

Abstract
Spike timing is an important factor in the modification of synaptic strength. Various forms of spike timing-dependent plasticity (STDP) occur in the brains of diverse species, from insects to humans. In unimodal STDP, only LTP or LTD occurs at the synapse, regardless of which neuron spikes first; the magnitude of potentiation or depression increases as the time between presynaptic and postsynaptic spikes decreases. This from of STDP may promote developmental strengthening or weakening of early projections. In bidirectional Hebbian STDP, the magnitude and the sign (potentiation or depression) of plasticity depend, respectively, on the timing and the order of presynaptic and postsynaptic spikes. In the rodent barrel cortex, multiple forms of STDP appear sequentially during development, and they contribute to network formation, retraction, or fine-scale functional reorganization. Hebbian STDP appears at L4-L2/3 synapses starting at postnatal day (P) 15; the synapses exhibit unimodal “all-LTP STDP” before that age. The appearance of Hebbian STDP at L4-L2/3 synapses coincides with the maturation of parvalbumin-containing GABA interneurons in L4, which contributes to the generation of L4-before-L2/3 spiking in response to thalamic input by producing fast feedforward suppression of both L4 and L2/3 cells. After P15, L4-L2/3 STDP mediates fine-scale circuit refinement, essential for the critical period in the barrel cortex. In this review, we first briefly describe the relevance of STDP to map plasticity in the barrel cortex, then look over roles of distinct forms of STDP during development. Finally, we propose a hypothesis that explains the transition from network formation to the initiation of the critical period in the barrel cortex.
Introduction
Spike timing-dependent plasticity (STDP) was first reported by Markram et al. (1997) in dual whole-cell patch recordings from neocortical L5 neurons. These authors discovered that LTP is induced when an EPSP is followed within 100 ms by postsynaptic action potentials (APs), whereas LTD is induced by reversing the order (i.e., when an EPSP occurs after a postsynaptic AP). By varying the time interval between EPSP and AP in cultured hippocampal cells, Bi and Poo (1998) subsequently revealed the close temporal association between the magnitude of potentiation and spike timing. The term “spike timing-dependent plasticity” was coined later, in a computational modeling study (Song et al., 2000).
Since those first studies, many others have found that STDP can be induced in various brain regions. Furthermore, multiple forms of STDP operate with different rules governing induction in different regions of the brain and in different cell types. For example, in the hippocampus, the time window for the induction of LTP and LTD ranges from 0 to 50 ms and −50 to 0 ms, respectively, which is symmetrical; for L4-L2/3 synapses in the barrel cortex, the LTP and LTD window is 0 to 20 ms and −200 to 0 ms, respectively, which is not symmetrical. Furthermore, STDP at some synapses only involves LTP (all-LTP STDP) (Itami and Kimura, 2012), whereas at other synapses it only involves LTD (all-LTD STDP) (Han et al., 2000; Lu et al., 2007; Safo and Regehr, 2008; Itami et al., 2016). In these cases, the magnitude of LTP and LTD changes depending on spike timing in presynaptic and postsynaptic cells, but the order of spiking does not matter.
The roles of STDP in physiological functions have also been reported in in vivo experiments, in which sensory stimuli are paired with postsynaptic spikes after a specific delay, resulting in altered sensory responses in retinotectal cells (Zhang et al., 1998, 2000; Mu and Poo, 2006), visual cortical cells (Schuett et al., 2001; Meliza and Dan, 2006), and in the barrel cortex (Jacob et al., 2007).
Several excellent reviews about general properties of STDP (Bi and Poo, 2001; Sjöström and Nelson, 2002), its diversity (Dan and Poo, 2006; Caporale and Dan, 2008), or even its history (Markram et al., 2011) are available. For a relatively recent and thorough discussion, Feldman (2012) would be recommended.
Here we discuss recent studies demonstrating that, during development, multiple distinct forms of STDP appear, disappear, or even sequentially change form, concurrent with changes in circuit function, such as synapse formation or retraction. In particular, we discuss the switch of STDP from a spike order-independent form to the classical, order-dependent, Hebbian STDP, which occurs simultaneously with the formation of a regulatory circuit of spike order of presynaptic and postsynaptic cells. Furthermore, we discuss evidence indicating that Hebbian STDP, which occurs at L4-L2/3 synapses in the barrel cortex, is a critical factor driving activity-dependent map plasticity (Feldman, 2009; Itami and Kimura, 2012). Based on this evidence, we hypothesize that the developmental switch of STDP at L4-L2/3 in the barrel cortex is highly relevant to the initiation of map plasticity. Thus, this Viewpoints article provides a likely mechanism for the initiation of experience-dependent plasticity in the neocortex, an explanation that has been sought since seminal studies demonstrated deprivation-induced plasticity in the sensory neocortex (Wiesel and Hubel, 1963; Simons and Land, 1987).
Relevance of STDP to map plasticity in the sensory cortex
Sensory map plasticity in the barrel cortex
Tactile experience causes robust changes in somatic representations in the somatosensory cortex (Kaas et al., 1981). The cellular mechanisms underlying this plasticity are best documented in the rodent barrel cortex. Trimming or plucking one or more whiskers rapidly lowers responsiveness to the deprived whisker(s) when it (they) regrow(s), and slowly increases responsiveness to the spared whiskers (Fox, 1992; Diamond et al., 1993; Glazewski and Fox, 1996). These two changes, strengthening and weakening of responsiveness, are distinct phenomena that show different dynamics, development, and genetic dependence; thus, they are thought to reflect distinct mechanisms of plasticity (Glazewski et al., 2000; Fox, 2002; Drew and Feldman, 2009).
Somatosensory plasticity is most robust during development and remains present in adult animals older than 6 months. The sites of deprivation-induced plasticity in the cortex change during development. In neonates (<P4-P5), plasticity most likely involves thalamocortical synapses (Schlaggar et al., 1993), although it may occur at sites other than layer 4, the main recipient of thalamocortical axons. At this age, the cortex is still undergoing differentiation, and L4 differentiates from the cortical plate at P3-P4. Thalamocortical synapses on cortical plate cells or L4 cells show plasticity at the cellular level (Crair and Malenka, 1995; Lu et al., 2001; Itami et al., 2016). However, these synapses rapidly lose plasticity after differentiation of L4 is complete (discussed in detail later in this Viewpoints article). In juvenile and adult animals, the synaptic sites of map plasticity move to intracortical synapses. Changes in sensory maps induced by whisker deprivation occur in both L2/3 and L5, but changes tend to occur more rapidly and extensively, and sometimes exclusively, in L2/3 (Diamond et al., 1994; Glazewski and Fox, 1996; Stern et al., 2001; Feldman and Brecht, 2005; Drew and Feldman, 2009).
