

Abstract
Chronic intermittent hypoxia (CIH) reduces afferent-evoked excitatory postsynaptic currents (EPSCs) but enhances basal spontaneous (s) and asynchronous (a) EPSCs in second-order neurons of nucleus tractus solitarii (nTS), a major area for cardiorespiratory control. The net result is an increase in synaptic transmission. The mechanisms by which this occurs are unknown. The N-type calcium channel and transient receptor potential cation channel TRPV1 play prominent roles in nTS sEPSCs and aEPSCs. The functional role of these channels in CIH-mediated afferent-evoked EPSC, sEPSC, and aEPSC was tested in rat nTS slices following antagonist inhibition and in mouse nTS slices that lack TRPV1. Block of N-type channels decreased aEPSCs in normoxic and, to a lesser extent, CIH-exposed rats. sEPSCs examined in the presence of TTX (miniature EPSCs) were also decreased by N-type block in normoxic but not CIH-exposed rats. Antagonist inhibition of TRPV1 reduced the normoxic and the CIH-mediated increase in sEPSCs, aEPSCs, and mEPSCs. As in rats, in TRPV1+/+ control mice, aEPSCs, sEPSCs, and mEPSCs were enhanced following CIH. However, none were enhanced in TRPV1−/− null mice. Normoxic tractus solitarii (TS)-evoked EPSC amplitude, and the decrease after CIH, were comparable in control and null mice. In rats, TRPV1 was localized in the nodose-petrosal ganglia (NPG) and their central branches. CIH did not alter TRPV1 mRNA but increased its protein in NPG consistent with an increased contribution of TRPV1. Together, our studies indicate TRPV1 contributes to the CIH increase in aEPSCs and mEPSCs, but the CIH reduction in TS-EPSC amplitude occurs via an alternative mechanism.
NEW & NOTEWORTHY This study provides information on the underlying mechanisms responsible for the chronic intermittent hypoxia (CIH) increase in synaptic transmission that leads to exaggerated sympathetic nervous and respiratory activity at baseline and in response to low oxygen. We demonstrate that the CIH increase in asynchronous and spontaneous excitatory postsynaptic currents (EPSCs) and miniature EPSCs, but not decrease in afferent-driven EPSCs, is dependent on transient receptor potential vanilloid type 1 (TRPV1). Thus TRPV1 is important in controlling nucleus tractus solitarii synaptic activity during CIH.
Keywords: autonomic nervous system, calcium channels, intermittent hypoxia, respiration, synaptic transmission
INTRODUCTION
Chronic intermittent hypoxia (CIH), a model for obstructive sleep apnea, elevates sympathetic nervous system activity and respiration under basal conditions and in response to a hypoxic stimulus (Iturriaga et al. 2014; Sica et al. 2000). These changes are largely due to increased activity of carotid body chemosensory receptors (Prabhakar et al. 2005), which send their sensory afferents into the nucleus of the solitary tract (nTS; Chitravanshi and Sapru 1995; Finley and Katz 1992). We previously demonstrated 10 days of CIH alters synaptic transmission between visceral sensory afferent fibers and second-order neurons in the nTS of the rat brain stem (Kline et al. 2007). The CIH-induced change in synaptic transmission has two opposing components, a decrease in the tractus solitarii (TS) evoked excitatory postsynaptic current (TS-EPSC) and an increase in nonevoked spontaneous release including both miniature synaptic currents (mEPSCs) and the asynchronous (aEPSCs) release following a train of stimuli to the afferent pathway. CIH also increased the number of nTS neurons expressing aEPSCs. The net result is an increase in excitatory synaptic activity leading to augmented action potential discharge. Similar observations in the nTS have been observed following 7–10 days of CIH, including increased mEPSCs and reduced TS-EPSCs in rat nTS slices (Almado et al. 2012). Although we have suggested alteration in one or more presynaptic calcium (Ca2+) handling processes in the CIH-modified synaptic transmission, the mechanisms responsible are largely unknown.
Neurotransmitter release is dependent on Ca2+ entry into the synaptic terminal, and thus we sought to determine the role of two prominent sources of Ca2+ entry at the peripheral afferent-nTS neuron synapse. The N-type voltage-gated calcium channel (Cav2.2) is largely responsible for afferent (TS) stimulus-evoked Ca2+ entry, release of glutamate, and the resulting synaptic current (Mendelowitz et al. 1995). aEPSCs in the nTS have also been shown to be dependent on N-type calcium channel activation during tract-evoked stimuli (Peters et al. 2010). In addition, transient receptor potential vanilloid type 1 (TRPV1), a Ca2+-permeable cation channel, is localized to nonmyelinated vagal sensory neurons and their sensory terminals in the nTS (Hermes et al. 2016; Sun et al. 2009). Of importance to the present study, TRPV1 is also present in the peripheral branches and cell bodies of carotid body chemosensory afferents (Roy et al. 2012). TRPV1 is implicated in spontaneous transmitter release from sensory afferents in nTS (Peters et al. 2010). Together, these data provided the rationale to examine the role of N-type and TRPV1 channels in generating the changes in synaptic transmission in response to CIH.
We show in rat brain slices that following CIH, pharmacological block of TRPV1, rather than N-type, channels suppressed all spontaneous events. Therefore, to further examine the role of TRPV1 in increased release in CIH, we compared the synaptic activity in response to CIH in brain slices from control mice expressing TRPV1 with those from TRPV1 null mice. The increase in mEPSC and aEPSC activity in response to CIH that occurs in TRPV1+/+ control mice was absent in TRPV1−/− null mice. However, the depression in the evoked EPSC after CIH remained in both groups and was, thus, independent of the mechanisms responsible for increased spontaneous and asynchronous release. Taken together, these studies suggest activation of TRPV1 is an important component of the synaptic changes following CIH.
MATERIALS AND METHODS
Animals and Ethical Approval
Animal protocols were approved by the Animal Care and Use Committees of Case Western Reserve University and University of Missouri and handled according to their guidelines. Male Sprague-Dawley rats between 3 and 7 wk of age and male TRPV1+/+ control and TRPV1−/− null mice 4–7 wk of age (B6.129X1-Trpv1tm1Jul/J; Jackson Laboratories) were used in the present study. Animals were housed in in-house animal facilities with a 12:12-h light-dark cycle with food and water available ad libitum.
Exposure to CIH
Standard rat or mouse cages containing unrestrained animals were placed in a commercially available hypoxic system (BioSpherix, Redfield, NY) for exposure to intermittent hypoxia. Exposure to CIH consisted of alternating cycles of 21% and 6% O2, 9 episodes per hour, 8 h per day, for 10 consecutive days (Kline et al. 2007). Ambient oxygen, carbon dioxide, temperature, and humidity levels were continuously monitored. Animals were subjected to intermittent hypoxia between 9:00 AM and 5:00 PM and to room air [normoxia (NORM), 21% O2] between 5:00 PM and 9:00 AM. Control normoxic animals consisted of rodents not exposed to CIH and were housed in standard rat cages placed in the vivarium, in the hypoxic chamber but only exposed to 21% O2 or, alternatively, on the top surface of the hypoxic chamber during littermate CIH exposure (Kline et al. 2007). All electrophysiology or tissue harvesting protocols were performed the morning after the last hypoxic exposure or the comparable normoxic time period.
Immunohistochemistry
Immunohistochemistry was performed as previously (Kline et al. 2005; Matott et al. 2016). Following NORM or 10-day CIH, rodents were anesthetized with isoflurane and transcardially perfused with heparinized 0.01 M PBS followed by 4% paraformaldehyde. The brain stem and nodose-petrosal ganglia (NPG) were removed and postfixed in 4% paraformaldehyde for 2 h. Brain stem sections containing the nTS were cut at 30 μm via a vibratome (model VT1000S; Leica, Wetzlar, Germany) and processed as free-floating sections. NPG were cryoprotected via overnight submersion in 30% sucrose in PBS. The NPG was sectioned at 10 μm on a cryostat, mounted on gelatin-coated slides, and processed. Immunohistochemistry for NORM and CIH sections was run in parallel. nTS and NPG sections were blocked with 10% normal donkey serum (NDS; catalog no. S30; Millipore) in 0.3% Triton-PBS. Tissue sections were rinsed and subsequently incubated with primary antibodies against TRPV1 (nTS: 1:2,000, catalog no. RA10110, Neuromics, Edina, MN; NPG: 1:250, catalog no. ACC-029, Alomone Laboratories, Jerusalem, Israel) in 3% NDS in 0.01 M PBS overnight. These antibodies have been previously utilized and validated in other studies (Goswami et al. 2010; Guo et al. 1999; Jeong et al. 2018). Ganglia sections were also incubated with tyrosine hydroxylase (TH; 1:2,000, catalog no. 22941; ImmunoStar, Hudson, WI). The next day, sections were rinsed and incubated for 2 h in fluorescent-conjugated secondary antibodies (1:200; Jackson Immunoresearch Laboratories, West Grove, PA) in 3% NDS and 0.3% Triton-PBS. Brain stem sections were rinsed, mounted on gelatin-coated slides, and air dried. nTS and NPG sections were coverslipped with Prolong Gold and then sealed with nail polish. One section per run was incubated without primary antibody and served as negative control; no fluorescent staining was present.
