

Abstract
Pretargeting offers a way to enhance target specificity while reducing off-target radiation dose to healthy tissue during payload delivery. We recently reported the development of an 18F-based pretargeting strategy predicated on the inverse electron demand Diels-Alder reaction as well as the use of this approach to visualize pancreatic tumor tissue in vivo as early as 1 h post-injection. Herein, we report a comprehensive structure- pharmacokinetic relationship study of a library of 25 novel radioligands that aims to identify radiotracers with optimal pharmacokinetic and dosimetric properties. This investigation revealed key relationships between molecular structure and in vivo behavior and produced 2 lead candidates exhibiting rapid tumor targeting with high target-to-background activity concentration ratios at early time points. We believe this knowledge to be of high value for the design and clinical translation of next-generation pretargeting agents for the diagnosis and treatment of disease.
Keywords: in vivo pretargeting, bioorthogonal click chemistry, tetrazine radioligand, pharmacokinetics, PET imaging
Graphical Abstract
Introduction
In vivo pretargeting for diagnostic1–6 and therapeutic7 applications has emerged over the last three decades as a powerful technology to enhance target specificity and reduce off-target effects.2,8 Generally speaking, pretargeting strategies strive to combine the inherent advantages of macromolecular targeting vectors and small molecules, specifically high target specificity and short organ and tissue residence times, respectively.1,4 To achieve this, the targeting vector is administered first and allowed to accumulate at the target site and clear from off-target organs prior to the injection of a small effector molecule carrying the payload of interest (e.g. radionuclide; Fig. 1).4,5,9 In order to enable their in vivo recombination, both entities are equipped with complementary functionalities that enable an in vivo ligation reaction.10,11 Appropriate pairs of reaction partners that have been employed in in vivo pretargeting approaches include streptavidinbiotin12, complementary oligonucleotide strands13, and click chemistry-based reaction pairs.10,14,15 While strategies based on streptavidin-biotin have shown somewhat deflating outcomes in the clinic16, the use of a bispecific, CEA-targeting antibody in combination with a radiolabeled hapten peptide has shown very promising clinical results.17
Figure 1.

(a) Schematic illustration of the pretargeting approach: a macromolecular targeting vector — in our case an antibody-TCO conjugate — is injected first and allowed to reach the target site while clearing slowly from systemic circulation. After a specific accumulation time, the small molecule effector probe (in this case a radiolabeled tetrazine probe) is administered systemically and undergoes bioorthogonal click reaction with the TCO-groups of the immunoconjugate at the target site. (b) Modular chemistry approach for radioligand design: radioligands consisted of a Tz moiety for in vivo click chemistry, a linker for altering the biodistribution, and a chelator for the attachment of the positron-emitting metal ions 68Ga3+ and [18F]-AlF2+.
One of the newer members of the click chemistry toolbox, the inverse electron-demand Diels-Alder (IEDDA) reaction between trans-cyclooctene (TCO) and tetrazine (Tz) has proven particularly well suited for in vivo pretargeting.2–5,7,10,11 The IEDDA ligation is selective and bioorthogonal, but its principal advantage for pretargeting compared to other click reactions — such as the Staudinger ligation18 or the strain-promoted alkyne-azide cycloaddition14 (SPAAC) — lies in its speed. To wit, the first-order rate constants of the Tz/TCO ligation lie in the realm of 104-105 M−1s−1, orders of magnitude faster than the rates of the Staudinger and SPAAC reactions (0.002 M−1s−1 and 0.07 M−1s−1, respectively).11 The in vivo feasibility of the IEDDA reaction between a TCO-modified monoclonal antibody (mAb) and a radiolabeled Tz has been demonstrated by various groups using a wide range of antigen-targeting immunoconjugates and tetrazines labeled with an array of radionuclides for imaging (including 111In, 64Cu, 99mTc, 18F, 68Ga, and 11C)1,4,5,19–21 and therapy (177Lu).7
In terms of pretargeted positron emission tomography (PET) imaging, Zeglis, et al. presented promising results in 2013 in pretargeting experiments using the gpA33-targeting mAb huA33-TCO and a 64Cu-labeled tetrazine radioligand.4 Shortly thereafter, our laboratories developed a second-generation Tz for 64Cu-based pretargeted PET imaging applications by integrating the sarcophagine chelator system into the radioligand structure.3 At the same time, our laboratories demonstrated that an 18F-labeled Tz-based radioligand in combination with the carbohydrate antigen 19.9 (CA19.9)-targeting fully human mAb 5B1-TCO22 allowed for the successful PET imaging of subcutaneous (s.c.) pancreatic cancer xenografts as early as 1 h post-injection (p.i.)1. Critically, this new pretargeting approach utilized the short-lived radionuclide 18F (t½ = 109 min), resulting in only a fraction of off-target radiation doses to healthy tissues compared to directly labeled immunoconjugates with long-lived isotopes (124I or 89Zr, t½ > 3 days). Despite clear delineation of tumor tissue at early imaging time points demonstrating the general feasibility of this approach, the relatively low tumor-to-background activity concentration ratios at even 4 h p.i., such as tumor-to-intestines (1.6 ± 0.1) and tumor-to-kidney (1.8 ± 0.4) ratios, inspired us to undertake a thorough investigation into the fundamental relationships between molecular structure, pharmacokinetics, and pretargeting performance.
This first-of-its-kind structure-pharmacokinetics relationship (SPR) study was further fueled by the increased popularity of IEDDA pretargeting strategy. However, current approaches lack fundamental insight into the relationship between physicochemical properties and pretargeting performance. In order to address those issues, the study at hand was designed with two main objectives: (1) to identify a radiopharmaceutical lead candidate suitable for clinical development and (2) to generate experimental evidence for a rational understanding of how molecular parameters such as overall molecular net charge, distribution coefficient, plasma half-life (PHL), and stability influence the in vivo performance of small molecule radioligands in pretargeting systems.
Tracer library and SPR study design
The synthesis of the radioligands library as the first step of this study was based on previously reported protocols.1,2,4 New reaction routes and radiolabeling procedures developed within this study are described in the supporting information (Supplementary Sections 2 and 3). Overall, the radioligands were designed to display structural variation in order to cover a broad spectrum of physicochemical properties, thereby enabling the study of the relationship between structure and in vivo behavior (Tab. 1). Each radioligand is composed of 3 different structural building blocks (Tab. 1; Fig. 2a). First, a Tz component (1–4) was selected for in vivo click chemistry. Second, a linker moiety consisting of polyethylene glycol [PEG7 (5) or PEG11 (6)], amino acids (AA) [AA = lysine (K, 7), histidine (H, 8), aspartate (R, 9), and arginine (D, 10)], or a combination of both is attached. Finally, a bifunctional chelator, either 1,4,7-triazacyclononane-1,4-diacetic acid (NODA, 11,12) or 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA, 13,14), was introduced, allowing for the installation of 18F and 68Ga radionuclides. Tz moieties 1–4 were selected based on their previously reported stability and reaction kinetics with TCO to ensure a wide range of properties.4,5,21,23 The use of PEG linkers to modulate in vivo PK of small molecules has previously been reported, including accelerated non-target organ clearance as well as increased renal clearance.1,2,24 We included them into our study in order to investigate their impact on radiotracer PK alone or in combination with AA linkers (which, to the best of our knowledge, have not yet been systematically reported in any SPR study). The amino acids lysine, arginine, histidine, and aspartate were regarded as useful structural components to significantly influence in vivo behavior and PK parameters. It was reasoned that their charged side chains should have a measureable effect on tracer PK, and would further allow us to establish a correlation between molecular net charge and PK parameters. Both bifunctional chelator moieties NODA and NOTA currently find broad application in preclinical25,26 and clinical27 research for the radiolabeling of biological macromolecules and small molecule targeting probes. Precursors 15–32 were synthesized in good overall chemical yields (18–37%) via 3–8-step syntheses, depending on the starting materials (Fig. 2b).
Table 1.
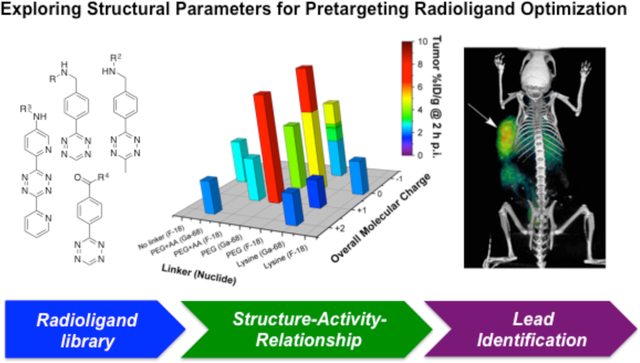
All 25 radioligands were employed in the first characterization process (green). Based on those results, 15 radioligands were selected for the next step of testing to investigate their performance in pretargeting experiments (blue). Finally, radioligands [18F]27 and [68Ga]27 were identified as lead compounds based on their overall tumoral uptake and tumor-to-NT activity concentration ratios (red).
| Tz | PEGx (x = 7,11) | AA (D, H, K, R) | Chelator | Precursor | Tracer |
|---|---|---|---|---|---|
| 1 | - | - | NODA | 15 | [18F]15 |
| 1 | - | K | NOTA | 16 | [18F]16 |
| 1 | 7 | - | NOTA | 17 | [18F]17 |
| 1 | 11 | - | NOTA | 18 | [18F]18 |
| 2 | - | K | NOTA | 19 | [18F]19 |
| [68Ga]19 | |||||
| 2 | - | K-K | NOTA | 20 | [18F]20 |
| [68Ga]20 | |||||
| 2 | - | K-K-K | NOTA | 21 | [18F]21 |
| [68Ga]21 | |||||
| 2 | - | K-K-K | NODA | 22 | [68Ga]22 |
| 2 | 11 | - | NOTA | 23 | [18F]23 |
| 3 | 11 | - | NOTA | 24 | [18F]24 |
| [68Ga]24 | |||||
| 4 | 7 | - | NODA | 25 | [18F]25 |
| 4 | 7 | - | NOTA | 26 | [68Ga]26 |
| 4 | 11 | - | NODA | 27 | [18F]27 |
| [68Ga]27 | |||||
| 4 | 11 | - | NOTA | 28 | [18F]28 |
| [68Ga]28 | |||||
| 4 | 7 | K | NODA | 29 | [18F]29 |
| 4 | 11 | H | NODA | 30 | [18F]30 |
| [68Ga]30 | |||||
| 4 | 11 | R | NODA | 31 | [18F]31 |
| 4 | 11 | D | NOTA | 32 | [18F]32 |
Figure 2.

Correlation diagrams showing how structural and physicochemical parameters (e.g. stability, linker, molecular charge, logD, PHL) influence each other. (a) Tracer stabilities after a 3 h incubation time in human serum are shown after tracers were divided into groups with the same linker and radionuclide. Serum stabilities were generally >60%, except for 68Ga-labeled compound 46. (b) LogD values (median shown, n = 3) summarized as a group diagram. (c) distribution coefficients in relation to molecular charge (under physiological conditions) showed a significant correlation. (d-f) in vivo PHLs plotted as a group diagram (d), in dependence of molecular net charge (e), as well as plotted against logD (f). Lysine-containing compounds showed overall fast clearance from circulation, with PEGylated compounds having 3-times longer PHLs. Interestingly, a positive correlation between a tracer’s PHL and its logD value was found.
All 18F- and 68Ga-labeled [t½ (68Ga) = 68 min] Tz-derived radioligands were furnished in high radiochemical yields [RCYs, >55% (18F); >83% (68Ga)], with specific activities (SAs) of >19 MBq/nmol and high radiochemical and radionuclidic purity (Supplementary Section 3). For in vitro analysis, purified tracers were incubated in human serum at 37 °C to analyze their stability under physiological conditions. Further, the distribution coefficient (logD, n = 3) of all radioligands in a 1:1 mixture of PBS:1-octanol was determined using the shake-flask method. Subsequently, the tracers (4–8 MBq, 0.5–1 nmol) were injected into healthy athymic nude mice via the lateral tail vein. At various time points (between 2–120 min p.i.), blood was drawn via the lateral tail vein or saphenous vein (n = 4) to calculate PHLs (all radioligands, n = 1) and plasma stabilities (n = 3). Tracers (11–14 MBq, 0.8–1.3 nmol) were injected into healthy athymic nude mice, and general biodistribution experiments for all tracers were performed using serial PET imaging hourly between 1–4 h p.i., unless stated otherwise (n = 4). Decay-corrected PET imaging data and reconstructed 3D images of the tracer distribution were then used to determine radioactivity concentrations (given as percent injected dose per gram, %ID/g) in the kidney and large intestine (quantitative ROI analysis). Additional ex vivo organ uptake values were determined for selected compounds and were found to be in line with image-derived uptake data, justifying the use of image-derived organ uptake data for the majority of compounds in order to reduce the number of animals euthanized. Data analysis led to the selection of 15 radioligands (Tab. 1, highlighted in blue) that were further tested in in vivo pretargeting experiments using athymic nude mice bearing subcutaneous pancreatic ductal adenocarcinoma (PDAC) xenografts (n = 4). A TCO-modified immunoconjugate of the CA19.9-targeting antibody 5B1 (5B1-TCO; 1.3 nmol, 200 μg per mouse) was injected 72 h prior to the injection of the small molecule radioligands (1.3–1.6 nmol, 0.6–1.4 μg, 1–1.2 eq.). PET images were acquired hourly between 1–4 h p.i., unless stated otherwise. In addition, ex vivo biodistribution experiments were conducted for selected tracers in order to obtain quantitative information of tracer distribution in up to 14 organs. Ultimately, this investigation identified two radioligands — [18F]27 and [68Ga]27 — as the most promising lead compounds (Tab. 1, highlighted in red).
Physicochemical and pharmacokinetic properties of Tz-derived PET tracer
Structural, in vitro, and pharmacokinetic (PK) data obtained for all 25 tracers were used to investigate potential correlations between physicochemical parameters. Tracer stabilities (given as %-intact, n = 3) in human serum (incubation for 4 h at 37 °C, Fig. 3c) ranged from >90% ([18F]23) to 54 ± 7% ([68Ga]24) (Fig. 3a, Supplementary Section 4). Considering all of the structural elements of the radioligands, we reasoned that both the tetrazine moiety and the metal complex would have an impact on in vitro (and presumably in vivo) stability. However, we found that instability was due primarily to the decomposition of the tetrazine moiety, and that the radioactive metal complexes were stable over the course of our experiments. The majority of the radioligands did not show any elevated protein binding (<2% of total radioactivity, Supplementary Section 4), with the exception of [18F]20, [18F]21, [68Ga]21, [68Ga]22, and [18F]29, each of which exhibited up to 16% protein-bound radioactivity. This result may be explained by the high lysine content residues and positive charge of these radioligands, prompting electrostatic interactions between the tracers and plasma proteins.24
Figure 3.

