

Abstract
Detailed glycan structural characterization is frequently achieved by collisionally activated dissociation (CAD) based sequential tandem mass spectrometry (MSn) analysis of permethylated glycans. However, it is challenging to implement MSn (n > 2) during online glycan separation, and this has limited its application to analysis of complex glycan mixtures from biological samples. Further, permethylation can reduce liquid chromatographic (LC) resolution of isomeric glycans. Here, we studied the electronic excitation dissociation (EED) fragmentation behavior of native glycans with a reducing-end fixed charge tag and identified key spectral features that are useful for topology and linkage determination. We also developed a de novo glycan sequencing software that showed remarkable accuracy in glycan topology elucidation based on the EED spectra of fixed charge-derivatized glycans. The ability to obtain glycan structural details at the MS2 level, without permethylation, via a combination of fixed charge derivatization, EED, and de novo spectral interpretation, makes the present approach a promising tool for comprehensive and rapid characterization of glycan mixtures.
The recent boom in -omics is largely catalyzed by the application of tandem mass spectrometry (MS/MS) methods to biopolymer sequencing.1,2 However, compared to the rapid growth of proteomics, progress in glycomics has been modest. This is, in part. due to the structural complexity of glycans and thus the necessity to determine their branching patterns, linkages, and stereochemical configurations to fully define their structures. The simultaneous presence of many isomeric glycans in biological samples adds another layer of challenge to structural glycomics, demanding analytical tools that can provide structural details and work well in tandem with various glycan separation methods, such as liquid chromatography (LC), capillary electrophoresis, and ion mobility spectrometry (IMS), for analysis of complex glycan mixtures.
To date, detailed glycan structural characterization is typically achieved by sequential tandem MS (MSn) analysis,3,4 as the conventional collisionally activated dissociation (CAD) method often fails to generate sufficient structural details in a single stage of MS/MS analysis. In MSn, a glycan structure can be identified when its gas-phase disassembly pathways are consistently observed. The presence of structural isomers is indicated by the observation of anomalous fragment ions; such ions can be further isolated and fragmented to deduce their structures. Comprehensive characterization of a glycome thus requires inspection of many fragmentation pathways for any given precursor ion mass and judicious choice of fragment ions at each stage for further fragmentation. The inherently lower throughput of the MSn approach and difficulty in its automation have hampered its effective implementation with online glycan separation. Meanwhile, radical-induced fragmentation methods, such as vacuum and extreme ultraviolet photodissociation,5–7 charge transfer dissociation,8 free radical-activated glycan sequencing (FRAGS),9,10 and a variety of electron-activated dissociation (ExD) techniques,11–17 can generate substantially more structural information than low-energy CAD, permitting topology deduction, and sometimes determination of linkage and stereochemical configurations, at the MS2 level. Several recent studies showed that the integration of radical-induced dissociation with LC or IMS separation can be a powerful approach for characterization of glycan mixtures, including those containing structural isomers.18–22
De novo sequencing of native glycans by tandem mass spectrometry is often complicated by gas-phase structural rearrangements23‘24 or the formation of fragments via loss of residues from more than one position (hereafter referred to as internal fragments). For native glycans, an internal fragment has the same mass as a terminal fragment with the same saccharide composition, and may be misinterpreted as such, leading to inaccurate structural determination. Permethylation is a common strategy that removes such ambiguity, since the terminal and internal fragments of permethylated glycans can be differentiated based on the number of free hydroxyl groups that represent scars left behind by glycosidic cleavages. Although permethylation offers several advantages for glycan analysis, it is sometimes desirable to characterize glycans without blocking all their free hydroxyl and amino groups, as not all glycans can be permethylated and some glycans contain naturally methylated residues. Moreover, LC separation of isomeric glycans can often be accomplished more easily when these polar groups are still present.
A recent report by Gao and Beauchamp presented a clever way to minimize interference from internal fragments in native glycans by conjugating to the reducing end a methylated (Me)-FRAGS reagent, a radical precursor with a quaternary amine fixed charge.10 Charge sequestration led to suppression of charge-induced dissociation during CAD of Me-FRAGS-labeled glycans, leaving radical-driven processes as the predominant fragmentation pathways. It also resulted in simplified tandem mass spectra by promoting detection of reducing-end fragments, while suppressing detection of internal fragments that did not contain the fixed charge tag. In CAD of glycans with a Me-FRAGS label, the only internal fragments observed were Z/Z-ions that still retain the fixed charge, formed at branching sites via radical-induced, sequential losses of two nonreducing-end branches. Finally, methylation at the pyridinyl nitrogen prevented protonation at this site and eradicated proton-mediated saccharide rearrangement that is detrimental to accurate glycan sequencing.
Despite its utility in glycan topology deduction, CAD of Me-FRAGS-labeled glycans does not produce many cross-ring fragments except for the linkage-independent 1,5X ions, and thus offers little value for linkage position determination. Meanwhile, these glycans with a single reducing-end fixed charge cannot be analyzed by either electron capture dissociation or electron transfer dissociation, since these methods would generate neutral, undetectable products. We have recently shown that singly charged and permethylated glycans can be effectively characterized by electronic excitation dissociation (EED).19,25 Here, we investigate the EED fragmentation behavior of unmethylated glycans with a fixed charge modification, and explore the potential of this approach for detailed glycan structural characterization.
EXPERIMENTAL SECTION
Materials and Sample Preparation.
Lacto-N-fucopen-taose I, II, and III (LNFP I, II, and III) were acquired from V-Laboratories, Inc. (Covington, LA). LNFP V, VI, laminarihexaose, maltohexaose, and isomaltohexaose were purchased from Carbosynth Limited (Berkshire, UK). HPLC grade water and acetonitrile were obtained from Fisher Scientific (Pittsburgh, PA). Iodomethane and acetic acid were purchased from Sigma-Aldrich (St. Louis, MO). The proton reagent for acid-catalyzed glycan sequencing (PRAGS, structure shown in Supporting Scheme S1) was synthesized at Dr. Gao’s laboratory, according to the procedure described in a previous report.9
For PRAGS labeling, 1 μg of glycan was dissolved in 10 μL of water containing 1% of acetic acid, followed by addition of 3 μL of 29.5 mM PRAGS solution in acetonitrile and incubation at 60 °C for 5 h. Solvent was removed by a SpeedVac concentrator (ThermoFisher Scientific) after reaction. Methylation of the PRAGS-labeled glycans was achieved by reaction with iodomethane in acetonitrile. To be consistent with the literature, the resultant tag with a fixed charge will be referred to as the methylated PRAGS, or Me-PRAGS (which is a misnomer, because this derivative does not require protonation). Me-PRAGS-labeled glycans were purified by size exclusion chromatography using PD MiniTrap G-10 columns (GE Healthcare, Buckinghamshire, UK).