Immunolabeling was examined with a conventional epifluorescence microscope (model BX51; Olympus). Appropriate filter sets and excitation wavelengths were used to visualize the different fluorophores. Digital images were acquired with a monochrome charge-coupled device camera (model ORCA-ER; Hamamatsu). Grayscale NORM and CIH nTS images were acquired at the same exposure times, and the magnitude of TRPV1 immunolabelling in the nTS was quantified using ImageJ (National Institutes of Health). A single 100 × 100-μm box medial to the TS was used to measure the grayscale intensity of each 8-bit nTS image at the subpostremal level (bregma, ~14.04 mm) an area comparable to our site of electrophysiological recordings and localization of chemosensory termination (Finley and Katz 1992). Grayscale pixel intensity was analyzed via the measure plugin of FIJI ImageJ and reported as grayscale units (g.u.). Published images were postprocessed for contrast and brightness and background subtracted (FIJI ImageJ).
Immunoblot Analysis
The relative presence of TRPV1 protein in NPG was examined by immunoblot analysis. NORM and 10-day CIH rats were anesthetized and decapitated, the NPG removed, and tissue samples prepared as previously described (Matott and Kline 2016). Ganglia were homogenized in extraction buffer [150 mM NaCl, 100 mM Tris-HCl, 1% Triton X, protease inhibitor cocktail (Complete Mini, EDTA-free; Roche Diagnostics, Indianapolis, IN)], centrifuged (15 min, 13,300 rpm, 4°C), and the supernatant collected. Protein concentrations were determined with the Bio-Rad Protein Assay Dye Reagent (Bio-Rad, Hercules, CA), and 20 µg of protein were separated in a 4–20% precast Mini-PROTEAN TGX gel (Bio-Rad) and transferred to an Immun-Blot polyvinylidene difluoride membrane (Bio-Rad). Membranes were subsequently incubated overnight at 4°C with primary antibody against TRPV1 (1:500, catalog no. RA10110; Neuromics) and tubulin (1:100, catalog no. 7291; Abcam), washed, and incubated with horseradish peroxidase-linked secondary antibodies (1:500 each, 2 h, 23°C; Jackson Immunoresearch Laboratories). Blots were developed with ImmunStar WesternC substrate (Bio-Rad) and imaged with an ChemiDoc XRS+ Imager using Image Laboratory Software (version 5.1; Bio-Rad). Intensity of bands was measured in ImageJ, and the relative amount of TRPV1 was normalized to tubulin and quantified.
Reverse Transcription Real-Time Polymerase Chain Reaction
The relative presence of TRPV1 mRNA in the NPG was examined via RT-PCR (Austgen et al. 2011). Briefly, NORM and 10-day CIH rats were anesthetized with isoflurane and decapitated, and the NPG were isolated, snap frozen, and stored at −80°C. RNA was isolated using the RNAqueous-Micro kit, following the manufacturer’s instructions (Ambion, Life Technologies, Grand Island, NY), and quantified (BioPhotometer Plus; Eppendorf, Hauppauge, NY). cDNA was generated from 100 ng of mRNA (oligo-dT primer set, SuperScript III; Invitrogen). Quantitative real-time PCR amplification of 2 µl of cDNA was performed using the SYBR Premix Ex Taq kit (Takara, Mountain View, CA), the SmartCycler System (Cepheid, Sunnyvale, CA), and the following primers: Trpv1 (forward: CAA CAG GAA GGG GCT CAC, reverse: TCT GGA GAA TGT AGG CCA AGA C, 10 µM; Fisher Scientific, Pittsburgh, PA) and the housekeeping gene β2-microglobulin (B2M) (forward: AGC AGG TTC CTC AAA CAA GG, reverse: TTC TGC CTT GGA GTC CTT TC, 10 µM; Fisher Scientific). The amount of Trpv1 mRNA was normalized to B2M using the 2ΔΔCT method (Livak and Schmittgen 2001).
Brain Stem nTS Slices
nTS slices were prepared from NORM or CIH-exposed rats and mice anesthetized with 5% isoflurane and decapitated. The brain stem was removed and placed in ice-cold low-Ca2+/high-Mg2+ artificial cerebral spinal fluid (aCSF) containing the following (in mM): 124 NaCl, 3 KCl, 1.2 NaH2PO4, 1.2 MgSO4, 25 NaHCO3, 11 d-glucose, 0.4 l-ascorbic acid, 2 MgCl2, and 1 CaCl2, saturated with 95% O2-5% CO2, pH 7.4 (300 mosM). Horizontal slices (rat, ~290 µm; mouse, ~200 µm) were cut with a vibrating microtome (Leica VT 1000S). The submerged sections were secured with nylon threads attached to a stainless steel harp and superfused at a flow rate of 3–4 ml/min with standard recording aCSF (in mM: 124 NaCl, 3 KCl, 1.2 NaH2PO4, 1.2 MgSO4, 25 NaHCO3, 11 d-glucose, 0.4 l-ascorbic acid, and 2 CaCl2, saturated with 95% O2-5% CO2, pH 7.4, 300 mosM) at 31–33°C. nTS neurons were visualized using an Olympus BX-51WI microscope with ×40 magnification, differential interface contrast, and an infrared-sensitive camera. Recording electrodes (3.5–5.0 MΩ, 7151 or 8250 glass type; King Precision Glass, Claremont, CA) were filled with a solution containing (in mM) 10 NaCl, 130 K-gluconate, 11 EGTA, 1 CaCl2, 10 HEPES, 1 MgCl2, 2 MgATP, and 0.2 NaGTP (pH 7.3, 295–300 mosM). The pipette was guided using a piezoelectric micromanipulator (ThorLabs, Newton, NJ). Recordings were made from medial and commissural nTS neurons. Cells with holding currents greater than −50 pA and a resting membrane potential less than −50 mV upon initial membrane rupture were not considered for further analysis.
Sensory afferents traveling through the TS were stimulated via a concentric bipolar stimulating electrode (200-µm outer diameter, 50-µm inner diameter; FHC, Bowdoinham, ME). Negative current pulses (0.01–0.3 mA, 0.1-ms duration) to the TS generated with an isolated programmable stimulator (AMPI, Jerusalem, Israel) elicited low onset variability TS-EPSCs (Fig. 1A). In experiments in which we recorded mEPSCs, the recording electrodes contained (in mM) 5 NaCl, 130 Cs-methanesulfonate, 10 CsCl, 11 EGTA, 1 CaCl2, 10 HEPES, 1 MgCl2, 2 MgATP, 0.2 NaGTP, and 5 QX-314 (pH 7.3), whereas the extracellular solution contained 1 µM tetrodotoxin (TTX) and 10 µM bicuculline. Data were recorded using a Molecular Devices Axopatch 200B or MultiClamp 700B amplifier, filtered at 2 kHz and sampled at 10 kHz using Clampex v9 or v10 programs (Molecular Devices, San Jose, CA). No leak subtractions, liquid junction potential corrections, or series resistance compensations were performed.
Fig. 1.
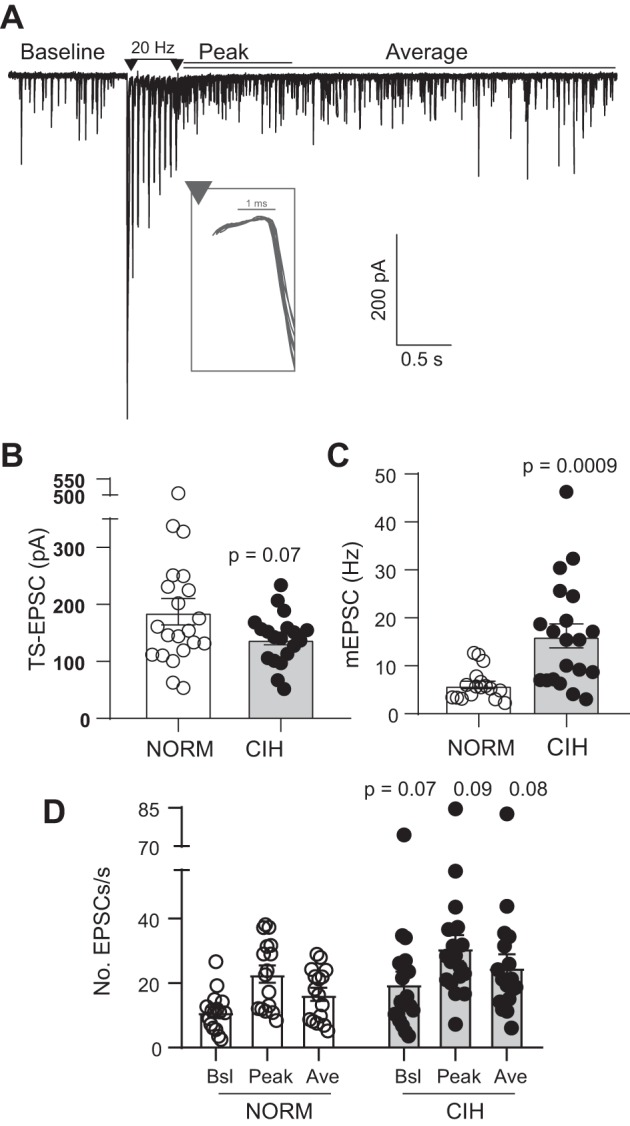
Representative example of excitatory postsynaptic currents (EPSCs) before and after a 20-Hz stimulation train: chronic intermittent hypoxia (CIH) alters glutamate signaling. A: baseline spontaneous EPSCs (sEPSCs) occur before the tractus solitarii (TS) stimulation. Events occurring during the 1 s following the stimulus train were classified as peak asynchronous EPSCs (aEPSCs), whereas the average aEPSCs include all events after the stimulus train. Example is from a single trace. Inset demonstrates low variability (i.e., jitter) in the first TS-EPSC (overlay of 10 traces). Stimulus artifacts are reduced for clarity. B–D: aggregate data of cells used in the current study. Compared with normoxia (NORM), CIH decreased TS-EPSC amplitude (B; NORM, n = 21; CIH, n = 19; t-test) and enhanced miniature EPSC (mEPSC) frequency (C; NORM, n = 17; CIH, n = 21; t-test) and asynchronous events (D; NORM, n = 16; CIH, n = 18) (2-way RM ANOVA with Fisher’s least significant difference post hoc: EPSC, P = 0.001; hypoxia, P = 0.08, EPSC × hypoxia, P = 0.94). Ave, average; Bsl, baseline.