Roles of STDP in map plasticity in the barrel cortex
Thalamocortical synapse
Thalamocortical (or thalamus-cortical plate) synapses exhibit all-LTP STDP in neonatal rodents (Itami et al., 2016), when L4 is not yet differentiated from the cortical plate. The magnitude of STDP is large at this time (as much as 500% of controls), but it rapidly decreases from P4 to P5. Subsequently, plasticity at thalamocortical synapses varies across target layers. Synapses between thalamic and L4 cells lose STDP. In contrast, thalamus-L2/3 synapses change to all-LTD STDP (Itami et al., 2016). Interestingly, map plasticity by whisker deprivation disappears in L4 around P4 (Schlaggar et al., 1993) in parallel with the disappearance of all-LTP STDP (Itami et al., 2016). The conventional form of LTP is induced during P3-P7, in which 100 stimuli are applied at 1 Hz, whereas postsynaptic cells are depolarized and held between −10 to 0 mV in current-clamp mode. This form of LTP also disappeared in the similar time course (after P7) (Crair and Malenka, 1995) at thalamus-L4 synapses. Thus, it is likely that, in neonates, LTP or all-LTP STDP serves as a mechanism for deprivation-induced map plasticity. Which of these two forms of plasticity better explains the rearrangement of synaptic connections? It has long been suggested that the rearrangement of synaptic connections during normal and deprived development is thought to be controlled by correlations in presynaptic and postsynaptic activity (Katz and Shatz, 1996). A favored model involves the postsynaptic detection of synchronously active afferents, and synapses are stabilized when presynaptic and postsynaptic activities are correlated. Under all-LTP STDP, correlated activity in presynaptic and postsynaptic cells occurring within ±50 ms (regardless of order) causes strengthening of synaptic connections; thus, only simultaneity, and not spiking order, is important in all-LTP STDP (Itami et al., 2016). From this point of view, it seems that all-LTP STDP better explain the rearrangement of synaptic connections. What about conventional LTP? Considering that spike timing is only one of the several factors governing induction of plasticity (Feldman, 2012), conventional paring protocol is supposed to induce LTP when all-LTP STDP could be induced.
L4-L2/3 synapses.
L4-L2/3 synapses exhibit Hebbian STDP (Feldman, 2000a; Bender et al., 2006; Nevian and Sakmann, 2006) after P13-P15 (Itami and Kimura, 2012); that is, LTP is induced by pre-before-post or L4-cell firing before L2/3-cell firing, whereas LTD is induced by post-before-pre or L2/3-cell firing before L4-cell firing. To exhibit Hebbian STDP at these synapses is of particular importance for driving sensory-input-dependent map plasticity, at least in the barrel cortex, because of the following two reasons. First, sensory input (from whisker deflection) causes spiking in L4 cells, which drives spiking in L2/3 cells (Simons, 1978; Armstrong-James et al., 1992; Armstrong-James, 1995; Brumberg et al., 1999; Celikel et al., 2004). Thus, every time sensory input arrives, L4-L2/3 synapses are potentiated. Second, when whisker(s) are deprived (but not all of the whiskers on one side), the sequence of spiking in L4 and L2/3 cells in the deprived column is suddenly reversed: L2/3 cells fire first, followed by L4-cell spikes (Allen et al., 2003; Celikel et al., 2004). This occurs partly because there are strong horizontal, cross-columnar projections among L2/3 cells; thus, when a spared whisker activates L2/3 cells in its target column, these cells drive spiking in L2/3 cells of the adjacent, deprived column (Allen et al., 2003; Celikel et al., 2004). (In these experiments, all whiskers are simultaneously deflected. Responses in a deprived column are obtained during deflections of all, except a single whisker, are deflected to reflect natural “deprivation.”) What drives firing of L4 cells in the deprived column is not well understood. Unlike L2/3, direct connections from L4 neurons in spared barrels are rare (Petersen and Sakmann, 2000). However, in electrophysiological experiments, stimulation of L2/3 induces monosynaptic EPSPs in the excitatory cells in the L4 barrel of the same column (Schubert et al., 2003; Ohshima et al., 2017). Thus, horizontal spread in L2/3 may ultimate induce spiking in L4 (Tanifuji et al., 1994), although anatomically, there are few, if any, direct connections from L2/3 to L4 in the same column (Burkhalter, 1989). The involvement of subcortical convergence from whisker to cortex may account for the responses of the deprived barrels in L4 (Goldreich et al., 1999). In any case, the spike latency of L4 cells in the deprived column is longer than that of L2/3 cells (Armstrong-James and Fox, 1987; Armstrong-James et al., 1992); thus, L2/3 firing is followed by L4 firing. This weakens the L4 to L2/3 synapses. Because of the LTD component of the Hebbian STDP rule, L4-L2/3 synapses in the deprived column are weakened every time the surrounding whisker(s) are stimulated. Whisker deprivation in vivo causes LTD in the deprived column as demonstrated in ex vivo slice preparations (Allen et al., 2003). Consequently, cells in the deprived column might gradually become driven more by neighboring spared whiskers, such that the receptive field gradually shifts to be dominated by spared whiskers. Plasticity of L2/3 horizontal connections may also play a role in whole-map changes, but Hebbian STDP at L4-L2/3 is the primary contributor to map plasticity.
Regulation of spike sequences at L4-L2/3 cells
Correct operation of Hebbian STDP at L4-L2/3 synapses requires that sensory input from whiskers leads to sequential activation of L4, then L2/3 neurons (Simons, 1978; Armstrong-James et al., 1992; Armstrong-James, 1995; Brumberg et al., 1999; Celikel et al., 2004). Simultaneous recording of L4 and L2/3 cells reveals that sensory stimulation does indeed produce the required sequence, with L4 cells spiking before L2/3 cells; reversed spike sequences are only infrequently observed (Allen et al., 2003; Celikel et al., 2004). Although L4 is a thalamo-recipient layer and L4 neurons project to L2/3 cells, it is difficult to see how this, by itself, can produce such precise sequential firing, Indeed, several factors are expected to reduce temporal precision. First, although the majority of thalamic afferents project to L4, some project directly to L2/3 cells, where they might evoke early spiking. Second, the excitation and conduction of neurons show substantial temporal variability, resulting in considerable temporal fluctuations in onset latencies for spikes, even for short latencies. Onset latencies from whisker stimulation to spikes in rat barrel cortex are 8.6 ± 1.0 ms (mean ± SD) for L4 cells, 10.2 ± 1.5 ms for L3 cells, and 13.0 ± 2.6 ms for L2 cells (Celikel et al., 2004). Thus, the difference in spike latency between L2 and L4 (4.4 ms) is only slightly larger than the sum of the SDs (3.6 ms), and the difference in spike latency between L3 and L4 (1.6 ms) is much less than the sum of the SD (2.5 ms). Third, spontaneous activity might occur as a result of spontaneous fluctuations in membrane potential or unidentified external inputs in cells in either layer. Finally, because the rise time of L2/3 cells' EPSPs is faster than those of L4 cells (Feldmeyer et al., 1999, 2002), L2/3 cells might be more likely to be activated by accidental inputs from thalamus or by spontaneous events.