Mass Spectrometry Analysis.
Derivatized glycans were dissolved in 50/50 (v/v) methanol/water solution to a concentration of 5 μM, and directly infused into a 12-T solariX hybrid Qh-Fourier transform ion cyclotron resonance (FTICR) mass spectrometer (Bruker Daltonics, Bremen, Germany) via a pulled glass capillary tip with ~1 μm orifice diameter. EED analyses were carried out with the cathode bias set between 12 and 20 V, and an electron irradiation time of half a second or less. All tandem mass spectra were internally calibrated with at least six fragment ions assigned with high confidence, including Y, Z, and 1,5X ions, resulting in a typical mass accuracy of 1 ppm or better for the majority of the assigned peaks. Peak picking was achieved by using the SNAP algorithm (Bruker Daltonics, Bremen, Germany),26 with the quality factor threshold set at 0.1.
RESULTS AND DISCUSSION
Two sets of isomeric oligosaccharide standards were used in this study, with their structures (in CFG graphic representation with linkage notation27) shown in Figure 1. The first set includes three linear hexasaccharide linkage isomers that consist of only glucose (Glc) residues: β1 → 3-linked laminarihexaose, α1 → 4-linked maltohexaose, and α1 → 6-linked isomaltohexaose. The second set includes five pentasaccharide isomers with either linear or branched structures. Among them, LNFP I, II, and V are topological isomers with their structures derived from lacto-N-tetraose (Galβ1 → 3GlcNAcβ1 → 3Galβ1 → 4Glc) via the addition of a fucose (Fuc) residue to the nonreducing-end galactose (Gal), N-acetylglucosamine (GlcNAc), or Glc residue, respectively. LNFP III and VI are related to lacto-N-neotetraose (Galβ → 4GlcNAcβ1 → 3Galβ1 → 4Glc), and they have the same topologies as LNFP II and V, respectively, but with different linkage configurations at the GlcNAc residue. All glycans were derivatized with the Me-PRAGS label at the reducing end. Unlike the FRAGS reagent, the PRAGS label does not contain a radical initiator, nor is it needed here because EED itself is a radical-generating process.
Figure 1.
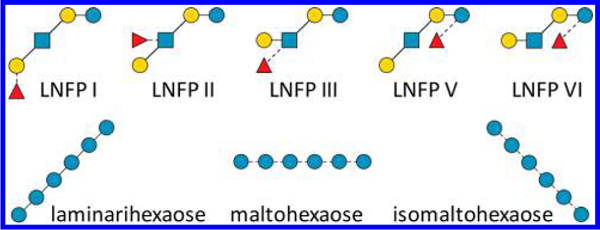
Glycan standards used in this study.
EED Tandem MS Analysis of Linear Hexasaccharides.
Figure 2 shows the EED (16 eV) spectra of Me-PRAGS-labeled hexaose isomers. As expected, EED of these glycans with a fixed charge on the reducing end produces predominantly reducing-end fragments, in particular the Z-, Y-, and 1,5X-ion series. The only nonreducing-end fragments observed are B-series that carry their own charge as oxonium ions. While 1,5X ions provide no linkage information, they do have value in de novo glycan sequencing. For example, a pair of Z and Y ions may be misinterpreted as a pair of B and C ions of the same saccharide composition, whereas a triplet of Z, Y, and 1,5X ions with the mass differences of 18.011 Da (H2O) and 27.995 Da (CO) between adjacent peaks can be easily differentiated from a triplet of 1,5A, B, and C ions with their spacing in reversed order. Complete series of Z, Y, and 1,5X triplets are present in EED spectra of all three linear hexaose isomers, allowing correct deduction of the glycan sequences.
Figure 2.
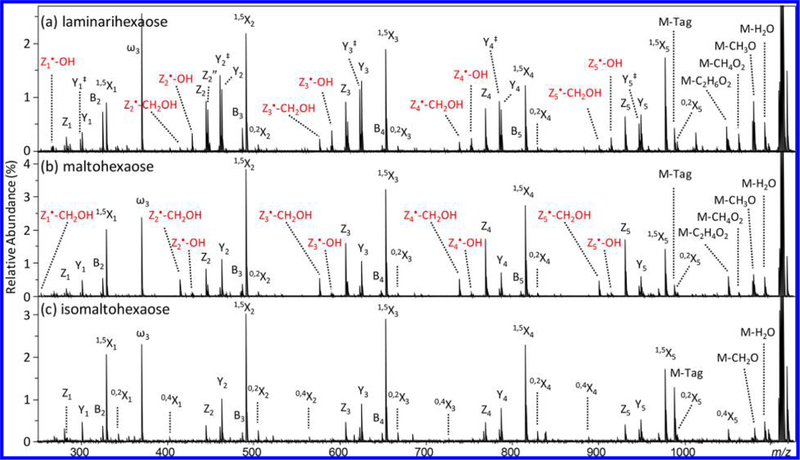
EED (16 eV) MS/MS spectra of Me-PRAGS-labeled (a) laminarihexaose, (b) maltohexaose, and (c) isomaltohexaose. Secondary fragments useful for linkage analysis are labeled in red.
