

SUMMARY
Long-term information storage in the brain requires continual modification of the neuronal transcriptome. Synaptic inputs located hundreds of micrometers from the nucleus can regulate gene transcription, requiring high-fidelity, long-range signaling from synapses in dendrites to the nucleus in the cell soma. Here, we describe a synapse-to-nucleus signaling mechanism for the activity-dependent transcription factor NFAT. NMDA receptors activated on distal dendrites were found to initiate L-type Ca2+ channel (LTCC) spikes that quickly propagated the length of the dendrite to the soma. Surprisingly, LTCC propagation did not require voltage-gated Na+ channels or back-propagating action potentials. NFAT nuclear recruitment and transcriptional activation only occurred when LTCC spikes invaded the somatic compartment, and the degree of NFAT activation correlated with the number of somatic LTCC Ca2+ spikes. Together, these data support a model for synapse to nucleus communication where NFAT integrates somatic LTCC Ca2+ spikes to alter transcription during periods of heightened neuronal activity.
In Brief
Signaling from synapse to nucleus can alter transcription and consolidate long-term changes in neuronal function. Wild et al. uncover a mechanism for rapid long-distance signaling from distal dendrites to the nucleus that utilizes L-type voltage-gated Ca2+ channel Ca2+ spikes to activate the transcription factor NFAT.
Graphical Abstract
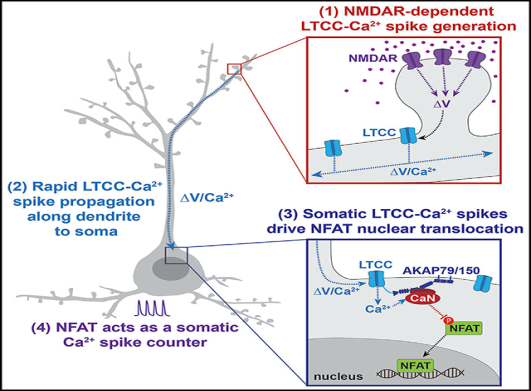
INTRODUCTION
Neuronal adaptations can be consolidated when membrane depolarizations are converted to molecular signals that are in turn relayed to the nucleus to alter transcription (Greer and Greenberg, 2008). This process, known as excitation-transcription (E-T) coupling, can be initiated at distal synaptic sites following repetitive synaptic stimulation (Deisseroth et al., 1996; Dudek and Fields, 2001; Frey et al., 1996; Zhao et al., 2005). Because dendritic architecture is complex, extending hundreds of micrometers from the soma, neurons face unique challenges in relaying signals from synapses on distal dendritic spines to the nucleus (Adams and Dudek, 2005; Greer and Greenberg, 2008; Panayotis et al., 2015). Three predominant models of synapseto-nucleus E-T coupling have been described, each requiring Ca2+ influx in distinct neuronal compartments. In the first, the “action potential model,” glutamate receptor–generated excitatory postsynaptic potentials (EPSPs) trigger back-propagating action potentials (bAPs) in the soma, somatic Ca2+ influx through voltage-gated Ca2+ channels, and rapid activation of somatonuclear signaling factors (Adams and Dudek, 2005; Bengtson et al., 2010; Dudek and Fields, 2002; Ma et al., 2014; Zhao et al., 2005). In the second, the “molecular translocation model,” signaling molecules activated postsynaptically translocate in mobile molecular signaling complexes from dendrites to the nucleus via diffusion or molecular transport mechanisms over much longer timescales (Ch’ng et al., 2012, 2015; Dinamarca et al., 2016; Panayotis et al., 2015; Zhai et al., 2013). In the third, the “Ca2+-wave model,” regenerative endoplasmic reticulum (ER) Ca2+ waves that are initiated near the synapse by ryanodine or IP3 receptors can spread to the soma via Ca2+-induced Ca2+ release (CICR) to elevate somatic Ca2+ and activate somato-nuclear signaling factors (Hagenston and Bading, 2011; Herbst and Martin, 2017; Panayotis et al., 2015; Ross, 2012). Each of these routes of communication are thought to be activated by different patterns of synaptic stimuli and may encode distinct temporal and source specific information, depending on both the subcellular origin of the molecular signals and the time taken for these signals to reach the nucleus.
Many nuclear signaling pathways depend critically on Ca2+ influx through L-type Ca2+ channels (LTCCs), which have a privileged role among voltage-gated Ca2+ channels in nuclear signaling (Ch’ng et al., 2012; Graef et al., 1999; Wang et al., 2017; Wheeler et al., 2012). Here, we focused our investigation on synapse-to-nucleus communication through the transcription factor nuclear-factor of activated T cells (NFAT) c3, one of four Ca2+-sensitive NFAT isoforms (NFATc1-c4) that has an important, yet not fully understood role in nervous system development and function (Kipanyula et al., 2016). Upon global neuronal depolarization, NFATc3 is dephosphorylated by the Ca2+-dependent phosphatase calcineurin (CaN) and rapidly imported into the nucleus (Murphy et al., 2014; Ulrich et al., 2012). This depends critically on LTCC activation and anchoring of CaN in the LTCC nanodomain by AKAP79/150 (Li et al., 2012; Murphy et al., 2014; Oliveria et al., 2007; Wild and Dell’Acqua, 2018). It is unclear, however, exactly how NFATc3 responds to synaptic activity and whether NFATc3 carries a molecular signal to the nucleus from distal synaptic sites or more proximal locations, such as the soma. This information is critical to properly understand the activity patterns that NFAT will respond to and the function of neuronal NFAT signaling. Here, using a combination of glutamate uncaging to mimic synaptic input to distal dendrites with imaging of cytosolic Ca2+ and NFATc3 localization, we found that the predominant mode of synapse-to-nucleus signaling through NFATc3 required NMDA receptor-dependent LTCC-Ca2+-spikes that propagate from dendrites to the soma to activate somatic NFATc3.
RESULTS
Ca2+ Signal Propagation to the Soma Is Required for NFAT Translocation
Previous studies found that 4-methoxy-7-nitroindolinyl-caged-L-glutamate (MNI-glu) uncaging near a distal dendrite or spine can promote synapse-to-nucleus molecular transport of cyclic AMP response element binding protein (CREB) regulators extracellular signal regulated kinase-2 (ERK2) and CREB-regulated transcriptional co-activator 1 (CRTC1), when Ca2+ influx is restricted to the distal spine and dendrite (Ch’ng et al., 2015; Zhai et al., 2013). To determine if NFATc3 can translocate to the nucleus following synaptic stimulation, we uncaged MNI-glu on distal spines of cultured hippocampal pyramidal neurons transfected with Ca2+ indicator j red genetically encoded calcium indicator for optical imaging 1a (jRGECO1a) and super enhanced GFP-nuclear factor of activated T cells cytoplasmic 3 (sGFP-NFATc3) (Figures 1A–1I). To prevent spontaneous network activity and reduce baseline sGFP-NFATc3 nuclear localization, tetrodotoxin (TTX; 1 μM) was included in all experiments unless otherwise stated. We observed two distinct Ca2+ signal signatures as a result of distal glutamate uncaging: 1 Hz glutamate uncaging (1 Hz-GU) for 60 pulses predominantly generated Ca2+ signals that propagated to the soma and throughout the dendritic tree (Figures 1B–1D; Video S1; 11/15 neurons), whereas 0.5 Hz-GU predominantly generated Ca2+ signals that were restricted to the activated spine and nearby dendrite (Figures 1F–1H; Video S2; 7/9 neurons). We found that 1 Hz-GU promoted robust sGFP-NFATc3 nuclear translocation when trains were repeated across 6 spines on a single branch (SB; Figures S1A, S1J, and S1K) or 6 spines on multiple branches (MBs; Figures 1E and S1B), while 2 Hz-GU on 5 separate branches also promoted a similar increase in nuclear sGFP-NFATc3 (Figures S1J and S1K). Conversely, 0.5 Hz-GU did not promote detectable sGFP-NFATc3 translocation when trains of uncaging were repeated across 6 spines on either a single branch (Figures S1J and S1K) or 7 spines across multiple branches (Figure 1I). Together these results indicate that Ca2+ signals must propagate beyond the activated dendrite to stimulate sGFP-NFATc3 nuclear translocation. In support of this idea, in a subpopulation of neurons where 1 Hz-GU generated dendrite-restricted Ca2+ signals (Figures S1D and S1E; 4/15 neurons), sGFP-NFATc3 nuclear accumulation was not detected (Figure S1F); meanwhile, in a subpopulation of neurons where distal stimulation with 0.5 Hz-GU generated propagating Ca2+ signals, rapid sGFP-NFATc3 translocation was observed (not shown). Prominent sGFP-NFATc3 translocation was also seen following 0.5 Hz-GU directly adjacent (~5 μm) to the soma (Figures S1C and S1G–S1I). Overall, we found that somatic Ca2+ influx was the best predictor of sGFP-NFATc3 nuclear translocation (Figures S1J and S1K), and the peak sGFP-NFATc3 signal correlated significantly with the integrated somatic, but not spine, Ca2+ influx (Figures S1L and S1M). Furthermore, the variability we observed in the distance that Ca2+ signals propagate following distal uncaging underscores the importance of our imaging approach, which monitors both Ca2+ and NFATc3 localization in all experiments.
Figure 1. Ca2+ Signal Propagation to the Soma Is Required for NFAT Translocation.

(A) Schematic for multiple spine stimulation.