Drugs
The TRPV1 antagonists SB-366791 and capsazepine were purchased from Tocris Bioscience (Minneapolis, MN) and used at 10 μM. The concentration was based on previous reports using similar concentrations (Grueter et al. 2010), including those in the nTS (Peters et al. 2010). Although both antagonists block TRPV1, SB-366791 is a more potent inhibitor than capsazepine (Gunthorpe et al. 2004; Julius and Clapham 2018), and thus both were used to delineate the role of TRPV1 and counter potential nonspecificity of each antagonist (Gunthorpe et al. 2004). The N-type calcium channel blocker ω-conotoxin GVIA (ω-CTx; Sigma-Aldrich, St. Louis, MO) was used at 1 μM. Blockers were bath applied. All other chemicals were purchased from Sigma-Aldrich and Fisher Scientific.
Data Analysis
Data were analyzed using Clampfit (Molecular Devices), MiniAnalysis (Synaptosoft), and Microsoft Excel. Second-order nTS neurons were identified in Clampfit by jitter analysis, defined as the standard deviation of the TS-EPSC onset latency from stimulus (Doyle and Andresen 2001; Kline et al. 2002). Only neurons with jitter values <250 µs, considered to be monosynaptic and directly connected to sensory TS neurons, were included in this study (Fig. 1, inset). A TS stimulus frequency of 0.5 Hz and 20Hz was chosen based on the physiological range of discharge of C-type sensory and chemosensory afferents (Andresen and Kunze 1994; Vidruk et al. 2001). Reported TS-EPSC amplitudes were an average of 10–20 events. Spontaneous EPSCs, including baseline, miniature and asynchronous, were identified in MiniAnalysis. Asynchronous EPSCs (aEPSCs) were designated as those events occurring after the 20-Hz TS-stimulation, relative to the baseline spontaneous EPSCs (sEPSCs) occurring before the train (Fig. 1A). Peak aEPSCs were classified as those occurring 1 s after the train. Average aEPSCs were designated as those events occurring 4 s (rat) or 3 s (mouse) after the train and were subsequently normalized as number of events per second. All events were manually confirmed. Peak and average aEPSCs were evaluated because these events are capable of eliciting action potential discharge over several seconds and induce prolonged discharge following CIH (Kline et al. 2007; Peters et al. 2010).
Statistical analysis was performed with GraphPad Prism 7.0 and 8.0 (GraphPad Software, La Jolla, CA), Excel with the Real Statistics using Excel Addin (http://www.real-statistics.com/), or Origin software (Origin Laboratories, Northampton, MA). Data were tested for normality via the Shapiro-Wilk test and subsequently by Student’s t-test or two-way ANOVA where appropriate. Effects of a particular pharmacological intervention on TS-EPSCs, sEPSCs, and aEPSCs were determined using repeated-measures (RM) ANOVA. Bonferroni multiple comparison or Fisher’s least significant difference (LSD) post hoc test identified individual differences. All data are means ± SE. Results were considered significantly different at P values <0.05.
RESULTS
Our previous study (Kline et al. 2007) demonstrated 10-day CIH induces significant changes in glutamate release compared with normoxic controls. Importantly, these responses develop between 3 and 10 days, remain consistent at 30 days of CIH, are reversible, and are calcium dependent. Consistent with these previous data, aggregate data from the current study demonstrate CIH decreases TS-EPSC amplitude (Fig. 1B), increases mEPSC frequency (Fig. 1C), and increases asynchronous events after a stimulus train (Fig. 1D). Therefore, the following study examined the contribution of N-type and TRPV1 calcium channels as a source for elevated spontaneous EPSCs and/or attenuated TS-EPSCs after 10 days of CIH.
N-Type Calcium Channels Do Not Contribute to Altered Spontaneous CIH Events
To understand the source of calcium involved in the changes in evoked, basal, and asynchronous EPSCs in response to 10-day CIH, we first addressed the role of N-type calcium channels using the selective blocker ω-conotoxin GVIA (ω-CTx). TS-evoked EPSCs at the nTS synapse are dependent primarily on Ca2+ influx through the N-type calcium channels (Mendelowitz et al. 1995). Such was the case in the present study where 1 µM ω-CTx reduced the peak amplitude of the sensory-evoked current. ω-CTx decreased NORM and CIH TS-EPSC amplitude (Fig. 2A). The percentage decrease of TS-EPSC amplitude by ω-CTx was not different between the groups (NORM, 59 ± 10% vs. CIH, 67 ± 8%, P = 0.51, unpaired t-test).
Fig. 2.

Effect of N-type calcium channel block with ω-conotoxin (CTx) on spontaneous, asynchronous and tractus solitarii (TS)-evoked activity. A: example of normoxic (NORM) and chronic intermittent hypoxia (CIH)-exposed TS-evoked excitatory postsynaptic currents (TS-EPSCs; overlay of 10 traces) in artificial cerebrospinal fluid (aCSF) and CTx. TS-EPSC amplitude decreased in CTx compared with their individual aCSF control (C) in NORM (n = 8) and CIH (n = 7) neurons (2-way RM ANOVA: drug, P = 0.006; hypoxia, P = 0.62, drug × hypoxia, P = 0.47). *P < 0.05; **P < 0.01, CTx vs. aCSF control, with Bonferroni multiple comparisons. B: example of NORM and CIH-exposed miniature EPSCs (mEPSCs; single traces) in aCSF and CTx. mEPSC frequency is reduced from aCSF control (C) by CTx in NORM (n = 6) slices but is unchanged in cells from CIH-treated (n = 5) animals (2-way RM ANOVA: drug, P = 0.001, hypoxia, P = 0.005, drug × hypoxia, P = 0.01). **P < 0.01, CTx vs. aCSF. $$P < 0.01, CIH vs. NORM with Bonferroni post hoc. C: diaries of events before and in response to 20-Hz TS stimulation in the absence (aCSF) and presence of CTx for individual NORM (left) and CIH-exposed (right) cells. Note the prominent decrease in asynchronous EPSCs by CTx in NORM. Events before TS stimulation are fit with a linear regression, whereas events following TS stimulation are fit via nonlinear curve. Events are binned per 100 ms. D: grouped data from experiments in C illustrate peak and average events are reduced to near baseline (pre-TS stimulation) in control (aCSF) for NORM (n = 7) cells, whereas the reduction of peak and average events by CTx is less pronounced in CIH (n = 7) cells (2-way RM ANOVA, NORM: drug, P = 0.10; EPSC, P = 0.001, drug × EPSC, P = 0.01; CIH: drug, P = 0.10; EPSC, P = 0.002; drug × EPSC, P = 0.0006). Dashed line indicates aCSF baseline events. Ave, average; Bsl, baseline. *P < 0.05; **P < 0.01; ***P < 0.001, peak or average vs. baseline (pre-TS stimulation) with Bonferroni post hoc. $$P < 0.01; $$$P < 0.001; $$$$P < 0.0001, CTx vs. aCSF control for each respective parameter with Bonferroni post hoc.
Previous studies have suggested asynchronous release to be dependent on N-type calcium channel activation (Peters et al. 2010). To confirm these results and determine whether the CIH-induced increase in aEPSCs is due to activation of N-type calcium channels, we examined the number of spontaneous EPSCs before and after a 20-Hz stimulus train (for example, see Fig. 1). Following a stimulus train, the peak and average number of events was elevated compared with those baseline events before stimulation. As shown in the individual event diary plot for aCSF and ω-CTx (Fig. 2C, left) and by grouped data (Fig. 2D, left), ω-CTx reduced the NORM peak and average number of events. After CIH, ω-CTx reduced baseline and asynchronous events, but to a lesser extent (Fig. 2, C and D, right). Comparing the peak and average aEPSCs with baseline events (Peak/Bsl and Ave/Bsl, respectively) demonstrated the magnitude of decrease by ω-CTx was greater in NORM (Peak/Bsl: NORM, −41.0 ± 9.5% vs. CIH, −3.9 ± 8.5%, P = 0.01, t-test; Ave/Bsl: NORM, −25.7 ± 8.6% vs. CIH, −4.5 ± 4.7%, P = 0.05, t-test), suggesting N-type channel inhibition was less effective in attenuating aEPSCs.
We also evaluated whether the mEPSCs recorded in TTX after CIH were due, in part, to N-type calcium channel involvement. Compared with NORM, mEPSCs were elevated in CIH (Fig. 2B; in aCSF: NORM, 7.5 ± 1.2 Hz vs. CIH, 18.0 ± 3.1 Hz, 2-way RM ANOVA). ω-CTx significantly reduced NORM mEPSC frequency by 32 ± 7% (in ω-CTx to 5.0 ± 0.7 Hz) but left mEPSC frequency unchanged in CIH (a 0.2 ± 2% decrease; 2-way RM ANOVA). NORM and CIH mEPSC amplitude was not altered in ω-CTx (data not shown).