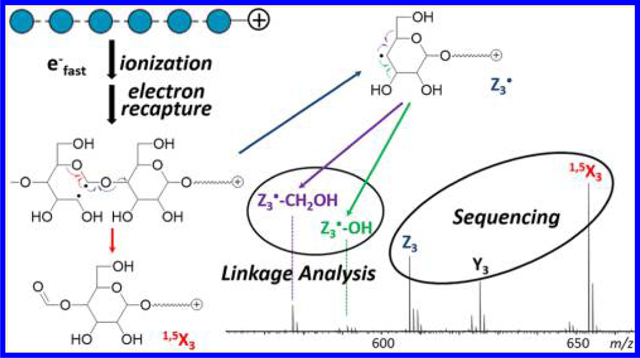
A recent study revealed that EED of metal-adducted glycans is initiated by ionization and electron recapture, with its fragmentation pattern influenced by the energetics of distonic ion intermediates and the stability of product ions.25 As the charge carrier does not directly participate in the EED process, EED of fixed-charge-derivatized glycans likely proceeds via a similar mechanism. Scheme 1 shows the proposed EED fragmentation pathways of Me-PRAGS-labeled maltohexaose for the formation of Y•, Z•, and 1,5X ions from various diradicals formed upon electron recapture. It was previously shown that ring opening by the C1–C2 bond cleavage is favored for sodium-adducted cellobiose due to the resonance stabilization of the C1 radical or cation by both O1 and O5. A C1/C2 diradical can undergo direct β-elimination to form a 1,5X ion (Scheme 1a) or a Z• ion (Scheme 1b). Because 1,5X ions are closed-shell species derived from the lowest-energy distonic ion, they are often the most abundant fragments in the glycan EED spectra, as also seen here. A Z• fragment may either lose a β-hydrogen or abstract a hydrogen from its complementary C fragment to form an even-electron Z or Z + 2H (hereafter denoted as Z”) ion, respectively. Since the radical on a Z• fragment is formed after the glycosidic bond cleavage, the extent of hydrogen abstraction by a Z• ion is influenced by the lifetime of the C + Z fragment pair. Glycosidic cleavage in the middle of the sequence is more likely to produce a long-lived complex, as both fragments contain an adequate number of polar groups with sufficient conformational flexibility to foster strong noncovalent interactions. The strength of interaction is slightly increased for a smaller Z• ion due to the presence of a fixed charge on its reducing end. Thus, the relative abundance of Z” ions is the highest in small- to moderate-sized Z ions, Z1, Z2, and Z3, but greatly diminishes in larger Z ions (Figure S1). Hydrogen transfer between complementary fragments before product separation is a well-known phenomenon in ECD of peptides, leading to the formation of z- and c•-type ions.28,29 For peptides, it has been determined that the relative abundance of c• and z ions is related to the lifetime of post-ECD complex, which can be measured by a double resonance (DR)-ECD experiment.30 In DR-ECD of peptides, the charge-reduced species, including c+z ion pairs, are resonantly ejected from the ICR cell during ECD, thus depleting the abundance of ions deriving from long-lived complexes. It is, however, not possible to measure the lifetime of the post-EED C+Z pair by DR, because EED results in no charge reduction, and a C + Z pair has the same m/z value as the precursor ion itself. Nonetheless, the hypothesis that formation of Z” ions results from post-EED, intracomplex hydrogen transfer is supported by the observation that the relative abundance of Z” ions drops as the electron energy is increased, presumably due to the disruption of noncovalent interactions at higher energies. Depletion of hydrogen transfer products was previously reported in AI-ECD of peptide ions,31 and used for differentiation of N- and C-terminal fragments.32
Scheme 1.
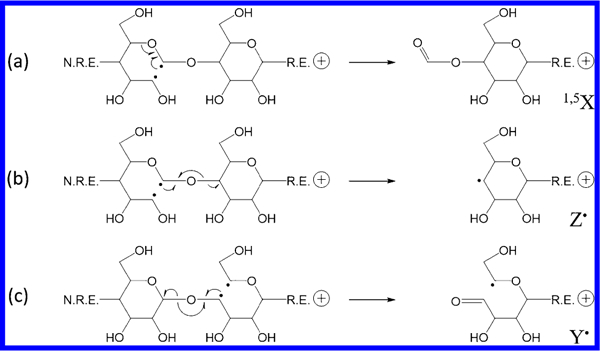
Proposed EED Mechanisms for the Formation of 1,5X, Z•, and Y• Ions
Formation of Y• ions likely originates from different open-ring diradicals, such as the C4/C5 diradical shown in Scheme 1c. A Y• fragment can also lose or gain a hydrogen to form an even-electron Y-2H (hereafter denoted as Y‡) or Y ion, respectively. Unlike in the case of Z• ions, hydrogen transfer to a Y• fragment can take place either before or after glycosidic bond cleavage as the radicals initially reside on the reducing-end residue in the B/Y fragmentation pathway. The relative abundance of Y ions to Y‡ ions appears to be significantly higher than that of Z” to Z ions. A Y ion may also be produced by vibrational excitation in closed-shell species, formed via electron recapture by a closed-ring radical cation, or through radical recombination in a singlet diradical. These alternative pathways do not involve hydrogen transfer, thus the relative abundance of Y to Y‡ ions does not follow the same trend as that of Z” to Z ions. There is no clear dependence on the size of the fragment, and the relative abundance of Y ions increases as the electron energy is increased (Figure S1), presumably because the higher energy input promotes fragmentation via vibrational excitation.
