused by impairment not only in presynaptic Erk signaling but also in postsynaptic LTP-IE.
Footnotes
This study was supported by grants from the National Research Foundation of Korea (Grant 2017R1A2B2006149) and Seoul National University Hospital.
The authors declare no competing financial interests.
References
- Amaral DG, Ishizuka N, Claiborne B (1990) Neurons, numbers and the hippocampal network. Prog Brain Res 83:1–11. 10.1016/S0079-6123(08)61237-6 [DOI] [PubMed] [Google Scholar]
- Berzhanskaya J, Urban NN, Barrionuevo G (1998) Electrophysiological and pharmacological characterization of the direct perforant path input to hippocampal area CA3. J Neurophysiol 79:2111–2118. 10.1152/jn.1998.79.4.2111 [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Lee CC, Colmers WF, Miller RJ (1998) Changes in mitochondrial function resulting from synaptic activity in the rat hippocampal slice. J Neurosci 18:4570–4587. 10.1523/JNEUROSCI.18-12-04570.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischofberger J, Engel D, Frotscher M, Jonas P (2006) Timing and efficacy of transmitter release at mossy fiber synapses in the hippocampal network. Pflugers Arch 453:361–372. 10.1007/s00424-006-0093-2 [DOI] [PubMed] [Google Scholar]
- Bodrikov V, Sytnyk V, Leshchyns'ka I, den Hertog J, Schachner M (2008) NCAM induces CaMKIIalpha-mediated RPTPalpha phosphorylation to enhance its catalytic activity and neurite outgrowth. J Cell Biol 182:1185–1200. 10.1083/jcb.200803045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt DT, Goerke A, Heuer M, Gimona M, Leitges M, Kremmer E, Lammers R, Haller H, Mischak H (2003) Protein kinase C delta induces src kinase activity via activation of the protein tyrosine phosphatase PTP alpha. J Biol Chem 278:34073–34078. 10.1074/jbc.M211650200 [DOI] [PubMed] [Google Scholar]
- Brautigan DL, Bornstein P, Gallis B (1981) Phosphotyrosyl-protein phosphatase. specific inhibition by zn. J Biol Chem 256:6519–6522. [PubMed] [Google Scholar]
- Cole TB, Wenzel HJ, Kafer KE, Schwartzkroin PA, Palmiter RD (1999) Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc Natl Acad Sci U S A 96:1716–1721. 10.1073/pnas.96.4.1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinney MJ 2nd, Reynolds IJ, Dineley KE (2005) Simultaneous detection of intracellular free calcium and zinc using fura-2FF and FluoZin-3. Cell Calcium 37:225–232. 10.1016/j.ceca.2004.10.003 [DOI] [PubMed] [Google Scholar]
- Gomez GA, McLachlan RW, Wu SK, Caldwell BJ, Moussa E, Verma S, Bastiani M, Priya R, Parton RG, Gaus K, Sap J, Yap AS (2015) An RPTPα/Src family kinase/Rap1 signaling module recruits myosin IIB to support contractile tension at apical E-cadherin junctions. Mol Biol Cell 26:1249–1262. 10.1091/mbc.E14-07-1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishin AA, Benquet P, Gerber U (2005) Muscarinic receptor stimulation reduces NMDA responses in CA3 hippocampal pyramidal cells via Ca2+-dependent activation of tyrosine phosphatase. Neuropharmacology 49:328–337. 10.1016/j.neuropharm.2005.03.019 [DOI] [PubMed] [Google Scholar]
- Gundlfinger A, Breustedt J, Sullivan D, Schmitz D (2010) Natural spike trains trigger short- and long-lasting dynamics at hippocampal mossy fiber synapses in rodents. PLoS One 5:e9961. 10.1371/journal.pone.0009961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman SJ, Schlögl A, Frotscher M, Jonas P (2016) Synaptic mechanisms of pattern completion in the hippocampal CA3 network. Science 353:1117–1123. 10.1126/science.aaf1836 [DOI] [PubMed] [Google Scholar]
- Haase H, Maret W (2003) Intracellular zinc fluctuations modulate protein tyrosine phosphatase activity in insulin/insulin-like growth factor-1 signaling. Exp Cell Res 291:289–298. 10.1016/S0014-4827(03)00406-3 [DOI] [PubMed] [Google Scholar]
- Helmchen F. (2011) Calibration of fluorescent calcium indicators. Cold Spring Harb Protoc 2011:923–930. 10.1101/pdb.top120 [DOI] [PubMed] [Google Scholar]