Because of the considerations described above, the relay order of excitatory connections (thalamus-L4-L2/3) is unlikely to be sufficient to create the precise spiking order required for appropriate Hebbian STDP at L4-L2/3 synapses. Strong recurrent excitatory coupling in L4, which creates strong synchronous discharges (Valiullina et al., 2016), may help to ensure appropriate spike timing. An inhibitory mechanism that actively regulates spiking in L4 and L2/3 might also be necessary. Ideally, such inhibition would also be driven by sensory inputs. Such a system has received little attention so far, except for in one study (Kimura et al., 2010), which is described in the next section.
Fast activation of feedforward parvalbumin-expressing (PV) inhibitory neurons by thalamic input and its relevance to L4-L2/3 spike sequences and STDP
Fast-spiking (FS), PV-expressing inhibitory neurons in L4 may play a crucial role in ensuring L4-leading-L2/3 firing sequences (Kimura et al., 2010). FS cells mature electrophysiologically and chemically (i.e., produce PV) at approximately the end of the second postnatal week (P14) (Itami et al., 2007). In mature animals, latency from the thalamus to FS cells in L4 is, on average, ∼0.7 ms shorter than that from the thalamus to excitatory cells in L4 (2.8 ± 0.1 ms vs 2.1 ± 0.1 ms for excitatory and FS cells, respectively) (Kimura et al., 2010). Although the difference is small, the slightly faster activation of FS cells is required for generating disynaptic IPSPs that regulate the spiking order of L4-L2/3 cells.
IPSPs appear in L4 excitatory relay cells as rapidly as 1.7 ± 0.1 ms after the onset of monosynaptic EPSPs induced by thalamic inputs. Because all thalamocortical projections are excitatory, the fastest IPSP induced by thalamocortical inputs to L4 must be at least disynaptic. The time required for AP generation and conduction within the FS cell together with the time required for synaptic transmission to the postsynaptic L4 excitatory cell add an extra delay for the IPSP relative to the EPSP in excitatory cells. Given that the generation of an AP in FS cells takes 1.43 ± 0.41 ms (from EPSP onset to the peak of AP), and the conduction of APs within FS axons together with synaptic transmission requires 0.87 ± 0.08 ms (from the peak of FS AP to IPSP onset in postsynaptic cells), one can calculate that ∼2.3 ms is required to produce disynaptic feedforward IPSPs. Thus, if L4 FS and excitatory cells receive simultaneous input from thalamocortical afferents, the IPSPs in L4 should occur at least 2.3 ms after the onset of thalamic EPSPs. However, the observed delay of IPSPs is only 1.7 ± 0.1 ms. To account for this difference (2.3 ms vs 1.7 ms), FS cells must be activated by thalamic input slightly earlier (∼0.7 ms) than excitatory cells (Fig. 1). Because of the rapid activation of L4 FS cells, generation of APs in L4 excitatory cells occurs specifically within the initial period of thalamic EPSPs (∼1.7 ms from onset).
Figure 1.

Schematic drawing of latency differences in feedforward PV cells and excitatory relay cells from the thalamus (left) and their influence on generating spike timing differences in L4 and L2/3 cells in the order “L4 followed by L2/3” spiking (right). Excitatory neurons in the thalamus, L4, and L2/3 (open circles), PV neuron in L4 (closed circles). Right, Lines indicate membrane potentials of L2/3 (top) and L4 (middle) excitatory cells, including monosynaptic EPSPs (in L4), disynaptic EPSPs (in L2/3), and disynaptic IPSPs (in L4 and L2/3). Dotted lines indicate APs in the excitatory cells. Bottom, Line indicates membrane potential of a PV cell. Faster (∼0.7 ms) activation of feedforward PV compared with excitatory relay cells in L4 causes the disynaptic IPSPs immediately after monosynaptic EPSPs in L4 and immediately before disynaptic EPSPs in L2/3 cells. This generates spike timing differences in L4, L2/3 cells. 1–5, Time differences. 1, Latency difference of PV and excitatory cell in L4 from thalamus (∼0.7 ms). 2, Time to generate APs in PV cell (1.43 ± 0.41 ms). 3, Time for conduction of an AP in PV to synaptic terminals and to produce IPSPs in L4 excitatory cells (0.84 ± 0.08 ms) and L2/3 excitatory cells. 4, Delay of disynaptic IPSP from monosynaptic EPSP from thalamus in L4 excitatory cell (∼1.7 ms). 5, 5′, Spike timing difference between L4 and L2/3 cells (1.6–4.4 ms). Based on data from Kimura et al. (2010) (1–4) and Celikel et al. (2004) (5).
In addition to inhibiting L4 excitatory neurons, L4 FS cells project to L2/3 (Itami et al., 2007) and help regulate the firing of L2/3 cells. Because thalamic input activates L4 FS cells before L4 excitatory cells, disynaptic feedforward IPSPs evoked in L2/3 cells by L4 FS cells should occur before disynaptic EPSPs from L4 excitatory cells, assuming comparable conduction of excitatory and FS inhibitory cells from L4 to L2/3 (Fig. 1). Indeed, in vivo sensory stimulation (Ferster and Lindström, 1983; Foeller et al., 2005) and thalamic stimulation in slice preparations (Kimura et al., 2010) produce disynaptic IPSPs that precede disynaptic EPSPs in L2/3. Thus, rapid activation of L4 FS cells guarantees the generation of the “L4-leading-L2/3” firing sequence both by restricting L4 firing to the initial part of monosynaptic EPSPs and by delaying L2/3 firing.