In addition to the 1,5X, Y, and Z ion series, several linkage-dependent cross-ring fragments are also observed. The presence of 0,2X ions in all three spectra can be used to rule out 1 → 2 linkages, whereas the presence of 0,4X ion series in the isomaltohexaose spectrum can be used to define its 1 → 6 linkages. Notably, the 3,5-cross-ring fragments that are critical for differentiating 1 → 4 linkages from 1 → 3 linkages are absent in the maltohexaose spectrum. This is in contrast to the previously reported EED spectrum of Na+-adducted, β1 → 4-linked cellobiose, where 3,5A2 ion was the most abundant fragment observed.25 For Me-PRAGS-labeled maltohexaose, 3,5A fragments are not detected because of charge sequestration at the reducing-end, whereas the complementary 3,5X ions are diradicals that easily undergo β-elimination to form stable 1,5X ions. Meanwhile, several secondary fragment ions generated by EED, for example, Z•-CH2OH and Z•-OH, may be useful for linkage analysis. These ions likely derive from Z• radical ions via β-elimination of the substituent at an adjacent carbon (Scheme 2). For the 1 → 4-linked maltohexaose, a Z• ion can lose either its C5 substituent to form a Z•-CH2OH ion or its C3 substituent to form a Z•-OH ion (Scheme 2a). Loss of CH2OH is energetically favored, because the C–C bond dissociation energy is generally lower than the C–O bond dissociation energy,33 and the Z•-CH2OH product is further stabilized by electron sharing between the newly formed double bond and the O5 atom. Consequently, Z•-CH2OH ions are more abundant than Z•-OH ions in the maltohexaose spectrum. For the 1 → 3-linked laminarihexaose, C/Z cleavage leaves the radical on C3, which can only form a Z•-OH ion via direct β-elimination (Scheme 2b), but may also lose its C5 substituent to form a Z•-CH2OH ion following radical migration to C4. Because 1,2-hydrogen migration is associated with a substantial barrier (>40 kcal/mol)34 whereas direct β-elimination is a barrierless process, formation of Z•-OH ions is kinetically favored. Both Z•-CH2OH and Z•-OH ions are observed in the laminarihexaose spectrum, with the latter series having the higher abundance, in contrast to the trend seen in the maltohexaose spectrum. For the 1 → 6 linked isomaltohexaose, consecutive α-cleavages from the C6 Z• radical eventually leads to formation of 1,5X ions (Scheme 2c). The EED spectrum of isomaltohexaose is thus characterized by higher-abundance 1,5X ions, lower-abundance Z ions, and no Z•-CH2OH or Z•-OH ions. These results show that the propensity to form Z•-CH2OH and Z•-OH ions can facilitate determination of glycan linkages.
Scheme 2.
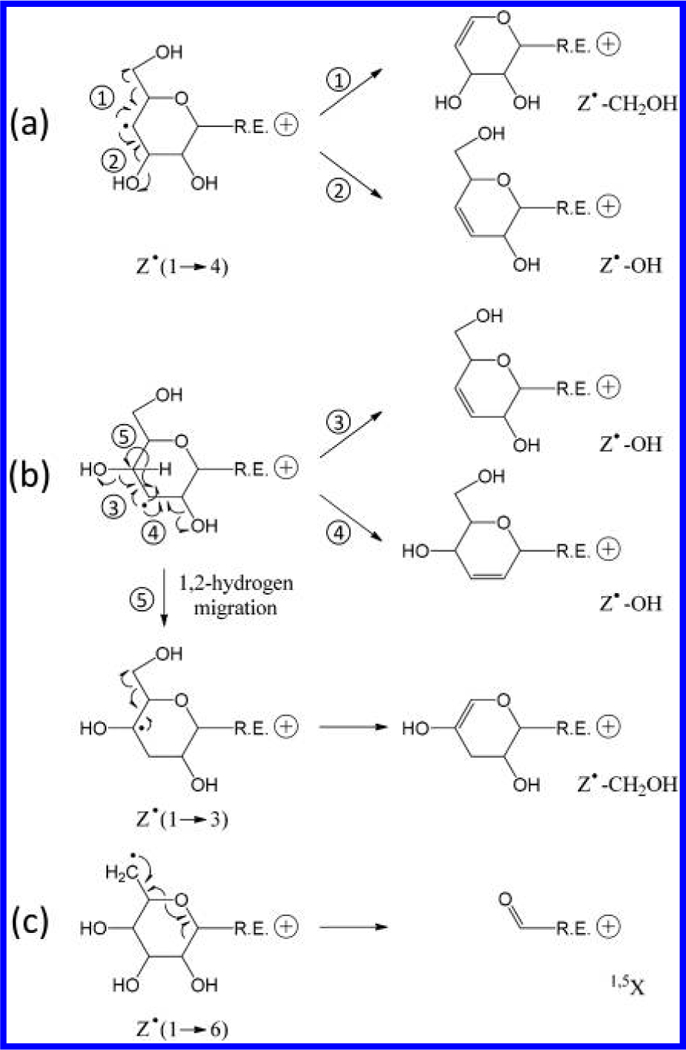
Proposed EED Mechanism for Formation of Secondary Fragments from Z• Ions at (a) 1 → 4, (b) 1 → 3, and (c) 1 → 6-Linked Sites
EED of LNFP Isomers at Lower Electron Energies (12 eV).
Figure 3 shows the EED (12 eV) spectra of Me-PRAGS-labeled LNFP isomers, where the majority of fragment ions are reducing-end fragments, including complete Y, Z, and 1,5X-ion series. Again, hydrogen transfer products are observed prominently in moderate-sized Z-type fragments: Z2” in LNFP I, II, and III spectra, and Z2α” in LNFP V and VI spectra, but exhibit negligible abundances for small or large Z ions. The only major nonreducing-end fragments are B-ions resulting from cleavage between the 1 → 3-linked GlcNAc and Gal residues: B3 for LNFP I, B2 for LNFP II and III, and B2α for LNFP V and VI. These B-ions can further lose a fucose, a 1 → 3-linked galactose, or, to a lesser degree, a 1 → 4-linked galactose residue to generate B/Y-type internal fragment ions: B3/Y4 in LNFP I, and B2/Y3α and B2/Y3β ions in LNFP II and III. Because these ions do not carry the reducing-end tag, they can be easily differentiated from Y- and Z-type ions by their nominal and accurate mass values. Therefore, the presence of B- and B/Y-type ions does not result in ambiguities in de novo glycan sequencing when only reducing-end fragments are utilized for topology deduction.
Figure 3.
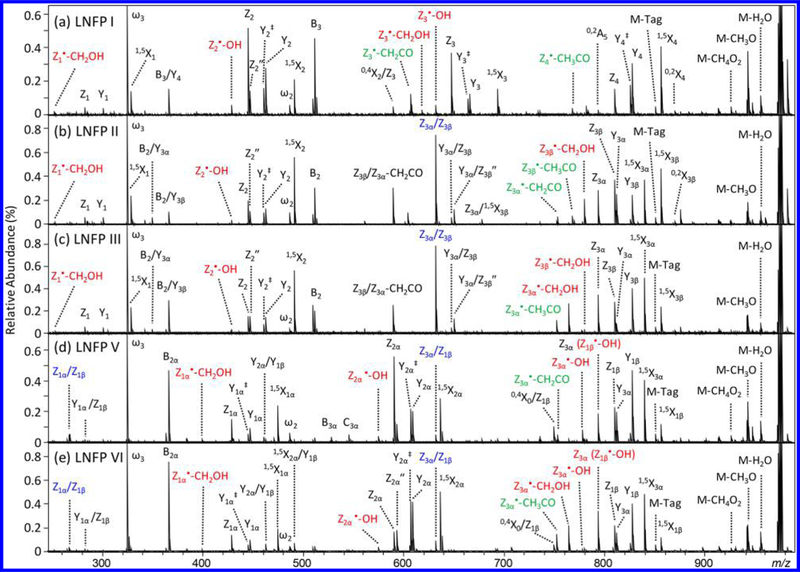
EED (12 eV) MS/MS spectra of Me-PRAGS-labeled (a) LNFP I, (b) LNFP II, (c) LNFP III, (d) LNFP V, and (e) LNFP VI. Secondary fragments useful for linkage analysis at hexose and GlcNAc sites are labeled in red and green, respectively. Internal ions are labeled in blue.
