

Abstract
Mutations in the lamin A/C gene (LMNA) encoding intermediate filament proteins associated with the inner nuclear membrane cause diseases known as laminopathies. Most LMNA mutations cause dilated cardiomyopathy with variable skeletal muscular dystrophy. Cell signaling abnormalities have been discovered in hearts of mouse models of cardiomyopathy caused by LMNA mutations that contribute to pathogenesis. These include abnormally increased signaling by extracellular signal-regulated kinase 1 and kinase 2 and other mitogen-activated protein kinases, protein kinase B/mammalian target of rapamycin complex 1 and transforming growth factor-β. Preclinical research suggests that specific inhibitors of these abnormally activated cell signaling pathways may be useful in treating human patients with this disease.
Introduction
The human lamin A/C gene (LMNA) encodes prelamin A, which is processed to lamin A, and lamin C [1]. Lamin A, lamin C and B-type lamins have structural similarity to intermediate filament proteins, but assemble into tetrameric filaments of 3.5 nm thickness at the inner aspect of the inner nuclear membrane in somatic cells [2–4]. Lamins interact with several integral proteins concentrated in the inner nuclear membrane as well as various nucleoplasmic proteins [5].
A-type lamins are expressed in most terminally differentiated cells. However, mutations in LMNA cause inherited diseases called laminopathies that are organ system selective. These can be broadly separated into disorders that primarily affect either (1) striated muscle, (2) adipose tissue, (3) peripheral nerve or (4) multiple organ systems usually with features of accelerated aging [6]. Among the laminopathies affecting striated muscle with onset from childhood to adulthood are Emery–Dreifuss muscular dystrophy, isolated dilated cardiomyopathy, a form of limb–girdle muscular dystrophy or variants of these entities that were originally defined based on clinical features [7–11]. Some LMNA mutations also cause a congenital muscular dystrophy presenting in very early childhood [12,13]. The inheritance pattern is almost always autosomal dominant, but rare compound heterozygous mutations have been described. The common and life-threatening feature of laminopathies primarily affecting striated muscle is dilated cardiomyopathy, which is generally associated with early onset atrioventricular conduction block. Cardiomyopathy caused by LMNA mutations has a relatively rapidly progressive course with sudden death from arrhythmias and the onset of heart failure occurring at earlier ages than most other inherited cardiomyopathies [14,15].
Signaling pathway abnormalities in cardiomyopathy caused by LMNA mutations
Several mouse lines have been generated with mutations in the lamin A/C (Lmna) gene. We have utilized LmnaH222P/H222P mice generated by Arimura et al. [16] to examine abnormalities in cell signaling pathways in the heart. These mice recapitulate most features of cardiomyopathy seen in humans with LMNA mutations; however, with the caveat that the mice are homozygous for the Lmna mutation, whereas in humans the disease is almost always dominant.
Bioinformatics analyses of transcriptomes from hearts of male LmnaH222P/H222P mice at 10 weeks of age, which is when symptoms of cardiomyopathy first appear, suggested abnormalities in several signaling pathways [17–19]. Prominent among these were mitogen-activated protein kinase (MAP kinase), protein kinase B (AKT)/mammalian target of rapamycin complex 1 (mTORC1) and transforming growth factor-β (TGF-β) signaling. TGF-β signaling had previously been shown to be hyperactivated in the original description of LmnaH222P/H222P mice [16]. WNT/β-catenin signaling was also suggested to be altered.
Biochemical interrogation of heart tissue from LmnaH222P/H222P mice showed that among the MAP kinase cascades, c-Jun N-terminal kinase ( JNK), p38α and extracellular signal-regulated kinases 1 and 2 (ERK1 and 2) were all hyperactivated [17–19]. Biochemical experiments also confirmed hyperactivation of the AKT/mTORC1 pathway [20]. Other investigators also showed mTORC1 signaling hyperactivation in hearts of Lmna−/− mice that develop cardiomyopathy at a younger age [21]. The abnormal increases in AKT/mTORC1 signaling were linked to decreased macroautophagy (autophagy) in heart muscle. ERK1/2 and AKT/mTORC1 signaling are hyperactivated in hearts of male LmnaH222P/H222P mice as early as 4 weeks of age, which is prior to the onset of any detectable pathology [17,20]. WNT/β-catenin signaling was later shown to be decreased in hearts of LmnaH222P/H222P mice starting at ∼12 weeks of age in male mice [22]. Similar alterations in ERK1/2, AKT/mTORC1, TGF-β and WNT/β-catenin signaling have been demonstrated in hearts from humans with cardiomyopathy caused by LMNA mutations, although tissues have only been available at late-disease stages [20,22,23,24]. Female LmnaH222P/H222P mice have a later onset of disease and, for practical purposes, were not used in most of these experiments; however, a research in progress has shown similar defects in ERK1/2 and AKT activities but at older ages.