Moreover, because the disynaptic IPSPs are driven by thalamus or sensory inputs, these IPSPs prevent L2/3 cells from firing by unexpected inputs each time immediately before the disynaptic EPSPs arrive at L2/3 cells. This is a great advantage, which becomes evident if one considers how the cells are activated by sensory inputs in vivo. In most cases, sensory stimuli will deflect multiple whiskers and therefore activate multiple barrels. Because there are strong horizontal connections among barrels in L2/3, an L2/3 cell receives inputs from adjacent barrels around the same time as it receives input from L4 cells in its own barrel. But because excitatory inputs from its own L4 are always preceded by inhibition, input from adjacent barrels is unlikely to generate an early spike in the L2/3 cell. In contrast, when a whisker is removed, there is no inhibition of either L4 or L2/3 cells in the deprived column. The deprived L2/3 is free to spike in response to input from L2/3 cells in the adjacent column that represents spared whisker. Therefore, reversed firings will take place without suppression. This would explain the expansion of spared barrels to cells in deprived columns.
Maturation of thalamus-FS circuitry and simultaneous switch of STDP at L4-L2/3 synapses
The activation of L4 FS cells before L4 excitatory cells by thalamic input, which, as described above, is thought to be crucial for generating “L4-leading-L2/3” spike sequences, is detected only after P12-P14 (Kimura et al., 2010). Therefore, one would predict that the “L4-leading-L2/3” sequence occurs only after P12-P14. This remains to be experimentally confirmed. Nonetheless, it has been shown that, at P8 and P11, the firing of L4 and L2/3 cells is not sequential but random, with substantial overlap as revealed by peristimulus-time histograms following whisker stimulation (Kimura et al., 2010).
In the rodent cortex, inhibitory synaptic transmission matures after birth. Activation of GABA receptors in neonates causes depolarization rather than hyperpolarization because a high intracellular concentration of chloride ions results in a relatively depolarized chloride reversal potential (Ben-Ari et al., 1989; Ben-Ari, 2002; Owens and Kriegstein, 2002). Hyperpolarization is seen only after P6-P7 (Daw et al., 2007) in the barrel cortex. Subsequently, the sizes of IPSCs continue to increase through P7-P9 (Daw et al., 2007). Thus, regulation of spike sequences by synaptic inhibition must occur after P9. However, cross-correlation analyses that determine when “L4-leading-L2/3” spike sequences are established are still required to determine whether this is the case.
Another important question regarding maturation of thalamus-FS circuitry is whether L4-L2/3 synapses exhibit Hebbian STDP earlier than P12-P14. This is especially important because, under Hebbian STDP, random firing of presynaptic and postsynaptic cells causes LTD (Feldman, 2000a). This is because the time window that causes the LTD component of STDP (post-pre timing, ∼200 ms) is much longer than that inducing the LTP component (pre-post timing, ∼20 ms). L4 axons initiate synapse formation with L2/3 cells between P6 and P7, and synaptogenesis continues at least throughout the second postnatal week (Bender et al., 2003). Therefore, L4-L2/3 synapses form while networks are still immature; therefore, the precise timing of the firing of presynaptic and postsynaptic cells cannot be expected. In other words, having the Hebbian type of STDP during synaptic formation would be disadvantageous and is therefore very unlikely.
A recent study investigated plasticity mechanisms in developing barrel cortex (Itami and Kimura, 2012) and found that, before P13, LTP occurs regardless of spike order. As in thalamocortical synapses before P7-P8, both pre-before-post and post-before-pre spike orders caused LTP, with the magnitude depending on the time interval: the shorter the delay, the larger the potentiation. The magnitude of potentiation induced by post-before-pre timing decreases gradually with age, and potentiation eventually changes into depression after P15. Thus, at L4-L2/3 synapses, STDP switches from all-LTP STDP to Hebbian STDP after P15.
Such a developmental switch of STDP has never been reported before even in any other area in the brain, which is intriguing for the following reasons:
The absence of the LTD component of STDP before P13 suggests that whisker deprivation at this age will not cause suppression at L4-L2/3 synapses. This was confirmed experimentally (Itami and Kimura, 2012). Notably, deprivation-induced suppression at L4-L2/3 synapse is thought to be the first important step for map plasticity, as well as the explanation for the eventual loss of representation of deprived whiskers. This suggests that the switch from all-LTP STDP to Hebbian STDP allows map plasticity to take place. Thus, this STDP switch, together with sequential firing of L4-leading-L2/3, due to faster activation of FS cells compared with that of excitatory cells in the thalamocortical projection, may account for the initiation of the critical period.
In the visual cortex (VC), the initiation of the critical period requires maturation of GABAergic inhibition (Hensch et al., 1998; Fagiolini and Hensch, 2000), which involves PV-positive inhibitory circuits (Fagiolini et al., 2004). However, why the maturation of inhibitory circuit is required to initiate the critical period in the VC is unknown. It sounds reasonable to conclude that the PV circuit is necessary for the regulation of spike sequences of L4, L2/3 cells in the visual and somatosensory cortices, provided that Hebbian STDP underlies ocular dominance plasticity in the VC. Increased inhibition may help change the temporal structure of neural responses through visual inputs, representing a possible mechanism that initiates the critical period (Feldman, 2000b).
If the presence of all-LTP STDP before P13 does not produce deprivation-induced suppression, what is its role? This form of plasticity is well suited for strengthening immature synapses, when timing is imprecise. Indeed, L4 cells initiate synapse formation with L2/3 from P6 to P7. Thus, the age at which L4-L2/3 synapses exhibit all-LTP STDP exactly matches that of synapse formation.
Role of all-LTD STDP in network reorganization
Whereas all-LTP STDP contributes to the formation of L4-L2/3 synapses, all-LTD STDP is important in the development of thalamocortical synapses. Thalamocortical axons invade the cortical plate at birth (P0) and form synapses with cells in the cortical plate, and these synapses exhibit all-LTP STDP. Although some axons approach the pia surface at P2-P4 (Senft and Woolsey, 1991; Rebsam et al., 2002), these axons begin to retract when L2/3 differentiates (P4-P5); and eventually, thalamocortical axons are confined below the border between L2/3 and L4. During this same period, the magnitude of all-LTP STDP decreases and is ultimately replaced by all-LTD STDP or loses STDP depending on the target layers. After P7-P8, all-LTD STDP can be consistently induced from thalamus-L2/3 synapses. Thalamus to L4 synapses lose STDP after P8 (Itami et al., 2016).
These observations suggest that “aberrant” thalamocortical projections that extend to the newly generated L2/3 area are forced to retract in an activity-dependent manner because of the all-LTD STDP rule. Consistent with this, activation of cannabinoid receptors (CB1R), on which all-LTD STDP depends, induces retraction of thalamocortical axons. Furthermore, CB1R-KO animals exhibit excessive invasion of L2/3 by thalamocortical axons (Itami et al., 2016), indicating that endogenous cannabinoid signaling regulates the formation of thalamocortical projections.