For branched structures, fragments corresponding to cleavages on more than one nonreducing-end branch may also be produced. These are primarily Z/Z-type fragments at the branching point, resulting from the elimination of an entire nonreducing-end branch at the β-position of a Z• radical (Supporting Scheme S2a): Z3α/Z3β for LNFP II and III, and Zlα/Z1β for LNFP V and VI, similar to what was previously reported in CAD of Me-FRAGS-labeled glycans. Here, loss of multiple residues is also observed at sites distant from each other, generating ions such as Z3α/Z1β for LNFP V and VI. Loss of distant residues likely occurs as a result of radical migration (Scheme S2b) and not via two consecutive EED processes (Scheme S2c). The latter would have produced fragment ions that are 2 Da lighter than the ones observed in the spectra. The presence of Z/Z-type ions can potentially lead to ambiguous interpretation, as they too, carry the reducing-end tag, and cannot be easily differentiated from reducing-end glycosidic fragments based on the accurate mass measurement alone. We recently demonstrated that fragment ion assignment can be assisted by examining its context, defined as a collection of neighboring peaks within a predetermined mass window. Classifying ions by their contextual features was shown to be an effective way to reduce ambiguity in topology reconstruction from EED spectra of permethylated glycans.35 Here, in EED of Me-PRAGS-labeled glycans, a true Z ion is almost always accompanied by its corresponding Y and 1,5X ions, showing up as a high-abundance triplet with well-characterized mass shifts. In contrast, Z/Z ions are generally not observed as part of such a triplet. The only exception is the Z3α/Z3β ion of LNFP II, occurring at the branching site, yet the Z/Z, Z/Y, and Z/1,5X-triplet has a very different abundance distribution than that of a typical Z, Y, 1,5X-triplet, allowing easy differentiation by a properly trained IonClassifier.
Accurate glycan topology analysis requires not only the presence of informative glycosidic fragments and the ability to differentiate terminal and internal fragments, but also elimination of undesirable gas-phase structural rearrangements, most commonly observed as fucose migration during CAD of native, reductively aminated, or even permethylated glycans, particularly for protonated precursors.23,24,36,37 Fucose migration can result in the formation of Z ions that have either lost or gained a fucose residue. The presence of the former, however, is not indicative of the occurrence of fucose migration, as it can also result from loss of multiple terminal residues as described above. For example, the ion at m/z 648.262, observed in the EED spectra of Me-PRAGS-labeled LNFP II and III, can be assigned as a Y3α/Z3β (or Y3β/Z3α) ion. Unlike the high-abundance Z3 ion of the same composition from LNFP I, Y/Z ions are not accompanied by their corresponding Y/Y and Y/1,5X ions, and can be easily ruled out as a simple Z ion during topology analysis. In these mass spectra, there are no Z ions with an unexpected addition of a fucose residue that would implicate fucose migration. For example, the Z(Hex2Fuc) ion (m/z 591.240) only exists in the EED spectra of LNFP V and VI (Z2α), but not in the EED spectra of LNFP I, II, and III. The fragment ion at m/z 590.255 in the LNFP II and III spectra is not related to Z(Hex2Fuc), and is tentatively assigned as Z3β/Z3α-CH2CO. Similarly, the Z(HexNAcHex2Fuc) ion (m/z 794.319) only exists in the EED spectra of LNFP II, III, V, and VI (Z3α), but not in the LNFP I spectrum. The low-abundance fragment ion at m/z 794.292 in the LNFP I spectrum is assigned as 0,2A5 based on its accurate mass.
At 12 eV, EED of LNFP isomers generates only a few low-abundance 0,2X ions. In contrast, linkage-informative secondary fragments, Z•-CH2OH and Z•–OH ions, are produced in much higher proportion, with their relative abundances following the same trend as that observed in the EED spectra of linear hexaose isomers. At 1 → 4-linked sites, Z•-CH2OH ions are more abundantly formed than Z•-OH ions, for example, Z1•-CH2OH for LNFP I, II, and III, Z1α•-CH2OH for LNFP V and VI, Z3β•-CH2OH for LNFP II, and Z3α•-CH2OH for LNFP III and VI. On the other hand, secondary fragmentation at 1 → 3-linked sites produces Z•-OH ions in higher abundance than Z•-CH2OH ions, for example, Z2•–OH for LNFP I, II, and III, Z2α•-OH for LNFP V and VI, Z3•-OH for LNFP I, and Z3α•-OH for LNFP V. Because the mass difference between a Z•-OH ion and a Z (i.e., Z•-H) ion is the same as that between a Fuc and a Gal residue, in branched structures containing both terminal Fuc and Gal residues, it is not possible to determine the abundance of a Z•-OH ion following the loss of a Fuc residue. Specifically, Z3β•-OH is isomeric to Z3α in LNFP II and III and Z1β•-OH is isomeric to Z3α in LNFP V and VI. In cases like this, it is important to recognize potential ambiguity in peak assignment, and look for other evidence that may assist in linkage analysis. For example, in LNFP III, the presence of a high-abundance Z3α•-CH2OH ion and the absence of a Z3α•-OH ion would place Gal at the C4 position of the GlcNAc residue, thus leaving only the C3 position as the possible linkage site for Fuc, since the C2 position of a GlcNAc residue is already occupied by an acetylamino group, and C6 substitution would have eliminated the Z•-CH2OH pathway altogether which contradicts with the observation of an abundant Z3α•-CH2OH ion.