(B) Neuron transfected with sGFP-NFATc3 and jRGECO1a (pseudocolored). Left: pre-uncaging. Right: during a 1 Hz glutamate uncaging. White crosses indicate sites of 1 Hz-GU. Scale bar, 20 μm.
(C) jRGECO1a ΔF/F0 traces from ROIs drawn for a dendritic spine and the soma as indicated by white arrow spines in (B).
(D) Sum integrated ΔF/F0 for spine (sp) and soma (so) following 1 Hz-GU.
(E) sGFP-NFATc3 nuclear and cytosolic intensity normalized to pre-uncaging values. Inset: images of soma for neuron in (B) with sGFP-NFATc3 distribution pre(start) and post-uncaging as indicated (n = 9; one-way ANOVA repeated-measures with Dunnett, ***p < 0.001).
(F) Multi-spine stimulation and jRGECO1a Ca2+ imaging as described in (B) but for 0.5 Hz-GU.
(G) Representative ΔF/F0 Ca2+ traces as described in (C) but for 0.5 Hz-GU.
(H) Integrated ΔF/F0 Ca2+ measurements as described in (D) but for 0.5 Hz-GU.
(I) sGFP-NFATc3 nuclear and cytosolic intensity measurements as described in (E) but for 0.5 Hz-GU (n = 8).
(J) Top: diagram of 3xNFAT-2xsGFPnls transcriptional reporter construct. Bottom: images of neuronal soma pre-uncaging and 5 h after uncaging with either 1 Hz-GU or 0.5 Hz-GU. Neurons transfected with the 3xNFAT/AP1–2xsGFPnls reporter (pseudocolored) and jRGECO1a (shown in white).
(K) Graph of 2xsGFPnls nuclear intensity (normalized to pre-uncaging values) for nonstimulated (NS, n = 5) and either 0.5 Hz-GU (n = 6) or 1Hz-GU (n = 5) on 7 different branches (one-way ANOVA repeated-measures with Dunnett, ***p < 0.001, *p < 0.05).
(L) Left: sum integrated somatic ΔF/F0 following distal 1 Hz-GU across 3 spines (MBs) with AKAP150-shRNAi and rescue with AKAP79WT (n = 5) or AKAP79ΔPIX (n = 6). Right: peak fold change in sGFP-NFATc3 nucleus/cytosol ratio following uncaging protocol described (unpaired t test, *p < 0.05).
See also Figure S1.
To determine if propagating Ca2+ signals are also required to induce transcriptional activity of endogenous NFATs, we transfected neurons with jRGECO1a and a reporter construct that expressed a tandem-sGFP with a nuclear localization sequence (nls) under the control of an NFAT/AP-1 promoter (3xNFAT/AP-1–2xsGFPnls; Figure 1J). We uncaged across 7 dendritic distal branches with either 0.5 Hz-GU or 1 Hz-GU, monitoring nuclear sGFP intensity for up to 5 hr. Importantly, sGFP nuclear fluorescence was only significantly increased following 1 Hz-GU (propagating Ca2+ signals) but not 0.5 Hz-GU (dendrite restricted Ca2+ signals; Figure 1K). We showed previously that following chemical neuronal depolarization, NFATc3 is dephosphorylated by CaN anchored to AKAP79/150 (Murphy et al., 2014; Oliveria et al., 2007). Here we employed a similar knock-down rescue approach (AKAP150 shRNAi with AKAP79WT or AKAP79ΔPIX rescue) to measure sGFP-NFATc3 translocation following 1 Hz-GU on 3 distal branches (Hoshi et al., 2005). Delocalizing CaN from the LTCC-AKAP79 complex with the AKAP79ΔPIX mutation significantly reduced sGFP-NFATc3 nuclear translocation, despite comparable somatic Ca2+ influx in both groups (Figure 1L). In summary, sGFP-NFATc3 translocation and NFAT-dependent transcription are not inherently dependent on the distribution (single versus multi-branch) or the frequency of uncaging pulses, but they are governed by the ability of a distal input to generate a long-distance propagating Ca2+ signal that reaches the soma.
LTCCs Are Required for Propagating Ca2+ Signals
The results presented so far indicate that NFATc3 translocation requires Ca2+ signals that propagate beyond the stimulated dendrite. Interestingly, Nav channels are not required for Ca2+ signal propagation, as TTX (1 μM) was included in all aforementioned experiments, excluding a role for bAPs in signal propagation. We next investigated the modality of Ca2+ signal spread, focusing on neurons that exhibited strongly propagating responses to 1Hz-GU for 20 pulses at a single distal spine. The distance of propagation (imaged with GCaMP6f; Chen et al., 2013) was determined under control conditions from integrated ΔF/F0 measurements (area under trace) made at 10 μm intervals along the dendrite from the activated spine to the soma. To assess the involvement of glutamate receptors, an NMDA receptor (NMDAR) antagonist (D-AP5; 50 μM) was added to the bath (5 min) and 1Hz-GU repeated (Figure S2A). D-AP5 prevented Ca2+ influx in the activated spine and along the dendrite, indicating that NMDARs are required for initiation of the propagating signal (Figure S2B). To determine if glutamate could diffuse to the soma following distal 1 Hz-GU, neurons were transfected with the fluorescent glutamate sensor iGluSnFR (Figure S2C) (Marvin et al., 2013). The iGluSnFR signal rapidly decayed along the dendrite, failing to reach the soma (λ = 15.9 μm, fit with a one-phase exponential decay; Figures S2D and S2E).
We next used the LTCC antagonist nimodipine (nim; 5 μM; 10 mins) to determine whether Cav1 L-type voltage-gated Ca2+ channels were required for propagating Ca2+ signals initiated by 1 Hz-GU (Figures 2A and 2B). Following addition of nim, the dendritic Ca2+ signal was restricted to the region near where MNI-glu was uncaged and Ca2+ influx in the soma (measured as somatic integrated ΔF/F0) was completely blocked (Figures 2C and 2D). Similar results were observed with a different class of LTCC antagonist, verapamil (25 μM), and an alternative dihydropyridine antagonist, isradipine (Figures 2E and 2F). To determine if Nav channels could alter the spatial spread of Ca2+ signals, we performed 1 Hz-GU without TTX in the bath (1 Hz-GU control, no TTX), and following the addition of nim (1 Hz-GU nim, no TTX). In the absence of TTX, the peak Ca2+ signal amplitude was enhanced in all compartments, with additional neuron-wide spontaneous events observed independent of MNI-glu uncaging (Figure 2G). Following addition of nim, Ca2+ signals were again spatially restricted to dendrites and did not reach the soma (λ = 19.8 μm fit with a one-phase exponential decay; Figures 2H and 2I), closely resembling both the integral ΔF/F0 and decay seen with nim in the presence of TTX (λ = 22.0 μm; Figure S2F). Both Cav1 (L-type) and Cav2 (P/Q- and N-type) channels are known to play a role in neuronal nuclear signaling (Wheeler et al., 2008). To test the role of Cav2 channels in Ca2+ signal propagation, 1 Hz-GU was performed under control conditions and then following sequential addition of P/Q- and N- type Ca2+-channel antagonist conotoxins (cTx; GVIA 1 μM + MVIIC 5 μM; 30 mins) and then nim (5 μM; 10 mins) (Figures S2H and S2I). The range of Ca2+ signal propagation was unaltered by cTx but was again substantially attenuated by nim (Figure S2J; Video S3), while somatic Ca2+ influx was unaffected by cTx and blocked by nim (Figure S2K).
Figure 2. LTCCs Are Required for Ca2+ Signal Propagation to the Soma and Are Activated Preferentially by Higher Intensity Distal Spine Stimulation.

(A) Neuron transfected with mCh and GCaMP6f (pseudocolored). Cross indicates location of uncaging. Left: pre-uncaging in control. Middle: 2 s after start of 1 Hz-GU (20 pulses) in control. Right: 2 s after start of 1 Hz-GU following 10 min incubation in nim (10 μM). Scale bar, 20 μm.
(B) Representative GCaMP6f ΔF/F0 traces after distal 1 Hz-GU in control and after addition of nim, with TTX (1 μM) in the bath. Measurements were made at uncaged spine and at intervals along the dendrite to the soma as indicated.
(C) Integrated GCaMP6f ΔF/F0 for ROIs along the dendrite every 10 μm from uncaged spine toward the soma for experiments described in (B) (n = 11).
(D) Somatic integrated GCaMP6f ΔF/F0 from ROI drawn on soma before and after nim (n = 11; paired t test, ***p < 0.001).
(E) As (D) but with verapamil (25 μM; n = 10; paired t test, **p < 0.01).
(F) As (D) but with isradipine (5 μM; n = 5; paired t test, *p < 0.05).
(G) Representative GCaMP6f ΔF/F0 traces following 1 Hz-GU in control and nim without TTX in the bath.
(H) Integrated GCaMP6f ΔF/F0 for experiments described in (G) (n = 9).
(I) Somatic integrated GCaMP6f ΔF/F0 before and after nim with no TTX (n = 9; paired t test, paired t test, **p < 0.01).
(J) Representative GCaMP6f ΔF/F0 traces following 0.5 Hz-GU in control and after addition of BayK (5 μM).
(K) Integrated GCaMP6f ΔF/F0 along the dendrite for experiments described in (J) (n = 9).
(L) Somatic integrated GCaMP6f ΔF/F0 before and after BayK (n = 9; paired t test, **p < 0.01).
(M) Graph of sGFP-NFATc3 nuclear intensity following 1 Hz-GU trains (60 pulses) at 6 spines on separate branches with nim (5 μM) in bath (n = 7). Inset: representative image of soma with sGFP-NFATc3 distribution before (start) and after uncaging as indicated.