Unidirectional versus bidirectional STDP
As indicated above, after P6-P7, L4 axons form synapses with L2/3 cells and these synapses exhibit all-LTP STDP, and at the same time thalamic axons retract their projections to L2/3 and thalamus-L2/3 synapses exhibit all-LTD STDP. Therefore, all-LTP and all-LTD STDP may be the mechanisms of activity-dependent network formation and retraction. This hypothesis further posits that all-LTP STDP at thalamus-cortical plate synapses during the first postnatal week provides a mechanism for synapse formation between the thalamus and cortical plate cells. Together, these potential mechanisms suggest that unidirectional STDP (all-LTP or all-LTD STDP), which depends on timing but not spike order, is suitable for driving activity-dependent network formation and retraction.
In contrast, bidirectional STDP (Hebbian STDP that produces both LTP and LTD), which is dependent on both timing and spike order, is best suited for mediating fine-scale circuit refinement after precise spiking is present. This also suggests that the developmental switch of STDP from unidirectional to bidirectional Hebbian STDP at approximately P13–P15, together with simultaneous maturation of PV inhibitory circuits in L4 to regulate L4-L2/3 spike sequences, represents a transition from circuit formation to input-dependent fine-scale refinement (i.e., the initiation of the critical period for map plasticity).
Specific convergence of distinct forms of STDP only during the second postnatal week: possible interaction of the two forms of STDP
During the second postnatal week, all-LTP STDP from L4 cells and all-LTD STDP from the thalamus converge to the target layer of L2/3. L4 is the canonical target layer of the thalamus. Thus, thalamic activity may activate presynaptic and postsynaptic cells in the L4-L2/3 projection, leading to strengthening under the all-LTP STDP rule that operates at this synapse (Fig. 2). Indeed, a study demonstrated that spike-timing stimulation applied to the thalamus and L2/3 induces LTP at L4-L2/3 synapses in concert with LTD at the thalamus-L2/3 synapses. Thus, converging STDP induces concurrent plasticity (Itami and Kimura, 2016), which has important and interesting implications. Strengthening of L4-L2/3 synapses by all-LTP STDP should occur exclusively in those L2/3 cells that are driven by thalamic input, input that is driving all-LTD STDP at the thalamus-L2/3 synapse. Thus, a replacement of afferent innervation should occur in that specific L2/3 cell; that is, thalamic input should be replaced by L4 input. Thus, the preceding thalamic innervation guides the L4 afferents to the proper site of synapse formation.
Figure 2.
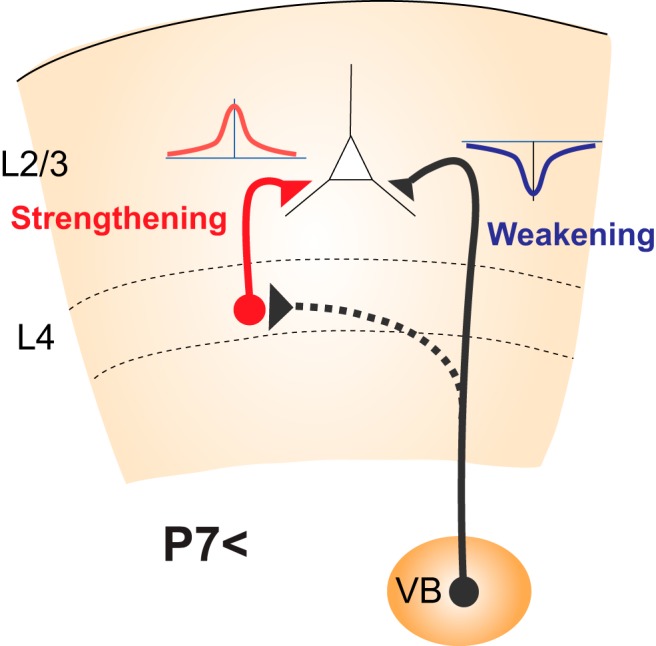
Interaction between two opposite forms of STDP converging on L2/3 pyramidal cells during the second postnatal week, when L4-to-L2/3 synapses exhibit all-LTP STDP, which strengthens this connection, whereas thalamus-L2/3 synapses exhibit all-LTD STDP, which weakens this connection. The canonical target of the thalamus is L4 (dotted line). Thus, the activity of the thalamus drives all-LTP STDP of the L4–L2/3 synapse, which then drives all-LTD STDP of the thalamus-L2/3 synapse, thus shaping the cortical network reorganization in a self-organizing manner.
This raises the interesting possibility that the early thalamic innervation of L2/3, which is induced by all-LTP STDP at synapses between the thalamus and cortical plate cells before L2/3 has differentiated but subsequently retracts, is not unproductive, excess innervation, but instead serves as an important guide for subsequent L4 innervation. Furthermore, thalamic innervation of L2/3 undergoes all-LTD STDP, the weakening of residual innervation would accelerate when L4 to L2/3 synapses become firmly established through all-LTP STDP. Thus, retraction of thalamic afferents from L2/3 and formation of L4 to L2/3 may be driven by thalamic activity in a self-organizing manner. From this point of view, the reorganization of thalamus-L2/3 and L4-L2/3 connections through mutual interaction of STDP is likely to be highly efficient.
STDP and experience-dependent plasticity in the VC
As an experience-dependent plasticity, monocular deprivation during a critical period causes a shift of ocular dominance of cortical cell responses in the VC (Wiesel and Hubel, 1963). The mechanism of this plasticity has also been widely studied, but the involvement of STDP is unclear. Nevertheless, several findings seem to imply that a similar mechanism underlies this form of plasticity. First, and notably, maturation of PV cells contributes to the initiation of the critical period in the VC (Fagiolini and Hensch, 2000; Fagiolini et al., 2004), although how the maturation of inhibitory circuit leads to an opening of the critical period is yet unknown. Second, monocular deprivation causes LTD at L4-L2/3 synapses, which is dependent on CB1R (Crozier et al., 2007; Liu et al., 2008). Whether this LTD possesses spike timing dependence has not been tested, but STDP leads to changes in response properties of neurons in the VC in vivo (Schuett et al., 2001; Meliza and Dan, 2006). Together, these fragmental findings support the possibility that the same mechanism that governs the opening of the critical period in the barrel cortex also operates in VC. A simple test of this hypothesis is to determine whether acute occlusion of one eye reverses the firing order of L4 followed by L2/3 cells.