- Henze DA, Wittner L, Buzsáki G (2002) Single granule cells reliably discharge targets in the hippocampal CA3 network in vivo. Nat Neurosci 5:790–795. 10.1038/nn887 [DOI] [PubMed] [Google Scholar]
- Huang X, Morielli A, Peralta E (1993) Tyrosine kinase-dependent suppression of a potassium channel by the G protein-coupled M1 muscarinic acetylcholine receptor. Cell Calcium 75:1145–1156. [DOI] [PubMed] [Google Scholar]
- Hunt DL, Puente N, Grandes P, Castillo PE (2013) Bidirectional NMDA receptor plasticity controls CA3 output and heterosynaptic metaplasticity. Nat Neurosci 16:1049–1059. 10.1038/nn.3461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun JH, Eom K, Lee KH, Ho WK, Lee SH (2013) Activity-dependent downregulation of D-type K+ channel subunit Kv1.2 in rat hippocampal CA3 pyramidal neurons. J Physiol 591:5525–5540. 10.1113/jphysiol.2013.259002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun JH, Eom K, Lee KH, Bae JY, Bae YC, Kim MH, Kim S, Ho WK, Lee S (2015) Kv1.2 mediates heterosynaptic modulation of direct cortical synaptic inputs in CA3 pyramidal cells. J Physiol 593:3617–3643. 10.1113/JP270372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegaya Y, Sasaki T, Ishikawa D, Honma N, Tao K, Takahashi N, Minamisawa G, Ujita S, Matsuki N (2013) Interpyramid spike transmission stabilizes the sparseness of recurrent network activity. Cereb Cortex 23:293–304. 10.1093/cercor/bhs006 [DOI] [PubMed] [Google Scholar]
- Jia Y, Jeng JM, Sensi SL, Weiss JH (2002) Zn2+ currents are mediated by calcium-permeable AMPA/Kainate channels in cultured murine hippocampal neurones. J Physiol 543:35–48. 10.1113/jphysiol.2002.020172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston D. (1981) Passive cable properties of hippocampal CA3 pyramidal neurons. Cell Mol Neurobiol 1:41–55. 10.1007/BF00736038 [DOI] [PubMed] [Google Scholar]
- Kay AR, Tóth K (2006) Influence of location of a fluorescent zinc probe in brain slices on its response to synaptic activation. J Neurophysiol 95:1949–1956. 10.1152/jn.00959.2005 [DOI] [PubMed] [Google Scholar]
- Kerchner GA, Canzoniero LM, Yu SP, Ling C, Choi DW (2000) Zn2+ current is mediated by voltage-gated Ca2+ channels and enhanced by extracellular acidity in mouse cortical neurones. J Physiol 528:39–52. 10.1111/j.1469-7793.2000.00039.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiedrowski L. (2011) Cytosolic zinc release and clearance in hippocampal neurons exposed to glutamate—the role of pH and sodium. J Neurochem 117:231–243. 10.1111/j.1471-4159.2011.07194.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Lee SH, Park KH, Ho WK, Lee SH (2003) Distribution of K+-dependent Na+/Ca2+ exchangers in the rat supraoptic magnocellular neuron is polarized to axon terminals. J Neurosci 23:11673–11680. 10.1523/JNEUROSCI.23-37-11673.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Poo MM (2004) Spike train timing-dependent associative modification of hippocampal CA3 recurrent synapses by mossy fibers. Neuron 41:445–454. 10.1016/S0896-6273(03)00873-0 [DOI] [PubMed] [Google Scholar]
- Kowalski J, Gan J, Jonas P, Pernía-Andrade AJ (2016) Intrinsic membrane properties determine hippocampal differential firing pattern in vivo in anesthetized rats. Hippocampus 26:668–682. 10.1002/hipo.22550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Kesner RP (2004) Encoding versus retrieval of spatial memory: double dissociation between the dentate gyrus and the perforant path inputs into CA3 in the dorsal hippocampus. Hippocampus 14:66–76. 10.1002/hipo.10167 [DOI] [PubMed] [Google Scholar]
- Lee SH, Schwaller B, Neher E (2000) Kinetics of Ca2+ binding to parvalbumin in bovine chromaffin cells implications for Ca2+ transients of neuronal dendrites. J Physiol 525:419–432. 10.1111/j.1469-7793.2000.t01-2-00419.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leutgeb JK, Leutgeb S, Moser MB, Moser EI (2007) Pattern separation in the dentate gyrus and CA3 of the hippocampus. Science 315:961–966. 10.1126/science.1135801 [DOI] [PubMed] [Google Scholar]
- Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman G, Rudy B, Schlessinger J (1995) Protein tyrosine kinase PYK2 involved in Ca2+-induced regulation of ion channel and MAP kinase functions. Nature 376:737–745. 10.1038/376737a0 [DOI] [PubMed] [Google Scholar]
- Li Y, Hough C, Frederickson C, Sarvey J (2001a) Induction of mossy fiber→Ca3 long-term potentiation requires translocation of synaptically released Zn2+. J Neurosci 21:8015–8025. 10.1523/JNEUROSCI.21-20-08015.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hough CJ, Suh SW, Sarvey JM, Frederickson CJ (2001b) Rapid translocation of Zn2+ from presynaptic terminals into postsynaptic hippocampal neurons after physiological stimulation. J Neurophysiol 86:2597–2604. 10.1152/jn.2001.86.5.2597 [DOI] [PubMed] [Google Scholar]
- Mackenzie PJ, Murphy TH (1998) Mckenzie High safety factor for action potential conduction along axons but not dendrites of cultured hippocampal and cortical neurons. J Neurophysiol 80:2089–2101. 10.1152/jn.1998.80.4.2089 [DOI] [PubMed] [Google Scholar]
- Marr D. (1971) Simple memory: a theory for archicortex. Philos Trans R Soc Lond B Biol Sci 262:23–81. 10.1098/rstb.1971.0078 [DOI] [PubMed] [Google Scholar]
- McMahon DB, Barrionuevo G (2002) Short- and long-term plasticity of the perforant path synapse in hippocampal area CA3. J Neurophysiol 88:528–533. 10.1152/jn.2002.88.1.528 [DOI] [PubMed] [Google Scholar]
- Mistry R, Dennis S, Frerking M, Mellor JR (2011) Dentate gyrus granule cell firing patterns can induce mossy fiber long-term potentiation in vitro. Hippocampus 21:1157–1168. 10.1002/hipo.20815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa K, Sun LD, Quirk MC, Rondi-Reig L, Wilson MA, Tonegawa S (2003) Hippocampal CA3 NMDA receptors are crucial for memory acquisition of one-time experience. Neuron 38:305–315. 10.1016/S0896-6273(03)00165-X [DOI] [PubMed] [Google Scholar]
- Neunuebel JP, Knierim JJ (2014) CA3 retrieves coherent representations from degraded input: direct evidence for CA3 pattern completion and dentate gyrus pattern separation. Neuron 81:416–427. 10.1016/j.neuron.2013.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohana E, Hoch E, Keasar C, Kambe T, Yifrach O, Hershfinkel M, Sekler I (2009) Identification of the Zn2+ binding site and mode of operation of a mammalian Zn2+ transporter. J Biol Chem 284:17677–17686. 10.1074/jbc.M109.007203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly RC, McClelland JL (1994) Hippocampal conjunctive encoding, storage, and recall: avoiding a trade-off. Hippocampus 4:661–682. 10.1002/hipo.450040605 [DOI] [PubMed] [Google Scholar]
- Pan E, Zhang XA, Huang Z, Krezel A, Zhao M, Tinberg CE, Lippard SJ, McNamara JO (2011) Vesicular zinc promotes presynaptic and inhibits postsynaptic long-term potentiation of mossy fiber-CA3 synapse. Neuron 71:1116–1126. 10.1016/j.neuron.2011.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Nairn AC, Wang P, Lombroso PJ (2003) NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat Neurosci 6:34–42. 10.1038/nn989 [DOI] [PubMed] [Google Scholar]
- Perez-Rosello T, Baker J, Ferrante M, Iyengar S, Ascoli G, Barrionuevo G (2011) Passive and active shaping of unitary responses from associational/commissural and perforant path synapses in hippocampal CA3 pyramidal cells. J Comput Neurosci 31:159–182. 10.1007/s10827-010-0303-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernía-Andrade A, Jonas P (2014) Theta-gamma-modulated synaptic currents in hippocampal granule cells in vivo define a mechanism for network oscillations. Neuron 81:140–152. 10.1016/j.neuron.2013.09.