(N) Graph of sGFP-NFATc3 nuclear intensity following a 0.5 Hz-GU (30 pulses) at 1 spine with BayK in the bath (n = 4). Inset: representative image of soma with sGFP-NFATc3 distribution before (start) and after uncaging as indicated (one-way ANOVA repeated-measures with Dunnett, ***p < 0.001).
See also Figure S2.
The spread of Ca2+ generated by 0.5 Hz-GU was not altered by nim, consistent with the idea that NMDARs were largely responsible for the observed Ca2+ signal and the 0.5 Hz-GU stimulus was subthreshold for initiation of LTCC-dependent Ca2+ propagation to the soma (Figure S2G). In an attempt to lower the threshold for Ca2+ signal propagation, the LTCC agonist Bay K 8644 (BayK; 5 μM) was added to the bath. Importantly, BayK converted the 0.5 Hz-GU evoked dendrite restricted Ca2+ signal to a propagating Ca2+ signal that reached the soma (Figures 2J–2L). To directly observe sub- and supra-threshold stimuli for propagating Ca2+ signals originating from the same dendritic spine, we sequentially uncaged at 0.5 Hz for 20 s, followed by a 7 s interval, then at 1 Hz for 20 s (Figure S2L). While the Ca2+ signal in response to 0.5 Hz-GU was dendrite-restricted, 1 Hz-GU generated a propagating Ca2+ signal that reached the soma, indicating that higher frequency distal uncaging trains have a higher likelihood of surpassing the threshold to trigger a propagating Ca2+ event (Figure S2M).
In neurons transfected with sGFP-NFATc3 and jRGECO1a, 1 Hz-GU dispersed across 6 branches was not able to promote sGFP-NFATc3 translocation in the presence of nim (Figure 2M), while 0.5 Hz-GU at a single spine with BayK added to the bath stimulated robust sGFP-NFATc3 translocation (Figure 2N). Together, these findings indicate that uncaging protocols that generate suprathreshold postsynaptic responses to activate LTCCs can trigger Ca2+ signal propagation to the soma, which in turn can promote rapid nuclear translocation of sGFP-NFATc3. Importantly, this form of rapid synapse-to-nucleus signaling does not require Nav channels or bAPs, and the observed Ca2+ signals propagate to the soma and throughout the dendrites rapidly within 100 ms (the time between 2 imaging frames).
Propagating Ca2+ Signals Are LTCC-Mediated Forward Propagating Ca2+ Spikes
To determine the site of initiation of propagating Ca2+ signals following 1 Hz-GU, we performed rapid acquisition (40 Hz) time-lapse Ca2+ imaging with GCaMP6f at 25°C to detect temporal differences in Ca2+ signal increases along the dendrite (Figures S3A–S3C). The uncaging laser power was tuned so that a single uncaging pulse was just above threshold to generate a propagating Ca2+ signal. Uncaging pulses were repeated in trains at 0.33 Hz to generate temporally separated Ca2+ responses. Measurements of ΔF/F0 were made at 25 μm intervals along the dendrite (Figure S3A; warm colors closest to site of uncaging; cool colors closest to soma) and were averaged across multiple iterations (33 responses from 3 neurons). Ca2+ signals rapidly propagated from the site of uncaging greater than 200 μm along the dendrite to arrive at the soma in <75 ms (Figures S3B and S3C). These results are supportive of rapid Ca2+ signals that originate in the distal dendrites and propagate toward the soma.
One proposed mechanism for synapse-to-nucleus communication is through regenerative ER Ca2+ waves, mediated by ryanodine and IP3 receptors that release Ca2+ from the ER (Hagenston and Bading, 2011; Herbst and Martin, 2017; Panayotis et al., 2015; Ross, 2012). We have shown here that 1 Hz-GU activates NMDARs and LTCCs, both of which we have previously shown to induce CICR in dendrites (Dittmer et al., 2017). To determine if propagating Ca2+ signals are a result of ER Ca2+ waves, we simultaneously monitored the spread of cytosolic Ca2+ (jRGECO1a) and ER Ca2+ depletion (D1ER; ratiometric ER Ca2+ sensor) following 1 Hz-GU on a distal spine (selecting for neurons with strongly propagating Ca2+ signals), before and after pharmacologic manipulation of CICR (Figures S3D–S3I). As seen previously, ER depletion following 1 Hz-GU was highest within 40 μm of the uncaged spine (Figure S3E) (Dittmer et al., 2017). While treatment with ryanodine (200 μM; 1 h) to block ryanodine receptors nearly eliminated dendritic ER depletion (Figure S3E), it only partially reduced cytosolic Ca2+ increases in dendrites (~50%; Figure S3F) and the soma (~25%; Figure S3J), and thus did not prevent Ca2+ signal propagation along the dendrite to the soma (Figures S3F and S3J). Intracellular IP3 production is predominantly achieved through activation of G-protein coupled receptors. Direct pharmacologic inhibition of IP3Rs is difficult, owing to a lack of highly selective antagonists. Therefore, we indirectly inhibited IP3 production following MNI-glu uncaging with broad-spectrum metabotropic glutamate receptor (mGluR) antagonist (S)-α-methyl-4-carboxyphenylglycine (S-MCPG; 250 μM; 10 min; Figure S3G). In contrast to ryanodine, mGluR blockade did not substantially alter either ER Ca2+ depletion or cytosolic Ca2+ along the dendrite (Figures S3H–S3I). However, because ryanodine modestly reduced the cytosolic Ca2+ signal generated in the soma following distal 1 Hz-GU (Figure S3J), we investigated if this RyR-mediated contribution to the somatic Ca2+ signal was important for NFAT activation. We found that ryanodine did not significantly reduce sGFP-NFATc3 translocation to the nucleus following 1 Hz-GU (60 pulses on 3 separate branches; Figure S3K). Together, these results indicate that although RyR-mediated CICR may be engaged along the length of the dendrite and in the soma to increase cytosolic Ca2+ following 1 Hz-GU, regenerative ER Ca2+ waves are not the mechanism of Ca2+ signal propagation, and CICR does not appreciably contribute to NFAT activation.
To determine the extent to which 1 Hz-GU and propagating Ca2+ signals depolarized the cell membrane, we used simultaneous current-clamp recording at the soma, Ca2+ imaging (GCaMP6f) and 1 Hz-GU on a distal dendritic spine in the presence of TTX to block Nav channels (Figure 3A). Two distinct responses were observed within the uncaging trains: dendrite-restricted Ca2+ signals associated with slowly decaying EPSPs in the voltage trace (Figure S3L) that reached a plateau after repeated stimuli, and propagating Ca2+ signals that spread throughout the entire neuron and soma associated with faster, higher amplitude spikes in the current trace (Figure 3B and 3C). Importantly, Ca2+ increases in the soma were highly temporally correlated with depolarizing spikes in the current clamp recording (Figure 3B; dashed lines). We next focused our attention on the spikes in the trace. Analysis of 24 spikes from 5 neurons showed that the mean threshold for spiking was −33.4 mV (Figure S3M), the mean spike amplitude was ΔV = 26.9 mV, with spikes depolarizing the membrane to <0 mV, and the width at half-maximum amplitude was 17.6 ms (Figure 3D). These properties closely resemble those of CaV spikes or “spikelets” previously observed ex vivo and in vivo, including in hippocampal CA1 pyramidal neurons, and which are a component of complex spikes (Grienberger et al., 2014; Liu et al., 2018; Remy and Spruston, 2007).
Figure 3. Propagating Ca2+ Signals Are LTCC-Mediated, Forward Propagating Ca2+ Spikes.

(A) Neuron transfected with GCaMP6f (inverted grayscale) indicating location of distal spine 1 Hz-GU (20 pulses; cross) during simultaneous whole-cell current clamp recording at the soma. Scale bar, 20 μm.
(B) Representative GCaMP6f ΔF/F0 traces taken from ROIs drawn at the uncaged spine (top) and soma (middle) with current clamp recording trace (bottom) following 1 Hz-GU (with 1 μM TTX in the bath).
(C) Expanded view of 3 responses in current clamp trace indicated with an asterisk (*) in (B).
(D) Averaged traces of 24 spikes from 5 neurons. THOLD, threshold (mV); ½ max, width at half maximum (ms); AMP, amplitude (mV).
(E) Representative traces following 1 Hz-GU from another neuron, as described in (A) and (B).
(F) GCaMP6f and current clamp traces from neuron shown in (E) following bath addition of nim (5 μM).
(G) Spine-integrated GCaMP6f ΔF/F0 before and after nim.
(H) ΔV plateau in current clamp trace before and after nim.
(I) Soma-integrated GCaMP6f ΔF/F0 before and after nim.
(J) Number of spikes before and after nim (G–J: n = 7; paired t test, *p < 0.05).
See also Figure S3.