Summary and model of STDP and its role in synaptic development in L4 and L2/3
Finally, it is worth summarizing the forms of STDP chronologically during the first 3 postnatal weeks in the barrel cortex (Fig. 3). At birth, thalamocortical axons invade the cortical plate, from which L2/3 and L4 subsequently differentiate. Synapses between thalamic axons and cortical plate cells exhibit robust all-LTP STDP, which contributes to synapse formation at thalamocortical connections. However, the magnitude of all-LTP STDP decreases rapidly as L4 and L2/3 differentiate from the cortical plate. At approximately the second postnatal week, thalamus-L4 synapses lose STDP, whereas thalamus-L2/3 synapses switch to all-LTD STDP after P7-P8. This rapid diminution of the magnitude of all-LTP STDP at thalamus-cortical plate cells and the ultimate switch to all-LTD STDP at aberrant projections to L2/3 areas likely explain the observations that only a limited number of thalamocortical axons extend to the pia surface and that the subsequent retraction occurs in the projections to the L2/3 area. From P6-P7, L4 cells start to extend their axons to form synapses with L2/3 cells, and synapses at L4-L2/3 exhibit all-LTP STDP, which drives synapse formation. Thus, during the second postnatal week, all-LTP STDP from L4 and all-LTD STDP from the thalamus converge onto L2/3 and may interact to self-reorganize the cortical network. At approximately the end of the second postnatal week (P12-P14), latency from the thalamus to cortical cells becomes shorter, but to different extents for excitatory and inhibitory cells: latency for inputs to inhibitory cells becomes slightly shorter than the latency for inputs to excitatory cells. Slightly faster activation of inhibitory cells contributes to the generation of sequential activation of “L4 followed by L2/3” cells by curtailing monosynaptic EPSPs from the thalamus in L4 cells and delaying EPSPs in L2/3 cells, which is disynaptic from the thalamus, by producing fast disynaptic IPSPs earlier than disynaptic EPSPs. At the same time, the axon terminals of L4 cells at L4-L2/3 synapses express CB1Rs, which enables the switch of STDP at this synapse from all-LTP STDP to Hebbian STDP. Thus, these cells begin to exhibit LTP in response to pre-post timing and LTD in response to post-pre timing, as well as in response to uncorrelated timing activity of presynaptic and postsynaptic cells. Initiation of Hebbian STDP at L4-L2/3 synapses provides the minimum conditions required for map plasticity, which may initiate a critical period for map plasticity.
Figure 3.

Chronological sequences of the appearance, switch, and interaction of STDP forms that induce network formation, retraction, and initiation of the critical period of map plasticity in barrel cortex during the first 3 postnatal weeks. The distinct forms of STDP, all-LTP STDP (red), all-LTD STDP (blue), and Hebbian STDP (red and blue), are shown.
Footnotes
This work was supported by Ministry of Education, Culture, Sports, Science and Technology 17K07057 to F.K., and Ministry of Education, Culture, Sports, Science and Technology 26430022 and Kawano Masanori Memorial Public Interest Incorporated Foundation for Promotion of Pediatrics to C.I.
The authors declare no competing financial interests.
References
- Allen CB, Celikel T, Feldman DE (2003) Long-term depression induced by sensory deprivation during cortical map plasticity in vivo. Nat Neurosci 6:291–299. 10.1038/nn1012 [DOI] [PubMed] [Google Scholar]
- Armstrong-James M. (1995) The nature and plasticity of sensory processing within adult rat barrel corex. In: Cerebral cortex (Jones EG, Diamond IT, eds), pp 333–373. New York: Plenum. [Google Scholar]
- Armstrong-James M, Fox K (1987) Spatiotemporal convergence and divergence in the rat S1 “barrel” cortex. J Cell Biol 263:265–281. 10.1002/cne.902630209 [DOI] [PubMed] [Google Scholar]
- Armstrong-James M, Fox K, Das-Gupta A (1992) Flow of excitation within rat barrel cortex on striking a single vibrissa. J Neurophysiol 68:1345–1358. 10.1152/jn.1992.68.4.1345 [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. (2002) Excitatory actions of GABA during development: the nature of the nurture. Nat Rev Neurosci 3:728–739. 10.1038/nrn920 [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL (1989) Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol 416:303–325. 10.1113/jphysiol.1989.sp017762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender KJ, Rangel J, Feldman DE (2003) Development of columnar topography in the excitatory layer 4 to layer 2/3 projection in rat barrel cortex. J Neurosci 23:8759–8770. 10.1523/JNEUROSCI.23-25-08759.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender KJ, Allen CB, Bender VA, Feldman DE (2006) Synaptic basis for whisker deprivation-induced synaptic depression in rat somatosensory cortex. J Neurosci 26:4155–4165. 10.1523/JNEUROSCI.0175-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi G, Poo M (1998) Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci 18:10464–10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi G, Poo M (2001) Synaptic modification by correlated activity: Hebb's postulate revisited. Annu Rev Neurosci 24:139–166. [DOI] [PubMed] [Google Scholar]
- Brumberg JC, Pinto DJ, Simons DJ (1999) Cortical columnar processing in the rat whisker-to-barrel system. J Neurophysiol 82:1808–1817. 10.1152/jn.1999.82.4.1808 [DOI] [PubMed] [Google Scholar]
- Burkhalter A. (1989) Intrinsic connections of rat primary visual cortex: laminar organization of axonal projections. J Comp Neurol 279:171–186. 10.1002/cne.902790202 [DOI] [PubMed] [Google Scholar]
- Caporale N, Dan Y (2008) Spike timing-dependent plasticity: a Hebbian learning rule. Annu Rev Neurosci 31:25–46. 10.1146/annurev.neuro.31.060407.125639 [DOI] [PubMed] [Google Scholar]
- Celikel T, Szostak VA, Feldman DE (2004) Modulation of spike timing by sensory deprivation during induction of cortical map plasticity. Nat Neurosci 7:534–541. 10.1038/nn1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crair MC, Malenka RC (1995) A critical period for long-term potentiation at thalamocortical synapses. Nature 375:325–328. 10.1038/375325a0 [DOI] [PubMed] [Google Scholar]