Taken together, these data suggest that TS-EPSCs are modulated by N-type calcium channels to the same degree in NORM and CIH, and that elevated sEPSCs, mEPSCs, and aEPSCs in CIH are due to enhanced involvement of non-ω-CTx-sensitive channels.
Contribution of TRPV1 Channels in CIH
In the next series of experiments, we focused on the underlying mechanisms for the ω-CTx-independent increase in basal, aEPSC, and mEPSC frequency following 10-day CIH. Previous reports demonstrated the presence of TRPV1 in carotid chemosensory (Roy et al. 2012) and TS sensory afferents within the nTS (Hermes et al. 2016).
Expression of TRPV1 in NORM and following CIH in sensory afferents and nTS.
We examined by immunohistochemistry, RT-PCR, and immunoblots the expression of TRPV1 in NORM and CIH-exposed rats. Figure 3A demonstrates the presence of TRPV1 in the rat NPG complex, including fibers coursing through the ganglia. A portion of these TRPV1 positive cells in the NPG colocalized with TH, a marker of chemoafferent neurons (Finley and Katz 1992). Compared with NORM, mRNA expression for TRPV1 in NPG after 10-day CIH was unchanged (Fig. 3B; P = 0.672, t-test). Immunoblot analysis of NORM and CIH ganglia demonstrated protein expression increased in CIH (Fig. 3C; P = 0.016, t-test). Immunohistochemistry was used to examine the distribution of the elevated NPG TRPV1 protein in the nTS. TRPV1 was localized to the NORM and CIH medial and commissural nTS (Fig. 3D), an area of dense chemoafferent termination (Finley and Katz 1992). We focused on TRPV1 labeling medial to the TS, the area in which we recorded the functional influence of this channel. Across NORM and CIH rats, although it tended to increase in CIH rats, there were no consistent changes in TRPV1 protein expression between the groups (NORM, 53.9 ± 7.8 g.u. vs. CIH, 59.8 ± 13.6 g.u.; P = 0.72, t-test, n = 3 each).
Fig. 3.
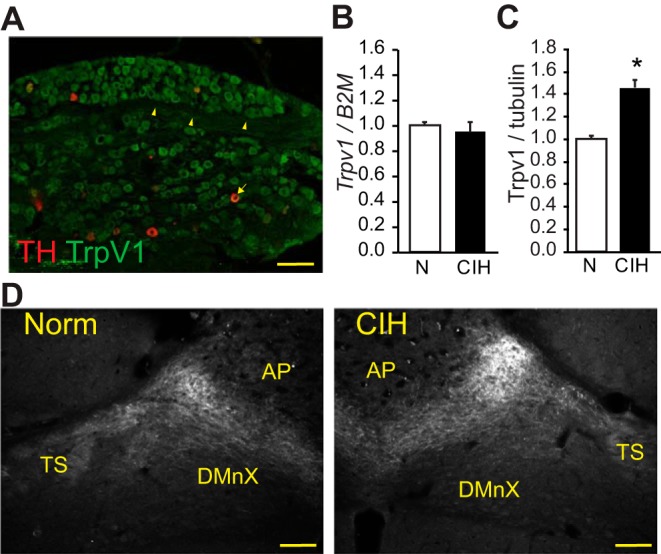
TRPV1 in sensory afferents and nucleus of the solitary tract (nTS) after chronic intermittent hypoxia (CIH). A: TRPV1 is localized in normoxic (NORM) sensory afferents of the nodose-petrosal ganglia (NPG; green) and colocalizes with tyrosine hydroxylase (TH; red). Arrowheads indicate TRPV1 fibers in NPG; arrows indicate the TRPV1 and TH co-labeled neuron. B: relative expression of TRPV1 mRNA in NPG from NORM and CIH-exposed rats (n = 3 each), as determined by the 2ΔΔCT method. Trpv1 was normalized to the housekeeping gene B2M. CIH did not alter Trpv1 mRNA. C: immunoblot analysis of TRPV1 protein from NPG tissue from NORM and CIH-exposed rats (n = 2–3). TRPV1 was normalized to tubulin. CIH elevated TRPV1 protein in NPG. *P < 0.05, t-test. D: expression of TRPV1 in the nTS of NORM and CIH-exposed rats. Note the expression of TRPV1 in the afferent-containing tractus solitarii (TS) and the tendency for increased immunofluorescence in CIH rats. Measurements were taken from 100 × 100-mm box adjacent to the TS. Bregma level ~14.04 mm. AP, area postrema; DMnX, dorsal motor nucleus of the vagus. Scale bars, 100 μm.
Block of TRPV1 inhibits the CIH-mediated increase in spontaneous release.
A TRPV1 antagonist, SB-366791 (SB), has been shown to attenuate the frequency of mEPSCs and aEPSCs at the nTS synapse (Peters et al. 2010; Shoudai et al. 2010), although TS-EPSCs were relatively spared (Peters et al. 2010). We examined the functional consequence of TRPV1 in CIH. Specifically, we looked at whether the decrease in TS-EPSCs or the increase in the frequency of aEPSCs and mEPSCs that occurs after CIH is affected to the same extent as NORM neurons using two TRPV1 antagonists.
The TRPV1 inhibitors capsazepine (CPZ; 10 μM) and SB (10 μM) were bath applied, and the change in amplitude of TS-EPSCs was examined in nTS neurons exposed to either NORM or CIH. As illustrated in the representative NORM and CIH neurons (Fig. 4, A and B), CPZ did not appreciably alter TS-EPSC amplitude compared with their aCSF control. Across all of the neurons examined, CPZ and SB had minimal effect on TS-EPSC amplitude after NORM or CIH exposure compared with their individual aCSF controls; one exception is the decrease in TS-EPSC amplitude with SB in NORM. However, the change in TS-EPSC amplitude in NORM and CIH with either CPZ (−25.3 ± 9.0% vs. −18.6 ± 10.7%) or SB (−25.3 ± 6.9% vs. −10.7 ± 8.8%) was comparable (P > 0.05, one-way ANOVA).
Fig. 4.

TRPV1 inhibitors do not alter tractus solitarii-evoked excitatory postsynaptic currents (TS-EPSCs) in normoxic (NORM) and chronic intermittent hypoxia (CIH)-exposed neurons of the nucleus of the solitary tract (nTS). A and B: example of TS-EPSCs in a NORM (A) and CIH-exposed (B) nTS neuron in artificial cerebrospinal fluid (aCSF) baseline and in the presence of the TRPV1 inhibitor capsazepine (CZP). Arrows depict time of TS stimulation. Note the minimal decrease in TS-EPSC with TRPV1 inhibition. Examples are overlay of 10 events each. C and D: group data demonstrating the TRPV1 inhibitors CPZ (C) and SB-366791 (SB; D) minimally altered TS-EPSCs compared with aCSF (2-way RM ANOVA, CPZ exposure: drug, P = 0.06; hypoxia, P = 0.27; drug × hypoxia, P = 0.33; SB exposure: drug, P = 0.01; hypoxia, P = 0.36; drug × hypoxia, P = 0.21). *P < 0.05 vs. aCSF with Bonferroni post hoc (CPZ: NORM and CIH, n = 4 each; SB: NORM, n = 9, CIH, n = 8).
We also examined the contribution of TRPV1 to the increase in aEPSCs that occurs after CIH. As shown in the representative examples, CPZ reduced peak and average aEPSC events in NORM (Fig. 5A) and CIH neurons (Fig. 5D). Event diaries shown from the examples in Fig. 5, A and D, further demonstrate the reduction in aEPSCs after TRPV1 antagonist (Fig. 5, B and E). As a group, in NORM neurons, the blocking of TPRV1 with CPZ reduced the peak and average of aEPSC events to a level comparable to baseline (Fig. 5C; 2-way RM ANOVA). In CIH cells (Fig. 5F), aEPSCs were also reduced after TRPV1 antagonist compared with aCSF controls.
Fig. 5.

TRPV1 inhibitor capsazepine (CPZ) reduces the increase in spontaneous activity in chronic intermittent hypoxia (CIH)-exposed rats. A: example of excitatory postsynaptic currents (EPSCs) from normoxic (NORM) rats during artificial cerebrospinal fluid (aCSF) control (top) and following TRPV1 inhibition (bottom) with CPZ (10 µM). Note the decrease in asynchronous EPSC events after TRPV1 inhibition. Horizontal bars depict time of 20-Hz tractus solitarii (TS) stimulation. Examples are overlays of 2 traces each. Insets are zoomed images of peak events. B: diary of events of NORM cell in A before and after response to TS stimulation. C: group data for NORM (n = 4) shows CPZ depression of peak and average events compared with aCSF control following stimulation (2-way RM ANOVA: EPSC, P = 0.009; drug, P = 0.02; EPSC × drug, P = 0.08). D: EPSCs from CIH-exposed rat during aCSF control (top) and following TRPV1 inhibition (bottom) with CZP. Examples are overlays of 2 traces each. Insets are zoomed images of peak events. E: diary of events of CIH cell in D. F: group data for CIH-exposed cells (n = 6), showing TRPV1 inhibition with CPZ attenuated baseline, peak, and average events (2-way RM ANOVA: EPSC, P = 0.0005; drug, P = 0.01; EPSC × drug, P = 0.03). In B and E, events before TS stimulation are fit with a linear regression, whereas events following TS stimulation are fit via nonlinear curve; data are plotted as events per 100 ms. In C and F, dashed lines indicates aCSF baseline events. Ave, average; Bsl, baseline. **P < 0.01; ***P < 0.001; ****P < 0.0001, peak or average vs. baseline (pre TS-stimulation) with Bonferroni multiple comparison. $P < 0.05; $$P < 0.01; $$$$P < 0.0001, CPZ vs. aCSF control with Bonferroni multiple comparison.