Because propagating Ca2+ signals evoked by 1 Hz-GU were inhibited by LTCC blockers (Figures 2A–2I), we next determined if the spikes in the trace were also sensitive to LTCC inhibition. Following distal 1 Hz-GU, propagating Ca2+ signals were again evoked that were associated with temporally correlated spikes in somatic Ca2+ and depolarizing spikes in the current clamp recording (Figure 3E). After addition of nim (5 μM) to the bath, the amplitude of dendritic Ca2+ signals and the plateau in the current-clamp trace evoked by 1 Hz-GU was not significantly altered (Figures 3F–3H). However, Ca2+ signals were spatially restricted to distal dendrites (as seen in Figures 2A–2C), and importantly, no spikes were observed in the current clamp recording made at the soma (Figures 3F, 3I, and 3J). In the absence of TTX, we observed that 1 Hz-GU evoked a combination of action potentials (APs) alone and complex spikes that included both APs and putative CaV spikes in the current clamp trace, with only complex spikes and somatic Ca2+ influx being sensitive to inhibition by bath application of nim (Figures S3N and S3O). Interestingly, while complex spikes were associated with large increases in cytosolic Ca2+ throughout the neuron, a burst of APs alone did not substantially increase cytosolic Ca2+ in any compartment (Figure S3N). Together, these results indicate that propagating Ca2+ signals are electrical in nature and are mediated by NMDAR-dependent depolarizing Ca2+ spikes that rapidly propagate from the activated branch to the soma. Because of the tight coupling between propagating Ca2+ signals (observed as neuron-wide Ca2+ events) and depolarizing Ca2+ spikes (recorded at the soma), and their sensitivity to nim, we refer to these events as LTCC-Ca2+ spikes. While CaV-spikes have been widely reported in the literature (Golding et al., 1999; Remy and Spruston, 2007; Takahashi and Magee, 2009; Young and Yang, 2004), their role in synapse-to-nucleus signaling has not been investigated. Thus, we next sought to better define the relationship between LTCC-Ca2+ spikes and NFAT signaling.
NFAT Activation Is Sensitive to the Number, Not Frequency, of Propagating Ca2+ Signals
LTCC channel density is thought to be highest on the soma and proximal dendrites of hippocampal neurons (Hell et al., 1993). To determine if Ca2+ spikes were initiated more readily at proximal sites, we measured somatic Ca2+ influx (jRGECO1a) following a single pulse of MNI-glu uncaging at spines that were spaced along the dendrite at ~30 μm intervals (Figures S4A and S4B). We found that the evoked propagating Ca2+ signal (measured as peak amplitude in the soma) increased as the region of interest (ROI) was moved closer to the soma. At a distance of 60 μm from the soma, a single uncaging pulse reliably evoked Ca2+ spikes that were significantly inhibited by nim (Figures S4A–S4F). Thus, these findings are consistent with a higher LTCC density, possibly allowing for more efficient Ca2+ spike initiation at proximal versus distal dendrites.
Transcriptional pathways regulated by ERK have been shown to be highly sensitive to both the pattern and duration of neuronal burst firing (Lee et al., 2017; Tyssowski et al., 2018). To gain a better understanding of the neuronal activity patterns that govern NFAT signaling, we next investigated if sGFP-NFATc3 translocation is sensitive to the number or frequency of MNI-glu driven LTCC-Ca2+ spikes, using Ca2+ influx in the soma as a readout of Ca2+ spike occurrence. We uncaged at a proximal spine (60 μm from the soma; Figure 4A) with either 1 Hz-GU or 0.1 Hz-GU and varied the number of uncaging pulses in the train (3, 8, 14, or 20 pulses) (Figures 4B–4D). The integrated somatic Ca2+ influx (measured with jRGECO1a) did not differ between the 2 frequencies for any number of pulses tested (Figures 4E and 4F). Similarly, the magnitude of sGFP-NFATc3 nuclear translocation did not show any frequency dependence (Figures 4E–4H). Conversely, the peak level of sGFP-NFATc3 translocation correlated with the number of uncaging pulses (Figure 4H) and integrated somatic Ca2+ signal (Figure S4I), indicating that NFATc3 translocation is sensitive to the total somatic Ca2+ signal, which better reflected the number of Ca2+ spikes rather than the frequency.
Figure 4. NFAT Translocation to the Nucleus Is Sensitive to the Number, Not Frequency, of Propagating Ca2+ Signals.

(A) Neuron transfected with jRGECO1a (pseudocolored) and sGFP-NFATc3. The cross indicates position of uncaging for experiments (~60 μm from soma) described in this figure. Arrows indicate where ROIs were drawn to make jRGECO1a ΔF/F0 measurements at uncaged spine and soma. Scale, 20 μm.
(B–D) Representative jRGECO1a ΔF/F0 traces following 3 (B), 14 (C), and 20 (D) uncaging pulses with 1 Hz-GU or 0.1 Hz-GU.
(E) Left: graph of somatic integrated jRGECO1a ΔF/F0 taken from ROI drawn on soma for experiments shown on the right. Right: sGFP-NFATc3 nucleus/cytosol ratio following 1 Hz-GU for 3 (n = 5), 8 (n = 6), 14 (n = 7), or 20 (n = 10) pulses.
(F) As (E) but with 0.1 Hz-GU for 3 (n = 9), 8 (n = 7), 14 (n = 8), or 20 (n = 8) pulses.
(G) Images of soma with sGFP-NFATc3 distribution before (start) and immediately after uncaging for pulse number and frequency as indicated.
(H) Peak sGFP-NFATc3 nuclear intensity versus number of uncaging pulses (unaveraged data analyzed with Pearson’s correlation).
(I) Left: graph of somatic integrated jRGECO1a ΔF/F0 taken from ROI drawn on soma for experiments shown on the right. Right: sGFP-NFATc4 nucleus/cytosol ratio following 1 Hz-GU for 3 (n = 6), 8 (n = 6), 14 (n = 7), 20 (n = 7), or 60 (n = 7) pulses.
(J) As (I) but with 0.1 Hz-GU for 3 (n = 4), 8 (n = 5), 14 (n = 5), 20 (n = 6), or 60 (n = 7) pulses.
(K) Images of soma with sGFP-NFATc4 distribution before (start) and immediately after uncaging for pulse number and frequency as indicated.
(L) Peak sGFP-NFATc4 nuclear intensity versus number of uncaging pulses (un-averaged data analyzed with Pearson’s correlation).
See also Figure S4.
To determine how an alternative NFATc isoform responds to the same MNI-glu evoked Ca2+ signals, we repeated the experiments with neurons co-transfected with sGFP-NFATc4 (Figures 4I–4L). Consistent with previous reports, NFATc4 was far less sensitive to stimulation than NFATc3 (Ulrich et al., 2012), with both frequencies of uncaging at 3, 8, 14, and 20 pulses failing to produce significant sGFP-NFATc4 translocation (Figures 4I–4L). Stimulation of significant sGFP-NFATc4 translocation required increasing the number of pulses to 60, which produced a modest increase in sGFP-NFATc4 nuclear occupancy that was higher with 0.1 Hz-GU than 1 Hz-GU, reflecting a larger integrated somatic Ca2+ influx with 0.1 Hz-GU (Figure 4I–4L). Similarly, sGFP-NFATc3 translocation was also higher following 60 pulses at 0.1 Hz than 1 Hz, confirming that the integrated Ca2+ influx in the soma is the best predictor of NFAT nuclear translocation for both isoforms (Figures S4G and S4H). Accordingly, for both NFATc isoforms the peak of NFATc translocation correlated significantly with the integrated soma Ca2+, with NFATc3 being most sensitive to increases in cytosolic Ca2+ (Figures S4I and S4J). Together, these results demonstrate that NFATc3 translocation responds linearly to the number of somatic Ca2+ spikes induced by short bursts (3–20 pulses) of proximal MNI-glu uncaging, independent of the train frequency or duration. Conversely, NFATc4 requires a far greater number of Ca2+ spikes for activation, and is largely inactive during lower pulse number trains, requiring prolonged stimuli and larger overall increases in somatic Ca2+ for nuclear translocation.
Somatic LTCC Ca2+ Spikes Are Required for NFAT Translocation
While somatic Ca2+ elevation following propagating Ca2+ spikes is a good predictor of sGFP-NFATc3 nuclear translocation, these Ca2+ signals concurrently spread throughout the soma and the entire dendritic arbor. To definitively determine which compartment (somatic, dendritic, or both) is the primary source of the Ca2+ that activates sGFP-NFATc3 following 1 Hz-GU, neurons were cultured in microfluidic devices to compartmentalize the soma and dendrites for pharmacologic manipulation (Taylor et al., 2010; Figures 5A and 5B). MNI-glu was continuously perfused into the dendritic compartment, while either control solution or nim (5 μM) was added to the soma compartment (Figures 5A and 5B). A dye (Cy5.5) was added to the MNI-glu solution to confirm the fluidic separation of the 2 compartments (Figures S5A and S5B). Neurons formed active synapses within the dendritic compartment, which were revealed by spontaneous spine Ca2+ transients likely resulting from quantal presynaptic glutamate release (Figure S5C). With control solution in the soma compartment, 1 Hz-GU on distal spines produced both Ca2+ spikes that propagated along the dendrite to the soma (Figure 5C) and sGFP-NFATc3 nuclear translocation (Figure 5E). When nim was included in the soma compartment, Ca2+ spikes propagated beyond the activated dendrite, but dissipated in the microgrooves and were extinguished at the soma compartment boundary (Figures 5D and S2D; Video S4). These findings highlight the dependence upon dendritic LTCCs to mediate Ca2+ spike propagation from synapse to soma. Strikingly, sGFP-NFATc3 nuclear translocation was not detected when somatic LTCCs were selectively blocked with nim (Figure 5F). These results demonstrate that for 1 Hz-GU, the bulk of sGFP-NFATc3 nuclear translocation depends upon Ca2+ influx through proximal/somatic rather than distal/dendritic LTCCs, and NFATc3 likely primarily translocates from soma to nucleus.