(a) Chemical structures and maximum intensity projections (MIPs) acquired 2 h and 4 h p.i. for 5 selected radioligands ([18F]19, [18F]20, [18F]21, [18F]27, and [18F]30) in healthy athymic nude mice (B = bladder, K = kidney, I = intestine). Tracers (11–14 MBq, 0.8–1.3 nmol) were administered via the lateral tail vein (t = 0). Notably, lysine-containing radioligands [18F]19, [18F]20, and [18F]21 showed fast renal clearance, whereas [18F]30 showed more predominantly uptake in the intestines. Lead compound [18F]27 showed clearance via both excretion routes. The increasing uptake and retention in the kidneys facilitated by an increasing number of lysine residues in the radioligand structure is well visible for [18F]19–21. (b) Kidney uptake of radioligands at 2 h p.i. is shown as a function of molecular net charge. (c) Kidney uptake values at 2 h p.i. dependent on the linker and radionuclide. Solely lysine containing radioligands exhibited elevated kidney uptake values of up to 16.0 ± 5.8 (2 h p.i.) and 21.1 ± 5.6 %ID/g (4 h p.i.) for [68Ga]22. Even one lysine residue had a dramatic effect on kidney uptake as shown for [18F]29, where a lysine residue was integrated between the PEG7 linker and the chelator moiety, increasing kidney uptake from 2.6 ± 0.3 ([18F]25, Tz-PEG7-NODA) to 8.3 ± 1.5 %ID/g ([18F]29, Tz-PEG7-Lysine-NODA). Substitution of lysine by other amino acids with either positively or negatively charged side chains reduced kidney uptake to below 4 %ID/g.
Not surprisingly, a significant negative correlation between the logD value of a tracer and its overall molecular charge under physiological conditions was found (p-value <0.0001, Supplementary Sections 4 and 9). Generally speaking, the closer the net charge of a tracer is to 0, the higher its partition coefficient. However, it is not quite that simple. For instance, radioligands possessing the same overall molecular charge but a different number of formally charged functional groups could still show significant differences in their distribution coefficients. One explanation could be enhanced solvation by water molecules through charge-dipole interactions: if the molecule exhibits a greater number of formal charges, it would lead to a higher degree of solvation.28 For example, radioligands with a net charge of 0 containing a PEG linker (or no linker) exhibited logD values between −1.0 and −1.5, while radioligands with the same overall charge but with lysine residues in the linker displayed logD values between −1.8 and −3.1. PHLs for the radioligands correlated (p-value <0.0001, Supplementary Sections 5 and 9) with their net charge. Tracers with lower (more negative) logD values and higher net charges possessed shorter PHLs (Fig. 3d,e). The replacement of a PEG linker by AAs or the incorporation of AA(s) to an already existing PEG linker resulted in reduced PHLs. PEG-containing tracers [18F]21–[68Ga]28 — even though ranging from −1 to +1 in terms of net charge — possessed calculated PHLs of >10 min. For instance, lead compounds [18F]27 and [68Ga]27 exhibited PHL of 17.1 and 15.1 min, respectively. In contrast, AA-containing radioligands exhibited PHLs <10 min. Tracers that solely contained lysine moieties as linkers displayed the shortest PHLs, values that decreased further as the number of lysine residues increased. For example, [68Ga]22 — with a charge of +4 and 3 lysine residues — possessed a PHL of 1.9 min, the shortest of all tracers. Longitudinal PET imaging experiments conducted in healthy animals revealed a significant difference in biodistribution patterns and clearance pathways between tracers (Supplementary Section 7). In general, lysine-containing radioligands — regardless of the presence of a PEG group — showed fast clearance from circulation and relatively high kidney radioactivity concentration of >5%ID/g as early as 1 h p.i. Radioactivity uptake and retention increased in a nearly linear fashion with the number of lysine residues per radioligand (Fig. 4a–c). High retention of the tracers containing lysine residues was likely due to reabsorption of those tracers in the proximal tubules of the kidney. In fact, recent studies have shown that peptides high in lysine residues are powerful kidney targeting agents, facilitating the uptake and retention of those constructs in the renal clearing organs.29,30 All other radioligands that did not contain lysine residues exhibited significantly lower kidney activity concentrations (<3.5 %ID/g, Fig. 4). Compound [68Ga]30, for instance, exhibited the lowest uptake values of 1.2 ± 0.3 %ID/g at 2 h p.i., though it also displayed higher liver (>1.8 %ID/g) and intestinal uptake (>3 %ID/g). Generally, tracers with overall net charges were cleared faster from circulation through globular filtration, whereas compounds with low or no net charge exhibited elongated circulation times and were predominantly cleared hepatically and excreted via the intestines.
Figure 4.

(a) MIPs and transverse image slices acquired in pretargeted PET imaging experiments 2 h after radioligand administration. White arrows indicate tumor tissue. To this end, 5B1-TCO (200 μg, 1.33 nmol, in 150 μL 0.9% saline) was injected into the lateral tail vein 72 h prior to the radioligand (1.3–1.6 nmol in 150 μL 0.9% saline, containing <5% v/v ethanol). [18F]19 exhibited slightly higher tumor uptake (2.2 ± 0.2 %ID/g, 1 lysine residue) than [18F]20 (1.7 ± 0.2 %ID/g, 2 lysine residues), where the tumor was not visible due to the high background of kidney. However, those uptake values were significantly lower compared to the PEGylated lead compounds [18F]27 (7.6 ± 1.8 %ID/g) and [68Ga]27 (6.6 ± 1.4 %ID/g). Incorporation of a lysine residue as for [18F]29 (PEG + Lysine) increased kidney uptake, while decreasing PHL and tumoral uptake, indicating a positive correlation. (b-d) Plots showing tumoral uptake in dependence of in vivo stability (2 h p.i.) and PHL (2, 4 h p.i.). In vivo stability (shown as %-intact tracer and determined at 2 h p.i.) of all radioligands employed in pretargeting experiments was >50%, except for [18F]24 with a measured stability of 31.6 ± 6.3%. In vivo stability had no measurable effect on the pretargeting performance, most likely because the majority of intact tracer had undergone click reaction at the target site within the first 2 h of the experiment. Tumor uptake of radioligands did however correlate with their PHLs: the longer the PHL the higher the overall tumoral uptake.
Evaluation of pretargeting performance in a PDAC xenograft model
Based on this in vitro and PK data, 15 radioligands were selected for in vivo pretargeting experiments to probe correlations between PK parameters and pretargeting performance. Compounds were selected in order to cover a broad range of structural and physicochemical diversity. Generally, we reasoned that PHL and the primary clearance pathway of a tracer should have significant impact on its pretargeting performance. PHL would determine how much tracer molecules reach the target site before clearance, and the clearance pathway should influence the target-to-background ratio, as renal clearance should reduce background noise more quickly than hepatic clearance.
All initial pretargeting experiments were carried out in mice bearing s.c. BxPC3 xenografts. Approximately 3–4 weeks after inoculation (2.5 × 106 cells), TCO-modified anti-CA19.9 mAb 5B1-TCO (1.33 nmol, 200 μg in 150 μL 0.9% saline) was injected via the tail vein. 72 hours later, Tz-derived radioligands (1.3–1.6 nmol, 1–1.2 eq., in 150 μL 0.9% saline, containing <5% v/v ethanol) were injected into the opposite lateral tail vein (t = 0).1,2
PET imaging and ex vivo biodistribution experiments were conducted at 2 h and 4 h p.i. Tumor uptake values of all the radioligands were determined via PET image analysis (ROI analysis) as well as via ex vivo biodistribution analysis for selected compounds (Supplementary Sections 6 and 7). The median tumoral activity concentration values at 2 h p.i. ranged from 7.6 ± 1.8 %ID/g for 18F-labeled lead compound [18F]27, to 1.7 ± 0.8 %ID/g for the fast-clearing tracer [18F]20 (Figs. 5 and 6). Lysine-containing radioligands generally possessed tumor uptake values of <3%ID/g at all time points, with tumor uptake values of <2 %ID/g for compounds solely containing a lysine linker. The highest tumor uptake values were observed for radioligands containing PEG linkers only. The introduction of AA linker led to accelerated plasma clearance, and thus reduced the tumoral uptake due to fewer tracer molecules capable of reaching the tumor site. As expected, the PHL of a radiotracer correlated (p-value <0.0001, Supplementary Section 9) with the tracer’s overall tumoral uptake, as illustrated for the 2 h and 4 h time points (Fig. 5c,d). Additionally, tracer stabilities in vivo were determined to be >50% at 3 h p.i. and did not correlate with tumoral uptake (Fig. 5b). In case of tracer [68Ga]24, possessing low stabilities in both human serum (66.9 ± 7.9%) and in vivo (31 ± 6.3%), stability may indeed explain the relatively poor tumoral uptake of 3.1 ± 0.7%ID/g at 2 h p.i., despite exhibiting a PHL (15.5 min) that would allow for higher accumulation at the target site over time.
Figure 5.

(a) 3D bar diagram summarizing tumoral uptake values (Z-axis) for all evaluated radioligands at 2 h p.i. (Compound numbers are given for selected compounds only). Uptake values are presented in relation to the employed linker moiety (X-axis) and molecular net charge (Y-axis). In opposition to kidney uptake data, tumoral uptake values of lysine containing and other, fast-clearing radioligands were generally low (<3.5 %ID/g for all time points). Tracer with PHLs of >10 min generally exhibited higher tumoral uptake, and the highest values were measured for PEGylated compounds based on Tz 4, including lead compounds [18F]27 and [68Ga]27. (b) Summary of tumor uptake values of all 15 radioligands tested in pretargeting experiments using the PDAC model.
Figure 6.

(a) Transverse (top) and coronal (middle) slices as well as MIPs (bottom) of pretargeted PET imaging experiments using lead compounds [18F]27 (left) and [68Ga]27 (center) in the BxPC3-based PDAC model. PET images show high tumor uptake of >6%ID/g 2 h p.i. In addition, the 68Ga-labeled tracer [68Ga]27 was tested in a pretargeting study using a subcutaneous CRC model (SW1222, right). The gpA33-targeting immunoconjugate huA33-TCO (0.95 nmol) was injected 48 h prior to tracer [68Ga]27. Good delineation of tumor tissue was achieved as early as 1 h p.i., with the tumor showing highest uptake at 3 h p.i. These data confirming results obtained from the PDAC model, and supported the overall promising performance of both radioligands. (b) Quantitative comparison of uptake values calculated for tumor, blood, large intestines, and kidneys. [18F]27 showed overall the highest tumor uptake (8.8 ± 1.7 %ID/g), but also the highest residual radioactivity in the intestines. [68Ga]27 showed highest tumor-to-clearance organ ratios in both models, but exhibited relatively high residual blood radioactivity concentrations, most likely due to residual antibody in the bloodstream at the time of tracer injection.
Identification of lead candidates
Taking all of the data into account, our study identified two lead candidates: [18F]27 and [68Ga]27. Both tracers possessed good stabilities in vitro as well as in vivo and exhibited PHLs (17.1 and 15.1 min, respectively) that allowed for a fast enough clearance from circulation without impairing tumor accumulation. [18F]27 showed tumor uptake values of 7.6 ± 1.8 %ID/g at 2 h p.i. and 8.8 ± 1.7 %ID/g after 4 h, as well as promising tumor-to-NT (non-target) activity concentration ratios, although it did display some uptake in the intestines (Fig. 8a,b). [68Ga]27 exhibited tumoral uptake values of 6.8 ± 1.4 %ID/g at 2 h p.i. and 7.1 ± 1.8 %ID/g at 4 h p.i. and boasted superior tumor-to-NT activity concentration ratios compared to its 18F-labeled counterpart, especially with regard to the large intestines (>3 at 2 h p.i. compared to ~1.2 for [18F]27; Supplementary Section 6).
However, [68Ga]27 showed elevated activity concentrations in the blood pool in both tumor models with tumor-to-blood ratios of 0.8 ± 0.2 and 1.7 ± 0.4 at 2 h and 4 h p.i., respectively, in the PDAC model, suggesting that a significant amount of 5B1-TCO was still present in circulation when the tracer was administered. High tumor uptake as well as similar residual blood radioactivity concentrations were observed when [68Ga]27 was tested in a s.c. colorectal cancer (CRC, SW1222 cell line, Supplementary Section 7) model, performed to demonstrate the versatility and modularity of this approach. To this end, the anti-A33 mAb huA33-TCO was injected 48 h prior to the radioligand.31,32 Despite a relatively low tumor-to-blood activity concentration ratio due to residual circulating mAb, [68Ga]27 showed excellent tumor uptake as early as 1 h p.i., with maximum uptake values of 7.7 ± 1.0 %ID/g at 4 h p.i. (Fig. 8a,b). In accordance to our data showing the influence of molecular net charge, [68Ga]27 (+1) showed significantly reduced uptake in the intestines in both in vivo models compared to [18F]27 (0). Instead, enhanced renal clearance was observed that led to an increased tumor-to-background signal. Furthermore, dosimetric calculations based on ex vivo biodistribution data gave whole-body effective doses of 0.009 mSv/MBq ([18F]27) and 0.013 mSv/MBq ([68Ga]27) for both lead compounds. Compared to approaches using longer-lived radionuclides (89Zr), this means a >70-fold reduction in radiation doses to healthy tissue (Supplementary Section 8). Ultimately, [18F]27 and [68Ga]27 showed the most favorable pretargeting PK with high tumor accumulation with minimal off-target radiation. As a result, both compounds are currently being evaluated to enter the clinical development phase.
Discussion
Since the advent of in vivo pretargeting as a diagnostic and therapeutic tool, clinical success so far has been limited. Despite the ability to successfully target tumor tissue, peripheral issues (such as the immunogenicity of streptavidin) have impaired clinical development, though recent results obtained with bispecific antibodies are extremely promising. The question whether click chemistry-based approaches — most notably those based on the IEDDA reaction — will have more success in the clinic has yet to be answered.
In recent years, the IEDDA reaction utilizing full-sized, TCO-modified mAbs in combination with small, Tz-derived imaging probes has attracted by far the most attention in the scientific community. Due to its nearly ideal properties such as speed, selectivity, and bioorthogonality, the IEDDA reaction is currently regarded as an approach with high clinical potential. Despite its popularity, limited efforts to investigate the influence of small molecule PK have been reported. Thus, a comprehensive investigation of how small molecule in vivo behavior would significantly improve our current understanding of how molecular parameters influence pretargeting performance, and increase the potential for designing and identifying clinical candidates. The feasibility of combining the pretargeting approach with short-lived (t½ < 2 h) radionuclides was only recently demonstrated.1, 21 Improving the small molecule’s PK in order to achieve high target-to-NT activity concentration ratios as early as possible after tracer administration would be highly valuable from a clinical perspective. In order to achieve this goal, effector molecules with the ability to accumulate quickly at the target site while simultaneously exhibiting short off-target residence times are needed. Taken together, these issues motivated us to pursue a comprehensive SPR study. The study at hand includes the in vitro and in vivo evaluation of 25 novel 18F- and 68Galabeled Tz-derived radioligands capable of in vivo click reaction with clinically validated mAbs as pretargeting constructs.