- Crozier RA, Wang Y, Liu CH, Bear MF (2007) Deprivation-induced synaptic depression by distinct mechanisms in different layers of mouse visual cortex. Proc Natl Acad Sci U S A 104:1383–1388. 10.1073/pnas.0609596104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan Y, Poo MM (2006) Spike timing-dependent plasticity: from synapse to perception. Physiol Rev 86:1033–1048. 10.1152/physrev.00030.2005 [DOI] [PubMed] [Google Scholar]
- Daw MI, Ashby MC, Isaac JT (2007) Coordinated developmental recruitment of latent fast spiking interneurons in layer IV barrel cortex. Nat Neurosci 10:453–461. 10.1038/nn1866 [DOI] [PubMed] [Google Scholar]
- Diamond ME, Armstrong-James M, Ebner FF (1993) Experience-dependent plasticity in adult rat barrel cortex. Proc Natl Acad Sci U S A 90:2082–2086. 10.1073/pnas.90.5.2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond ME, Huang W, Ebner FF (1994) Laminar comparison of somatosensory cortical plasticity. Science 265:1885–1888. 10.1126/science.8091215 [DOI] [PubMed] [Google Scholar]
- Drew PJ, Feldman DE (2009) Intrinsic signal imaging of deprivation-induced contraction of whisker representations in rat somatosensory cortex. Cereb Cortex 19:331–348. 10.1093/cercor/bhn085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagiolini M, Hensch TK (2000) Inhibitory threshold for critical-period activation in primary visual cortex. Nature 404:183–186. 10.1038/35004582 [DOI] [PubMed] [Google Scholar]
- Fagiolini M, Fritschy JM, Löw K, Möhler H, Rudolph U, Hensch TK (2004) Specific GABAA circuits for visual cortical plasticity. Science 303:1681–1683. 10.1126/science.1091032 [DOI] [PubMed] [Google Scholar]
- Feldman DE. (2000a) Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron 27:45–56. 10.1016/S0896-6273(00)00008-8 [DOI] [PubMed] [Google Scholar]
- Feldman DE. (2000b) Inhibition and plasticity. Nat Neurosci 3:303–304. 10.1038/73849 [DOI] [PubMed] [Google Scholar]
- Feldman DE. (2009) Synaptic mechanisms for plasticity in neocortex. Annu Rev Neurosci 32:33–55. 10.1146/annurev.neuro.051508.135516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE. (2012) The spike-timing dependence of plasticity. Neuron 75:556–571. 10.1016/j.neuron.2012.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE, Brecht M (2005) Map plasticity in somatosensory cortex. Science 310:810–815. 10.1126/science.1115807 [DOI] [PubMed] [Google Scholar]
- Feldmeyer D, Egger V, Lubke J, Sakmann B (1999) Reliable synaptic connections between pairs of excitatory layer 4 neurones within a single 'barrel' of developing rat somatosensory cortex. J Physiol 521:169–190. 10.1111/j.1469-7793.1999.00169.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmeyer D, Lübke J, Silver RA, Sakmann B (2002) Synaptic connections between layer 4 spiny neurone-layer 2/3 pyramidal cell pairs in juvenile rat barrel cortex: physiology and anatomy of interlaminar signalling within a cortical column. J Physiol 538:803–822. 10.1113/jphysiol.2001.012959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferster D, Lindström S (1983) An intracellular analysis of geniculo-cortical connectivity in area 17 of the cat. J Physiol 342:181–215. 10.1113/jphysiol.1983.sp014846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foeller E, Celikel T, Feldman DE (2005) Inhibitory sharpening of receptive fields contributes to whisker map plasticity in rat somatosensory cortex. J Neurophysiol 94:4387–4400. 10.1152/jn.00553.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox K. (1992) A critical period for experience-dependent synaptic plasticity in rat barrel cortex. J Neurosci 12:1826–1838. 10.1523/JNEUROSCI.12-05-01826.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox K. (2002) Anatomical pathways and molecular mechanisms for plasticity in the barrel cortex. Neuroscience 111:799–814. 10.1016/S0306-4522(02)00027-1 [DOI] [PubMed] [Google Scholar]
- Glazewski S, Fox K (1996) Time course of experience-dependent synaptic potentiation and depression in barrel cortex of adolescent rats. J Neurophysiol 75:1714–1729. 10.1152/jn.1996.75.4.1714 [DOI] [PubMed] [Google Scholar]
- Glazewski S, Giese KP, Silva A, Fox K (2000) The role of alpha-CaMKII autophosphorylation in neocortical experience-dependent plasticity. Nat Neurosci 3:911–918. 10.1038/78820 [DOI] [PubMed] [Google Scholar]
- Goldreich D, Kyriazi HT, Simons DJ (1999) Functional independence of layer IV barrels in rodent somatosensory cortex. J Neurophysiol 82:1311–1316. 10.1152/jn.1999.82.3.1311 [DOI] [PubMed] [Google Scholar]
- Han VZ, Grant K, Bell CC (2000) Reversible associative depression and nonassociative potentiation at a parallel fiber synapse. Neuron 27:611–622. 10.1016/S0896-6273(00)00070-2 [DOI] [PubMed] [Google Scholar]
- Hensch TK, Fagiolini M, Mataga N, Stryker MP, Baekkeskov S, Kash SF (1998) Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science 282:1504–1508. 10.1126/science.282.5393.1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itami C, Kimura F (2012) Developmental switch in spike timing-dependent plasticity at layers 4–2/3 in the rodent barrel cortex. J Neurosci 32:15000–15011. 10.1523/JNEUROSCI.2506-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itami C, Kimura F (2016) Concurrently induced plasticity due to convergence of distinct forms of spike timing-dependent plasticity in the developing barrel cortex. Eur J Neurosci 44:2984–2990. 10.1111/ejn.13431 [DOI] [PubMed] [Google Scholar]
- Itami C, Kimura F, Nakamura S (2007) Brain-derived neurotrophic factor regulates the maturation of layer 4 fast-spiking cells after the second postnatal week in the developing barrel cortex. J Neurosci 27:2241–2252. 10.1523/JNEUROSCI.3345-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itami C, Huang JY, Yamasaki M, Watanabe M, Lu HC, Kimura F (2016) Developmental switch in spike timing-dependent plasticity and cannabinoid-dependent reorganization of the thalamocortical projection in the barrel cortex. J Neurosci 36:7039–7054. 10.1523/JNEUROSCI.4280-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob V, Brasier DJ, Erchova I, Feldman D, Shulz DE (2007) Spike timing-dependent synaptic depression in the in vivo barrel cortex of the rat. J Neurosci 27:1271–1284. 10.1523/JNEUROSCI.4264-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaas S, Nelson R, Sur M, Merzenich MM (1981) Organization of somatosensory cortex in primates. In: The organization of the cerebral cortex (Schmitt F, Worden F, Adelman G, Dennis S, eds), pp 237–262. Cambridge, MA: Massachusetts Institute of Technology. [Google Scholar]