In contrast to the linear hexaose isomers consisting solely of hexose residues, LNFP glycans also contain GlcNAc residues that can give rise to additional fragmentation pathways. Specifically, CH2CO loss is often observed at 1 → 3-linked GlcNAc sites following the C/Z cleavage, for example, Z3•-CH2CO in LNFP I and Z3α•-CH2CO in LNFP II and V, whereas CH3CO loss from a Z• ion is observed at all 1 → 4 linked GlcNAc sites, for example, Z3β•-CH3CO in LNFP II and Z3α•-CH3CO in LNFP III and VI. Thus, although differing in mass by only that of a single hydrogen atom, these secondary fragmentation pathways can provide additional information that facilitates linkage analysis at GlcNAc sites. Scheme S3 shows the proposed mechanisms for these GlcNAc-specific fragmentation pathways.
EED of LNFP Isomers at Higher Electron Energies (16 eV).
Figure S2 shows the EED spectra of Me-PRAGS-labeled LNFP isomers, acquired with the cathode bias set at 16 V. Irradiation with higher-energy electrons leads to more efficient EED, as evidenced by the increased abundance and improved signal-to-noise ratio of most fragment ions. It also opens up new fragmentation channels. Some of the fragments that are only observed in higher-energy EED spectra are highlighted. In particular, EED at 16 eV leads to formation of doubly charged fragment ions. Generation of fragment ions in charge states one higher than that of the precursor ion was previously reported in electron ionization dissociation (EID) of peptide cations.38 Since ionization itself does not cause dissociation of the resultant radical, it was suggested that EID proceeds via tandem ionization followed by electron capture, forming an electronically excited, charge-increased radical cation that subsequently dissociates. The EID process may also have contributed to the formation of doubly charged fragments here. The presence of GlcNAc in LNFP glycans appears to be important for the EID process, as no doubly charged fragment ions are observed in the 20 eV EED spectra of Me-PRAGS-labeled hexaoses.
Importantly, formation of cross-ring fragments is significantly boosted at higher electron energies. For example, all four 0,2X ions are observed in the 16 eV EED spectrum of LNFP I, whereas only one such ion, 0,2X4, is identified in its 12 eV EED spectrum. Linkage-informative secondary fragments are also more abundantly formed, especially for those derived from smaller Z• ions. For example, Z1•-CH2OH (m/z 253.118) is observed just above the noise level in the 12 eV EED spectra of LNFP I, II, and III, but easily identified in their respective 16 eV EED spectra. Similarly, Z2•-OH (m/z 429.187) of LNFP I, II, and III, and Z2α•-OH (m/z 575.245) of LNFP V and VI, are on average four times more abundant in the 16 eV EED spectra than in the 12 eV spectra. Thus, linkage analysis may be more easily achieved with higher-energy EED spectral data.
On the other hand, enhanced secondary fragmentation at higher electron energies could potentially result in ambiguity in topology determination. For example, EED of LNFP I at 16 eV produces a fragment ion at m/z 591.239 that is not identified in its 12 eV spectrum (Figure S3a, b). This ion is likely formed via loss of the C2 substituent (CH3CONH) from the Z3• ion (theoretical m/z 591.2396, Scheme S4), and can be easily resolved from the A+1 isotope peak of 0,4X2/Z3 (m/z 591.259). However, Z3•-CH3CONH has the same m/z value as that of Z(Hex2Fuc), and may be mistaken as a supporting ion to infer the presence of LNFP V (or VI). Notably, the true Z2α ion of LNFP V is nearly 20 times more abundant than Z3•-CH3CONH of LNFP I, and is accompanied by its corresponding Y2α (m/z 609.250) and 1,5X2α (m/z 637.245) ions (Figure S3c), which are not present in the EED spectra of LNFP I. Thus, erroneous interpretation of secondary fragments as glycosidic fragments is not expected to affect the accuracy of topology reconstruction, so long as one takes into account the abundance and context of supporting peaks when evaluating candidate topologies.
At 16 eV, EED also produces more internal fragments. The presence of internal fragments with the reducing-end label could potentially complicate topology analysis. However, triplets of internal fragments are rarely observed, and such fragments typically have an order of magnitude lower abundance when compared to simple Z, Y, and 1,5X ions, even in higher energy EED spectra. Further, these internal fragments, except for the Z/Z fragments at the branching sites, are either absent or have very low abundances in the lower-energy EED spectra. Thus, it may be advantageous to perform topology analysis based on lower-energy EED spectra, and use higher-energy EED spectral data for linkage determination.
De Novo Topology Elucidation from EED of Native Glycans with a Fixed Charge Tag.
The EED spectra of glycans are significantly more complex than their CAD spectra, and difficult to interpret manually, especially for unknown structures. We have recently developed a de novo glycan sequencing software, named GlycoDeNovo, which can efficiently deduce and accurately rank the glycan topologies from their tandem mass spectra.35 GlycoDeNovo identifies potential B- and C-type glycosidic fragments sequentially, by attempting to interpret a heavier fragment as a combination of a monosaccharide (root) and one or more previously identified, lighter nonreducing-end glycosidic fragments (branches), eventually leading to the interpretation of the precursor ion. The candidate topologies can then be ranked either by the number of supporting peaks (SPN) or, more accurately, by the cumulative IonClassifier scores of supporting peaks. IonClassifier is a measure of confidence in peak assignment, and obtained via machine learning from tandem MS data of glycan standards.
GlycoDeNovo was initially written for analysis of tandem mass spectra of permethylated glycans. Here, the algorithm has been modified to accommodate the mass difference between unmethylated and permethylated glycans. In addition, because EED of Me-PRAGS-labeled glycans produces predominantly reducing-end fragments while GlycoDeNovo builds candidate topologies from the nonreducing end, complementary peaks are artificially added to the peak list before analysis. IonClassifier was retrained with the EED tandem mass spectra of Me-PRAGS-labeled glycans, as they produce spectral features different from those of permethylated glycans. IonClassifier training involves boosting39 the decision tree classifier40 using the experimental tandem mass spectra of known glycan standards. Each decision tree utilizes one or several contextual features of a peak to decide probabilistically if the peak is a B/C ion. The features include both the mass shifts of the neighboring peaks with respect to the peak of interest, and the abundance of those neighboring peaks. The final score is the weighted sum of the output from all decision trees. The weight of each tree is automatically learned by the boosting procedure from the training data. The number of trees is capped at 100 in the present implementation.