Figure 5. Selective Block of Somatic LTCCs Prevents NFATc3 Translocation Following Distal Spine Stimulation.

(A) Schematic of neurons grown in microfluidic culture device. The perfusion channel and somatic compartment are pharmacologically isolated due to hydrostatic pressure across microgrooves preventing solution exchange. LTCC antagonist nim (5 μM) or control solution were added to the soma compartment and MNI-glu and cy5.5 were added to the perfusion channel.
(B) Cultured hippocampal neuron grown in a microfluidic device and transfected with jRGECO1a (pseudocolored) and sGFP-NFATc3. Scale, 20 μm.
(C) jRGECO1a ΔF/F0 traces from control neuron taken at various distances from uncaged spine following 1 Hz-GU.
(D) jRGECO1a ΔF/F0 traces from neuron in (B) with nim (5 μM) in soma compartment.
(E) sGFP-NFATc3 nuclear intensity following distal spine uncaging for neuron grown in a microfluidic device with control solution in soma compartment (n = 3; one-way ANOVA repeated-measures with Dunnett, ***p < 0.001, **p < 0.01, *p < 0.05). Inset: images of soma showing sGFP-NFATc3 distribution.
(F) As in (E) but with nim in soma compartment (n = 5). Inset: image of soma showing sGFP-NFATc3 distribution for neuron in (B).
See also Figure S5.
NFAT Does Not Translocate from Distal Dendrites to the Nucleus after a Brief Uncaging Stimulus
While the previously described results suggest that the majority of NFATc3 activation takes place in the soma, it is possible that a small amount of NFATc3 may reach the nucleus from distal sites, but this amount could be below the detection threshold when imaging sGFP-NFATc3. Therefore, we used photo-convertible mEos3.2 (green to red) tagged to NFATc3 (mEos3.2-NFATc3) to track movement of the red photo-converted species from distal sites with or without simultaneous MNI-glu uncaging (+MNI-glu or −MNI-glu; 80 μM; Figures 6A–6F). We first photoconverted mEos3.2-NFATc3 in distal dendrites (>60 μm from the soma; Figure 6A) on up to 5 dendritic branches. Because preconversion red background fluorescence is very low, this method should enable detection of very small increases in nuclear red mEos3.2-NFATc3 arriving from distal sites. The photo-conversion/uncaging stimulus was first tested in neurons transfected with GCaMP6f with MNI-glu in the bath, revealing prolonged Ca2+ influx in both the dendrites and soma lasting ~1 min (Figures S6A and S6B). In neurons transfected with mEos3.2-NFATc3 without MNI-glu in the bath (−MNI-glu), no changes in mEos3.2-NFATc3 nucleus/cytosol ratios were observed for either species for up to 90 min after branch photo-conversion (Figures 6A–6C). With MNI-glu in the bath, a robust mEos3.2-NFATc3 nuclear translocation was observed for the green species (non–photo-converted) that showed similar kinetics to sGFP-NFATc3 nuclear translocation previously reported, while the red species (distally photo-converted) was not significantly different from control (Figures 6D–6F). The total length of dendrite photo-converted was not significantly different between the −MNI-glu and +MNI-glu groups (Figure S6C). Photo-conversion of a large region within the soma (Figure 6G) in control conditions led to no change in the nuc/cyto ratio of the green species but to a significant decrease in the nuc/cyto for the red species, as would be expected following a large postconversion increase in the soma population that could not be transported into the nucleus (Figures 6H and 6I). With MNI-glu in the bath, soma photo-conversion generated almost identical translocation profiles for the green and red species (Figures 6J–6L). These results further support the notion that the majority of NFATc3 that reaches the nucleus following a neuron-wide propagating Ca2+ signal originates within the soma.
Figure 6. NFATc3 Does Not Translocate from Distal Dendrites to the Nucleus after a Brief Uncaging Stimulus.

(A) Representative neuron transfected with mEos3.2-NFATc3 with preconversion green and red species and red species after conversion within distal dendrites indicated with dotted lines on the left.
(B) Images of soma showing mEos3.2-NFATc3 distribution before (pre) and after photo-conversion in control conditions (−MNI-glu). Top: green species; bottom: red species. Images are from the same neuron in (A).
(C) mEos3.2-NFATc3 nuc/cyto normalized to pre-uncaging values following branch photoconversion in 6–7 ROIs without MNI-glu in the bath. Purple arrows indicate photoconversion within a dendritic ROI (n = 6).
(D–F) As in (A)–(C) but from experiments with MNI-glu in the bath (+MNI-glu; n = 7; one-way ANOVA repeated-measures with Dunnett, ***p < 0.001).
(G–I) As in (A)–(C) but following photoconversion within a single ROI drawn on the soma (−MNI-glu; n = 5; one-way ANOVA repeated-measures with Dunnett, ***p < 0.001, **p < 0.01, *p < 0.05).
(J–L) As in (G)–(I) but following photoconversion within a single ROI drawn on the soma with MNI-glu in the path (+MNI-glu; n = 5; one-way ANOVA repeated-measures with Dunnett, ***p < 0.001, **p < 0.01). Scale bars = 20 μm.
See also Figure S6.
DISCUSSION
Rapid Communication between Synapses and the Nucleus
The results presented in this study show that synapseto-nucleus signaling through NFAT relies upon rapid somatic propagation of LTCC-Ca2+ spikes initiated in dendrites to drive soma-to-nucleus translocation of NFAT, whereas slow molecular NFAT transport from distal sites in dendrites was not observed. Previous proposed models of fast electrochemical synapse-to-nucleus signaling include the action potential and the Ca2+ wave models. In the Ca2+ spike mechanism reported here, NMDARs are required for the initiation of distal LTCC-dependent Ca2+ spikes, but neither Nav-dependent bAPs nor ER Ca2+ waves are needed for their somatic propagation and communication with the nucleus.
While our findings reveal a role for Ca2+-spikes in synapseto-nucleus signaling, other studies have demonstrated their importance in other forms of neural processing. Spontaneous and evoked responses that generate nonlinear depolarizations in dendrites and the soma have been observed both in vitro and in vivo, and are referred to most commonly as complex spikes (Apostolides et al., 2016; Golding et al., 1999; Grienberger et al., 2014; Larkum and Zhu, 2002) or plateau potentials (Bittner et al., 2015; Takahashi and Magee, 2009). Complex spikes arise from several depolarizing conductances, including NMDAR EPSPs, NaV-APs and Ca2+ spikes or spikelets, and have been demonstrated to be required for several forms of dendritic and spine LTP (Bittner et al., 2017; Remy and Spruston, 2007; Takahashi and Magee, 2009). Furthermore, in vivo studies in hippocampal CA1 neurons report that complex spikes are involved in the generation of high-frequency burst firing patterns (Grienberger et al., 2014) and the establishment and maintenance of place fields during visual learning tasks (Bittner et al., 2015, 2017; Sheffield and Dombeck, 2015; Sheffield et al., 2017).
Importantly, Ca2+-spikes within a complex spike can persist when NaV channels are blocked, and these isolated Ca2+-spikes share many similarities with the Ca2+-spikes recorded in this study (Grienberger et al., 2014; Remy and Spruston, 2007). In the experiments presented here, we identify these TTX-insensitive spike depolarizations to be LTCC-Ca2+ spikes. In addition, we observed complex spikes that were sensitive to LTCC antagonism when recording without TTX in the bath. While some studies have reported that other CaV channels are engaged during complex spikes (Remy and Spruston, 2007; Takahashi and Magee, 2009; Tsay et al., 2007), our results utilizing LTCC agonists and antagonists strongly implicate a critical role for LTCCs in spike generation. Similar LTCC spikes have been observed in prefrontal cortical neurons during blockade of NaV channels (Young and Yang, 2004), and LTCCs have been found to be the main source of Ca2+ entry during burst firing in cultured hippocampal neurons (Bengtson et al., 2013). In nonmammalian neurons that do not fire NaV-APs, LTCCs are able to support CaV-spikes or “calcium APs” (Liu et al., 2018). Intriguingly, these Ca2+-APs are abolished by pharmacologic block or genetic deletion of CaV1 and also have remarkably similar properties to the LTCC spikes we recorded here, including similar amplitude, half-width, and threshold. In sum, we present evidence for a model of synapse-to-nucleus signaling in which LTCC-Ca2+ spikes rapidly relay information from distal synapses to the soma to control transcription.
The Importance of Somatic LTCCs for NFAT Activation
The mechanisms of synapse-to-nucleus signaling have been characterized for several pathways. Slow synapto-nuclear transport has been demonstrated for such molecules as Jacob and importin-α, which are located in distal synapses, activated by NMDARs, and translocated slowly (>30 min) to the nucleus following brief chemical stimulation (Dieterich et al., 2008; Thompson et al., 2004). Photo-convertible protein tagging studies of these and other signaling proteins show that distally converted molecules can reach the nucleus following chemical or glutamate uncaging stimuli (Ch’ng et al., 2012, 2015; Dinamarca et al., 2016; Karpova et al., 2013; Lai et al., 2008). Conversely, LTCC-dependent nuclear translocation of CaMKIIγ occurs in <5 min following chemical treatment, a time-course consistent with soma-to-nucleus transport (Ma et al., 2014), but yet to be explicitly determined. ERK is thought to be capable of both slow synapse-to-nucleus molecular transport (Karpova et al., 2013; Zhai et al., 2013) and more rapid soma-to-nucleus transport (Dudek and Fields, 2001, 2002; Zhao et al., 2005), depending on the type of stimulation, with these two modes of communication likely having implications for the transcriptional outputs downstream of ERK (Tyssowski et al., 2018). Many of these nuclear signaling factors, including ERK, CRTC1, and CaMKIIα, are strongly activated by LTCCs and would therefore be predicted to respond well to somatic Ca2+ spikes.