Molecular and structural parameters were correlated with in vitro and in vivo results in order to investigate relationships between structure and in vivo behavior. Overall, four major trends were observed. First, the overall molecular charge of the radioligand had a significant impact on in vivo behavior, as high net charge reduced lipophilicity and shortened the PHL. Second, the introduction of amino acid residues into the radioligand structure did not only lead to reduced circulation times but also influenced whether the tracer was eliminated renally or hepatically. Charged molecules were predominantly cleared through glomerular filtration in the kidneys, whereas compounds without an overall net charge displayed higher uptake in the liver and intestines. Importantly, the introduction of lysine residues into the radioligand structure dramatically decreased the tracer’s PHL (<6 min), while it was quickly and significantly taken up and retained in the proximal tubules of the kidneys. Kidney uptake values of these radioligands were up to 20 %ID/g at 3 h p.i. ([68Ga]22), while their tumoral uptake values were dramatically reduced to <2%ID/g. Those results appeared to be in line with recently published data demonstrating that peptide-sequences high in lysine residues are powerful delivery agents to specifically target the proximal tubules of the kidneys.29,30 Third, the substitution of lysine by other amino acids (such as arginine) resulted in a significant reduction of kidney targeting ability. Those results are consistent with data presented in this study, showing that kidney uptake was decreased >4-fold when lysine was substituted by an arginine or histidine. Most notably, the incorporation of a lysine residue into the structure of [18F]25 to from radioligand [18F]29 dramatically changed in vivo behavior. Compared to [18F]25, [18F]29 exhibited a significantly reduced PHL and a lower tumor uptake value of 2.9 ± 0.3%ID/g at 2 h p.i., with the majority of radioactive tracer being accumulated in the kidneys (8.3 ± 2.1%ID/g, Fig. 6a, Supplementary Section 7). And finally, tumor uptake was mainly determined by the PHL of the radioligand. We found a significant and positive correlation between a radioligand’s tumor uptake and PHL (p-value <0.0001), explaining the observations made in our in vivo studies. The PHL of a radioligand had to be sufficiently long — approximately >10 min — to enable effective tumor accumulation.
Overall, we observed that the incorporation of amino acid residues — and with that the alteration of molecular net charge — had a more profound effect on tracer PK then the introduction of PEG-linkers alone. Although PEG-linker increased the kidney-to-intestines uptake ratios as expected, no change in PHL could be observed. Instead, our results suggest that the molecular net charge had the most significant impact on radioligand PK.
Lead compounds [18F]27 and [68Ga]27 contained PEG11-linkers, exhibited PHLs of approximately 15 min, and possessed net molecular charges of 0 and +1, respectively. Even though the 18F-labeled tracer [18F]27 was predominantly excreted via the intestines, it showed the highest overall tumoral uptake of all radioligands investigated, with 8.8 ± 1.7 %ID/g at 4 h p.i. The 68Ga-labeled lead compound [68Ga]27 was tested in 2 tumor models with different targeting vectors and showed high tumor uptake values in both models at 4 h p.i.: 6.9 ± 1.8%ID/g (PDAC, BxPC3) and 7.7 ± 1 %ID/g (CRC, SW1222). However, as for all other radioligands that were employed in pretargeting experiments, both lead compounds showed significant residual blood radioactivity concentrations that cannot be explained by the clearance of the tracer alone (Supplementary Section 7). Instead, we believe that residual mAb circulating in the blood at the time of tracer injection and subsequent click chemistry in circulation led to elevated blood radioactivity concentrations. We applied the established accumulation intervals between the mAb-TCO and tracer injections of several days (2 and 3 days for huA33 and 5B1, respectively) to allow mAb accumulation at the target site and clear from circulation. Due to the inherent instability of the TCO group (formation of the more stable cis-isomer over time)3,31 and internalization of some antibodies (e.g. 5B1) the accumulation interval cannot be extended infinitely. Instead, a mAb-dependent time interval between the injections of mAb-TCO and the Tz probe has to be determined.3 Indeed, pretargeting studies conducted by our groups that used longer accumulation intervals for 5B1-TCO did not lead to dramatically improved tumor-to-blood activity concentration ratios.2 Although a decreased antibody concentration in the blood pool can be achieved, the increased decomposition of TCO at the tumor site led to overall reduced tumoral uptake and TTB values.2 In case of non-internalizing huA33-TCO, however, longer intervals of up to 120 h yielded slightly higher TTB ratios.3 New technologies such as innovative clearing agents33 or the use of smaller antibody fragments have the potential to further improve this strategy by reducing residual activity concentrations in the blood.
We initially speculated that the pretargeting performance of a radioligand could be improved by solely increasing the renal-to-gut excretion ratio. However, our data suggest that it is not that simple. In order to facilitate sufficient delivery of radioligand to the tumor site a compromise, or “sweet spot”, between the excretion pathways, PHL, and tumor signal needs to be identified, and that achieving high on one of those parameters will most likely have a negative impact on the others. As an example, short PHL and fast organ clearance had a negative effect on tumor accumulation. On the other hand, molecules with no net charge or low overall net charge exhibited longer PHLs and higher tumoral uptake values but also higher background noise levels due to a slower excretion predominantly via the intestines.
At this point of the discussion, it is important to highlight the focus of this study to develop radioligands specifically for pretargeting approaches utilizing short-lived radionuclides with physical half-lives <2 h. Short physical half-lives, however, limit the time window for high-quality PET image acquisition to a maximum of 6–8 h after tracer injection (preclinically). Thus, radioligands have to possess PK properties that allow for successful tumor visualization with high tumor-to-background ratios at early time points, preferably between 1–4 h p.i. Conversely, approaches that apply longerlived radionuclides such as 64Cu (t½ = 12.7 h) allow image acquisition at later time points (up to 48 h p.i.), and are thus less sensitive to early radioligand PK. At the same time it is important to note that time restraints are easily justified and out-weighed by the dosimetric advantages that the use of short-lived radionuclides entails. In addition, the ability of virtually any major hospital and cancer center to produce 68Ga- and 18F-labeled clinical-grade radiopharmaceuticals on site and on a daily basis is a tremendous logistical advantage and thus justifies the development of these tracers.
Ultimately, this SPR study revealed fundamental relationships between the structure and in vivo behavior of tetrazine-based radioligands, allowing for a more rational approach to design next-generation radiopharmaceuticals for in vivo pretargeting. As discussed earlier, the second goal of this study was to generate candidates worthy of clinical translation. We believe that with lead compounds [18F]27 and [68Ga]27 we have identified 2 promising candidates that show high tumor targeting abilities and favorable dosimetries, justifying an IND application in the near future.
Experimental Section
General Information
All reactions involving tetrazine moieties were carried out using aluminum foil covered reaction vials due to light sensitivity of the tetrazine structure. Further, all reaction mixtures were purified using preparative HPLC with the following gradient: 5–95% of MeCN in H2O over 20 min, 8 mL/min. The final purity of all molecules was determined using analytical HPLC (see Table 2 for precursors 15–32, gradient: 5–95% of MeCN in H2O over 20 min, 1 mL/min) to achieve purities of >95%. Retention times (Rt) were assigned to all compounds. Upon purification and freeze-drying, all final precursor compounds 15–32 were aliquoted into 25 μL (50 nmol) fractions using metal-free, anhydrous DMSO. It is worth noting that reaction conditions, in particular for the formation of amide bonds, had to be designed with respect to the well-known sensitivity of tetrazines to harsh synthetic conditions, such as high pH and elevated temperatures23,34. For instance, the presence of bases such as pyridine, triethylamine and reagents such as N’,N’-dicylcohexylcarbodiimide and N-hydroxysuccinimide used for in situ carboxylic acid activation, led to decomposition of the tetrazine moiety, which was easily visible by eye due to loss of purple color and the absence of absorption at 525 nm. To overcome this limitation, we found that amide bonds could be formed in high yields and without impairing the tetrazine moiety by using 2 equivalents of both 1-ethyl-3-(3-demethylamunopropyl)carbodiimide and hydroxybenzotriazole in dry DMF or DMSO. The procedures and analytical data for the synthesis of all precursor molecules can be found in the Supplementary Information (Sections 2 and 3). Summarized below (Table 2) are the yield and purity data obtained for all precursor and tracer molecules. Further, the general 18F and 68Ga-labeling procedures that were developed for this study are presented.
Table 2.
Summary of purities and chemical yields (last step of synthesis) obtained for all Precursor molecules 15–32. On the right hand site are the radiochemical (RC) purity and the decay-corrected (d.c.) RC yields that were observed for all tracers that were evaluated in this study (all n = 3).
| Precursor | Purity [%] | Yield [%] | Tracer | RC Purity [%] | d.c. RC Yield [%] | SA [MBq/nmol] |
|---|---|---|---|---|---|---|
| 15 | >97 | 58 | [18F]15 | >98 | 44 ± 8 | 23 ± 3 |
| 16 | >96 | 90 | [18F]16 | >98 | 49 ± 5 | 28 ± 2 |
| 17 | >96 | 83 | [18F]17 | >99 | 64 ± 7 | 38 ± 4 |
| 18 | >97 | 73 | [18F]18 | >98 | 56 ± 4 | 31 ± 7 |
| 19 | >99 | 98 | [18F]19 | >97 | 38 ± 5 | 29 ± 3 |
| [68Ga]19 | >98 | 85 ± 7 | 24 ± 5 | |||
| 20 | >98 | 77 | [18F]20 | >98 | 51 ± 3 | 33 ± 5 |
| [68Ga]20 | >98 | 91 ± 4 | 26 ± 4 | |||
| 21 | >97 | 86 | [18F]21 | >97 | 62 ± 8 | 37 ± 5 |
| [68Ga]21 | >98 | 78 ± 12 | 27 ± 3 | |||
| 22 | >97 | 94 | [68Ga]22 | >98 | 88 ± 6 | 22 ± 2 |
| 23 | >98 | 89 | [18F]23 | >97 | 38 ± 4 | 29 ± 5 |
| 24 | >96 | 81 | [18F]24 | >96 | 60 ± 6 | 44 ± 7 |
| [68Ga]24 | >99 | 87 ± 5 | 29 ± 5 | |||
| 25 | >95 | 77 | [18F]25 | >97 | 44 ± 9 | 39 ± 4 |
| 26 | >98 | 45 | [68Ga]26 | >98 | 78 ± 4 | 36 ± 5 |
| 27 | >98 | 73 | [18F]27 | >98 | 66 ± 9 | 46 ± 4 |
| [68Ga]27 | >98 | 86 ± 3 | 36 ± 5 | |||
| 28 | >97 | 73 | [18F]28 | >97 | 49 ± 11 | 48 ± 3 |
| [68Ga]28 | >98 | 77 ± 3 | 38 ± 3 | |||
| 29 | >97 | 81 | [18F]29 | >97 | 62 ± 9 | 46 ± 6 |
| 30 | >98 | 91 | [18F]30 | >98 | 51 ± 3 | 41 ± 4 |
| [68Ga]30 | >99 | 90 ± 5 | 37 ± 2 | |||
| 31 | >95 | 75 | [18F]31 | >95 | 61 ± 9 | 39 ± 6 |
| 32 | >97 | 74 | [18F]32 | >97 | 42 ± 8 | 40 ± 4 |
Synthesis of precursor molecules 15–32
4-(1,2,4,5-tetrazin-3-yl)benzoic acid
The title compound was synthesized according to the previously published procedure from Karver, et al.23 Obtained analytical data were consistent with the reported data. 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.66 (s, 1H), 8.62 (d, J = 8.3 Hz, 2H), 8.22 (d, J = 8.3 Hz, 2H). MS (ESI) m/z [M + Cl]−: 239.2.
2,2′-(7-(4-(4-(1,2,4,5-tetrazin-3-yl)benzamido)benzyl)-1,4,7-triazonane-1,4-diyl)diacetic acid (Tz-1-NODA, 15)
4-(1,2,4,5-tetrazin-3-yl)benzoic acid (26.7 μmol) was dissolved in DMSO (0.5 mL) before p-Bn-NODA-NCS (26.7 μmol) and triethylamine (5 μL) were added. The pink reaction mixture was stirred at room temperature for 1 h (general procedure for the isothiocyanate-amine addition reaction). After completion of the reaction (monitored by LC-MS), precursor 15 was purified (Rt = 12.4 min). The collected fraction was concentrated under reduced pressure and dried over night under high vacuum. Precursor 15 (purity >97%) was obtained as pink oil (8.9 mg, 58%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.32 (s, 1H), 8.41 (d, J = 8.2 Hz, 2H), 8.29 (d, J = 8.3 Hz, 2H), 7.62 (d, J = 7.8Hz, 2H), 7.37 (d, J = 7.5 Hz, 2H), 4.16 (s, 2H), 3.12–3.04 (m, 12H), 2.42–2.11 (m, 5H). MS (ESI) m/z [M + H]+: 535.4. HRMS (ESI) m/z calcd. for C26H30N8NaO5 [M + Na]+: 557.3256 found: 557.3348.
N6-(4-(1,2,4,5-tetrazin-3-yl)benzoyl)-N2-(tert-butoxycarbonyl)lysine (Tz-1-Lysine-Boc)
Tz 1 (5 mg, 26.7 μmol) was dissolved in dry DMF (0.5 mL) under nitrogen atmosphere before Boc-Lysine (5.8 mg, 29 μmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (10.7 mg, 60 μmol), and hydroxybenzotriazole (6.8 mg, 60 μmol) were added to the pink solution. The reaction mixture was stirred at room temperature for 4–6 h until all starting material was converted according to LC-MS (general amide coupling conditions). The mixture was purified (Rt = 14.1 min) and the product solution was subsequently concentrated and dried to yield the above shown compound as pink oil (6.4 mg, 58%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.41 (s, 1H), 8.52 (t, J = 7.9 Hz, 1H), 8.40 (d, J = 7.9 Hz, 2H), 7.76 (s, 1H), 7.54 (d, J = 8.0 Hz, 2H), 6.99 (d, J = 7.7 Hz, 1H), 3.00 (s, 2H), 2.84–2.73 (m, 3H), 1.70–1.49 (m, 4H), 1.42 (s, 9H). (ESI) m/z [M + H]+: 431.5.
2,2′,2′′-(2-(4-(6-(4-(1,2,4,5-tetrazin-3-yl)benzamido)-2-((tert-butoxycarbonyl)amino)-hexanamido)-benzyl)-1,4,7-triazonane-1,4,7-triyl)triacetic acid (Tz-1-Lysine-Boc-NOTA)
The title compound was obtained using Tz-1-Lysine-Boc (5 mg, 12.1 μmol) and an equimolar amount of p-Bn-NOTA-NCS, as well as the general conditions for the isothiocyanate-amine addition reaction. The product was purified (Rt = 16.6 min) and dried under reduced pressure and furnished as pink oil (8.2 mg, 78%). 1H NMR (500 MHz, Chloroform-d) δ (ppm): 10.20 (s, 1H), 8.53 (d, J = 8.0 Hz, 2H), 7.73 (d, J = 7.9 Hz, 2H), 7.56 (d, J = 8.2 Hz, 2H), 7.47 (d, J = 7.9 Hz, 2H), 7.37 (t, J = 7.9 Hz, 3H), 7.31 – 7.25 (m, 7H), 6.72 (s, 1H), 5.52 (s, 1H), 4.49 (dd, J = 9.8, 6.8 Hz, 7H), 4.16 (d, J = 21.1 Hz, 2H), 3.12–3.09 (m, 4H), 2.02–1.71 (m, 9H), 1.41 (s, 9H). (ESI) m/z [M + H]+: 821.7.