In addition, using the TRPV1 antagonist SB and as shown in the representative diary traces, a decrease in aEPSC events also occurred within cells from NORM (Fig. 6A) and CIH animals (Fig. 6C). Quantitatively, SB reduced NORM aEPSCs (Fig. 6B) and nearly eliminated aEPSCs in CIH (Fig. 6D, 2-way RM ANOVA).
Fig. 6.

TRPV1 inhibitor SB-36679 (SB) reduces the increase in spontaneous and asynchronous activity in chronic intermittent hypoxia (CIH)-exposed rats. A and C: diary of events for individual normoxic (NORM; A) and CIH-exposed (C) cells before and after TRPV1 inhibition with SB (10 µM). Note the decrease in asynchronous excitatory postsynaptic currents (EPSCs) after TRPV1 inhibition. Events before tractus solitarii (TS) stimulation are fit with a linear regression. Events following TS stimulation are fit via nonlinear curve (aCSF, artificial cerebrospinal fluid) or linear regression (SB). Data are plotted as events per 100 ms. B and D: grouped data for NORM (B) and CIH (D) show depression of baseline, peak, and average events following stimulation [2-way RM ANOVA, NORM (n = 5): EPSC, P = 0.003; drug, P = 0.02; EPSC × drug, P = 0.05; CIH (n = 5): EPSC, P = 0.007; drug, P = 0.08; EPSC × drug, P = 0.01]. Dashed line in indicates aCSF baseline events. Ave, average; Bsl, baseline. **P < 0.01; ***P < 0.001; ****P < 0.0001 vs. baseline (pre TS-stimulation) with Bonferroni multiple comparison. $P < 0.05; $$P < 0.01; $$$P < 0.001; $$$$P < 0.0001, SB vs. aCSF control with Bonferroni multiple comparison.
CZP inhibition of representative mEPSCs traces is illustrated in Fig. 7, A and B. As shown, TRPV1 inhibition did not alter mEPSCs in NORM-exposed rats, yet decreased events after CIH. Quantitatively, TRPV1 inhibition with either CZP (Fig. 7C) or SB (Fig. 7D) did not change in NORM, whereas following CIH, mEPSC frequency was significantly decreased. As shown, after application of TRPV1 antagonist with SB, mEPSC frequency after CIH returned to nearly NORM levels.
Fig. 7.

TRPV1 inhibitors attenuate the elevated miniature excitatory postsynaptic currents (mEPSCs) after chronic intermittent hypoxia (CIH). A and B: examples of reduction in mEPSC events after TRPV1 inhibition with capsazepine (CPZ) in normoxic (NORM; A) and CIH-exposed (B) cells. Examples shown are a single trace. aCSF, artificial cerebrospinal fluid. C and D: mean mEPSC frequency data demonstrating CPZ (C) and SB-36679 (SB; D) tended to attenuate mEPSCs from aCSF control (C) in NORM neurons. Following CIH, CPZ and SB significantly decreased mEPSCs and returned their frequency to near NORM levels (2-way RM ANOVA, CPZ application: drug, P = 0.002; hypoxia, P = 0.24; drug × hypoxia, P = 0.11; SB application: drug, P = 0.0003; hypoxia, P = 0.06; drug × hypoxia × drug, P = 0.06). *P < 0.05, CPZ or SB vs. aCSF baseline with Bonferroni multiple comparison (CPZ: NORM, n = 6; CIH, n = 8; SB: NORM, n = 5; CIH, n = 7).
Collectively, these data illustrate that after CIH, TRPV1 antagonists minimally alter TS-EPSC amplitude but that TRPV1 contributes to the increase in mEPSCs and aEPSCs.
The increase in aEPSCs in response to CIH is not present in TRPV1 null mice whereas the decrease in TS-EPSC remains intact.
To further investigate the role of TRPV1 and circumvent possible nonspecific effects of the TRPV1 antagonists CPZ and SB, we compared the effect of CIH with NORM on TS-EPSCs, mEPSCs, sEPSCs, and aEPSCs in mice expressing (TRPV1+/+) with that in mice lacking (TRPV1−/−) TRPV1 expression. As shown in the examples in Fig. 8, A and B, NORM TS-EPSCs evoked at 0.5 Hz were comparable in TRPV1+/+ and TRPV1−/− mice, and CIH decreased TS-EPSC amplitude in both groups. Across the TRPV1+/+ neurons, CIH decreased TS-EPSC amplitude compared with NORM (Fig. 8C). Likewise, the amplitude of TS-EPSCs in TRPV1−/− mice was significantly reduced by CIH compared with NORM. The CIH-induced decrease in TS-EPSC amplitude was comparable between TRPV1+/+ (52 ± 7%) and TRPV1−/− mice (47 ± 12%, P = 0.72, t-test).
Fig. 8.
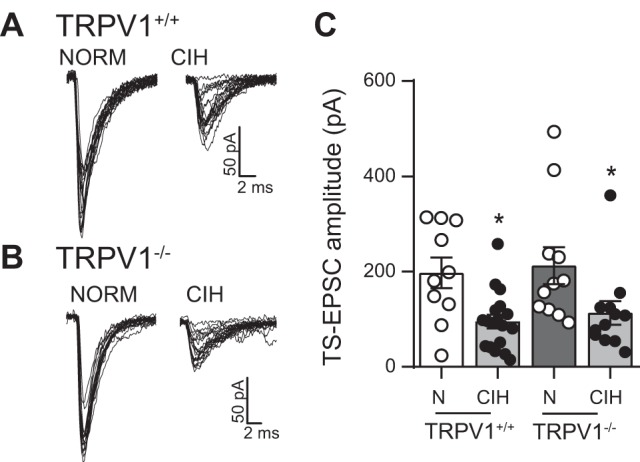
Tractus solitarii-evoked excitatory postsynaptic currents (TS-EPSCs) of TRPV1+/+ and TRPV1−/− neurons of nucleus tractus solitarii (nTS) are similar under normoxia (NORM) and both decrease following chronic intermittent hypoxia (CIH). A and B: TS-EPSCs in NORM were similar in TRPV1+/+ and TRPV1−/− mice. However, in both mice, the amplitude of the evoked response is reduced by CIH exposure. Examples are overlays of 20 traces each. C: mean data of TS-EPSCs in TRPV1+/+ and TRPV1−/− mice in NORM (N) and following CIH. As shown in our example, CIH decreased TS-EPSC amplitude to a comparable amplitude (2-way ANOVA: hypoxia, P = 0.004; mouse, P = 0.53; hypoxia × mouse, P = 0.96). *P < 0.05, CIH vs. NORM with Bonferroni post hoc (TRPV1+/+: NORM, n = 10; CIH, n = 18; TRPV1−/−: NORM, n = 11; CIH, n = 12).
We next examined the induction of aEPSCs in TRPV1 control and null mice after NORM and CIH exposure. Representative traces from NORM and CIH slices are shown in Fig. 9A for TRPV1+/+ mice and in Fig. 9D for TRPV1−/− mice, with their event diaries shown in Fig. 9, B and E, respectively. As illustrated, CIH induced an increase in aEPSCs in control but not null mice. As a group, TRPV1+/+ mice showed the expected increase in aEPSC events with CIH (Fig. 9C). However, there was no significant increase in aEPSCs above baseline in the TRPV1−/− mice subjected to CIH compared with their NORM controls (Fig. 9F; 2-way RM ANOVA).
Fig. 9.

Asynchronous excitatory postsynaptic current (aEPSC) events do not increase in TRPV1−/− mice after chronic intermittent hypoxia (CIH). CIH increases aEPSC in TRPV1+/+ (A) but not TRPV1−/− (D) mice. Examples are overlays of 2 traces each. Insets are zoomed images of peak events. B and E: diary plots of the events shown in representative examples (events/100 ms). Events before tractus solitarii (TS) stimulation are fit with a linear regression, whereas events following TS stimulation are fit via nonlinear curve. C and F: TRPV1+/+ mice increased peak and average aEPSC events after CIH (2-way ANOVA: EPSC, P = 0.001; hypoxia, P = 0.09; EPSC × hypoxia, P = 0.61). In NORM TRPV1−/− mutant mice, TS stimulation (20 Hz) increased peak events, which were eliminated after CIH (2-way ANOVA: EPSC, P = 0.001; hypoxia, P = 0.04; EPSC × hypoxia, P = 0.02). Ave, average; Bsl, baseline. *P < 0.05; **P < 0.01; ****P < 0.0001 vs. baseline within NORM or CIH group with Bonferroni post hoc. $P < 0.05, CIH vs. NORM with Bonferroni post hoc (TRPV1+/+: NORM, n = 9; CIH, n = 12; TRPV1−/−: NORM, n = 14; CIH, n = 15).