Table 1 shows the topology analysis result by GlycoDeNovo on the 16 eV EED spectra of Me-PRAGS-labeled glycans. The challenge of de novo glycan sequencing is evidenced by the significantly higher number of peaks interpretable as non-reducing-end glycosidic fragments than could be generated by the actual structures. For example, the pentasaccharide LNFP VI can produce a maximum of four B ions and four C ions, but 40 peaks are interpretable as B or C ions. Fortunately, most of these peaks do not lead to eventual interpretation of the precursor ion, and coincidental matches can be further identified by IonClassifier. As glycans can assume branched structures, even a small number of interpretable peaks can lead to prediction of many candidate topologies. For the three linear hexasaccharides consisting of only hexose residues, 11 candidate topologies (shown in Figure S4) are deduced when the search is limited to bifurcated structures. GlycoDeNovo correctly ranks the linear topology as the top candidate based on the number of its supporting peaks, including glycosidic fragments with one to five hexose residues. In contrast, only a subset of these fragments may be used to support other candidate topologies.
Table 1.
Topology Analysis by GlycoDeNovo on EED (16 eV) Spectra of Me-PRAGS-Labeled Glycansa
| glycan | no. Peaks | no. interpretable | no. candidates | rank by SPN | rank by IC | IC score no. 1 | IC score no. 2 |
| LNFP I | 186 (77) | 30 | 16 | 1 (1) | 1 (0) | 273.56 | 240.73 |
| LNFP II | 203 (81) | 35 | 23 | 1(11) | 1 (0) | 303.01 | 244.55 |
| LNFP III | 157 (67) | 26 | 16 | 1 (4) | 1 (0) | 313.55 | 262.32 |
| LNFP V | 216 (85) | 35 | 16 | 1 (9) | 1 (0) | 213.68 | 156.47 |
| LNFP VI | 199 (81) | 40 | 16 | 1 (4) | 1 (0) | 189.68 | 93.89 |
| LamHex | 227 (98) | 13 | 11 | 1 (0) | 1 (0) | 337.98 | 318.02 |
| MalHex | 251 (105) | 71 | 11 | 1 (0) | 1 (0) | 328.99 | 274.13 |
| IsomalHex | 152 (67) | 10 | 11 | 1 (0) | 1 (0) | 264.93 | 241.41 |
The “no. peaks” column lists the peak number in each enriched spectrum with the number of complementary peaks inside parentheses. The “no. interpretable” column lists the number of peaks that are interpreted as non-reducing-end glycosidic fragments. The “no. candidates” column lists the number of reconstructed topology candidates. The “rank by SPN” and “rank by IC” columns list the ranks of the true topologies among all candidates by their supporting peak counts and by IonClassifier, respectively. Numbers in parentheses indicate the number of candidate structures that are ranked as high as the true topology. The last two columns list the IonClassifier scores of the top two-ranked candidate topologies.
For branched structures, LNFP II, III, V, and VI, however, ranking by SPN alone is often insufficient for identifying the correct topology among several co-ranked candidate structures. This is perhaps not surprising, as the analysis was performed on spectral data generated by higher-energy EED, which promotes formation of secondary and internal fragments. In particular, Z/Z-, Y/Z-, and Y/Y-type ions, as well as secondary ions that still contain the reducing end, can be easily misinterpreted as Y- or Z-type ions, even with the reducing-end tagging. Nevertheless, these ions lack the features displayed in actual sequence ions, including, but not limited to the prominent 1,5X, Y, and Z triplets, and are therefore assigned with much lower IonClassifier scores. When ranked by IonClassifier, the true topology is always identified as the top candidate by itself in each case.
Accurate de novo glycan sequencing is no simple task, especially without permethylation. By combining the power of reducing-end fixed charge tagging, EED, and a well-designed glycan sequencing algorithm, this study represents a significant step toward that goal.
CONCLUSIONS
In this study, we examined the EED fragmentation behavior of fixed-charge-labeled, otherwise unmodified glycans by employing two sets of isomeric glycans representing both linear and branched structures with a variety of linkage configurations. EED spectra of these glycans are characterized by complete, prominent 1,5X, Y, and Z ion series, as well as many linkage-informative cross-ring and secondary fragments. Although the EED efficiency may be improved by raising the electron energy, higher-energy electron irradiation leads to formation of more secondary and internal fragment ions. Nonetheless, these ions can be easily recognized as nonsequence ions by an IonClassifier, and their presence does not negatively affect the accuracy of topology elucidation by GlycoDeNovo. Our results showed that accurate, automated glycan structural determination can be achieved based on EED tandem MS analysis of unmethylated glycans with a reducing-end fixed charge tag, thus paving the way for LC-MS/MS-based, high-throughput, de novo glycan sequencing.
Supplementary Material
ACKNOWLEDGMENTS
This research is supported by the NIH grants P41 GM104603, R21 GM122635, and S10 RR025082. The contents are solely the responsibility of the authors and do not represent the official views of the awarding offices.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.7b04077.