2,2′,2′′-(2-(4-(6-(4-(1,2,4,5-tetrazin-3-yl)benzamido)-2-aminohexanamido)benzyl)-1,4,7-triazonane-1,4,7-triyl)triacetic acid (16)
Tz-1-Lysine-Boc-NOTA (7 μmol) was dissolved in DCM (0.5 mL) before TFA (0.3 mL) was added dropwise to the solution under vigorous stirring. The mixture was stirred at room temperature for 45 min before DCM was removed under reduced pressure (General TFA-deprotection procedure). The remaining crude product was purified (Rt = 12.3 min, >96%), yielding precursor 14 as pink oil (4.8 mg, 90%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.10 (s, 1 H), 8.42 (d, J = 8.1 Hz, 2 H), 7.78 (d, J = 8.1 Hz, 2 H), 7.43 (d, J = 9.1 Hz, 2 H), 7.34 (d, J = 8.9 Hz, 2 H), 6.72 (s, 1 H), 5.52 (s, 1 H), 4.49 (m, 1 H), 4.16 (m, 2 H), 3.82–3.69 (m, 6 H), 3.12–3.09 (m, 9 H), 2.02–1.71 (m, 15 H); MS (ESI) m/z [M + H]+: 721.4. HRMS (ESI) calcd. for C34H44N14NaO8 [M + Na]+ m/z 743.3381 found: 743.3373.
tert-butyl-(1-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-1-oxo-5,8,11,14,17,20,23-heptaoxa-2-azapentacosan-25-yl)carbamate (Tz-1-PEG7-NHBoc)
Tz-1 (10 mg, 0.05 mmol) was dissolved in anhydrous dimethylsulfoxide (DMSO, 0.5 mL) before O-(2-aminoethyl)-O-[2-(boc-amino)ethyl]hexaethylene glycol (0.0375 mmol) and TEA (0.0057 mL, 0.0375 mmol) were added. The reaction mixture was stirred at room temperature for 1 h (general NHS ester coupling procedure). After completion of the reaction (monitored by LC-MS) the product was isolated and purified using preparative HPLC (Rt = 15.3 min). 1H NMR (500 MHz, Chloroform-d) δ (ppm): 10.21 (s, 1H), 8.58 (d, J = 8.2 Hz, 2H), 7.54 (d, J = 8.2 Hz, 2H), 6.89 (m, 2H), 6.45 (m, 3H), 5.08–4.88 (m, 4H), 4.56 (d, J = 8.0 Hz, 2H), 3.63 (d, J = 8.0 Hz, 2H), 3.54 (dt, J = 10.4, 4.9 Hz, 6H), 3.42 (q, J = 5.2 Hz, 3H), 3.33–3.27 (m, 5H), 2.37 (t, J = 10.2 Hz, 2H), 2.29 (d, J = 6.9 Hz, 2H), 2.01 (q, J = 7.1 Hz, 3H), 1.44 (s, 9H). MS (ESI) m/z [M + H]+: 653.6.
N-(23-amino-3,6,9,12,15,18,21-heptaoxatricosyl)-4-(1,2,4,5-tetrazin-3-yl)benzamide (Tz-1-PEG7-NH2)
The title compound was obtained from the Boc-protected starting material Tz-1-PEG7-NHBoc (5 mg, 6.7 μmol) using the general TFA deprotection procedure. After purification (Rt = 10.7 min) and lyophilization the product was furnished as a pink oil (3.9 mg, 89%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.17 (s, 1H), 8.47 (d, J = 8.1 Hz, 2H), 7.52 (d, J = 8.2 Hz, 2H), 6.58 (m, 2H), 6.32–6.12 (m, 3H), 5.11–5.02 (m, 4H), 4.46 (d, J = 6.0 Hz, 2H), 3.76 (d, J = 6.0 Hz, 2H), 3.42 (dt, J = 10.4, 4.9 Hz, 6H), 3.21 (q, J = 5.2 Hz, 3H), 3.33–3.27 (m, 6H), 2.37 (t, J = 10.2 Hz, 2H), 2.29 (d, J = 6.9 Hz, 2H), 2.01 (q, J = 7.1 Hz, 3H). MS (ESI) m/z [M + H]+: 553.8.
2,2′,2′′-(2-(4-(3-(1-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-1-oxo-5,8,11,14,17,20,23-heptaoxa-2-azapenta-cosan-25-yl)thioureido)benzyl)-1,4,7-triazonane-1,4,7-triyl)triacetic acid (17)
Precursor 17 was obtained using the starting material Tz-1-PEG7-NH2 (3.5 mg, 5.4 μmol) and p-Bn-NOTA-NCS (2.7 mg, 6 μmol) using the general isothiocyanate-amide addition reaction conditions as described above (see precursor 15). HPLC purification (>96%) and subsequent lyophilization furnished precursor 15 as a pink oil in high purities and good yield (5.1 mg, 83%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.51 (s, 1H), 8.39 (d, J = 7.7 Hz, 2H), 7.46 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 8.1 Hz, 2H), 7.12 (d, J = 8.1 Hz, 2H), 4.32 (d, J = 5.9 Hz, 3H), 3.92 (d, J = 17.9 Hz, 3H), 3.74 (d, J = 18.0 Hz, 3H), 3.56 (d, J = 5.8 Hz, 2H), 3.48 (d, J = 5.5 Hz, 2H), 3.33 (t, J = 8.2 Hz, 3H), 3.21–3.08 (m, 16H), 3.01–2.90 (m, 18H), 2.11 (t, J = 7.9 Hz, 2H), 2.03 (t, J = 5.4 Hz, 2H), 1.69 (p, J = 7.2 Hz, 3H). MS (ESI) m/z [M + H]+: 1004.1. HRMS (ESI) m/z calcd. for C45H66N10NaO14S [M + Na]+: 1026.5165 found: 1026.5156.
tert-butyl-(1-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-1-oxo-5,8,11,14,17,20,23,26,29,32,35-undecaoxa-2-azaheptatriacontan-37-yl)carbamate (Tz-1-PEG11-NHBoc)
The title compound was prepared according to the preparation of Tz-1-PEG7-NHBoc using O-(2-Aminoethyl)-O-[2-(Boc-amino)ethyl]decaethylene glycol (24.2 mg, 0.0375 mmol) instead. After completion of the reaction (monitored by HPLC, 5% MeCN/H to 95% MeCN over 20 min, Rt = 14.2 min, 1 mL/min) the product was purified using preparative HPLC (5% MeCN/H to 95% MeCN over 20 min, Rt = 14.5 min, 8 mL/min) with purity >95%. The product was furnished as a pink solid (18.2 mg, 96%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.65 (s, 1H), 8.77 (t, J = 6.4 Hz, 1H), 8.65–8.57 (m, 2H), 8.16–8.12 (m, 2H), 6.77–6.73 (m, 7H), 3.62–3.42 (m, 37 H), 3.38 (t, J = 6.3 Hz, 3H), 3.07 (q, J = 5.8 Hz, 2H), 1.38 (s, 9H). MS (ESI) m/z [M + H]+: 829.8.
N-(35-amino-3,6,9,12,15,18,21,24,27,30,33-undecaoxapentatriacontyl)-4-(1,2,4,5-tetrazin-3-yl)-benzamide (Tz-1-PEG11-NH2)
The title compound was furnished using Tz-1-PEG11-NHBoc (16.3 mg, 0.0216 mmol) and the standard TFA reaction conditions (see Tz-1-PEG7-NH2). The solvent was removed under reduced pressure before the deprotected product was purified via preparative HPLC (5% MeCN/H to 95% MeCN over 20 min, Rt = 11.6 min, 8 mL/min) with purity >97%. The product was furnished as a pink solid (14.4 mg, 92%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.59 (s, 1H), 8.74 (t, J = 5.2 Hz, 1H), 8.61–8.55 (m, 2H), 8.12–8.09 (m, 2H), 6.79–6.74 (m, 1H), 3.68–3.36 (m, 42H), 3.34 (t, J = 6.1 Hz, 4H), 3.10 (q, J = 6.0 Hz, 2H). MS (ESI) m/z [M+H]+: 829.7. HRMS (ESI) m/z calcd for C33H57N6O12 [M + H]+:729.7719 found: 729.7742.
2,2′,2′′-(2-(4-(3-(1-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-1-oxo-5,8,11,14,17,20,23,26,29,32,35-undecaoxa-2-azaheptatriacontan-37-yl)thioureido)benzyl)-1,4,7-triazonane-1,4,7-triyl)triacetic acid (Tz-1-PEG11-NOTA, 18)
Precursor 18 was obtained using Tz-1-PEG11-NH2 (13.5 mg, 0.0216 mmol) and NOTA-Bn-NCS (20.2 mg, 0.036 mmol) under the general conditions for isothiocyanate-amine addition reaction. After completion of the reaction (monitored by HPLC, 5% MeCN/H to 95% MeCN over 20 min, Rt = 13.5 min, 1 mL/min) the product was purified using preparative HPLC (5% MeCN/H2O to 95% MeCN over 30 min, Rt = 13.6 min, 8 mL/min) with purity >97%. The product was furnished as a pink solid (20.2 mg, 73%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.59 (s, 1 H), 8.47 (d, J = 7.3 Hz, 2 H), 7.87 (t, J = 7.2 Hz, 3 H), 7.55 (d, J = 7.6 Hz, 3 H), 7.43 (d, J = 7.5 Hz, 3 H), 7.20 (d, J = 7.7 Hz, 2 H), 4.41 (d, J = 5.8 Hz, 3 H), 4.00 (d, J = 17.5 Hz, 2 H), 3.82 (d, J = 17.9 Hz, 4 H), 3.51 (s, 49 H), 2.20 (t, J = 7.4 Hz, 3 H), 2.12 (t, J = 5.5 Hz, 3 H), 1.78–1.68 (m, 4 H). MS (ESI) m/z [M+H]+: 1180.9. HRMS (ESI) m/z calcd for C53H81N10O18S [M − H]−: 1178.2335 found: 1178.2345.
tert-butyl-(6-amino-1-((4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)amino)-1-oxohexan-2-yl)carbamate (Tz-2-Lysine-Boc)
The title compound was synthesized following the general amide coupling procedure as described for compound Tz-1-Lysine-NHBoc and identical amounts of material. After full conversion of the starting material (monitored by LC-MS), the product was purified using preparative HPLC (Rt = 14.4 min) and subsequently dried over night on the lyophilizer. The title compound was furnished as pink oil (6.8 mg, 59%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.52 (t, J = 5.9 Hz, 1H), 8.40 (d, J = 5.9 Hz, 2H), 7.76 (s, 2H), 7.54 (d, J = 8.0 Hz, 2H), 6.99 (d, J = 7.9 Hz, 1H), 3.00 (s, 3H), 2.84–2.73 (m, 2H), 1.70–1.49 (m, 3H), 1.42 (s, 6H), 1.35 (s, 9H). MS (ESI) m/z [M+H]+: 430.5.
2,2′,2′′-(2-(4-(3-(5-((tert-butoxycarbonyl)amino)-6-((4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)amino)-6-oxohexyl)thioureido)benzyl)-1,4,7-triazonane-1,4,7-triyl)triacetic acid (Tz-2-Lysine-Boc)
The title compound was synthesized according to the general isothiocyanate-amine addition procedure using Tz-2-Lysine-Boc (5 mg, 11.7 μmol) and p-Bn-NOTA-NCS (6.6 mg, 12 μmol). The target compound was purified (Rt = 15.1 min) and concentrated to yield the product as pink solid (8.8 mg, 86%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.50 (t, J = 6.1 Hz, 1H), 8.41 (d, J = 6.2 Hz, 1H), 7.98–7.88 (m, 2H), 7.56 (dd, J = 12.6, 8.2 Hz, 2H), 7.45 (d, J = 8.3 Hz, 2H), 6.98 (d, J = 12.8 Hz, 1H), 4.42 (d, J = 5.9 Hz, 1H), 4.32 (s, 1H), 3.96 (d, J = 5.9 Hz, 2H), 3.50 (s, 2H), 3.47 (s, 2H), 3.34 (s, 2H), 3.31 (s, 1H), 3.22–3.11 (m, 1H), 3.05 (dd, J = 13.5, 5.9 Hz, 1H), 2.99 (s, 1H), 2.82 (d, J = 8.2 Hz, 2H), 2.7–2.60 (m, 7H), 1.71–1.64 (m, 9H), 1.62–1.51 (m, 8H), 1.41 (s, 1H), 1.35 (s, 9H). MS (ESI) m/z [M + H]+: 881.1.
2,2′,2′′-(2-(4-(3-(5-amino-6-((4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)amino)-6-oxohexyl)thioureido)-benzyl)-1,4,7-triazonane-1,4,7-triyl)triacetic acid (19)
Deprotection of MeTz-Lysine-Boc-NOTA was performed under standard TFA deprotection conditions to yield precursor 19 as pink oil in quantitative yield (>98%) and high purity (>99%). compound was purified (Rt = 15.1 min) and concentrated to yield the product as pink solid (8.8 mg, 86%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.45 (t, J = 6.0 Hz, 1H), 8.40 (d, J = 6.1 Hz, 1H), 7.98–7.82 (m, 2H), 7.56 (dd, J = 12.5, 7.4 Hz, 2H), 7.41 (d, J = 12.2 Hz, 2H), 6.98 (d, J = 7.4 Hz, 1H), 4.42 (d, J = 5.9 Hz, 1H), 4.32 (s, 1H), 3.96 (d, J = 5.9 Hz, 2H), 3.70 (s, 2H), 3.57 (s, 2H), 3.44 (s, 2H), 3.36 (s, 1H), 3.32–3.21 (m, 1H), 3.12 (dd, J = 13.5, 5.9 Hz, 1H), 2.99 (s, 1H), 2.92 (d, J = 8.1 Hz, 2H), 2.70–2.51 (m, 7H), 2.21–1.94 (m, 9H), 1.92–1.81 (m, 10H). MS (ESI) m/z [M + H]+: 780.9. HRMS (ESI) m/z calcd for C36H49N11NaO7S [M + Na]+: 802.8435: 802.8431.
tert-butyl-(11-(4-aminobutyl)-15,15-dimethyl-1-(4-(6-methyl-1,2,4,5-tetrazin-3-yl)phenyl)-3,10,13-trioxo-14-oxa-2,9,12-triazahexadecan-4-yl)carbamate [Tz-2-(Lysine-Boc)2]
The title compound was synthesized using the general amide coupling procedure as presented for Tz-2-Lysine-Boc. In order to attach two lysine moieties instead of one, the molar ratios of all the reagents except the tetrazine were doubled as follows: (4-(1,2,4,5-Tetrazin-3-yl)phenyl)methanamine (5.2 mg, 30 μmol), Boc-Lysine (11.8 mg, 60 μmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (21.4 mg, 120 μmol) and hydroxybenzotriazole (13.6 mg, 120 μmol). After full conversion of the tetrazine starting material, the desired product was isolated and concentrated to yield the title compound as pink oil (7.4 mg, 38%). Note: the relatively low yield can be explained by the simultaneous formation of the mono- and tri-lysine compounds MeTz-Lysine-Boc-NH2 (3.1 mg, 34%) and MeTz-(Lysine-Boc)3-NH2 (4.2 mg, 28%), respectively. 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.47 (t, J = 6.0 Hz, 1H), 8.40 (d, J = 8.2 Hz, 1H), 7.78 (t, J = 6.1 Hz, 1H), 7.66 (s, 1H), 7.53 (d, J = 8.2 Hz, 1H), 6.94 (d, J = 8.0 Hz, 1H), 6.76 (d, J = 8.0 Hz, 1H), 4.41 (s, 1H), 3.97–3.89 (m, 10H), 3.87– 3.80 (m, 4H), 3.11 (s, 3H), 3.00 (s, 1H), 2.76 (q, J = 7.7, 6.4 Hz, 1H), 1.64–1.47 (m, 2H), 1.41 (s, 3H), 1.38 (s, 4H), 1.32–1.21 (m, 15H). MS (ESI) m/z [M + H]+: 658.8.