The frequency of mEPSCs was monitored in TRPV1+/+ and TRPV1−/− mice in NORM and following CIH. As shown in the representative traces, after CIH, mEPSC frequency did not increase in TRPV1−/− mice (Fig. 10B) as it did in TRPV1+/+ mice (Fig. 10A). As measured across these cells, a pairwise comparison of TRPV1+/+ mice exhibited the expected increase in mEPSC frequency from NORM after CIH (P = 0.02, t-test). On the other hand, the frequency of mEPSCs was unchanged in slices from TRPV1−/− mice from NORM following CIH (Fig. 10C). Interestingly, the mEPSC frequency under NORM was not different between the TRPV1+/+ and TRPV1−/− cells (P = 0.64, 2-way ANOVA).
Fig. 10.

The increase in miniature excitatory postsynaptic currents (mEPSCs) is eliminated in TRPV1−/− mice after chronic intermittent hypoxia (CIH). A and B: examples of mEPSCs under normoxia (NORM) and CIH in TRPV1+/+ (A) and TRPV1−/− (B) mice. Note the increase in mEPSCs after CIH in TRPV1+/+ mice but not TRPV1−/− mice. Examples are from a single trace. C: compared with NORM (N), mEPSCs increased in TRPV1+/+ mice after CIH. On the other hand, mEPSCs did not increase in TRPV1−/− mice after CIH (2-way ANOVA: mouse, P = 0.34; hypoxia, P = 0.29; mouse × hypoxia, P = 0.11). *P < 0.05, CIH vs. NORM with Bonferroni post hoc (TRPV1+/+: NORM, n = 10; CIH, n = 17; TRPV1−/−: NORM, n = 8; CIH, n = 4).
Taken together, these data indicate TRPV1 is not involved in the reduction in TS-EPSC with CIH. Moreover, this indicates the presence of TRPV1 is necessary for the CIH increase in mEPSCs and asynchronous activity following stimulation. The remaining baseline activity implicates additional channels in the generation of sEPSCs in the absence of TRPV1.
DISCUSSION
In the present study we examined the contribution of N-type calcium and TRPV1 channels in the CIH-induced spontaneous and afferent-driven synaptic currents in the rat and mouse nTS. Our data demonstrate N-type channels modulate TS-EPSC amplitude similarly in NORM and CIH. Regarding aEPSCs, in NORM, ω-CTx reduced peak and average EPSCs, returning them to baseline levels, and attenuated mEPSCs frequency. However, after CIH, although N-type block did attenuate aEPSCs, its effectiveness was not as great, nor did it alter mEPSC frequency. The two opposing effects elicited by CIH (increased aEPSC and mEPSC frequency vs. decrease in TS-EPSC amplitude) can now be separated on the basis of the role of TRPV1. Both the pharmacological and mouse studies support the role for TRPV1 in the increased spontaneous and aEPSCs activity induced by CIH, but not the attenuated TS-EPSC amplitude in CIH.
N-Type Calcium Channels Contribute to TS-EPSCs and to aEPSCs in NORM and CIH
The N-type calcium channel is thought to be responsible for the 60–70% of the Ca2+ influx on depolarization of the afferent terminal (Mendelowitz et al. 1995; Peters et al. 2010). Although the amplitude of TS-EPSCs is reduced after CIH, it remains similarly dependent on the N-type channel. In the present study, ω-CTx-sensitive channels contributed ~70% of presynaptic Ca2+ influx, whereas the remaining ~30% was carried by other voltage-dependent calcium channels. If the reduced amplitude of the TS-EPSC in CIH were due to a reduced contribution of ω-CTx-sensitive influx while the remaining contribution by other channels was unchanged, the effect of ω-CTx would be less in CIH. Because TS-EPSCs are a prerequisite for the asynchronous activity that follows stimulation, asynchronous activity is, at least indirectly, dependent on N-type calcium channels. Our present CZP and SB data confirm the additional dependence of aEPSCs on the TRPV1 channels. The relative contributions of N-type and TRPV1 channels to asynchronous activity are, however, difficult to separate.
It has been argued that the decay of the N-type calcium current during an action potential is sufficiently fast that Ca2+ influx through the voltage-gated calcium channels during stimulation contributes primarily to the early part of the asynchronous discharge (Peters et al. 2010). Our data showing a decrease in peak aEPSCs in NORM (Fig. 2D) by ω-CTx confirm this notion. The decrease in the prolonged, average aEPSCs is likely related to the reduced peak currents that were included in this parameter. Paradoxically, although the amplitude of the TS-EPSCs in CIH was significantly reduced, potentially decreasing Ca2+ influx, the peak and average asynchronous discharge was elevated. We have suggested that CIH modifies intracellular Ca2+ buffering, extrusion, or channel activity (Kline et al. 2007); the current study suggests the latter, a greater role for the TRPV1-mediated calcium influx.
Interestingly, after CIH, although CTx decreased the amplitude of TS-EPSCs and had a moderate effect on aEPSCs, it did not alter mEPSCs. This lack of effect by CTx on mEPSCs after CIH suggests upregulation of either a lower expressed voltage-gated calcium channel (Mendelowitz et al. 1995) or TRPV1.
TRPV1 Underlies the Increase in Spontaneous Activity and Asynchronous Activity That Occurs Following CIH
Acute inhibition of TRPV1 in NORM rats with SB or CZP reduces spontaneous and asynchronous glutamate release but not TS-evoked release, as previously reported for SB (Peters et al. 2010; Shoudai et al. 2010). After 10-day CIH, the usual CIH-induced increase in spontaneous and asynchronous activity is substantially reduced following TRPV1 antagonist inhibition, implicating TRPV1 in these changes. On further analysis, within baseline, peak, or average event groups, block of TRPV1 attenuated events similarly (~30%) regardless of the event type, suggesting in the rat that TRPV1 primarily contributes to overall spontaneous currents, including the increased currents in CIH. The prominent role of TRPV1 in modulating basal EPSCs is consistent with other studies (Andresen et al. 2012). The more robust decrease in mEPSCs in CIH after TRPV1 block further indicates a greater role of TRPV1 in spontaneous release compared with asynchronous.
When TRPV1 was genetically eliminated, the NORM TRPV1−/− mice still exhibited baseline and asynchronous activity, consistent with a previous study where TRPV1+/+ mice and a subgroup of TRPV1−/− mice expressed similar baseline spontaneous activity (Fenwick et al. 2014). In the present study, TRPV1+/+ mice showed the expected profile in response to CIH. However, when TRPV1−/− mice were subjected to 10-day CIH, there was no CIH-induced increase in mEPSC frequency or in baseline, peak, or average aEPSC events. The absence of an increase in spontaneous and asynchronous events after 10-day CIH in TRPV1−/− mice implicates TRPV1 in the increased activity following CIH.
Increased Expression of TRPV1 in CIH May Underlie Increased Spontaneous and Asynchronous Activity
Several mechanisms, not mutually exclusive, may contribute to the greater contribution of TRPV1 in CIH synaptic transmission. One of these is the increased expression of TRPV1. As shown in the present study and by others (Hermes et al. 2016; Roy et al. 2012; Sun et al. 2009), TRPV1 is readily expressed in small- and medium-sized neurons that project to the nTS, including TH+ chemoafferents. CIH may increase expression of TRPV1 within these sensory neurons, augment their expression at the membrane, or perhaps elevate TRPV1 in neuronal subtypes that do not typically contain TRPV1 (Zhang et al. 2008). Consistent with this idea, we have shown that TRPV1 protein, but not mRNA, is elevated in the NPG after 10-day CIH. It is likely that TRPV1 mRNA increased during the development of CIH for greater protein expression; however, we cannot rule out reduced TPRV1 turnover due to enhanced protein stabilization and/or reduced degradation. Although in our immunohistochemical study TRPV1 tended to be enhanced in nTS in the area in which we functionally tested its role, it did not reach the level anticipated from the increased NPG protein after CIH. The mechanism by which TRPV1 increased expression, or perhaps its localization, requires further study.
Other Potential Signals Modifying TRPV1 Activity in the CIH-Induced Changes
We have shown that CIH activates and phosphorylates Ca2+/calmodulin-dependent protein kinase II (CaMKII; Kline et al. 2007). Whereas our initial studies demonstrated inhibition of CaMKII returns CIH TS-EPSCs to NORM levels, CaMKII may also modulate spontaneous EPSCs via altered TRPV1 function. Phosphorylation of TRPV1 by CaMKII regulates ligand binding to TRPV1 and its activation, whereas dephosphorylation leads to desensitization of TRPV1 (Jung et al. 2004). TRPV1-activated (capsaicin) elevation of dorsal root ganglion cell intracellular Ca2+ is mediated by activation of CaMKII (Zhang et al. 2011). Interestingly, exposure to capsaicin induces phosphorylation of CaMKII (Price et al. 2005), suggesting a circular process that may elevate sEPSCs following CIH.
CIH exposure to reduced Po2 may also activate TRPV1 via either hypoxic exposure and/or the elevated neuronal activity and depolarization. Overnight exposure to hypoxia sensitizes TRPV1 in HEK-293T cells and rat sensory neurons, increasing inward current amplitude in response to capsaicin but not TRPV1 protein expression (Kim et al. 2012; Ristoiu et al. 2011). This would perhaps require a chronic but reversible increase in an endogenous activator of TRPV1 in response to a decrease in O2. A number of endogenous TRPV1 ligands (i.e., endovanilloids) are formed and released in an activity-dependent manner (Van Der Stelt and Di Marzo 2004), including several that have been shown to be functionally active in the NPG (Wang et al. 2005) or nTS (Sharkey et al. 2007), although not all studies agree (Fenwick et al. 2017). For instance, a number of endovanilloid lipoxygenase by-products are produced during inflammation (Van Der Stelt and Di Marzo 2004), a consequence of obstructive sleep apnea and CIH (de Lima et al. 2016; Del Rio et al. 2012). Thus it is conceivable that elevated TRPV1 contribution in CIH may be due to greater endovanilloid production, although future studies would require its identification.