Using our integrated imaging approach, which combined MNI-glu uncaging with Ca2+ imaging to simultaneously monitor Ca2+ compartmentalization and sGFP-NFATc3 translocation, we were able to comprehensively characterize the spatiotemporal dynamics of synapse-to-nucleus NFAT signaling to reveal a requirement for somatic Ca2+ elevation. Utilizing microfluidic neuron culture as a method to study the mechanism of synapse-to-nucleus signaling, we yielded a central finding of this study: selective block of somatic LTCCs prevented sGFP-NFATc3 nuclear translocation. Furthermore, by photo-converting mEos3.2-NFATc3 in either the dendrites or the soma with simultaneous uncaging of MNI-glu, we only observed significant increases in nuclear NFATc3 when it was photo-converted and activated within the soma. While labeling of endogenous NFATc3 is hampered by a lack of specific antibodies, our overexpression studies here would indicate that even if a large amount of NFATc3 is localized to distal dendrites, very little will be transported to the nucleus following either dendrite localized Ca2+ transients or repeated LTCC Ca2+ spikes. Conversely, somatic NFATc3 is readily imported into the nucleus following LTCC Ca2+ spikes at the soma. Together, these findings strongly support a soma-to-nucleus model of NFATc3 translocation.
We found previously that NFATc3/c4 translocation and NFAT-dependent transcription were reduced by mutations in the scaffolding protein AKAP79/150 that prevent its binding of CaN (Li et al., 2012; Murphy et al., 2014; Oliveria et al., 2007). Here we show that delocalizing CaN from the LTCC-AKAP79/150 complex also significantly diminishes sGFP-NFATc3 nuclear signaling after 1 Hz-GU, and thus we propose that the majority of LTCC-AKAP79/150-CaN-NFATc3 signaling takes place within the soma. Furthermore, we found that reducing CICR in the soma with ryanodine treatment did not alter sGFP-NFATc3 nuclear translocation, consistent with LTCCs serving as the primary Ca2+ source that activates NFATc3. Our findings further highlight the importance of LTCC nano-domain Ca2+ signaling machinery in transduction from the plasma membrane to the nucleus (Ma et al., 2014; Wheeler et al., 2012).
The Patterns of Activity that Activate NFAT
We found that NFATc3 translocation was governed by the integrated somatic Ca2+ signal, which was largely a function of the total number but not the frequency of somatic Ca2+ events. These findings are consistent with the activation properties of CaN, which functions as an input number counter and signal integrator rather than a frequency sensor (Fujii et al., 2013). Importantly, we show here that these properties of CaN also influence downstream signaling to the nucleus via NFATc3 and NFATc4 that could effect long-term changes in neuronal function. We propose that NFAT acts as a signaling factor that can relay information to the nucleus regarding the number of Ca2+ spikes that spread throughout the neuron to increase Ca2+ in the soma. While we use NFATc3/c4 nuclear occupancy as readouts of pathway activation, it is important to consider if NFAT-dependent transcription will also follow these spike counting rules. Owing to weak DNA binding affinity, NFAT transcription requires the binding of co-transcription factors, such as AP-1, MEF2 and GATA4 (Kipanyula et al., 2016). NFAT therefore acts as a coincidence detector for multiple nuclear signaling pathways, and NFAT transcriptional output will depend on not only the nuclear occupancy and DNA binding of NFAT, but also the extent to which the co-transcription factors have been activated. A full understanding of the transcriptional output of NFATs will require a detailed study of the activation properties and interactions between each of these pathways following a given stimulus.
Our findings indicate that NFAT will respond linearly to the integrated Ca2+ influx in the soma and will therefore reflect the number and/or duration of Ca2+ spikes/complex spiking. If so, what function might this serve? Several studies have demonstrated that various NFAT isoforms mediate transcriptional control of voltage-gated K+ (KV) channels, including KV7.2/3 in sympathetic neurons and KV4.2 in cerebellar and cortical neurons (Yao et al., 2016; Zhang and Shapiro, 2012). Thus, NFATc3 may function as an integrator of hippocampal neuronal activity, such that repetitive somatic Ca2+ spikes would best stimulate robust NFAT transcriptional activity, perhaps in cooperation with co-transcription factors that are similarly regulated, to alter neuronal excitability or manifest changes in intrinsic plasticity (Lin et al., 2011). Furthermore, NFAT-transcription has been linked to conditions associated with excessive neuronal depolarization that are predicted to strongly activate somatic LTCCs, including seizures and ischemia (Shioda et al., 2007; Zhang and Shapiro, 2012). However, the potential interactions of NFATs with other transcription factors to alter KV channel expression require more detailed investigation.
In summary, we define an LTCC-Ca2+ spike model for NFAT nuclear signaling following synaptic stimulation that not only provides further insight into the specific function of NFAT E-T coupling in neurons but also expands our general understanding of how neuronal signals can be relayed over long intracellular distances from synapse-to-nucleus.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mark Dell’Acqua (mark.dellacqua@ucdenver.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal procedures were approved by the University of Colorado Denver Institutional Animal Care and Use Committee in accordance with National Institutes of Health (NIH)/United States Public Health Service guidelines. Primary hippocampal neuron cultures were prepared from Sprague Dawley rats. Timed pregnant dams (~day 16) were obtained from Charles River Laboratories and housed under standard conditions.
Primary Hippocampal Cultures
Dissociated neurons prepared from P0-P1 male and female pups were plated onto poly-D-lysine coated coverglass (25 mm) at medium density (175,000 cells/ml) in MEM (Life Technologies) supplemented with BSA, penicillin/streptomycin (Life Technologies) and maintained at 37°C with 5% CO2. The following day media was exchanged to serum-free Neurobasal-A (Life Technologies) medium supplemented with GlutaMAX (Life Technologies), penicillin/streptomycin and B-27 (Invitrogen). Mitotic inhibitors (uridine + fluoro deoxyuridine; Sigma) were added at 5 Days in Vitro (DIV) and neurons were fed every 5–6 days thereafter. Experiments were performed at 15–17 DIV.
METHOD DETAILS
Plasmid DNA constructs
jRGECO1a and GCaMP6f were inserted using standard cloning techniques into a custom chick beta-actin promoter (pCAG) mammalian expression vector which was a gift from Matthew Kennedy (University of Colorado Denver). sGFP2-NFATc3 (referred to as sGFP-NFATc3) was described previously (Dittmer et al., 2017), and consisted of a C-terminally truncated human NFATc3 fragment (Hoey et al., 1995; Murphy et al., 2014) with an N-terminal sGFP2 tag. AKAP79WT and AKAP79ΔPIX were inserted into an mTq2_N1 vector using standard cloning techniques. To generate the tandem sGFP2 for the NFAT reporter construct (3xAP1-NFAT-2xsGFPnls), the open reading frame of sGFP2-C1 was amplified by PCR (forward 5′-GCGCTACCGTACGCCACCATG-3′; reverse 5′-CCGTCGACTGCAGAATTCGAA-3′) with a mismatch in the forward primer to introduce a restriction site for BsiWI (which produces BsrGI compatible overhangs). The PCR fragment was gel purified and digested with BsiWI and XhoI, then ligated into sGFP2-C1 vector cut with BsrGI and XhoI. The tandem sGFP2 open reading frame was then amplified by PCR (forward 5′-AGATCCGCTAGATCTACCGGTCGC-3′; reverse 5′-GAGTCCGGACTTGTACAGCTC-3′) to introduce a BglII restriction site with the forward primer. The product was gel purified, digested with BglII and BsrGI and ligated into a 3xAP1-NFAT-eCFPnls reporter construct (Li et al., 2012; Murphy et al., 2014) generating the final 3xAP1-NFAT-2xsGFPnls construct.
To generate the sGFP-NFATc4 construct, mNFATc4 (Mojsa et al., 2015) was PCR amplified with primers (forward 5′-CGACTCGA GGAGGGGCCGCAAGCTGCG-3′ and reverse 5′-CGATGAATTCTCAGGCAGGAGGCTCTTCTCC-3′) to introduce a 5′ XhoI site and a 3′ EcoRI site to the resulting product, which was then gel purified. The mNFATc4 insert and pSGFP2-C1 backbone were digested with XhoI and EcoRI-HF (NEB) in dual restriction digests, gel purified and ligated with T4 DNA ligase (NEB). To generate the mEos3.2-NFATc3 construct, the HindIII restriction site within the mEos coding sequence of mEos3.2-C1 was deleted with a site-directed mutagenesis PCR reaction (forward 5′-GTATTCGTGGGAACGAAGTTTGA-3′; reverse 5′- CCGTCTTCGAAAGTCAAACTTCG-3′). The NFATc3 cloning fragment used previously and the ΔHindIII mEos3.2-C1 were then digested with HindIII and ApaI, gel purified and ligated. Transformations were performed with DH5α competent cells.
Cell Transfection
Hippocampal neurons grown in standard culture conditions were transfected at DIV 13–15, 48 hr prior to imaging with Lipofectamine 2000 (Invitrogen). DNA (0.5 – 5 μg) and Lipofectamine 2000 (6 μl) were mixed in 500 μL un-supplemented neurobasal A and incubated at room temperature for 20 min. The transfection solution was added to the neurons for 90 min. Following transfection, all neurons were then returned to the original medium supplemented with 50% fresh neurobasal A for 48 h at 37°C with 5% CO2.