2,2′,2′′-(2-(4-(3-(5-((tert-butoxycarbonyl)amino)-6-((5-((tert-butoxycarbonyl)amino)-6-((4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)amino)-6-oxohexyl)amino)-6-oxohexyl)thioureido)benzyl)-1,4,7-triazonane-1,4,7-triyl)triacetic acid [Tz-2-(Lysine-Boc)2-NOTA]
The title compound was obtained using Tz-2-(Lysine-Boc)2 (5 mg, 7.6 μmol) and the general isothiocyanate-amine addition procedure as described above. Upon purification, the title compound was obtained as pink solid (6.4 mg, 76%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.42 (t, J = 6.1 Hz, 1H), 8.40 (d, J = 6.2 Hz, 1H), 7.88– 7.72 (m, 2H), 7.56 (dd, J = 12.5, 7.4 Hz, 2H), 7.41 (d, J = 7.2 Hz, 2H), 6.98 (d, J = 12.6 Hz, 1H), 4.42 (d, J = 5.7 Hz, 1H), 4.32 (s, 1H), 3.96 (d, J = 5.7 Hz, 2H), 3.70 (s, 2H), 3.57 (s, 2H), 3.44 (s, 2H), 3.36 (s, 1H), 3.32–3.21 (m, 1H), 3.12 (dd, J = 13.5, 5.9 Hz, 1H), 2.99 (d, J = 13.4 Hz, 1H), 2.91 (d, J = 5.9 Hz, 1H), 2.60–2.48 (m, 8H), 2.34–2.03 (m, 10H), 1.82–1.71 (m, 15H), 1.53 (s, 9H), 1.47 (s, 10H). MS (ESI) m/z [M + H]+: 1109.4.
2,2′,2′′-(2-(4-(3-(5-amino-6-((5-amino-6-((4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)amino)-6-oxohexyl)-amino)-6-oxohexyl)thioureido)benzyl)-1,4,7-triazonane-1,4,7-triyl)triacetic acid (20)
Starting from MeTz-(Lysine-Boc)2–NOTA (6.3 mg, 5.6 μmol), precursor 20 was obtained as a pink oil (3.9 mg, 77%, purity >98%) by applying the general TFA-based deprotection procedure. 1H NMR (500 MHz, DMSO-d6) δ (ppm): 9.11 (t, J = 5.9 Hz, 1H), 8.47 (d, J = 5.9 Hz, 1H), 8.23–8.18 (m, 2H), 8.16–8.07 (m, 2H), 7.74 (s, 1H), 7.59 (s, 1H), 4.62–4.41 (m, 1H), 3.90–3.79 (m, 8H), 3.74–3.64 (m, 5H), 3.10 (q, J = 6.6 Hz, 1H), 3.01 (s, 1H), 2.81–2.69 (m, 2H), 1.83–1.73 (m, 2H), 1.72–1.65 (m, 10H), 1.57–1.49 (m, 12H), 1.48–1.42 (m, 4H), 1.39–1.21 (m, 8H). MS (ESI) m/z [M+H]+: 909.4. HRMS (ESI) m/z calcd for C42H61N13NaO8S [M + Na]+: 931.4384 found: 931.4379.
tert-butyl-(18-(4-aminobutyl)-11-((tert-butoxycarbonyl)amino)-22,22-dimethyl-1-(4-(6-methyl-1,2,4,5-tetrazin-3-yl)phenyl)-3,10,17,20-tetraoxo-21-oxa-2,9,16,19-tetraazatricosan-4-yl)carbamate [Tz-2-(Lysine-Boc)3]
The title compound was obtained in the process of synthesizing Tz-2-(Lysine-Boc)2 as described above and was furnished as pink oil (4.2 mg, 28%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.47 (t, J = 6.0 Hz, 1H), 8.40 (d, J = 8.0 Hz, 2H), 7.76 (d, J = 6.1 Hz, 2H), 7.63 (s, 3H), 7.53 (d, J = 8.0 Hz, 2H), 6.93 (d, J = 7.9 Hz, 1H), 6.74 (d, J = 8.0, 2H), 4.41–4.28 (m, 2H), 3.92 (d, J = 7.6 Hz, 1H), 3.82 (d, J = 7.7 Hz, 2H), 3.00 (s, 7H), 2.83–2.70 (m, 6H), 1.67–1.44 (m, 13H), 1.53–1.40 (m, 27H). MS (ESI) m/z [M + H]+: 887.2.
2,2′,2′′-(2-(4-(3-(13,20-bis((tert-butoxycarbonyl)amino)-2,2-dimethyl-6-((4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)carbamoyl)-4,12,19-trioxo-3-oxa-5,11,18-triazatetracosan-24-yl)thioureido)benzyl)-1,4,7-triazonane-1,4,7-triyl)triacetic acid [Tz-2-(Lysine-Boc)3-NOTA]
The title compound was obtained using MeTz-(Lysine-Boc)3-NH2 (4.1 mg, 4.7 μmol) and the general isothiocyanate-amine addition procedure as described above. Upon purification, the title compound was obtained as pink oil (3.7 mg, 60%). 1H NMR (500 MHz, Chloroform-d) δ (ppm): 8.47 (t, J = 6.0 Hz, 1H), 8.40 (d, J = 8.0 Hz, 2H), 7.76 (d, J = 6.1 Hz, 2H), 7.63 (s, 3H), 7.53 (d, J = 8.0 Hz, 2H), 6.93 (d, J = 7.9 Hz, 1H), 6.74 (dd, J = 19.2, 8.0 Hz, 2H), 4.66 (s, 2H), 4.41 (d, J = 5.9 Hz, 2H), 3.92 (d, J = 7.6 Hz, 1H), 3.82 (d, J = 7.7 Hz, 2H), 3.00 (s, 7H), 3.56–3.49 (m, 10H) 2.83–2.70 (m, 6H), 2.53–2.41 (m, 13H), 1.67–1.44 (m, 13H), 1.53–1.40 (m, 27H). MS (ESI) m/z [M + H]+: 1337.8.
2,2′,2′′-(2-(4-(3-(5-amino-6-((5-amino-6-((5-amino-6-((4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)amino)-6-oxohexyl)amino)-6-oxohexyl)amino)-6-oxohexyl)thioureido)benzyl)-1,4,7-triazonane-1,4,7-triyl)triacetic acid (21)
The title compound was obtained using MeTz-(Lysine-Boc)3-NOTA (3.7 mg, 2.8 μmol) and the general isothiocyanate-amine addition procedure as described above. Upon purification, precursor 19 was obtained as pink oil (2.5 mg, 86%, purity >97%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.61 (s, J = 6.5 Hz, 1H), 8.32 (d, J = 8.0 Hz, 2H), 7.76 (d, J = 6.4 Hz, 2H), 7.53–7.49 (m, 2H), 7.47 (d, J = 8.0 Hz, 2H), 6.81 (d, J = 7.9 Hz, 1H), 6.74 (dd, J = 19.2, 8.0 Hz, 2H), 4.66 (s, 2 H), 4.32 (m, 2H), 3.92 (d, J = 7.1 Hz, 1H), 3.82 (d, J = 7.1 Hz, 2H), 3.51–3.40 (m, 7H), 3.32–3.29 (m, 18H) 2.72–2.60 (m, 5H), 2.63–2.44 (m, 12H), 1.61–1.42 (m, 11H). MS (ESI) m/z [M + H]+: 1037.3. HRMS (ESI) m/z calcd for C48H73N15NaO9S [M + Na]+: 1058.5334 found: 1058.5330.
2′-(7-(4-(3-(13,20-bis((tert-butoxycarbonyl)amino)-2,2-dimethyl-6-((4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)carbamoyl)-4,12,19-trioxo-3-oxa-5,11,18-triazatetracosan-24-yl)thioureido)benzyl)-1,4,7-triazonane-1,4-diyl)diacetic acid [Tz-2-(Lysine-Boc)3-NODA]
The title compound was obtained using MeTz-(Lysine-Boc)3-NH2 (3.5 mg, 2.9 μmol) and the general isothiocyanate-amine addition procedure using p-Bn-NODA-NCS as described above. Upon purification, the title compound was obtained as pink oil (3.6 mg, 68%). 1H NMR (500 MHz, Chloroform-d) δ (ppm): 8.47 (t, J = 6.0 Hz, 1H), 8.40 (d, J = 8.0 Hz, 2H), 7.76 (d, J = 6.1 Hz, 2H), 7.63 (s, 3H), 7.53 (d, J = 8.0 Hz, 2H), 6.93 (d, J = 7.9 Hz, 1H), 6.74 (dd, J = 19.2, 8.0 Hz, 2H), 4.66 (s, 2H), 4.41 (d, J = 5.9 Hz, 2H), 3.92 (d, J = 5.9 Hz, 1H), 3.82 (d, J = 7.7 Hz, 2H), 3.00 (s, 7 H), 3.56–3.49 (m, 9H) 2.89–2.70 (m, 10H), 2.53–2.41 (m, 6H), 1.77–1.71 (m, 11H), 1.67–1.32 (m, 29H). MS (ESI) m/z [M + H]+: 1279.8.
2,2′-(7-(4-(3-(5-amino-6-((5-amino-6-((5-amino-6-((4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)amino)-6-oxohexyl)amino)-6-oxohexyl)amino)-6-oxohexyl)thioureido)benzyl)-1,4,7-triazonane-1,4-diyl)diacetic acid (22)
The title compound was obtained using MeTz-(Lysine-Boc)3-NODA (3.5 mg, 2.7 μmol) and the general isothiocyanate-amine addition procedure as described above. Upon purification, precursor 20 was obtained as pink oil (2.5 mg, 94%, purity >97%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.79 (t, J = 5.4 Hz, 2H), 8.60 (d, J = 8.4 Hz, 2H), 8.13 (d, J = 8.4 Hz, 2H), 7.43 (d, J = 5.3 Hz, 2H), 7.20 (d, J = 8.3 Hz, 1H), 7.11 (d, J = 8.2 Hz, 1H), 4.00 (d, J = 17.6 Hz, 2H), 3.81 (d, J = 17.4 Hz, 3H), 3.64 (s, 3H), 3.57–3.45 (m, 39H), 3.31–3.20 (m, 1H), 3.05–2.94 (m, 5H), 2.86–2.74 (m, 8H). MS (ESI) m/z [M + H]+: 988.4.
4-((4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)amino)-4-oxobutanoic acid (Tz-2-succinate)
The title compound was obtained using Tz-2 amine (10 mg, 49 μmol), succinic anhydride (5 mg, 50 μmol), and TEA (5 μL) in dry DMF. The mixture was stirred at room temperature for 4 h and monitored by LC-MS. The product was then purified as pink solid (11.6 mg, 79%) using preparative HPLC and subsequent drying. 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.60 (d, J = 8.4 Hz, 2H), 8.13 (d, J = 8.4 Hz, 2H), 7.73 (s, 1 H, NH), 4.21 (s, 2H), 3.81–3.77 (m, 2H), 3.57–3.49 (m, 2H), 2.74 (s, 3H). MS (ESI) m/z [M + H]+: 302.4.
tert-butyl-(1-(4-(6-methyl-1,2,4,5-tetrazin-3-yl)phenyl)-3,6-dioxo-10,13,16,19,22,25,28–31,34,37,40-undecaoxa-2,7-diazadotetracontan-42-yl)carbamate (Tz-2-PEG11-NHBoc)
The title compound was obtained from Tz-2-succinate (10 mg, 30.1 μmol) and NH2-PEG11-NHBoc (1.2 eq.) using the amide coupling conditions requiring in situ acid activation. Upon purification and lyophilization, the title compound was obtained as pink solid (19 mg, 68%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.79 (t, J = 5.4 Hz, 2H), 8.60 (d, J = 5.4 Hz, 2H), 4.21 (s, 2H), 4.08–4.02 (m, 5H), 3.81–3.77 (m, 2H), 3.57–3.49 (m, 2H), 3.47–3.33 (m, 24H) 2.74 (s, 3H), 2.67–2.46 (m, 24H), 1.45 (s, 7H). MS (ESI) m/ 929.2 [M + H]+: 929.2.
N1-(35-amino-3,6,9,12,15,18,21,24,27,30,33-undecaoxapentatriacontyl)-N4-(4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)succinamide (Tz-2-PEG11-NH2)
The title compound was obtained using Tz-2-PEG11-NHBoc (17 mg, 18.3 μmol) as starting material and the standard TFA-mediated deprotection conditions, furnishing the title compounds as pink oil (11.5 mg, 76%); 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.58 (d, J = 5.9 Hz, 2H), 8.67 (d, J = 5.9 Hz, 2H), 4.21 (s, 2H), 4.18–4.12 (m, 4H), 3.77–3.69 (m, 9H), 3.52–3.32 (m, 8H), 3.27–3.13 (m, 21H) 2.71 (s, 3H), 2.44–2.26 (m, 14H). MS (ESI) m/z [M + H]+: 828.9.
2,2′,2′′-(2-(4-(3-(1-(4-(6-methyl-1,2,4,5-tetrazin-3-yl)phenyl)-3,6-dioxo-10,13,16,19,22–25,28,31–34,37,40-undecaoxa-2,7-diazadotetracontan-42-yl)thioureido)benzyl)-1,4,7-triazonane-1,4,7-triyl)triacetic acid (23)
The title compound was obtained using Methyltetrazine-PEG12-NHS ester (6.8 mg, 7.5 μmol) and p-Bn-NOTA-NH2 (5.6 mg, 10 μmol) and the general NHS ester coupling procedure as described for the synthesis of Tz-PEG7-NHBoc. Precursor 23 was furnished as pink oil (7.1 mg, 89%, purity >98%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.43 (d, J = 5.7 Hz, 2H), 7.54 (d, J = 5.7 Hz, 2H), 7.23 (d, J = 8.6 Hz, 2H), 7.17 (d, J = 8.5 Hz, 2H), 7.11 (s, 1H), 6.56 (s, 2H), 4.27–4.23 (m, 7H), 3.83–3.79 (m, 8H), 3.70 (t, J = 6.3 Hz, 7H), 3.65–3.61 (m, 4H), 3.59–3.49 (m, 49H), 2.97–2.76 (m, 6H). MS (ESI) m/z [M + H]+: 1279.5. HRMS (ESI) m/z calcd for C58H91N11NaO19S [M + Na]+: 1301.5836 found: 1301.5831.