Limitations
Our study suggests TRPV1 contributes to overall spontaneous activity, especially following CIH. One limitation of our expression studies is the sample size. Although not large, the suggestion of an increase in TRPV1 is in agreement with our electrophysiology studies. Although both TRPV1 antagonist and genetic studies support this interpretation, there are limitations to both. For instance, CZP acts on other ion channels known to be present in the terminals of the sensory afferents (Gill et al. 2004; Wladyka and Kunze 2006; Zuo et al. 2013). Whereas SB is more selective, it is also reported to act on receptors present in sensory afferents that may influence glutamate release (Gunthorpe et al. 2004) or function (Lappin et al. 2006). This may account, in part, for the decrease in TS-EPSC amplitude by SB in NORM rats. In addition, studies using genetically altered mice to study the role of an individual ion channel have the potential to lead to alterations in expression of other channels or channel modulators as compensation, for example, in this case, the deletion of TRPV1. The appearance of a higher than expected frequency of spontaneous events in the NORM TRPV1−/− mouse as previously reported may have its basis in upregulated expression of other Ca2+-permeable channels present in these sensory neurons. Despite these limitations, inhibition of spontaneous activity by the two antagonists and the observation that the increase in spontaneous and asynchronous activity associated with CIH did not occur in the TRPV1−/− mouse provide support for a role for TRPV1.
Although we are recording from neurons in the region of the nTS that receives chemosensory input, we have not labeled that pathway specifically. Therefore, though we interpret the data in terms of the carotid body chemosensory reflex pathway, we have not specifically identified the input to the postsynaptic cell from which we are recording or its projection in the reflex pathway. Future studies will need to identify the contribution of TRPV1 in chemoafferent fibers that innervate nTS neurons that project to chemoreflex pathways, such as the rostral ventrolateral medulla or ventral respiratory group.
Physiological Role of TRPV1 in CIH
Our data demonstrate that TRPV1 contributes to the spontaneous and aEPSCs increase in synaptic transmission after CIH. Our focus was on the medial and commissural nTS, the primary site of carotid body chemosensory input into the brain stem (Finley and Katz 1992). Overactivation of the carotid body and its reflex arc, such as occurs during CIH or long-term facilitation (LTF), can induce prolonged sympathoexcitation and hypertension. Relevant to the current work, TRPV1 has been identified in chemosensory afferents (Roy et al. 2012) and significantly contributes to in vitro chemoafferent LTF and in vivo sympathetic nerve activity LTF (Roy et al. 2018). These authors further demonstrated that chemosensory TRPV1 primarily contributes to the maintenance of LTF rather than the hypoxic responses (Roy et al. 2018). Our data support and extend these results. Specifically, we and others (Peters et al. 2010) have demonstrated that TRPV1 has limited influence on TS-EPSC amplitude in normoxia or CIH but does contribute to sEPSCs and aEPSCs following a stimulus train. Furthermore, we have shown that aEPSCs are capable of inducing prolonged action potential discharge in nTS neurons (Kline et al. 2007). The observed TRPV1-mediated elevation of synaptic events in nTS is consistent with the hypothesized central plasticity that contributes to sympathetic LTF (Roy et al. 2018). Thus it is conceivable that TRPV1 contributes to chemosensory afferent activity and subsequent triggering of second-order nTS to induce sympathetic LTF. This may occur via direct projections from nTS to the rostral ventral lateral medulla (Kline et al. 2010). These data demonstrate a powerful physiological role of TRPV1 in the nTS in maintaining or modulating cardiorespiratory plasticity normally and following intermittent hypoxic challenges.
GRANTS
This work was funded by National Heart, Lung, and Blood Institute Grants R01 HL128454 (to D. D. Kline) and R01 HL090886 (to D. L. Kunze).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.D.K., S.W., and D.L.K. conceived and designed research; D.D.K., S.W., and D.L.K. performed experiments; D.D.K., S.W., and D.L.K. analyzed data; D.D.K. and D.L.K. interpreted results of experiments; D.D.K. and D.L.K. prepared figures; D.D.K. and D.L.K. drafted manuscript; D.D.K. and D.L.K. edited and revised manuscript; D.D.K., S.W., and D.L.K. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the late Christine Schramm for assistance in RT-PCR and immunoblot analysis, and Heather A. Dantzler and Pat Glazebrook for assistance in immunohistochemistry.
Present address for S. Wang: Dept. of Physiology, Hebei Medical University, Shijiazhuang, Hebei Province 050017, China.
REFERENCES
- Almado CE, Machado BH, Leão RM. Chronic intermittent hypoxia depresses afferent neurotransmission in NTS neurons by a reduction in the number of active synapses. J Neurosci 32: 16736–16746, 2012. doi: 10.1523/JNEUROSCI.2654-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andresen MC, Hofmann ME, Fawley JA. The unsilent majority-TRPV1 drives “spontaneous” transmission of unmyelinated primary afferents within cardiorespiratory NTS. Am J Physiol Regul Integr Comp Physiol 303: R1207–R1216, 2012. doi: 10.1152/ajpregu.00398.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andresen MC, Kunze DL. Nucleus tractus solitarius—gateway to neural circulatory control. Annu Rev Physiol 56: 93–116, 1994. doi: 10.1146/annurev.ph.56.030194.000521. [DOI] [PubMed] [Google Scholar]
- Austgen JR, Hermann GE, Dantzler HA, Rogers RC, Kline DD. Hydrogen sulfide augments synaptic neurotransmission in the nucleus of the solitary tract. J Neurophysiol 106: 1822–1832, 2011. doi: 10.1152/jn.00463.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitravanshi VC, Sapru HN. Chemoreceptor-sensitive neurons in commissural subnucleus of nucleus tractus solitarius of the rat. Am J Physiol 268: R851–R858, 1995. [DOI] [PubMed] [Google Scholar]
- de Lima FF, Mazzotti DR, Tufik S, Bittencourt L. The role inflammatory response genes in obstructive sleep apnea syndrome: a review. Sleep Breath 20: 331–338, 2016. doi: 10.1007/s11325-015-1226-7. [DOI] [PubMed] [Google Scholar]
- Del Rio R, Moya EA, Parga MJ, Madrid C, Iturriaga R. Carotid body inflammation and cardiorespiratory alterations in intermittent hypoxia. Eur Respir J 39: 1492–1500, 2012. doi: 10.1183/09031936.00141511. [DOI] [PubMed] [Google Scholar]
- Doyle MW, Andresen MC. Reliability of monosynaptic sensory transmission in brain stem neurons in vitro. J Neurophysiol 85: 2213–2223, 2001. doi: 10.1152/jn.2001.85.5.2213. [DOI] [PubMed] [Google Scholar]
- Fenwick AJ, Fowler DK, Wu SW, Shaffer FJ, Lindberg JE, Kinch DC, Peters JH. Direct anandamide activation of TRPV1 produces divergent calcium and current responses. Front Mol Neurosci 10: 200, 2017. doi: 10.3389/fnmol.2017.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenwick AJ, Wu SW, Peters JH. Isolation of TRPV1 independent mechanisms of spontaneous and asynchronous glutamate release at primary afferent to NTS synapses. Front Neurosci 8: 6, 2014. doi: 10.3389/fnins.2014.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley JC, Katz DM. The central organization of carotid body afferent projections to the brainstem of the rat. Brain Res 572: 108–116, 1992. doi: 10.1016/0006-8993(92)90458-L. [DOI] [PubMed] [Google Scholar]
- Gill CH, Randall A, Bates SA, Hill K, Owen D, Larkman PM, Cairns W, Yusaf SP, Murdock PR, Strijbos PJ, Powell AJ, Benham CD, Davies CH. Characterization of the human HCN1 channel and its inhibition by capsazepine. Br J Pharmacol 143: 411–421, 2004. doi: 10.1038/sj.bjp.0705945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami C, Rademacher N, Smalla KH, Kalscheuer V, Ropers HH, Gundelfinger ED, Hucho T. TRPV1 acts as a synaptic protein and regulates vesicle recycling. J Cell Sci 123: 2045–2057, 2010. doi: 10.1242/jcs.065144. [DOI] [PubMed] [Google Scholar]
- Grueter BA, Brasnjo G, Malenka RC. Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat Neurosci 13: 1519–1525, 2010. doi: 10.1038/nn.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunthorpe MJ, Rami HK, Jerman JC, Smart D, Gill CH, Soffin EM, Luis Hannan S, Lappin SC, Egerton J, Smith GD, Worby A, Howett L, Owen D, Nasir S, Davies CH, Thompson M, Wyman PA, Randall AD, Davis JB. Identification and characterisation of SB-366791, a potent and selective vanilloid receptor (VR1/TRPV1) antagonist. Neuropharmacology 46: 133–149, 2004. doi: 10.1016/S0028-3908(03)00305-8. [DOI] [PubMed] [Google Scholar]
- Guo A, Vulchanova L, Wang J, Li X, Elde R. Immunocytochemical localization of the vanilloid receptor 1 (VR1): relationship to neuropeptides, the P2X3 purinoceptor and IB4 binding sites. Eur J Neurosci 11: 946–958, 1999. doi: 10.1046/j.1460-9568.1999.00503.x. [DOI] [PubMed] [Google Scholar]
- Hermes SM, Andresen MC, Aicher SA. Localization of TRPV1 and P2X3 in unmyelinated and myelinated vagal afferents in the rat. J Chem Neuroanat 72: 1–7, 2016. doi: 10.1016/j.jchemneu.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iturriaga R, Andrade DC, Del Rio R. Enhanced carotid body chemosensory activity and the cardiovascular alterations induced by intermittent hypoxia. Front Physiol 5: 468, 2014. doi: 10.3389/fphys.2014.00468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong JH, Lee DK, Liu SM, Chua SC Jr, Schwartz GJ, Jo YH. Activation of temperature-sensitive TRPV1-like receptors in ARC POMC neurons reduces food intake. PLoS Biol 16: e2004399, 2018. doi: 10.1371/journal.pbio.2004399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julius C, Clapham DE. Transient Receptor Potential channels: TRPV1 (Online). http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=507. 2018. [19 June 2018].