Confocal Microscopy
Imaging was performed on a Zeiss Observer.Z1 inverted microscope equipped with a Yokogawa CSU-X1 spinning disk unit; 405 nm/50 mW, 445nm/40 mW, 488 nm/40 mW, 515 nm/50 mW, 561 nm/50 mW and 640 nm/50 mW lasers; an EC Plan-Neofluar 40x (1.30 NA) oil immersion objective lens; an mSAC unit for correction of spherical ablation (Intelligent Imaging Innovations); a Vector live specimen scanner (Intelligent Imaging Innovations) for positioning of the photo-uncaging laser; a Photometrics PVCAM (Dynamic) EMCCD camera with 16-bit dynamic range and an mSwitcher ms optical switching unit (Intelligent Imaging Innovations) that enabled simultaneous use of the CSU-X 1 and Vector units. Instrumentation was controlled by SlideBook 6.0 software (Intelligent Imaging Innovations). Data acquisition and analysis were performed with Slidebook 6.0 software and ImageJ software.
MNI-glutamate uncaging
Neurons were incubated in artificial CSF (aCSF) containing the following in mM: 135 NaCl, 5 KCl, 25 HEPES, 10 glucose, 3 CaCl2, 0.03 glycine, and 0.001 TTX, pH 7.4 in all experiments unless otherwise stated. Following identification of neurons to be tested, the imaging aCSF was supplemented with 4-methoxy-7-nitroindolinyl (MNI)-caged L-glutamate (Tocris Bioscience) to a concentration of 1.3 mM. An ROI (1 μm x 1 μm) was selected near a dendritic spine of interest. Glutamate was photo-uncaged from MNI-glutamate in this region using 1 ms pulses from a 405 nm/50 mW laser (50 – 100 μW pulses; 1 ms dwell time), according to the laser pulse patterns as indicated for each experiment.
Live-cell Ca2+ imaging and sGFP2-NFATc3 imaging
Experiments were performed in an incubator maintained at 34°C. A dichroic mirror (445/515/640; Semrock) and two different filter sets (RFP, 561 nm excitation, 617/73 nm emission; GFP, 488 nm excitation, 525/50 nm emission) were used to capture images at a 40 ms exposure for jRGECO1a or GCaMP6f time-lapse imaging. Single z-plane images of a portion of the dendritic arbor and soma covering a 170 3 170 μm field of view were acquired at 10 Hz.
To measure the peak and integrated jRGECO1a, GCaMP6f and iGluSnFR signal following MNI-glu uncaging, regions of interest (ROIs) were drawn within the spine, dendrite, soma and background area outside of the neuron. The mean background-corrected fluorescence within each ROI was measured. A baseline average of 10 frames was measured prior to MNI-glu uncaging and each frame was normalized to this average to generate measurements of ΔF/F0. The area under the ΔF/F0 trace was measured for each compartment to generate the integrated ΔF/F0. Where multiple time-lapse images were acquired from the same neuron, the sum integrated ΔF/F0 was calculated as the total integrated ΔF/F0 from all time-lapse acquisitions. For pre- and post-treatment experiments using GCaMP6f, the same regions were analyzed pre- and post-treatment. For all BayK experiments, a sub-population of neurons that generated spontaneous neuron wide Ca2+ transients upon addition of BayK (5 μM) to the bath were excluded from analysis. Baseline sGFP-NFATc3 images were taken immediately before and 5 min after addition of BayK (5 μM) to the bath to ensure BayK alone did not promote sGFP-NFATc3 translocation, and post uncaging values were normalized to an average of the two pre uncaging values.
For sGFP2-NFATc3 imaging of the nucleus and soma, a total of 15 xy images were collected within a 7 μm z stack, at 0.5 μm intervals with a 100 ms exposure. Z stack images of sGFP2-NFATc3 were collected at −5, −1, 1, 5, 10 and then every 10 min up to 70 min from initiation of glutamate uncaging. To calculate normalized sGFP2-NFATc3 intensity, regions of interest (ROI) were drawn around the nucleus, cytosolic area in the neuronal soma, and a background area outside the neuron to determine background-corrected mean sGFP2 fluorescence intensity in the nucleus and cytosol. The nucleus was identified as the region within the soma lacking red fluorescence, as jRGECO1a includes a nuclear export signal (NES). Background-corrected nuclear and cytosolic mean fluorescence intensities were normalized to pre-uncaging values.
For experiments using the 3xAP1-NFAT-2xsGFPnls NFAT reporter construct, neurons were transfected with 3xAP1-NFAT-2xsGFPnls (0.2 μg) and jRGECO1a (2 μg). TTX (1 μM) was added to the bath after 36 hr, and neurons were imaged at 48 hr post transfection. Two-color z stack images (GFP and RFP) of the soma were collected pre-uncaging and at 2, 4 and 5 hours post-uncaging. For measurement of sGFP intensity changes ROIs were drawn over the nucleus and a background area and background-corrected nuclear fluorescence intensities were normalized to pre-uncaging values.
For the AKAP79/150 knock-down rescue experiments, neurons were transfected with jRGECO1a, sGFP-NFATc3, AKAP150 shRNAi (Hoshi et al., 2005) to knock down endogenous AKAP150 (rodent isoform), and either AKAP79WT-mTurqoise2 (mTq2) (human isoform) or AKAP79ΔPIX-mTq2. The AKAP79ΔPIX mutation deletes the CaN binding site within AKAP79. Expression of mTq2 was monitored using CFP epi-fluorescence. Experiments were then performed as previously described.
Simultaneous ER and cytosolic Ca2+ imaging
For simultaneous D1ER imaging of ER Ca2+ and jRGECO1a imaging of cytosolic Ca2+, a dichroic mirror (445/515/561/640; Semrock) was used. jRGECO1a images were collected every second using 100 ms exposure with the RFP filter set (561 nm excitation, 617/73 nm emission) and ΔF/F0 values were calculated as above. To measure changes in [Ca2+]ER, FRET and donor (CFP) images of D1ER-transfected neurons were captured sequentially every 15 s using 200–400 ms exposure to 445 nm/40 mW laser excitation with 525/50 nm emission for FRET and 200–400 ms exposure of a 445 nm/40 mW laser excitation with 482/35 nm emission for donor CFP fluorescence. Intramolecular FRET ratio (R) for D1ER was calculated from background-corrected (BKG) FRET and CFP fluorescence image intensities (INT) for each time point before and after uncaging as: R = (FRETINT – FRETBKG)/(CFPINT – CFPBKG). We then generated D1ER R/R0 – 1 time traces and integrated R/R0 – 1 values for specific distances along the dendrite from the uncaged spine to the soma as done above for integrated ΔF/F0 measurements.
Electrophysiology
Simultaneous current clamp recording/Ca2+ imaging/glutamate uncaging of dissociated neurons was carried out on an Olympus IX71 equipped with a spinning disc scan head (Yokogawa). Excitation illumination was delivered from an acousto-optic tunable filter (AOTF) controlled laser launch (Andor). Images were acquired using a 40x Plan Apochromat 1.35 NA objective, and collected on a 1024×1024 pixel Andor iXon EM-CCD camera. Neurons were incubated in aSCF supplemented with MNI-glu as described above. Individual dendritic spines were focally stimulated using galvanometric mirrors (FRAPPA, Andor Technologies) to steer a diffraction-limited 405 nm spot. An AOTF was used to gate a 500 μsec pulse of 405 nm light, with the laser intensity adjusted to 8%–10% of total laser power from a 100 mW 405 nm laser that was fiber coupled to the FRAPPA laser scanning unit. Intracellular solution contained (in mM): 137 potassium gluconate, 5 potassium chloride, 4 Na2ATP, 0.5 Na3GTP, 10 phosphocreatine, 10 HEPES (287 mOsm). The pH was adjusted to 7.25. Data were collected using a multiclamp 700b amplifier and digitized using a National Instruments DAQ board at 10 KHz and filtered at 2 KHz (single pole Bessel filter) and collected with WinLTP software (University of Bristol). EPSPs and Ca2+ spikes were identified using template searches in ClampFit. The Ca2+ spike threshold for each trace was estimated from the inflection point in the 1st derivative calculated from the averaged Ca2+ spike waveform (Figure S3K). The amplitude and half-width of each response were then measured using the threshold as a baseline. All EPSP and Ca2+ spike analyses were performed in ClampFit.
Microfluidic Neuron Culture, transfection and experimental procedures
Dissociated hippocampal neurons were prepared as stated previously, and seeded at a density of 4 × 106 cells/ml into both compartments of non-plasma bonded microfluidic devices (uLP; Xona Microfluidics), as described in (Taylor et al., 2010). Neurons were left to adhere to the coverglass for 20 – 30 min, then MEM was added to the four cell compartment inlets. Devices were then placed on an elevated platform in 6 cm covered dishes containing ~0.3 mL of sterile DI H2O to maintain humidity. Cultures were maintained as described above for standard culture conditions. Neurobasal-A media was added to the three perfusion channel inlets on DIV 1. All microfluidic experiments were performed at DIV 14.
Hippocampal neurons grown in microfluidic neuron culture were transfected with jRGECO1a and sGFP-NFATc3 using lipofectamine2000 (Invitrogen). DNA (3 μg) and lipofectamine (6 μl) were mixed in 100 μL un-supplemented neurobasal A and incubated at room temperature for 20 min. Media was removed from the two wells flanking to the soma compartment and 100 μL solution was added to the top well and allowed to flow into the soma compartment. The transfection solution was redistributed from the bottom well to the top well every 10 min for a total of 40 min. Following transfection, all neurons were then returned to the original medium supplemented with 50% fresh neurobasal A for 48 h at 37°C with 5% CO2.