Connecting signaling abnormalities to pathology
Given the abnormal elevations in MAP kinase, AKT/mTORC1 and TGF-β signaling in hearts of LmnaH222P/H222P mice, we hypothesized that reducing them would have beneficial effects on cardiac function. If so, it would link these cell signaling defects to cardiac pathology. We initially tested PD98059, one of the first synthetic inhibitors of mitogen-activated protein kinase kinases 1 and 2 (MEK1/2), the enzyme upstream of ERK1/2 that activates it by phosphorylation [25]. The treatment of male LmnaH222P/H222P mice with PD98059 starting at 8 weeks of age, prior to the onset of significant cardiac dysfunction, reduced ERK1/2 phosphorylation and delayed the development of left ventricular dilatation. At 16 weeks of age, the drug-treated mice demonstrated normal left ventricular ejection fraction, whereas the placebo-treated mice demonstrated a 30% reduction. We similarly treated male LmnaH222P/H222P mice starting at 8 weeks of age with SP600125, an inhibitor of all mammalian isoforms of JNK [26]. Treatment with SP600125 inhibited JNK phosphorylation in the heart with no detectable effect on ERK1/2, delayed the development of left ventricular dilatation, prevented decreases in cardiac ejection fraction and decreased fibrosis at 16 weeks of age.
Perhaps more relevant to potential treatment of human subjects is the benefit of blocking ERK1/2 or JNK activity after the onset of detectable cardiac dysfunction. Unless their preventive effects were exceptionally powerful, it would be imprudent to use such drugs as prophylactic treatment in asymptomatic humans with LMNA mutations. We, therefore, treated male LmnaH222P/H222P with PD98059 or SP600125 to respectively reduce ERK1/2 or JNK signaling, starting at 16 weeks of age when they already have decreased left ventricular fractional shortening. Treatment with either of these inhibitors led to decreased left ventricular end-systolic dilatation and increased left ventricular ejection fraction after 4 weeks of treatment compared with mice receiving placebo [27]. There was also a decrease in cardiac fibrosis compared with placebo-treated mice. Notably, fibrosis is prominent in cardiomyopathy caused by LMNA mutations, which not only contributes to left ventricular stiffness, but at earlier stages may underlie the development of atrioventricular conduction block and ventricular arrhythmias [28–31].
We next tested if the inhibition of p38α signaling would have impact on the deterioration of left ventricular function in LmnaH222P/H222P mice. We blocked p38α signaling using ARRY-371797, an orally available inhibitor of p38α. After the treatment for 4 weeks in male LmnaH222P/H222P mice starting at 16 weeks of age, ARRY-371797 reduced phosphorylated p38α in hearts that was associated with significantly decreased left ventricular diameters and increased left ventricular fractional shortening compared with placebo [19]. However, ARRY-371797 did not significantly reduce the expression of genes encoding collagens, suggesting that it did not have a major effect on cardiac fibrosis. Nonetheless, this study showed that abnormally increased p38α signaling contributed to cardiac pathology in LmnaH222P/H222P mice.