6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-amine (3)
The title compound was synthesized according to the previously published procedure from Devaraj, et al.1 Obtained yield and analytical data were consistent with the reported data. 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.89 (s, 1H), 8.53 (s, 1H), 8.58–8.51 (m, 2H), 7.99–7.92 (m, 2H), 7.61 (d, J = 8.5 Hz, 1H), 7.48 (m, 1H), 7.10–6.99 (dd, J = 8.6 Hz, 2.7 Hz, 1H), 5.78 (s, 2H).
4-oxo-4-((6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)amino)butanoic acid (Tz-3-succinate)
The title compound was obtained using the same procedure as stated for Tz-2-succinate (same molarities). Tz-2-succinate was furnished as pink solid (9.6 mg, 55%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.95 (s, 1H), 8.79 (t, J = 5.4 Hz, 1H), 8.60 (t, J = 8.4 Hz, 1H), 8.33 (d, J = 8.4 Hz, 1H), 8.29 (dd, J = 5.4 Hz, 1H), 8.10 (dd, J = 8.6 Hz, 1H), 7.52–7.44 (m, 2H), 7.13–7.10 (m, 1H), 2.72–2.63 (m, 4H). MS (ESI) m/z [M + H]+: 352.4.
tert-butyl-(37,40-dioxo-40-((6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)amino)-3,6,9,12,15,18,–21,24,27,30,33-undecaoxa-36-azatetracontyl)carbamate (Tz-3-PEG11-NHBoc)
The title compound was obtained using the general amide bond formation procedure. The title compound was then obtained as pink oil (4.5 mg, 4.6 μmol). 1H NMR (500 MHz, Chloroform-d) δ (ppm): 10.16 (s, 1H), 9.09–9.03 (m, 2H), 8.98 (d, J = 5.6 Hz, 1H), 8.72 (dd, J = 11.1, 8.3 Hz, 2H), 8.55 (dd, J = 8.4, 2.4 Hz, 1H), 8.01 (dd, J = 8.3, 2.2 Hz, 1H), 7.60–7.56 (m, 2H), 3.68–3.56 (m, 37H), 3.55– 3.51 (m, 4H), 3.48 (t, J = 7.7 Hz, 3H), 3.30 (s, 4H), 2.84 (t, J = 7.6 Hz, 3H), 2.71–2.67 (m, 1H), 1.44 (s, 9H). MS (ESI) m/z [M + H]+: 979.3.
N1-(35-amino-3,6,9,12,15,18,21,24,27,30,33-undecaoxapentatriacontyl)-N4-(6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)succinamide (Tz-3-PEG11-NH2)
The title compound was obtained from Tz-3-PEG11-NHBoc (4.3 mg, 4.4 μmol) using the standard TFA deprotection protocol. The title compound was obtained as pink oil (3.8 mg, 98%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 8.62 (dd, J = 15.9, 8.2 Hz, 1H), 8.42 (dd, J = 8.2, 2.3 Hz, 1H), 8.19–8.14 (m, 4H), 7.99 (t, J = 7.4 Hz, 1H), 7.74 (dd, J = 7.5, 2.7 Hz, 2H), 3.62–3.37 (m, 49H), 3.22 (q, J = 5.6 Hz, 2H), 2.99 (t, J = 5.5 Hz, 2H), 2.68 (t, J = 5.8 Hz, 1H). MS (ESI) m/z [M + H]+: 878.9.
2,2′,2′′-(2-(4-(3-(37,40-dioxo-40-((6-(6-(pyridin-2-yl)-1,2,4,5-tetrazin-3-yl)pyridin-3-yl)amino)-3,6,9,12,15,–18,21,24,27,30,33-undecaoxa-36-azatetracontyl)thioureido)benzyl)-1,4,7-triazonane-1,4,7-triyl)triacetic acid (24)
The title compound was obtained from Tz-3-PEG11-NH2 (3.6 mg, 4.1 μmol) and p-Bn-NOTA-NCS using the standard isothiocyanate-amine addition procedure. Precursor 24 was obtained as pink oil (4.4 mg, 81%, purity >96%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.64 (s, 1H), 9.07 (s, 1H), 8.94 (d, J = 4.8 Hz, 2H), 8.61 (dd, J = 15.5, 8.9 Hz, 1H), 8.42 (dd, J = 9.0, 4.8 Hz, 1H), 8.16 (t, J = 8.0 Hz, 1H), 7.99 (t, J = 6.0 Hz, 1H), 7.74 (dd, J = 7.9, 5.9 Hz, 2H), 7.43 (d, J = 8.3 Hz, 1H), 7.20 (d, J = 8.4 Hz, 1H), 4.34–4.21 (m, 16H), 4.00 (d, J = 18.2 Hz, 1H), 3.81 (d, J = 18.3 Hz, 1H), 3.58–3.47 (m, 45H), 3.42–3.32 (m, 5H), 3.23–3.28 (m, 6H), 2.67–2.54 (m, 3H). MS (ESI) m/z [M+H]+: 1329.7. HRMS (ESI) m/z calcd for C60H89N13NaO19S [M + Na]+: 1351.6016 found: 1351.6012.
tert-butyl-(1-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-1-oxo-5,8,11,14,17,20,23-heptaoxa-2-azapentacosan-25-yl)carbamate (Tz-4-PEG7-NHBoc)
The title compound was obtained using the NHS-activated Tz 4 (6.1 mg, 30 μmol), NH2-PEG7-NHBoc (12.4 mg, 26.4 μmol), and TEA (4.5 μL, 3 eq.) in anhydrous DMSO (400 μL). The resulting pink mixture was stirred at room temperature for 1 h. The desired compound was furnished as pink oil (14.3 mg, 85%), and all spectral data were in line with previously published reports.2 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.21 (s, 1H), 8.43–8.31 (m, 2H), 7.75–7.71 (m, 1H), 7.54 (s, 2H), 6.69–6.54 (m, 1H), 4.33–4.28 (m, 3H), 3.48–3.39 (m, 22H), 3.33–3.28 (m, 2H), 3.39–3.19 (m, 14H), 1.29 (s, 9H).
N1-(4-(1,2,4,5-tetrazin-3-yl)benzyl)-N5-(23-amino-3,6,9,12,15,18,21-heptaoxatricosyl)-glutaramide
The title compound (9.6 mg, 79%) was obtained using the standard TFA deprotection conditions and in accordance to previously published reports. All spectral data were in line with previously published results.2 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.21 (s, 1H), 8.76–8.72 (m, 1H), 7.72–7.69 (m, 2H), 7.51 (s, 2H), 6.62–6.52 (m, 2H), 4.30–4.24 (m, 4H), 3.58–3.51 (m, 18H), 3.48–3.42 (m, 3H), 3.31–3.22 (m, 16H).
2,2′-(7-(4-(3-(1-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-1-oxo-5,8,11,14,17,20,23-heptaoxa-2-azapentacosan-25-yl)thioureido)benzyl)-1,4,7-triazonane-1,4-diyl)diacetic acid (Tz-4-PEG7-NODA, 25)
Precursor 25 (6.6 mg, 77%, purity >95%) was obtained as pink solid using Tz-4-PEG7-NH2 (5 mg, 9.1 μmol) and p-Bn-NODA-NCS as well as the standard isothiocyanate-amine addition conditions. 1H NMR (500 MHz, DMSO-d6) δ (ppm): 9.91 (s, 1H), 8.50 (d, J = 5.9 Hz, 1H), 8.42 (d, J = 5.9 Hz, 1H), 7.74–7.69 (m, 2H), 7.54 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 8.1 Hz, 2H), 4.41 (d, J = 5.9 Hz, 2H), 3.99 (d, J = 5.8 Hz, 2H), 3.83 (d, J = 5.0 Hz, 1H), 3.79 (s, 1H), 3.68 (dd, J = 11.0, 4.9 Hz, 2H), 3.55–3.44 (m, 31H), 3.43–3.24 (m, 5H), 3.18 (s, 15H), 2.41–2.32 (m, 5H). MS (ESI) m/z [M + H]+: 1045.4. HRMS (ESI) m/z calcd for C48H73N11NaO13S [M + Na]+: 1067.4324 found: 1067.4318.
2,2′,2′′-(2-(4-(3-(1-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-3,7-dioxo-11,14,17,20,23,26,29-heptaoxa-2,8-diaza-hentriacontan-31-yl)thioureido)benzyl)-1,4,7-triazonane-1,4,7-triyl)-triacetic acid (26)
Precursor 26 (4.1 mg, 45%, purity >98%) was obtained as pink solid using Tz-4-PEG7-NH2 (5 mg, 9.1 μmol) and p-Bn-NOTA-NCS as well as the standard isothiocyanate-amine addition conditions. All obtained spectral data was in line with previously published reports.2 1H NMR (500 MHz, DMSO-d6) δ (ppm) 10.51 (s, 1H), 9.50 (s, 1H), 8.40–8.27 (m, 3H), 7.79–7.68 (m, 1H), 7.62–7.59 (m, 1H), 7.47 (d, J = 5.9 Hz, 2H), 7.35 (d, J = 5.8 Hz, 2H), 7.03–6.92 (m, 2H), 4.43 (d, J = 7.4 Hz, 2H), 4.00–3.20 (m, 48H), 3.12–3.02 (m, 6H), 2.11–2.01 (m, 6 H).
tert-butyl-(1-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-3,7-dioxo-11,14,17,20,23,26,29,32,35,38,41-undecaoxa-2,8-diazatritetracontan-43-yl)carbamate (Tz-4-PEG11-NHBoc)
The title compound was synthesized according to the PEG7 derivative (identical stoichiometry) and obtained as pink oil (15.7 mg, 63%). All spectral data were in line with previously published reports.1 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.65 (s, 1H), 8.77 (t, J = 5.4 Hz, 1H), 8.65–8.57 (m, 2H), 8.16–8.12 (m, 2H), 6.77–6.73 (m, 1H), 3.62–3.42 (m, 46H), 3.38 (t, J = 6.1 Hz, 3H), 3.07 (q, J = 5.8 Hz, 2H), 1.38 (s, 9H).
N1-(4-(1,2,4,5-tetrazin-3-yl)benzyl)-N5-(35-amino-3,6,9,12,15,18,21,24,27,30,33-undecaoxapenta-triacontyl)-glutaramide (Tz-4-PEG11-NH2)
The title compound was synthesized according to the PEG7 derivative using the TFA deprotection conditions and was obtained as pink oil (10.9 mg, 89%). All spectral data were in line with previously published reports.1 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.59 (s, 1H), 8.74 (t, J = 5.2 Hz, 1H), 8.61–8.55 (m, 2H), 8.12–8.09 (m, 2H), 6.79–6.74 (m, 1H), 3.68–3.36 (m, 46H), 3.34 (t, J = 6.1 Hz, 3H), 3.10 (q, J = 5.8 Hz, 2H).
2,2′-(7-(4-(3-(1-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-3,7-dioxo-11,14,17,20,23,26,29,32,35–38,41-undecaoxa-2,8-diazatritetracontan-43-yl)thioureido)benzyl)-1,4,7-triazonane-1,4-diyl)-diacetic acid (27)
Precursor 27 was synthesized according to the PEG7 derivative using the standard isothiocyanate-amine addition conditions and isolated as pink solid (12.2 mg, 73%, purity >98%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.66 (s, 1H), 8.78 (t, J = 8.2 Hz, 1H), 8.60 (d, J = 8.2 Hz, 1H), 8.14 (d, J = 8.2 Hz, 2H), 7.67–7.61 (m, 3H), 7.43 (d, J = 8.1 Hz, 2H), 7.20 (d, J = 8.4 Hz, 2H), 4.00 (d, J = 18.2 Hz, 1H), 3.82 (d, J = 18.1 Hz, 2H), 3.61–3.43 (m, 63H), 3.36–3.21 (m, 1H), 3.13–2.99 (m, 6H), 2.87–2.72 (m, 2H), 2.64 (d, J = 12.4 Hz, 2H). MS (ESI) m/z [M + H]+: 1207.5. HRMS (ESI) m/z calcd for C56H89N10NaO17S [M + Na]+: 1229.5341 found: 1229.5333.
2,2′,2′′-(2-(4-(3-(1-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-3,7-dioxo-11,14,17,20,23,26,29,32–35,38,41-undecaoxa-2,8-diazatritetracontan-43-yl)thioureido)benzyl)-1,4,7-triazonane-1,4,7-triyl)triacetic acid (28)
Precursor 28 was synthesized as recently reported.1 Analytical data matched previously established protocols. 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.59 (s, 1H), 8.47 (d, J = 7.3 Hz, 3H), 7.87 (t, J = 5.2 Hz, 3H), 7.55 (d, J = 7.6 Hz, 3H), 7.43 (d, J = 8.1 Hz, 3H), 7.20 (d, J = 7.7 Hz, 2H), 4.41 (d, J = 5.8 Hz, 3H), 4.00 (d, J = 17.5 Hz, 2H), 3.82 (d, J = 17.9 Hz, 4H), 3.51 (s, 53H), 2.20 (t, J = 7.4 Hz, 3H), 2.12 (t, J = 7.5 Hz, 5H), 1.78 (dt, J = 14.4, 7.2 Hz, 4H).
tert-butyl-(1-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-38-amino-3,7,33-trioxo-11,14,17,20,23,26,29-heptaoxa-2,8,32-triazaoctatriacontan-34-yl)carbamate (Tz-4-PEG7-Lysine-Boc)
The title compound was obtained using Tz-4-PEG7-NH2 (8.6 mg, 15.6 μmol) and the standard amide coupling conditions using Boc-lysine (5.0 mg, 20 μmol). The title compound was furnished as pink oil (9.2 mg, 76%). 1H NMR (500 MHz, DMSO-d6) δ (ppm): 9.91 (s, 1H), 8.50 (d, J = 5.9 Hz, 1H), 8.42 (d, J = 5.9 Hz, 1H), 7.54 (d, J = 7.9 Hz, 1H), 7.18 (d, J = 8.0 Hz, 1H), 4.41 (d, J = 5.9 Hz, 2H), 3.99–3.77 (m, 6H), 3.83 (d, J = 5.9 Hz, 2H), 3.79 (s, 1H), 3.68 (dd, J = 11.0, 4.7 Hz, 2H), 3.55–3.44 (m, 34H), 3.43–3.24 (m, 5H), 3.18 (s, 1H), 2.44 (t, J = 4.6 Hz, 2H), 1.67 (s, 9H). MS (ESI) m/z [M + H]+: 881.1.