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Noebels JL (2005) Visualization of transmitter release with zinc fluorescence detection at the mouse hippocampal mossy fibre synapse. J Physiol 566:747–758. 10.1113/jphysiol.2005.089276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollenhagen A, Sätzler K, Rodríguez E, Jonas P, Frotscher M, Lübke J (2007) Structural determinants of transmission at large hippocampal mossy fiber synapses. J Neurosci 27:10434–10444. 10.1523/JNEUROSCI.1946-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumschik SM, Nydegger I, Zhao J, Kay AR (2009) The interplay between inorganic phosphate and amino acids determines zinc solubility in brain slices. J Neurochem 108:1300–1308. 10.1111/j.1471-4159.2009.05880.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachidhanandam S, Blanchet C, Jeantet Y, Cho YH, Mulle C (2009) Kainate receptors act as conditional amplifiers of spike transmission at hippocampal mossy fiber synapses. J Neurosci 29:5000–5008. 10.1523/JNEUROSCI.5807-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MW, Kalia LV (2004) Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 5:317–328. 10.1038/nrn1368 [DOI] [PubMed] [Google Scholar]
- Sensi SL, Canzoniero LM, Yu SP, Ying HS, Koh JY, Kerchner GA, Choi DW (1997) Measurement of intracellular free zinc in living cortical neurons routes of entry. J Neurosci 17:9554–9564. 10.1523/JNEUROSCI.17-24-09554.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sensi SL, Ton-That D, Sullivan PG, Jonas EA, Gee KR, Kaczmarek LK, Weiss JH (2003) Modulation of mitochondrial function by endogenous Zn2+ pools. Proc Natl Acad Sci U S A 100:6157–6162. 10.1073/pnas.1031598100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindreu C, Palmiter R, Storm D (2011) Zinc transporter ZnT-3 regulates presynaptic erk signaling and hippocampus-dependent memory. Proc Natl Acad Sci U S A 108:3366–3370. 10.1073/pnas.1019166108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda A, Fuke S, Minami A, Oku N (2007) Role of zinc influx via AMPA/kainate receptor activation in metabotropic glutamate receptor-mediated calcium release. J Neurosci Res 85:1310–1317. 10.1002/jnr.21233 [DOI] [PubMed] [Google Scholar]
- Treves A, Rolls ET (1992) Computational constraints suggest the need for two distinct input systems to the hippocampal CA3 network. Hippocampus 2:189–199. 10.1002/hipo.450020209 [DOI] [PubMed] [Google Scholar]
- Tsai W, Morielli A, Cachero T, Peralta E (1999) Receptor protein tyrosine phosphatase alpha participates in the m1 muscarinic acetylcholine receptor-dependent regulation of Kv1.2 channel activity. EMBO J 18:109–118. 10.1093/emboj/18.1.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsay D, Dudman JT, Siegelbaum SA (2007) HCN1 channels constrain synaptically evoked Ca2+ spikes in distal dendrites of CA1 pyramidal neurons. Neuron 56:1076–1089. 10.1016/j.neuron.2007.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto M, Yasui T, Yamada MK, Nishiyama N, Matsuki N, Ikegaya Y (2003) Mossy fibre synaptic NMDA receptors trigger non-hebbian long-term potentiation at entorhino-CA3 synapses in the rat. J Physiol 546:665–675. 10.1113/jphysiol.2002.033803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergnano AM, Rebola N, Savtchenko LP, Pinheiro PS, Casado M, Kieffer BL, Rusakov DA, Mulle C, Paoletti P (2014) Zinc dynamics and action at excitatory synapses. Neuron 82:1101–1114. 10.1016/j.neuron.2014.04.034 [DOI] [PubMed] [Google Scholar]
- Wilson M, Hogstrand C, Maret W (2012) Picomolar concentrations of free zinc(II) ions regulate receptor protein-tyrosine phosphatase beta activity. J Biol Chem 287:9322–9326. 10.1074/jbc.C111.320796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Bertoglio BA, Gee KR, Kay AR (2008) The zinc indicator FluoZin-3 is not perturbed significantly by physiological levels of calcium or magnesium. Cell Calcium 44:422–426. 10.1016/j.ceca.2008.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]