We and others also tested the effects of inhibitors of mTORC1 activity in mouse models of cardiomyopathy caused by LMNA mutations. In a trial lasting only 2 weeks, treatment of male LmnaH222P/H222P mice with the rapamycin analog temsirolimus starting at 14 weeks of age after the onset of cardiac pathology reduced phosphorylated mTOR, a protein kinase component of mTORC1, in hearts; this was associated with significantly smaller left ventricular diameters and significantly increased left ventricular fractional shortening compared with mice treated with placebo [20]. In this short-term experiment, however, the expressions of genes encoding collagens and fibronectin involved in fibrosis were not reduced. Treatment with an MEK1/2 inhibitor also reduced phosphorylation of mTOR in hearts of LmnaH222P/H222P mice, suggesting that ERK1/2 hyperactivation contributes to increased mTORC1 signaling. Rapamycin treatment of Lmna−/− mice, which have a median survival of only 6–8 weeks and develop heart disease early in life, had beneficial effects on heart function and prolonged survival [21]. Temsirolimus and rapamycin also partially reversed the reduction in autophagy in heart muscle of these Lmna mutant mice, correlating defects in this cellular process with cardiac pathology [20,21].
Given the enhanced TGF-β signaling in hearts of LmnaH222P/H222P mice and its known role in fibrosis, we tested if reducing its signaling activity would attenuate myocardial fibrosis and improve cardiac function. We treated male LmnaH222P/H222P mice with SB-431543, an inhibitor of the TGF-β type I receptor kinase, starting at 16 weeks of age. After 4 weeks of treatment, these mice had reduced myocardial fibrosis along with decreased left ventricular diameters and increased left ventricular fractional shortening compared with mice treated with placebo [24]. The treatment of LmnaH222P/H222P mice with an MEK1/2 inhibitor also led to a decrease in nuclear phosphorylated Smad2 and Smad3, mediators of TGF-β signaling, compared with placebo-treated mice. This suggests that hyperactivation of ERK1/2 signaling contributes to TGF-β-mediated myocardial fibrosis in LmnaH222P/H222P mice.
Toward drug therapy for cardiomyopathy caused by LMNA mutations
The work on cell signaling abnormalities in cardiomyopathy caused by LMNA mutations provides a starting point for human clinical investigation. However, there are several large steps from experimental studies in genetically modified mice to human therapy. While the mTORC1 signaling inhibitors rapamycin and temsirolimus are used clinically in human subjects for different indications, the MAP kinase and TGF-β inhibitors used in the studies described above are not all ideal drug candidates. Only ARRY-371797 is currently in active clinical development.
Most of the studies described above have been relatively short term and used relatively small numbers of mice. The most extensively studied class of drugs for this inherited cardiomyopathy in model mice has been the MEK1/2 inhibitors that reduce ERK1/2 activity. Multiple studies with different drugs in this class have shown reproducible improvements in left ventricular function, decreased cardiac fibrosis and prolonged survival in LmnaH222P/H222P mice [23,25,27,32]. One of these utilized selumetinib, an MEK1/2 inhibitor, currently in advanced clinical trials for cancer [23]. More recently, we have shown that CIP-137401, a novel macrocyclic preclinical MEK1/2 inhibitor with excellent drug-like properties, appears safe, improves left ventricular function, decreases cardiac fibrosis and prolongs survival [32].
We propose a model in which an interplay between abnormal signaling pathways leads from LMNA mutations to cardiomyopathy (Figure 1). This model proposes that alterations in A-type lamins, such as those in LmnaH222P/H222P mice, lead to activation of ERK1/2 and AKT. One missing piece of the puzzle is exactly how alterations in A-type lamins activate these two signaling pathways. One study has shown that phosphorylated ERK1/2 interacts with lamin A at the inner nuclear membrane [33]. However, it remains unknown how this leads to ERK1/2 activation, especially in striated muscle, when A-type lamins are altered. Regardless of the mechanism behind ERK1/2 and AKT activation in striated muscle, these protein kinases activate mTORC1 signaling, depressing autophagy, which contributes to disease pathogenesis. ERK1/2 also synergizes with TGF-β to induce fibrosis in the heart. ERK1/2, mTORC1 and TGF-β signaling have all been blocked with small-molecule inhibitors in LmnaH222P/H222P mice and all of these interventions have had some beneficial impact. However, reducing signaling of each of these pathways alone in LmnaH222P/H222P mice is not definitively curative. Future progress may depend on appropriately reducing the activity of more than one of these pathways simultaneously with combinations of safe and effective drugs.
Figure 1. Model showing interplay between cell signaling pathways leading from LMNA mutations to cardiomyopathy.