2,2′-(7-(4-(3-(1-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-34-((tert-butoxycarbonyl)amino)-3,7,33-trioxo-11,14,17,20–23,26,29-heptaoxa-2,8,32-triazaoctatriacontan-38-yl)thioureido)benzyl)-1,4,7-triazonane-1,4-diyl)diacetic acid (Tz-4-PEG7-Lysine-Boc-NODA)
The title compound (1.9 mg, 72%) was obtained as pink solid using Tz-4-PEG7-Lysine-Boc-NH2 (1.5 mg, 2 μmol), p-Bn-NODA-NCS (1.3 mg, 3 μmol), as well as the general isothiocyanate-amine addition conditions. 1H NMR (500 MHz, DMSO-d6) δ (ppm): 9.82 (s, 1H), 8.43 (d, J = 5.6 Hz, 2H), 8.41 (d, J = 5.6 Hz, 2H), 7.24 (d, J = 7.9 Hz, 1H), 7.16 (d, J = 8.0 Hz, 2H), 6.9 (d, J = 8.4 Hz, 2H), 4.55–4.51 (m, 1H), 3.92 (d, J = 17.8 Hz, 2H), 3.83 (d, J = 5.0 Hz, 2H), 3.79–3.70 (m, 15H), 3.68 (dd, J = 17.6, 4.9 Hz, 2H), 3.55–3.44 (m, 47H), 3.41–3.34 (m, 3H), 2.44–2.38 (m, 2H), 1.67 (s, 9H). MS (ESI) m/z [M + H]+: 1273.6.
2,2′-(7-(4-(3-(1-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-34-amino-3,7,33-trioxo-11,14,17,20,23–26,29-heptaoxa-2,8,32-triazaoctatriacontan-38-yl)thioureido)benzyl)-1,4,7-triazonane-1,4-diyl)diacetic acid (29)
Precursor 29 (1.3 mg, 81%, purity >97%) was obtained as pink solid from the previous title compound (1.9 mg, 1.5 μmol) using standard TFA deprotection conditions. 1H NMR (500 MHz, DMSO-d6) δ (ppm): 9.91 (s, 1H), 8.50 (d, J = 5.9 Hz, 1H), 8.42–8.35 (m, 2H), 7.54 (d, J = 5.9 Hz, 1H), 7.18 (d, J = 8.1 Hz, 2H), 6.89 (d, J = 8.1 Hz, 2H), 3.99 (d, J = 17.8 Hz, 2H), 3.83 (d, J = 4.4 Hz, 2H), 3.79–3.70 (m, 15H), 3.68 (dd, J = 17.5, 4.2 Hz, 2H), 3.55–3.44 (m, 43H), 3.43–3.24 (m, 4H), 2.44–2.33 (m, 8H); MS (ESI) m/z [M + H]+: 1273.6. HRMS (ESI) m/z calcd for C54H85N13NaO14S [M + Na]+: 1195.5273 found: 1195.5263. tert-butyl-(1-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-47-(1H-imidazol-4-yl)-3,7,45-trioxo-11,14,17,20,23,26–29,32,35,38,41-undecaoxa-2,8,44-triazaheptatetracontan-46-yl)carbamate (Tz-4-PEG11-His-Boc). The title compound (12.7 mg, 72%) was obtained as pink solid using Tz-4-PEG11-NH2 (10 mg, 18 μmol), Boc-Histidine (5.9 mg, 25 μmol), and the general reaction conditions for amide bond formations. 1H NMR (500 MHz, Chloroform-d) δ (ppm): 12.53 (s, 1H), 9.66 (s, 1H), 8.50 (d, J = 5.9 Hz, 1H), 8.42 (d, J = 5.8 Hz, 1H), 8.35 (s, 1H), 7.54 (d, J = 7.9 Hz, 1H), 7.41 (s, 1H), 7.18 (d, J = 8.0 Hz, 1H), 7.09–7.01 (m, 2H), 4.23 (dd, J = 17.8, 6.5 Hz, 1H), 4.08 (dd, J = 17.5, 6.2 Hz, 1H), 3.99 (d, J = 17.8 Hz, 2H), 3.92 (dd, J = 16.3, 6.1 Hz, 1H), 3.83 (d, J = 5.0 Hz, 2H), 3.79–3.70 (m, 15H), 3.68 (dd, J = 16.1, 4.9 Hz, 2H), 3.51–3.35 (m, 27H), 3.33–3.21 (m, 4H), 2.42–2.23 (m, 6H), 1.82 (s, 9H). MS (ESI) m/z [M + H]+: 1066.4.
N1-(4-(1,2,4,5-tetrazin-3-yl)benzyl)-N5-(38-amino-39-(1H-imidazol-4-yl)-37-oxo-3,6,9,12,15,18,21,24–27,30,33-undecaoxa-36-azanonatriacontyl)glutaramide (Tz-4-PEG11-His)
The title compound (8.8 mg, 87%) was obtained as pink solid using Tz-4-PEG11-Histidine-Boc-NH2 (12.7 mg, 13 μmol) and the general TFA deprotection procedure. 1H NMR (500 MHz, DMSO-d6) δ (ppm): 12.71 (s, 1H), 9.34 (s, 1H), 8.53 (d, J = 8.0 Hz, 1H), 8.33 (d, J = 8.0 Hz, 1H), 8.18 (s, 1H), 7.55 (d, J = 7.9 Hz, 1H), 7.45 (s, 1H), 7.21 (d, J = 7.9 Hz, 1H), 7.11–7.02 (m, 3H), 4.56 (dd, J = 17.8, 6.5 Hz, 1H), 4.22–4.13 (m, 2H), 4.01 (dd, J = 17.6, 6.3 Hz, 1H), 3.89 (d, J = 17.8 Hz, 2H), 3.84 (dd, J = 17.6, 6.3 Hz, 1H), 3.78 (d, J = 6.2 Hz, 2H), 3.71–3.60 (m, 13H), 3.52 (dd, J = 17.4, 6.2 Hz, 2H), 3.41–3.35 (m, 26H), 3.29–3.21 (m, 5H), 2.42–2.23 (m, 6H). MS (ESI) m/z [M + H]+: 966.2.
2,2′-(7-(4-(3-(1-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-41-(1H-imidazol-4-yl)-1,39-dioxo-5,8,11,14,17–20,23,26-,29,32,35-undecaoxa-2,38-diazahentetracontan-40-yl)thioureido)-benzyl)-1,4,7-triazonane-1,4-diyl)diacetic acid (Tz-4-PEG11-His-NODA, 30)
Precursor 30 (6.8 mg, 91%, purity >98%) was furnished as a pink oil using Tz-4-PEG11-Histidine-NH2 (5 mg, 5.8 μmol), p-Bn-NODA-NCS, and the established isothiocyanate-amine addition procedure. 1H NMR (500 MHz, DMSO-d6) δ (ppm): 12.61 (s, 1H), 9.71 (s, 1H), 8.74 (d, J = 5.3 Hz, 1H), 8.51 (d, J = 5.2 Hz, 1H), 8.01 (s, 1H), 7.49 (d, J = 7.5 Hz, 1H), 7.41 (s, 1H), 7.33 (d, J = 7.6 Hz, 2H), 7.20 (d, J = 7.9 Hz, 1H), 7.14 (m, 4H), 6.89 (d, J = 7.7 Hz, 2H), 4.78 (dd, J = 15.3, 6.2 Hz, 1H), 4.22–4.04 (m, 8H), 4.01 (dd, J = 15.1, 6.2 Hz, 1H), 3.89 (d, J = 17.8 Hz, 2H), 3.84 (dd, J = 17.5, 5.1 Hz, 1H), 3.78 (d, J = 5.0 Hz, 2H), 3.71–3.60 (m, 13H), 3.58 (dd, J = 10.0, 4.2 Hz, 2H), 3.52–3.44 (m, 11H) 3.41–3.35 (m, 25H), 3.29–3.21 (m, 4H), 2.12–2.02 (m, 10H). MS (ESI) m/z [M + H]+: 1358.7. HRMS (ESI) m/z calcd for C62H96N14NaO18S [M + Na]+: 1380.7961 found: 1380.7955.
tert-butyl-(51-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-1-amino-1-imino-7,45,49-trioxo-11,14,17,20–23,26–29,32,35,38,41-undecaoxa-2,8,44,50-tetraazahenpentacontan-6-yl)carbamate (Tz-4-PEG11-Arg-Boc)
The title compound (6.8 mg, 80%) was obtained as pink solid using Tz-4-PEG11-NH2 (5 mg, 9 μmol), Boc-Arginine (5 mg, 13 μmol), and the general reaction conditions for amide bond formations. 1H NMR (500 MHz, DMSO-d6) δ (ppm): 9.88 (s, 1H), 8.45 (d, J = 6.1 Hz, 1H), 8.34 (d, J = 6.2 Hz, 1H), 8.24 (s, 1H), 7.54 (d, J = 7.9 Hz, 1H), 7.19 (d, J = 7.9 Hz, 1H), 7.12 (s, 1H), 4.19 (d, J = 5.9 Hz, 2H), 3.91 (d, J = 17.8 Hz, 2H), 3.83 (d, J = 5.8 Hz, 2H), 3.79 (s, 1H), 3.68 (dd, J = 11.0, 4.7 Hz, 2H), 3.55–3.44 (m, 38H), 3.41–3.23 (m, 19H), 3.17 (s, 1H), 2.33–2.28 (m, 2H), 1.52 (s, 9H). MS (ESI) m/z [M + H]+: 1085.4.
N1-(4-(1,2,4,5-tetrazin-3-yl)benzyl)-N5-(1,6-diamino-1-imino-7-oxo-11,14,17,20,23,26,29–32,35,38,41-undecaoxa-2,8-diazatritetracontan-43-yl)glutaramide (Tz-4-PEG11-Arg)
The title compound (4.7 mg, 84%) was obtained as pink solid using Tz-4-PEG11-Arginine-Boc-NH2 (6.6 mg, 6.3 μmol) and the general TFA deprotection procedure. 1H NMR (500 MHz, DMSO-d6) δ 10.21 (s, 1H), 8.69 (d, J = 6.7 Hz, 1H), 8.52 (d, J = 6.8 Hz, 1H), 8.42 (s, 1H), 7.83 (d, J = 7.2 Hz, 1H), 7.52 (d, J = 7.3 Hz, 1H), 7.11 (s, 1H), 7.03 (s, 1H), 4.19 (d, J = 5.9 Hz, 2H), 4.08–3.98 (m, 4H), 3.91 (d, J = 13.2 Hz, 2H), 3.83 (d, J = 5.8 Hz, 2H), 3.79 (s, 1H), 3.68 (dd, J = 13.2, 3.9 Hz, 2H), 3.55–3.44 (m, 35H), 3.41–3.23 (m, 11H), 3.17 (s, 1H), 2.33–2.24 (m, 8H). MS (ESI) m/ 985.2 [M + H]+: 985.2.
2,2′-(7-(4-(3-(51-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-1-amino-1-imino-7,45,49-trioxo-11,14,17–20,23–26,29,32,35,38,41-undecaoxa-2,8,44,50-tetraazahenpentacontan-6-yl)thioureido)benzyl)-1,4,7-triazonane-1,4-diyl)diacetic acid (31)
Precursor 31 (5.1 mg, 75%, purity >95%) was furnished as a pink oil using Tz-4-PEG11-Arginine-NH2 (4.7 mg, 5.3 μmol), p-Bn-NODA-NCS, and the established isothiocyanate-amine addition procedure. 1H NMR (500 MHz, DMSO-d6) δ (ppm): 12.61 (s, 1H), 9.71 (s, 1H), 8.74 (d, J = 8.2 Hz, 1H), 8.66 (s, 1H), 8.51 (d, J = 8.2 Hz, 1H), 8.01 (s, 1H), 7.49 (d, J = 7.5 Hz, 1H), 7.41 (s, 1H), 7.33 (d, J = 7.5 Hz, 2H), 7.20 (d, J = 7.9 Hz, 1H), 7.14–7.04 (m, 4H), 6.89 (d, J = 6.7 Hz, 2H), 4.74 (dd, J = 6.6, 13.3 Hz, 1H), 4.22–4.14 (m, 9H), 4.09 (dd, J = 12.8, 6.3 Hz, 1H), 3.92 (d, J = 12.9 Hz, 2H), 3.84 (dd, J = 6.2, 16.1 Hz, 1H), 3.76 (d, J = 6.2 Hz, 2 H), 3.73–3.64 (m, 11H), 3.62 (dd, J = 9.6, 6.1 Hz, 2H), 3.58–3.54 (m, 5H) 3.51–3.45 (m, 17H), 3.39–3.31 (m, 9H), 2.31–2.02 (m, 24H). MS (ESI) m/z [M + H]+: 1377.7. HRMS (ESI) m/z calcd for C62H101N15NaO18S [M + Na]+: 1399.6383 found: 1399.6371.
1-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-46-((tert-butoxycarbonyl)amino)-3,7,45-trioxo-11,14,17,20,23–26,29,32,35,38,41-undecaoxa-2,8,44-triazaoctatetracontan-48-oic acid (Tz-4-PEG11-Asp-Boc)
The title compound (6.2 mg, 73%) was obtained as pink solid using Tz-4-PEG11-NH2 (5 mg, 9 μmol), Boc-Aspartate (2.5 mg, 10 μmol), and the general reaction conditions for amide bond formations. 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.21 (s, 1H), 8.61 (d, J = 5.7 Hz, 1H), 8.47 (d, J = 8.3 Hz, 1H), 7.81 (d, J = 5.6 Hz, 1H), 7.42 (d, J = 8.4 Hz, 1H), 4.41 (d, J = 5.9 Hz, 2H), 3.99 (d, J = 5.8 Hz, 2H), 3.83–3.74 (m, 7H), 3.79 (s, 1H), 3.68 (dd, J = 11.0, 4.7 Hz, 2H), 3.55–3.44 (m, 39H), 3.43–3.24 (m, 5H), 3.18 (s, 1H), 2.44 (t, J = 6.3 Hz, 5H), 1.56 (s, 9H); MS (ESI) m/z 1044.3 [M+H]+.
1-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-46-amino-3,7,45-trioxo-11,14,17,20,23,26,29,32,35,38,41-undecaoxa-2,8,44-triazaoctatetracontan-48-oic acid (Tz-4-PEG11-Asp)
The title compound (5.1 mg, 92%) was obtained as pink solid using Tz-4-PEG11-Aspartate-Boc-NH2 (6.2 mg, 6.6 μmol) and the general TFA deprotection procedure. 1H NMR (500 MHz, DMSO-d6) δ (ppm): 11.04 (s, 1H), 8.73 (d, J = 5.8 Hz, 1H), 8.44 (d, J = 5.7 Hz, 1H), 7.88 (d, J = 7.2 Hz, 1H), 7.51 (d, J = 7.1 Hz, 1H), 4.41 (d, J = 7.2 Hz, 2H), 4.28 (d, J = 14.1 Hz, 2H), 3.98–3.91 (m, 5H), 3.87 (s, 1H), 3.78 (dd, J = 11.0, 4.7 Hz, 2H), 3.71–3.54 (m, 39H), 3.43–3.24 (m, 8H), 2.32–2.21 (m, 5H). MS (ESI) m/ 944.1 [M + H]+: 944.1.