- Jung J, Shin JS, Lee SY, Hwang SW, Koo J, Cho H, Oh U. Phosphorylation of vanilloid receptor 1 by Ca2+/calmodulin-dependent kinase II regulates its vanilloid binding. J Biol Chem 279: 7048–7054, 2004. doi: 10.1074/jbc.M311448200. [DOI] [PubMed] [Google Scholar]
- Kim KS, Yoo HY, Park KS, Kim JK, Zhang YH, Kim SJ. Differential effects of acute hypoxia on the activation of TRPV1 by capsaicin and acidic pH. J Physiol Sci 62: 93–103, 2012. doi: 10.1007/s12576-011-0185-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD, Buniel MC, Glazebrook P, Peng YJ, Ramirez-Navarro A, Prabhakar NR, Kunze DL. Kv1.1 deletion augments the afferent hypoxic chemosensory pathway and respiration. J Neurosci 25: 3389–3399, 2005. doi: 10.1523/JNEUROSCI.4556-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD, King TL, Austgen JR, Heesch CM, Hasser EM. Sensory afferent and hypoxia-mediated activation of nucleus tractus solitarius neurons that project to the rostral ventrolateral medulla. Neuroscience 167: 510–527, 2010. doi: 10.1016/j.neuroscience.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD, Ramirez-Navarro A, Kunze DL. Adaptive depression in synaptic transmission in the nucleus of the solitary tract after in vivo chronic intermittent hypoxia: evidence for homeostatic plasticity. J Neurosci 27: 4663–4673, 2007. doi: 10.1523/JNEUROSCI.4946-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline DD, Takacs KN, Ficker E, Kunze DL. Dopamine modulates synaptic transmission in the nucleus of the solitary tract. J Neurophysiol 88: 2736–2744, 2002. doi: 10.1152/jn.00224.2002. [DOI] [PubMed] [Google Scholar]
- Lappin SC, Randall AD, Gunthorpe MJ, Morisset V. TRPV1 antagonist, SB-366791, inhibits glutamatergic synaptic transmission in rat spinal dorsal horn following peripheral inflammation. Eur J Pharmacol 540: 73–81, 2006. doi: 10.1016/j.ejphar.2006.04.046. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔCT) method. Methods 25: 402–408, 2001. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Matott MP, Kline DD. Activation of 5-hyrdoxytryptamine 7 receptors within the rat nucleus tractus solitarii modulates synaptic properties. Brain Res 1635: 12–26, 2016. doi: 10.1016/j.brainres.2016.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matott MP, Ruyle BC, Hasser EM, Kline DD. Excitatory amino acid transporters tonically restrain nTS synaptic and neuronal activity to modulate cardiorespiratory function. J Neurophysiol 115: 1691–1702, 2016. doi: 10.1152/jn.01054.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelowitz D, Reynolds PJ, Andresen MC. Heterogeneous functional expression of calcium channels at sensory and synaptic regions in nodose neurons. J Neurophysiol 73: 872–875, 1995. doi: 10.1152/jn.1995.73.2.872. [DOI] [PubMed] [Google Scholar]
- Peters JH, McDougall SJ, Fawley JA, Smith SM, Andresen MC. Primary afferent activation of thermosensitive TRPV1 triggers asynchronous glutamate release at central neurons. Neuron 65: 657–669, 2010. doi: 10.1016/j.neuron.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhakar NR, Peng YJ, Jacono FJ, Kumar GK, Dick TE. Cardiovascular alterations by chronic intermittent hypoxia: importance of carotid body chemoreflexes. Clin Exp Pharmacol Physiol 32: 447–449, 2005. doi: 10.1111/j.1440-1681.2005.04209.x. [DOI] [PubMed] [Google Scholar]
- Price TJ, Jeske NA, Flores CM, Hargreaves KM. Pharmacological interactions between calcium/calmodulin-dependent kinase II alpha and TRPV1 receptors in rat trigeminal sensory neurons. Neurosci Lett 389: 94–98, 2005. doi: 10.1016/j.neulet.2005.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristoiu V, Shibasaki K, Uchida K, Zhou Y, Ton BH, Flonta ML, Tominaga M. Hypoxia-induced sensitization of transient receptor potential vanilloid 1 involves activation of hypoxia-inducible factor-1 alpha and PKC. Pain 152: 936–945, 2011. doi: 10.1016/j.pain.2011.02.024. [DOI] [PubMed] [Google Scholar]
- Roy A, Farnham MMJ, Derakhshan F, Pilowsky PM, Wilson RJ. Acute intermittent hypoxia with concurrent hypercapnia evokes P2X and TRPV1 receptor-dependent sensory long-term facilitation in naïve carotid bodies. J Physiol 596: 3149–3169, 2018. doi: 10.1113/JP275001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Mandadi S, Fiamma MN, Rodikova E, Ferguson EV, Whelan PJ, Wilson RJ. Anandamide modulates carotid sinus nerve afferent activity via TRPV1 receptors increasing responses to heat. J Appl Physiol (1985) 112: 212–224, 2012. doi: 10.1152/japplphysiol.01303.2010. [DOI] [PubMed] [Google Scholar]
- Sharkey KA, Cristino L, Oland LD, Van Sickle MD, Starowicz K, Pittman QJ, Guglielmotti V, Davison JS, Di Marzo V. Arvanil, anandamide and N-arachidonoyl-dopamine (NADA) inhibit emesis through cannabinoid CB1 and vanilloid TRPV1 receptors in the ferret. Eur J Neurosci 25: 2773–2782, 2007. doi: 10.1111/j.1460-9568.2007.05521.x. [DOI] [PubMed] [Google Scholar]
- Shoudai K, Peters JH, McDougall SJ, Fawley JA, Andresen MC. Thermally active TRPV1 tonically drives central spontaneous glutamate release. J Neurosci 30: 14470–14475, 2010. doi: 10.1523/JNEUROSCI.2557-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica AL, Greenberg HE, Ruggiero DA, Scharf SM. Chronic-intermittent hypoxia: a model of sympathetic activation in the rat. Respir Physiol 121: 173–184, 2000. doi: 10.1016/S0034-5687(00)00126-2. [DOI] [PubMed] [Google Scholar]
- Sun H, Li DP, Chen SR, Hittelman WN, Pan HL. Sensing of blood pressure increase by transient receptor potential vanilloid 1 receptors on baroreceptors. J Pharmacol Exp Ther 331: 851–859, 2009. doi: 10.1124/jpet.109.160473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Stelt M, Di Marzo V. Endovanilloids. Putative endogenous ligands of transient receptor potential vanilloid 1 channels. Eur J Biochem 271: 1827–1834, 2004. doi: 10.1111/j.1432-1033.2004.04081.x. [DOI] [PubMed] [Google Scholar]
- Vidruk EH, Olson EB Jr, Ling L, Mitchell GS. Responses of single-unit carotid body chemoreceptors in adult rats. J Physiol 531: 165–170, 2001. doi: 10.1111/j.1469-7793.2001.0165j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Miyares RL, Ahern GP. Oleoylethanolamide excites vagal sensory neurones, induces visceral pain and reduces short-term food intake in mice via capsaicin receptor TRPV1. J Physiol 564: 541–547, 2005. doi: 10.1113/jphysiol.2004.081844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wladyka CL, Kunze DL. KCNQ/M-currents contribute to the resting membrane potential in rat visceral sensory neurons. J Physiol 575: 175–189, 2006. doi: 10.1113/jphysiol.2006.113308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Lin RL, Wiggers M, Snow DM, Lee LY. Altered expression of TRPV1 and sensitivity to capsaicin in pulmonary myelinated afferents following chronic airway inflammation in the rat. J Physiol 586: 5771–5786, 2008. doi: 10.1113/jphysiol.2008.161042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Daugherty SL, de Groat WC. Activation of CaMKII and ERK1/2 contributes to the time-dependent potentiation of Ca2+ response elicited by repeated application of capsaicin in rat DRG neurons. Am J Physiol Regul Integr Comp Physiol 300: R644–R654, 2011. doi: 10.1152/ajpregu.00672.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo GF, Li MH, Zhang JX, Li B, Wang ZM, Wang Q, Xiao H, Chen SL. Capsazepine concentration dependently inhibits currents in HEK 293 cells mediated by human hyperpolarization-activated cyclic nucleotide-gated 2 and 4 channels. Exp Biol Med (Maywood) 238: 1055–1061, 2013. doi: 10.1177/1535370213498973. [DOI] [PubMed] [Google Scholar]