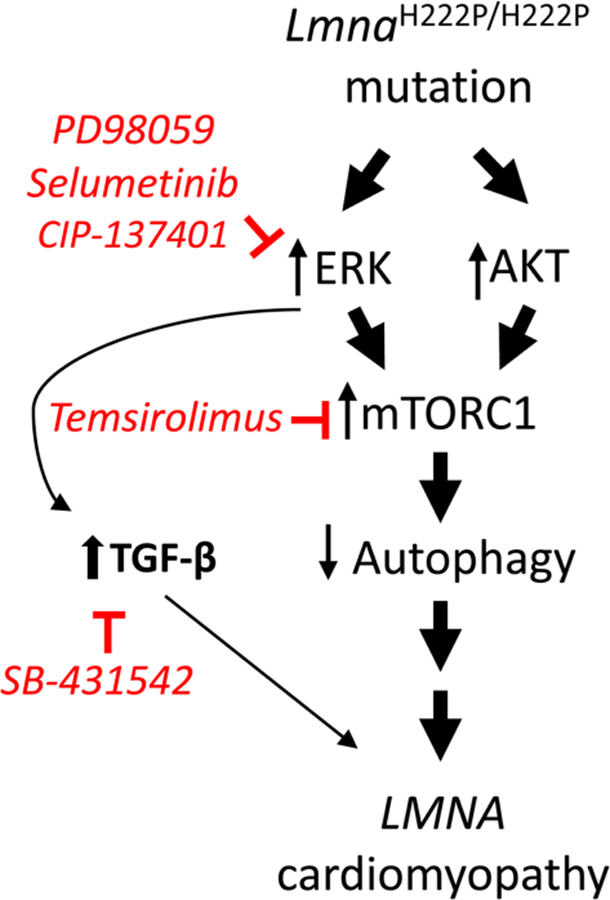
Most of the data upon which this model is based have come from studies in LmnaH222P/H222P mice, a small animal model of ‘LMNA cardiomyopathy’ in humans. Drugs shown to inhibit each signaling pathway in LmnaH222P/H222P mice are shown in red. See the text for a more detailed explanation.
Acknowledgements
The research reviewed in the present study was carried out in collaboration with many colleagues. Antoine Muchir, Jason Choi and Wei Wu played especially key roles. Takuro Arimura, Gisèle Bonne, Maria Chatzifrangkeskou, Jean-Claude Courvalin, William Dauer, Gregg Gundersen, Barry Hart, Alan Herron, Sunichi Homma, Uday Khire, Michael Lawlor, Caroline Le Dour, Feng Lin, Jonathan Lu, John Morrow, Cecilia Östlund, Paul Pavlides, Ji-Yeon Shin and Yuexia Wang also made significant contributions.
Funding
Research discussed in this review was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases under award number [R01AR048997] and the National Center for Advancing Translational Sciences under award number [R41TR001008] of the United States National Institutes of Health. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health. The Muscular Dystrophy Association also supported some of this research.
Competing Interests
H.J.W. has received research support from, is a member of the Scientific Advisory Board of and owns equity in AlloMek Therapeutics, which is developing the MEK1/2 inhibitor CIP-137401. He is also an inventor on a pending patent application related to work discussed in this manuscript.