- Katz LC, Shatz CJ (1996) Synaptic activity and the construction of cortical circuits. Science 274:1133–1138. 10.1126/science.274.5290.1133 [DOI] [PubMed] [Google Scholar]
- Kimura F, Itami C, Ikezoe K, Tamura H, Fujita I, Yanagawa Y, Obata K, Ohshima M (2010) Fast activation of feedforward inhibitory neurons from thalamic input and its relevance to the regulation of spike sequences in the barrel cortex. J Physiol 588:2769–2787. 10.1113/jphysiol.2010.188177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CH, Heynen AJ, Shuler MG, Bear MF (2008) Cannabinoid receptor blockade reveals parallel plasticity mechanisms in different layers of mouse visual cortex. Neuron 58:340–345. 10.1016/j.neuron.2008.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HC, Gonzalez E, Crair MC (2001) Barrel cortex critical period plasticity is independent of changes in NMDA receptor subunit composition. Neuron 32:619–634. 10.1016/S0896-6273(01)00501-3 [DOI] [PubMed] [Google Scholar]
- Lu JT, Li CY, Zhao JP, Poo MM, Zhang XH (2007) Spike-timing-dependent plasticity of neocortical excitatory synapses on inhibitory interneurons depends on target cell type. J Neurosci 27:9711–9720. 10.1523/JNEUROSCI.2513-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Sakmann B (1997) Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275:213–215. 10.1126/science.275.5297.213 [DOI] [PubMed] [Google Scholar]
- Markram H, Gerstner W, Sjöström PJ (2011) A history of spike-timing-dependent plasticity. Front Synaptic Neurosci 3:4. 10.3389/fnsyn.2011.00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meliza CD, Dan Y (2006) Receptive-field modification in rat visual cortex induced by paired visual stimulation and single-cell spiking. Neuron 49:183–189. 10.1016/j.neuron.2005.12.009 [DOI] [PubMed] [Google Scholar]
- Mu Y, Poo MM (2006) Spike timing-dependent LTP/LTD mediates visual experience-dependent plasticity in a developing retinotectal system. Neuron 50:115–125. 10.1016/j.neuron.2006.03.009 [DOI] [PubMed] [Google Scholar]
- Nevian T, Sakmann B (2006) Spine Ca2+ signaling in spike-timing-dependent plasticity. J Neurosci 26:11001–11013. 10.1523/JNEUROSCI.1749-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima M, Itami C, Kimura F (2017) The alpha2A-adrenoceptor suppresses excitatory synaptic transmission to both excitatory and inhibitory neurons in layer 4 barrel cortex. J Physiol 595:6923–6937. 10.1113/JP275142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DF, Kriegstein AR (2002) Is there more to GABA than synaptic inhibition? Nat Rev Neurosci 3:715–727. 10.1038/nrn919 [DOI] [PubMed] [Google Scholar]
- Petersen CC, Sakmann B (2000) The excitatory neuronal network of rat layer 4 barrel cortex. J Neurosci 20:7579–7586. 10.1523/JNEUROSCI.20-20-07579.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebsam A, Seif I, Gaspar P (2002) Refinement of thalamocortical arbors and emergence of barrel domains in the primary somatosensory cortex: a study of normal and monoamine oxidase a knock-out mice. J Neurosci 22:8541–8552. 10.1523/JNEUROSCI.22-19-08541.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safo P, Regehr WG (2008) Timing dependence of the induction of cerebellar LTD. Neuropharmacology 54:213–218. 10.1016/j.neuropharm.2007.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaggar BL, Fox K, O'Leary DD (1993) Postsynaptic control of plasticity in developing somatosensory cortex. Nature 364:623–626. 10.1038/364623a0 [DOI] [PubMed] [Google Scholar]
- Schubert D, Kötter R, Zilles K, Luhmann HJ, Staiger JF (2003) Cell type-specific circuits of cortical layer IV spiny neurons. J Neurosci 23:2961–2970. 10.1523/JNEUROSCI.23-07-02961.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuett S, Bonhoeffer T, Hübener M (2001) Pairing-induced changes of orientation maps in cat visual cortex. Neuron 32:325–337. 10.1016/S0896-6273(01)00472-X [DOI] [PubMed] [Google Scholar]
- Senft SL, Woolsey TA (1991) Growth of thalamic afferents into mouse barrel cortex. Cereb Cortex 1:308–335. 10.1093/cercor/1.4.308 [DOI] [PubMed] [Google Scholar]
- Simons DJ. (1978) Response properties of vibrissa units in rat SI somatosensory neocortex. J Neurophysiol 41:798–820. 10.1152/jn.1978.41.3.798 [DOI] [PubMed] [Google Scholar]
- Simons DJ, Land PW (1987) Early experience of tactile stimulation influences organization of somatic sensory cortex. Nature 326:694–697. 10.1038/326694a0 [DOI] [PubMed] [Google Scholar]
- Sjöström PJ, Nelson SB (2002) Spike timing, calcium signals and synaptic plasticity. Curr Opin Neurobiol 12:305–314. 10.1016/S0959-4388(02)00325-2 [DOI] [PubMed] [Google Scholar]
- Song S, Miller KD, Abbott LF (2000) Competitive Hebbian learning through spike-timing-dependent synaptic plasticity. Nat Neurosci 3:919–926. 10.1038/78829 [DOI] [PubMed] [Google Scholar]
- Stern EA, Maravall M, Svoboda K (2001) Rapid development and plasticity of layer 2/3 maps in rat barrel cortex in vivo. Neuron 31:305–315. 10.1016/S0896-6273(01)00360-9 [DOI] [PubMed] [Google Scholar]
- Tanifuji M, Sugiyama T, Murase K (1994) Horizontal propagation of excitation in rat visual cortical slices revealed by optical imaging. Science 266:1057–1059. 10.1126/science.7973662 [DOI] [PubMed] [Google Scholar]
- Valiullina F, Akhmetshina D, Nasretdinov A, Mukhtarov M, Valeeva G, Khazipov R, Rozov A (2016) Developmental changes in electrophysiological properties and a transition from electrical to chemical coupling between excitatory layer 4 neurons in the rat barrel cortex. Front Neural Circuits 10:1. 10.3389/fncir.2016.00001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesel TN, Hubel DH (1963) Single-cell responses in striate cortex of kittens deprived of vision in one eye. J Neurophysiol 26:1003–1017. 10.1152/jn.1963.26.6.1003 [DOI] [PubMed] [Google Scholar]
- Zhang LI, Tao HW, Holt CE, Harris WA, Poo M (1998) A critical window for cooperation and competition among developing retinotectal synapses. Nature 395:37–44. 10.1038/25665 [DOI] [PubMed] [Google Scholar]
- Zhang LI, Tao HW, Poo M (2000) Visual input induces long-term potentiation of developing retinotectal synapses. Nat Neurosci 3:708–715. 10.1038/76665 [DOI] [PubMed] [Google Scholar]