2,2′,2′′-(2-(4-(3-(1-(4-(1,2,4,5-tetrazin-3-yl)phenyl)-47-carboxy-3,7,45-trioxo-11,14,17,20,23,26–29,32,35,38,41-undecaoxa-2,8,44-triazaheptatetracontan-46-yl)thioureido)-benzyl)-1,4,7-triazonane-1,4,7-triyl)triacetic acid (32)
Precursor 32 (5.8 mg, 74%, purity >97%) was furnished as a pink oil using Tz-4-PEG11-Aspartate-NH2 (5.1 mg, 6.0 μmol), p-Bn-NOTA-NCS, and the established isothiocyanate-amine addition procedure. 1H NMR (500 MHz, DMSO-d6) δ (ppm): 11.22 (s, 1H), 9.38 (s, 1H), 9.12 (s, 1H), 8.81 (d, J = 5.9 Hz, 1H), 8.42 (d, J = 5.8 Hz, 1H), 7.92 (d, J = 7.5 Hz, 1H), 7.72 (d, J = 7.4 Hz, 1H), 7.56 (s, 1H), 6.88 (s, 1H), 4.62–4.52 (m, 13H), 4.38 (d, J = 15.5 Hz, 2H), 4.29 (d, J = 15.3 Hz, 2H), 4.18–3.97 (m, 10H), 3.82 (s, 1H), 3.78 (dd, J = 11.0, 4.7 Hz, 2H), 3.63–3.49 (m, 43H), 3.41–3.22 (m, 8H), 2.12 (t, J = 6.1 Hz, 5H). MS (ESI) m/z [M+H]+: 1394.6. HRMS (ESI) m/z calcd for C62H96N12NaO22S [= M + Na]+: 1416.5696 found: 1416.5687.
Supplementary Material
Scheme 1.

Reaction scheme exemplifying the synthesis of precursor molecules using Tz 1 and Tz 4 via two different synthetic routes, both of which furnish final compounds in good overall yields and high chemical purities. For Tz starting materials such as Tz 1, in situ activation of the carboxylic acid group under mild conditions was performed using 1-ethyl-3-(3-dimethylamino-propyl)carbodiimide (EDC) and 1-hydroxybenzotriazole (HOBt), enabling subsequent amide coupling with the terminal amine of an appropriate linker moiety. NHS-activated, commercially available starting materials such as Tz 4 could be coupled to the linker moiety under weakly basic conditions with high conversion rates and without the need for further activation reagents. After TFA-mediated deprotection of the terminal Boc-group (if applicable), terminal amines (or carboxylic acids) were reacted with a bifunctional NODA or NOTA chelator construct that possessed a complementary functional group for chemical ligation.
Acknowledgements
The authors gratefully acknowledge the MSKCC Small Animal Imaging Core Facility as well as the Radiochemistry and Molecular Imaging Probe core, which were supported in part by NIH grant P30 CA08748. The authors also would like to thank the NIH (F32 CA210396, JPM; R00 CA1440138, BMZ). We also thank Wolfgang W. Scholz and MabVax Therapeutics (San Diego, CA) for providing the fully human monoclonal antibody 5B1. We gratefully acknowledge Mr. William H. and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research and The Center for Experimental Therapeutics of Memorial Sloan Kettering Cancer Center.
Abbreviations used
- IEDDA
inverse electron-demand Diels-Alder
- Tz
tetrazine
- TCO
trans-cyclooctene
- PET
positron emission tomography
- SPR
structure-pretargeting relationship
- %ID/g
percent injected dose per gram
- SA
specific activity
- PDAC
pancreatic ductal adenocarcinoma
- CA19.9
carbohydrate antigen 19.9
- PHL
plasma half-life
- TTB
tumor-tobackground
- PK
pharmacokinetics
Footnotes
Competing financial interests
The authors have no financial interests to disclose.
Supporting Information
Supporting information includes further general experimental and equipment information. Chemical structures and additional data of all precursor molecules. Representative radio-HPLC traces. Physicochemical data obtained for all radioligands and detailed procedures for their determination. Ex vivo biodistribution data and additional PET imaging data. Details on statistical analyses performed.
References
- 1.Meyer JP; Houghton JL; Kozlowski P; Abdel-Atti D; Reiner T; Pillarsetty NV; Scholz WW; Zeglis BM; Lewis JS (18)F-Based Pretargeted PET Imaging Based on Bioorthogonal Diels-Alder Click Chemistry. Bioconjugate Chem. 2016, 27, 298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Houghton JL; Zeglis BM; Abdel-Atti D; Sawada R; Scholz WW; Lewis JS Pretargeted Immuno-PET of Pancreatic Cancer: Overcoming Circulating Antigen and Internalized Antibody to Reduce Radiation Doses. J. Nucl. Med 2016, 57, 453–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeglis BM; Brand C; Abdel-Atti D; Carnazza KE; Cook BE; Carlin S; Reiner T; Lewis JS Optimization of a Pretargeted Strategy for the PET Imaging of Colorectal Carcinoma via the Modulation of Radioligand Pharmacokinetics. Mol. Pharmaceutics 2015, 12, 3575–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeglis BM; Sevak KK; Reiner T; Mohindra P; Carlin SD; Zanzonico P; Weissleder R; Lewis JS A Pretargeted PET Imaging Strategy based on Bioorthogonal Diels-Alder Click Chemistry. J. Nucl. Med 2013, 54, 1389–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rossin R; Verkerk PR; van den Bosch SM; Vulders RC; Verel I; Lub J; Robillard MS In vivo Chemistry for Pretargeted Tumor Imaging in live Mice. Angew. Chem. Int. Ed. Engl 2010, 49, 3375–3378. [DOI] [PubMed] [Google Scholar]
- 6.Sharkey RM; Cardillo TM; Rossi EA; Chang CH; Karacay H; McBride WJ; Hansen HJ; Horak ID; Goldenberg DM Signal Amplification in Molecular Imaging by Pretargeting a Multivalent, Bispecific Antibody. Nat. Med 2005, 11, 1250–1255. [DOI] [PubMed] [Google Scholar]
- 7.Houghton JL; Membreno R; Abdel-Atti D; Cunanan KM; Carlin S; Scholz WW; Zanzonico PB; Lewis JS; Zeglis BM Establishment of the In Vivo Efficacy of Pretargeted Radioimmunotherapy Utilizing Inverse Electron Demand Diels-Alder Click Chemistry. Mol. Cancer Ther 2017, 16, 124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Houghton JL; Zeglis BM; Abdel-Atti D; Aggeler R; Sawada R; Agnew BJ; Scholz WW; Lewis JS Site-specifically Labeled CA19.9-targeted Immunoconjugates for the PET, NIRF, and Multimodal PET/NIRF Imaging of Pancreatic Cancer. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 15850–15855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McBride WJ; Sharkey RM; Karacay H; D’Souza CA; Rossi EA; Laverman P; Chang CH; Boerman OC; Goldenberg DM A Novel Method of 18F Radiolabeling for PET. J. Nucl. Med 2009, 50, 991–998. [DOI] [PubMed] [Google Scholar]
- 10.Blackman ML; Royzen M; Fox JM Tetrazine Ligation: Fast Bioconjugation based on Inverse-Electrondemand Diels-Alder Reactivity. J. Am. Chem. Soc 2008, 130, 13518–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyer JP; Adumeau P; Lewis JS; Zeglis BM Click Chemistry and Radiochemistry: The First 10 Years. Bioconjugate Chem. 2016, 27, 2791–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paganelli G; Magnani P; Zito F; Villa E; Sudati F; Lopalco L; Rossetti C; Malcovati M; Chiolerio F; Seccamani E; Siccardi AG; Fazio F Three-step Monoclonal Antibody Tumor Targeting in Carcinoembryonic Antigen-positive Patients. Cancer Res. 1991, 51, 5960–5966. [PubMed] [Google Scholar]
- 13.Bos ES; Kuijpers WH; Meesters-Winters M; Pham DT; de Haan AS; van Doornmalen AM; Kaspersen FM; van Boeckel CA; Gougeon-Bertrand F In vitro Evaluation of DNA-DNA Hybridization as a Two-step Approach in Radioimmunotherapy of Cancer. Cancer Res. 1994, 54, 3479–3486. [PubMed] [Google Scholar]
- 14.Gordon CG; Mackey JL; Jewett JC; Sletten EM; Houk KN; Bertozzi CR Reactivity of Biarylazacyclooctynones in Copper-free Click Chemistry. J. Am. Chem. Soc 2012, 134, 9199–9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hein JE; Fokin VV Copper-catalyzed Azide-alkyne Cycloaddition (CuAAC) and Beyond: New Reactivity of Copper(I) Acetylides. Chem. Soc. Rev 2010, 39, 1302–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knox SJ; Goris ML; Tempero M; Weiden PL; Gentner L; Breitz H; Adams GP; Axworthy D; Gaffigan S; Bryan K; Fisher DR; Colcher D; Horak ID; Weiner LM Phase II Trial of Yttrium-90-DOTA-biotin Pretargeted by NR-LU-10 Antibody/Streptavidin in Patients with Metastatic Colon Cancer. Clin. Cancer Res 2000, 6, 406–414. [PubMed] [Google Scholar]
- 17.Schoffelen R; Boerman OC; Goldenberg DM; Sharkey RM; van Herpen CM; Franssen GM; McBride WJ; Chang CH; Rossi EA; van der Graaf WT; Oyen WJ Development of an Imaging-guided CEA-pretargeted Radionuclide Treatment of Advanced Colorectal Cancer: First Clinical Results. Br. J. Cancer 2013, 109, 934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Berkel SS; van Eldijk MB; van Hest JC Staudinger Ligation as a Method for Bioconjugation. Angew. Chem., Int. Ed 2011, 50, 8806–8827. [DOI] [PubMed] [Google Scholar]
- 19.Yazdani A; Bilton H; Vito A; Genady AR; Rathmann SM; Ahmad Z; Janzen N; Czorny S; Zeglis BM; Francesconi LC; Valliant JF A Bone-Seeking Trans-Cyclooctene for Pretargeting and Bioorthogonal Chemistry: A Proof of Concept Study Using 99mTc- and 177Lu-Labeled Tetrazines. J. Med. Chem 2016, 59, 9381–9389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Denk C; Svatunek D; Mairinger S; Stanek J; Filip T; Matscheko D; Kuntner C; Wanek T; Mikula H Design, Synthesis, and Evaluation of a Low-Molecular-Weight (11)C-Labeled Tetrazine for Pretargeted PET Imaging Applying Bioorthogonal in Vivo Click Chemistry. Bioconjugate Chem. 2016, 27, 1707–1712. [DOI] [PubMed] [Google Scholar]
- 21.Evans HL; Nguyen QD; Carroll LS; Kaliszczak M; Twyman FJ; Spivey AC; Aboagye EO A Bioorthogonal (68)Ga-labelling Strategy for Rapid in vivo Imaging. Chem. Commun. (Cambridge, U. K.) 2014, 50, 9557–9560. [DOI] [PubMed] [Google Scholar]
- 22.Viola-Villegas NT; Rice SL; Carlin S; Wu X; Evans MJ; Sevak KK; Drobjnak M; Ragupathi G; Sawada R; Scholz WW; Livingston PO; Lewis JS Applying PET to Broaden the Diagnostic Utility of the Clinically Validated CA19.9 Serum Biomarker for Oncology. J. Nucl. Med 2013, 54, 1876–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karver MR; Weissleder R; Hilderbrand SA Synthesis and Evaluation of a Series of 1,2,4,5-Tetrazines for Bioorthogonal Conjugation. Bioconjugate Chem. 2011, 22, 2263–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi HS; Gibbs SL; Lee JH; Kim SH; Ashitate Y; Liu F; Hyun H; Park G; Xie Y; Bae S; Henary M; Frangioni JV Targeted Zwitterionic Near-infrared Fluorophores for Improved Optical Imaging. Nat. Biotechnol 2013, 31, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vilche M; Reyes AL; Vasilskis E; Oliver P; Balter H; Engler H (68)Ga-NOTA-UBI-29–41 as a PET Tracer for Detection of Bacterial Infection. J. Nucl. Med 2016, 57, 622–627. [DOI] [PubMed] [Google Scholar]
- 26.Chatalic KL; Franssen GM; van Weerden WM; McBride WJ; Laverman P; de Blois E; Hajjaj B; Brunel L; Goldenberg DM; Fehrentz JA; Martinez J; Boerman OC; de Jong M Preclinical Comparison of Al18F- and 68Ga-labeled Gastrin-releasing Peptide Receptor Antagonists for PET Imaging of Prostate Cancer. J. Nucl. Med 2014, 55, 2050–2056. [DOI] [PubMed] [Google Scholar]
- 27.Skovgaard D; Persson M; Brandt-Larsen M; Christensen C; Madsen J; Klausen TL; Holm S; Andersen FL; Loft A; Berthelsen AK; Pappot H; Brasso K; Kroman N; Hoejgaard L; Kjaer A Safety, Dosimetry and Tumor Detection Ability of 68Ga-NOTA-AE105: First-in-Humans Study of a Novel Radioligand for uPAR PET Imaging. J. Nucl. Med 2017, 58, 379–386. [DOI] [PubMed] [Google Scholar]
- 28.Bao K; Nasr KA; Hyun H; Lee JH; Gravier J; Gibbs SL; Choi HS Charge and Hydrophobicity Effects of NIR Fluorophores on Bone-specific Imaging. Theranostics 2015, 5, 609–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wischnjow A; Sarko D; Janzer M; Kaufman C; Beijer B; Brings S; Haberkorn U; Larbig G; Kubelbeck A; Mier W Renal Targeting: Peptide-Based Drug Delivery to Proximal Tubule Cells. Bioconjugate Chem. 2016, 27, 1050–1057. [DOI] [PubMed] [Google Scholar]
- 30.Janzer M; Larbig G; Kubelbeck A; Wischnjow A; Haberkorn U; Mier W Drug Conjugation Affects Pharmacokinetics and Specificity of Kidney-Targeted Peptide Carriers. Bioconjugate Chem. 2016, 27, 2441–2449. [DOI] [PubMed] [Google Scholar]
- 31.Cook BE; Adumeau P; Membreno R; Carnazza KE; Brand C; Reiner T; Agnew BJ; Lewis JS; Zeglis BM Pretargeted PET Imaging Using a Site-Specifically Labeled Immunoconjugate. Bioconjugate Chem. 2016, 27, 1789–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adumeau P; Carnazza KE; Brand C; Carlin SD; Reiner T; Agnew BJ; Lewis JS; Zeglis BM A Pretargeted Approach for the Multimodal PET/NIRF Imaging of Colorectal Cancer. Theranostics 2016, 6, 2267–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rossin R; van Duijnhoven SM; Lappchen T; van den Bosch SM; Robillard MS Trans-cyclooctene tag with improved properties for tumor pretargeting with the diels-alder reaction. Mol. Pharmaceutics 2014, 11, 3090–3096. [DOI] [PubMed] [Google Scholar]
- 34.Zeglis BM; Emmetiere F; Pillarsetty N; Weissleder R; Lewis JS; Reiner T Building Blocks for the Construction of Bioorthogonally Reactive Peptides via Solid-Phase Peptide Synthesis. ChemistryOpen 2014, 3, 48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.