Abbreviations
- AKT
protein kinase B
- autophagy
macroautophagy
- ERK1/2
extracellular signal-regulated kinases 1 and 2
- JNK
c-Jun N-terminal kinase
- LMNA
human lamin A/C gene
- Lmna
mouse lamin A/C gene
- MAP kinase
mitogen-activated protein kinase
- MEK1/2
mitogen-activated protein kinase kinases 1 and 2
- mTOR
mammalian target of rapamycin
- mTORC1
mammalian target of rapamycin complex 1
- TGF-β
transforming growth factor-β
References
- 1.Lin F and Worman HJ (1993) Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J. Biol. Chem 268, 16321–16326 [PubMed] [Google Scholar]
- 2.Fisher DZ, Chaudhary N and Blobel G (1986) cDNA sequencing of nuclear lamins A and C reveals primary and secondary structural homology to intermediate filament proteins. Proc. Natl Acad. Sci. U.S.A 83, 6450–6454 10.1073/pnas.83.17.6450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKeon FD, Kirschner MW and Caput D (1986) Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature 319, 463–468 10.1038/319463a0 [DOI] [PubMed] [Google Scholar]
- 4.Turgay Y, Eibauer M, Goldman AE, Shimi T, Khayat M, Ben-Harush K et al. (2017) The molecular architecture of lamins in somatic cells. Nature 543, 261–264 10.1038/nature21382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson KL and Foisner R (2010) Lamin-binding proteins. Cold Spring Harb. Perspect. Biol 2, a000554 10.1101/cshperspect.a000554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dauer WT and Worman HJ (2009) The nuclear envelope as a signaling node in development and disease. Dev. Cell 17, 626–638 10.1016/j.devcel.2009.10.016 [DOI] [PubMed] [Google Scholar]
- 7.Bonne G, Di Barletta MR, Varnous S, Bécane H-M, Hammouda E-H, Merlini L et al. (1999) Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet 21, 285–288 10.1038/6799 [DOI] [PubMed] [Google Scholar]
- 8.Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M et al. (1999) Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med 341, 1715–1724 10.1056/NEJM199912023412302 [DOI] [PubMed] [Google Scholar]
- 9.Bonne G, Mercuri E, Muchir A, Urtizberea A, Bécane HM, Recan D et al. (2000) Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol 48, 170–180 [DOI] [PubMed] [Google Scholar]
- 10.Brodsky GL, Muntoni F, Miocic S, Sinagra G, Sewry C and Mestroni L (2000) Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation 101, 473–476 10.1161/01.CIR.101.5.473 [DOI] [PubMed] [Google Scholar]
- 11.Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA et al. (2000) Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum. Mol. Genet 9, 1453–1459 10.1093/hmg/9.9.1453 [DOI] [PubMed] [Google Scholar]
- 12.Quijano-Roy S, Mbieleu B, Bönnemann CG, Jeannet P-Y, Colomer J, Clarke NF et al. (2008) De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann. Neurol 64, 177–186 10.1002/ana.21417 [DOI] [PubMed] [Google Scholar]
- 13.Prigogine C, Richard P, Van den Bergh P, Groswasser J and Deconinck N (2010) Novel LMNA mutation presenting as severe congenital muscular dystrophy. Pediatr. Neurol 43, 283–286 10.1016/j.pediatrneurol.2010.05.016 [DOI] [PubMed] [Google Scholar]
- 14.Taylor MRG, Fain PR, Sinagra G, Robinson ML, Robertson AD, Carniel E et al. (2003) Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J. Am. Coll. Cardiol 41, 771–780 10.1016/S0735-1097(02)02954-6 [DOI] [PubMed] [Google Scholar]
- 15.van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB et al. (2005) Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death? J. Mol. Med 83, 79–83 10.1007/s00109-004-0589-1 [DOI] [PubMed] [Google Scholar]
- 16.Arimura T, Helbling-Leclerc A, Massart C, Varnous S, Niel F, Lacène E et al. (2005) Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum. Mol. Genet 14, 155–169 10.1093/hmg/ddi017 [DOI] [PubMed] [Google Scholar]
- 17.Muchir A, Pavlidis P, Decostre V, Herron AJ, Arimura T, Bonne G et al. (2007) Activation of MAPK pathways links LMNA mutations to cardiomyopathy in Emery-Dreifuss muscular dystrophy. J. Clin. Invest 117, 1282–1293 10.1172/JCI29042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muchir A, Pavlidis P, Bonne G, Hayashi YK and Worman HJ (2007) Activation of MAPK in hearts of EMD null mice: similarities between mouse models of X-linked and autosomal dominant Emery–Dreifuss muscular dystrophy. Hum. Mol. Genet 16, 1884–1895 10.1093/hmg/ddm137 [DOI] [PubMed] [Google Scholar]