PRAGS structure, proposed EED mechanism for various fragmentation pathways, extent of hydrogen transfer between glycosidic fragments as a function of the fragment size and electron energy, 16 eV EED spectra of LNFP isomers, secondary fragmentation at a GlcNAc residue, candidate topologies reconstructed by GlycoDeNovo for linear hexasaccharides, and lists of assigned peaks (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Zaia J Chem. Biol. 2008, 15, 881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Nilsson T; Mann M; Aebersold R; Yates JR III; Bairoch A; Bergeron J J. Nat. Methods 2010, 7, 681–685. [DOI] [PubMed] [Google Scholar]
- (3).Ashline D; Singh S; Hanneman A; Reinhold V Anal. Chem. 2005, 77, 6250–6262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ashline DJ; Lapadula AJ; Liu Y-H; Lin M; Grace M; Pramanik B; Reinhold VN Anal. Chem. 2007, 79, 3830–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Devakumar A; Thompson MS; Reilly JP Rapid Commun. Mass Spectrom. 2005, 19, 2313–2320. [DOI] [PubMed] [Google Scholar]
- (6).Devakumar A; Mechref Y; Kang P; Novotny MV; Reilly JP J. Am. Soc Mass Spectrom. 2008, 19, 1027–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ropartz D; Lemoine J; Giuliani A; Bittebière Y; Enjalbert Q; Antoine R; Dugourd P; Ralet M-C; Rogniaux H Anal. Chim. Acta 2014, 807, 84–95. [DOI] [PubMed] [Google Scholar]
- (8).Ropartz D; Li P; Fanuel M; Giuliani A; Rogniaux H; Jackson GP J. Am. Soc. Mass Spectrom. 2016, 27, 1614–1619. [DOI] [PubMed] [Google Scholar]
- (9).Gao J; Thomas DA; Sohn CH; Beauchamp J J. Am. Chem. Soc. 2013, 135, 10684–10692. [DOI] [PubMed] [Google Scholar]
- (10).Desai N; Thomas DA; Lee J; Gao J; Beauchamp J Chem. Sci. 2016, 7, 5390–5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Budnik BA; Haselmann KF; Elkin YN; Gorbach VI; Zubarev RA Anal. Chem. 2003, 75, 5994–6001. [DOI] [PubMed] [Google Scholar]
- (12).Adamson JT; Hakansson K Anal. Chem. 2007, 79, 2901–2910. [DOI] [PubMed] [Google Scholar]
- (13).Wolff JJ; Amster IJ; Chi L; Linhardt RJ J. Am. Soc. Mass Spectrom. 2007, 18, 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhao C; Xie B; Chan SY; Costello CE; O’Connor PB J Am. Soc. Mass Spectrom. 2008, 19, 138–150. [DOI] [PubMed] [Google Scholar]
- (15).Wolff JJ; Leach FE; Laremore TN; Kaplan DA; Easterling ML; Linhardt RJ; Amster I J. Anal. Chem. 2010, 82, 3460–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Han L; Costello CE J. Am. Soc. Mass Spectrom. 2011, 22, 997–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yu X; Huang Y; Lin C; Costello CE Anal. Chem. 2012, 84, 7487–7494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhu F; Lee S; Valentine SJ; Reilly JP; Clemmer DE J. Am. Soc. Mass Spectrom. 2012, 23, 2158–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Yu X; Jiang Y; Chen Y; Huang Y; Costello CE; Lin C Anal. Chem. 2013, 85, 10017–10021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Pu Y; Ridgeway ME; Glaskin RS; Park MA; Costello CE; Lin C Anal. Chem. 2016, 88, 3440–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ropartz D; Giuliani A; Fanuel M; Hervé C; Czjzek M; Rogniaux H Anal. Chim. Acta 2016, 933, 1–9. [DOI] [PubMed] [Google Scholar]
- (22).Morrison KA; Clowers BH J. Am. Soc. Mass Spectrom. 2017, 28, 1236–1241. [DOI] [PubMed] [Google Scholar]
- (23).Brüll L; Kovácik V; Thomas-Oates J; Heerma W; Haverkamp J Rapid Commun. Mass Spectrom. 1998, 12, 1520–1532. [DOI] [PubMed] [Google Scholar]
- (24).Harvey DJ; Mattu TS; Wormald MR; Royle L; Dwek RA; Rudd PM Anal. Chem. 2002, 74, 734–740. [DOI] [PubMed] [Google Scholar]
- (25).Huang Y; Pu Y; Yu X; Costello CE; Lin C J. Am. Soc. Mass Spectrom. 2016, 27, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Koster C; Holle A Presented in part at ASMS annual conference, Dallas, TX, 1999. [Google Scholar]
- (27).Perez S; Aoki-Kinoshita KF In A Practical Guide to Using Glycomics Databases; Aoki-Kinoshita KF, Ed.; Springer: Tokyo, Japan, 2017; pp 7–25. [Google Scholar]
- (28).O’Connor PB; Lin C; Cournoyer JJ; Pittman JL; Belyayev M; Budnik BA J. Am. Soc. Mass Spectrom. 2006, 17, 576–585. [DOI] [PubMed] [Google Scholar]
- (29).Savitski MM; Kjeldsen F; Nielsen ML; Zubarev RA J. Am. Soc. Mass Spedrom. 2007, 18, 113–120. [DOI] [PubMed] [Google Scholar]
- (30).Lin C; Cournoyer JJ; O’Connor PB J. Am. Soc. Mass Spectrom. 2006, 17, 1605–1615. [DOI] [PubMed] [Google Scholar]
- (31).Lin C; Cournoyer JJ; O’Connor PB J. Am. Soc. Mass Spectrom. 2008, 19, 780–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Tsybin YO; He H; Emmett MR; Hendrickson CL; Marshall AG Anal. Chem. 2007, 79, 7596–7602. [DOI] [PubMed] [Google Scholar]
- (33).Park J; Zhu R; Lin M J. Chem. Phys. 2002, 117, 3224–3231. [Google Scholar]
- (34).Huang Y; Pu Y; Yu X; Costello CE; Lin C J. Am. Soc. Mass Spectrom. 2014, 25, 1451–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Hong P; Sun H; Sha L; Pu Y; Khatri K; Yu X; Tang Y; Lin C J. Am. Soc. Mass Spectrom. 2017, 28, 2288–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Brüll L; Heerma W; Thomas-Oates J; Haverkamp J; Kovácik V; Kovác P J. Am. Soc. Mass Spectrom. 1997, 8, 43–49. [Google Scholar]
- (37).Wuhrer M; Deelder AM; van der Burgt YE Mass Spectrom. Rev. 2011, 30, 664–680. [DOI] [PubMed] [Google Scholar]
- (38).Fung YE; Adams CM; Zubarev RA J. Am. Chem. Soc. 2009, 131, 9977–9985. [DOI] [PubMed] [Google Scholar]
- (39).Freund Y; Schapire RE J. Comput. Syst. Sci. 1997, 55, 119–139. [Google Scholar]
- (40).Breiman L; Friedman JH; Olshen RA; Stone CJ Classification and Regression Trees; Chapman and Hall/CRC, 1984. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.