- 19.Muchir A, Wu W, Choi JC, Iwata S, Morrow J, Homma S et al. (2012) Abnormal p38α mitogen-activated protein kinase signaling in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet 21, 4325–4333 10.1093/hmg/dds265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi JC, Muchir A, Wu W, Iwata S, Homma S, Morrow JP et al. (2012) Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med 4, 144ra102 10.1126/scitranslmed.3003875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramos FJ, Chen SC, Garelick MG, Dai D-F, Liao C-Y, Schreiber KH et al. (2012) Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med 4, 144ra103 10.1126/scitranslmed.3003802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le Dour C, Macquart C, Sera F, Homma S, Bonne G, Morrow JP et al. (2017) Decreased WNT/β-catenin signalling contributes to the pathogenesis of dilated cardiomyopathy caused by mutations in the lamin a/C gene. Hum. Mol. Genet 26, 333–343 10.1093/hmg/ddw389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muchir A, Reilly SA, Wu W, Iwata S, Homma S, Bonne G et al. (2012) Treatment with selumetinib preserves cardiac function and improves survival in cardiomyopathy caused by mutation in the lamin A/C gene. Cardiovasc. Res 93, 311–319 10.1093/cvr/cvr301 [DOI] [PubMed] [Google Scholar]
- 24.Chatzifrangkeskou M, Le Dour C, Wu W, Morrow JP, Joseph LC, Beuvin M et al. (2016) ERK1/2 directly acts on CTGF/CCN2 expression to mediate myocardial fibrosis in cardiomyopathy caused by mutations in the lamin A/C gene. Hum. Mol. Genet 25, 2220–2233 10.1093/hmg/ddw090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muchir A, Shan J, Bonne G, Lehnart SE and Worman HJ (2009) Inhibition of extracellular signal-regulated kinase signaling to prevent cardiomyopathy caused by mutation in the gene encoding A-type lamins. Hum. Mol. Genet 18, 241–247 10.1093/hmg/ddn343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu W, Shan J, Bonne G, Worman HJ and Muchir A (2010) Pharmacological inhibition of c-Jun N-terminal kinase signaling prevents cardiomyopathy caused by mutation in LMNA gene. Biochim. Biophys. Acta, Mol. Basic Dis 1802, 632–638 10.1016/j.bbadis.2010.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu W, Muchir A, Shan J, Bonne G and Worman HJ (2011) Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin A/C gene. Circulation 123, 53–61 10.1161/CIRCULATIONAHA.110.970673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raman SV, Sparks EA, Baker PM, McCarthy B and Wooley CF (2007) Mid-myocardial fibrosis by cardiac magnetic resonance in patients with lamin A/C cardiomyopathy: possible substrate for diastolic dysfunction. J. Cardiovasc. Magn. Reson 9, 907–913 10.1080/10976640701693733 [DOI] [PubMed] [Google Scholar]
- 29.van Tintelen JP, Tio RA, Kerstjens-Frederikse WS, van Berlo JH, Boven LG, Suurmeijer AJ et al. (2007) Severe myocardial fibrosis caused by a deletion of the 5’ end of the lamin A/C gene. J. Am. Coll. Cardiol 49, 2430–2439 10.1016/j.jacc.2007.02.063 [DOI] [PubMed] [Google Scholar]
- 30.Holmström M, Kivistö S, Heliö T, Jurkko R, Kaartinen M, Antila M et al. (2011) Late gadolinium enhanced cardiovascular magnetic resonance of lamin A/C gene mutation related dilated cardiomyopathy. J. Cardiovasc. Magn. Reson 13, 30 10.1186/1532-429X-13-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fontana M, Barison A, Botto N, Panchetti L, Ricci G and Milanesi M (2013) CMR-verified interstitial myocardial fibrosis as a marker of subclinical cardiac involvement in LMNA mutation carriers. JACC Cardiovasc. Imaging 6, 124–126 10.1016/j.jcmg.2012.06.013 [DOI] [PubMed] [Google Scholar]
- 32.Wu W, Chordia MD, Hart BP, Kumarasinghe ES, Ji MK, Bhargava A et al. (2017) Macrocyclic MEK1/2 inhibitor with efficacy in a mouse model of cardiomyopathy caused by lamin A/C gene mutation. Bioorg. Med. Chem 25, 1004–1013 10.1016/j.bmc.2016.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.González JM, Navarro-Puche A, Casar B, Crespo P and Andrés V (2008) Fast regulation of AP-1 activity through interaction of lamin A/C, ERK1/ 2, and c-Fos at the nuclear envelope. J. Cell Biol 183, 653–666 10.1083/jcb.200805049 [DOI] [PMC free article] [PubMed] [Google Scholar]