For microfluidic experiments, either control or nim (5 μM) supplemented aCSF was added to the soma compartment 30 min before the start of the experiment, while MNI-glu solution (1.3 mM) and cy5.5 (5 μM) were added to the perfusion channel inlets and continuously drawn through the perfusion channel with a syringe-pump (NE-1000; New Era Pump Systems Inc.) at a rate of 100 μl/hour. Cy5.5 was imaged with a 640 nm laserline to ensure MNI-glu perfusion into the perfusion channel and fluidic compartmentalization. MNI-glu was uncaged at spines located within the dendritic compartment as described previously and sGFP2-NFATc3 z stacks were taken at intervals before and after uncaging as indicated.
Photoconversion experiments
mEos3.2-NFATc3 was photo-converted within ROIs drawn on the dendrites or soma using 1 s pulses from a 405 nm/50 mW laser (50 – 100 μW pulses; 1 s dwell time; raster 150) during a two-color time-lapse acquisition of the green and red species. For imaging of the nucleus and soma, a total of 20 dual color xy images were collected within a 10 μm z stack, at 0.5 μm intervals with a 100 ms exposure. Z stack images of mEos3.2-NFATc3 were collected at −5, −1, 1, 5, 10 and then every 10 min up to 90 min from the end of the photo-conversion stimulus. To calculate normalized mEos3.2-NFATc3 intensity, regions of interest (ROI) were drawn around the nucleus, cytosolic area in the neuronal soma, and a background area outside the neuron to determine background-corrected nucleus/cytosol ratio. When MNI-glu was included in the bath, its concentration was reduced from that used in other experiments to 80 μM due to the prolonged length of the photoconversion stimulus. Excitation with 405 nm light increased fluorescence of the green species outside of the photo-converted ROI in many experiments (likely due to fluorophore transitions from the triplet to ground to excited state in response 405 nm light), therefore nucleus/cytosol ratio measurements were used.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification details for both time-lapse and z stack image analyses described in methods above. Statistical analyses (statistical test used and significance) for each dataset are reported in the figure legends. Data are presented as mean ± SEM “n” values for represent the number of neurons imaged and are reported for in the figure legends. Prism 5.0 (Graph Pad, USA) software was used to test statistical significance. Student’s t tests (either paired, unpaired or one-sample) were used as indicated. sGFP-NFATc3 or mEos3.2-NFATc3 translocation time course data were analyzed with One-way ANOVA repeated-measures with post hoc Dunnett tests (versus pre-uncaging control values). In Figures 2 and S2, individual distance decay curves were fit with a one-phase decay exponential in Prism to generate length constant λ values, with mean λ values reported. Pearson’s linear correlation was used for correlation analysis. Where averaged data are presented in Figures 4H and 4L, correlation analysis was performed on un-averaged data. For all statistical analyses used, *p < 0.05, **p < 0.01, ***p < 0.001.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| MNI-caged-L-glutamate | Tocris | 1490 |
| Nimodipine (nim) | Sigma | N149 |
| (S)-(-)-Bay K 8644 (BayK) | Tocris | 1546 |
| ω-Conotoxin GVIA | Sigma | C9915 |
| ω-Conotoxin MVIIC | Sigma | C4188 |
| D-AP5 | Tocris | 0106 |
| Sulfo-Cyanine5.5 carboxylic acid | Lumiprobe | 17390 |
| Tetrodotoxin (TTX) | Tocris | 1078 |
| S-MCPG | Tocris | 0337 |
| Ryanodine | Tocris | 1329 |
| Experimental Models: Organisms/Strains | ||
| Rat, Sprague Dawley | Charles River | Crl:CD(SD)BR; RRID: RGD_734476 |
| Recombinant DNA | ||
| pGP-CMV-NES-jRGECO1a (jREGECO1a) | Dana et al., 2016 | Addgene #61563 |
| pGP-CMV-GCaMP6f (GCaMP6f) | Chen et al., 2013 | Addgene #40755 |
| pCMV(MinDis).iGluSnFR (iGluSnFR) | Marvin et al., 2013 | Addgene #41732 |
| pmTurquoise2-N1 (mTq2) | Goedhart et al., 2012 | Addgene #60561 |
| sGFP2-C1 (sGFP) | Kremers et al., 2007 | Addgene #22881 |
| pSilencer | Hoshi et al., 2005 | N/A |
| mEos3.2-C1 | Zhang et al., 2012 | Addgene #54550 |
| pcDNA3-D1ER | Palmer et al., 2004 | Addgene #36325 |
| Software and Algorithms | ||
| Prism 5 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
| Slidebook 6.0 | Intelligent Imaging Innovations | https://www.intelligent-imaging.com/slidebook |
| ClampFit 10.6 | Molecular Devices | http://mdc.custhelp.com/app/answers/detail/a_id/18779 |
| Other | ||
| Microfluidic Culture devices uLP | Xona Microfluidics | μLP |
Highlights.
Distal dendritic stimuli that elevate soma Ca2+ drive NFAT nuclear translocation
LTCC-Ca2+ spikes rapidly signal from dendrite to soma to activate NFAT
NFAT translocates from soma to nucleus in response to soma LTCC-Ca2+ spikes
The number of somatic Ca2+ spikes governs the extent of NFAT translocation
ACKNOWLEDGMENTS
We thank Tyler Martinez for technical assistance with plasmid construction for sGFP-NFATc4. Some imaging experiments were performed in the University of Colorado Anschutz Medical Campus Advanced Light Microscopy Core, supported in part by Rocky Mountain Neurological Disorders NIH/NINDS Core Center Grant P30NS048154. This work was supported by the AHA postdoctoral fellowship 17POST33630148 to A.R.W., NIH/NINDS predoctoral fellowship F31NS100403 to B.L.S., NIH/NIMH grant R01MH102338 to M.L.D., and NIH/NINDS grant R01NS107554 to M.J.K. The contents are the authors’ sole responsibility and do not necessarily represent official AHA or NIH views.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found with this article online at https://doi.org/10.1016/j.celrep.2019.03.005.
REFERENCES
- Adams JP, and Dudek SM (2005). Late-phase long-term potentiation: getting to the nucleus. Nat. Rev. Neurosci 6, 737–743. [DOI] [PubMed] [Google Scholar]
- Apostolides PF, Milstein AD, Grienberger C, Bittner KC, and Magee JC (2016). Axonal Filtering Allows Reliable Output during Dendritic Plateau-Driven Complex Spiking in CA1 Neurons. Neuron 89, 770–783. [DOI] [PubMed] [Google Scholar]
- Bengtson CP, Freitag HE, Weislogel J-M, and Bading H (2010). Nuclear calcium sensors reveal that repetition of trains of synaptic stimuli boosts nuclear calcium signaling in CA1 pyramidal neurons. Biophys. J 99, 4066–4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtson CP, Kaiser M, Obermayer J, and Bading H (2013). Calcium responses to synaptically activated bursts of action potentials and their synapse-independent replay in cultured networks of hippocampal neurons. Biochim. Biophys. Acta 1833, 1672–1679. [DOI] [PubMed] [Google Scholar]
- Bittner KC, Grienberger C, Vaidya SP, Milstein AD, Macklin JJ, Suh J, Tonegawa S, and Magee JC (2015). Conjunctive input processing drives feature selectivity in hippocampal CA1 neurons. Nat. Neurosci 18, 1133–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner KC, Milstein AD, Grienberger C, Romani S, and Magee JC (2017). Behavioral time scale synaptic plasticity underlies CA1 place fields. Science 357, 1033–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch’ng TH, Uzgil B, Lin P, Avliyakulov NK, O’Dell TJ, and Martin KC (2012). Activity-dependent transport of the transcriptional coactivator CRTC1 from synapse to nucleus. Cell 150, 207–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch’ng TH, DeSalvo M, Lin P, Vashisht A, Wohlschlegel JA, and Martin KC (2015). Cell biological mechanisms of activity-dependent synapse to nucleus translocation of CRTC1 in neurons. Front. Mol. Neurosci 8, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T-W, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana H, Mohar B, Sun Y, Narayan S, Gordus A, Hasseman JP, Tsegaye G, Holt GT, Hu A, Walpita D, et al. (2016). Sensitive red protein calcium indicators for imaging neural activity. eLife 5, e12727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisseroth K, Bito H, and Tsien RW (1996). Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron 16, 89–101. [DOI] [PubMed] [Google Scholar]
- Dieterich DC, Karpova A, Mikhaylova M, Zdobnova I, König I, Landwehr M, Kreutz M, Smalla KH, Richter K, Landgraf P, et al. (2008). Caldendrin-Jacob: A protein liaison that couples NMDA receptor signalling to the nucleus. PLoS Biol. 6, 0286–0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinamarca MC, Guzzetti F, Karpova A, Lim D, Mitro N, Musardo S, Mellone M, Marcello E, Stanic J, Samaddar T, et al. (2016). Ring finger protein 10 is a novel synaptonuclear messenger encoding activation of NMDA receptors in hippocampus. eLife 5, e12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer PJ, Wild AR, Dell’Acqua ML, and Sather WA (2017). STIM1 Ca2+ Sensor Control of L-type Ca2+-Channel-Dependent Dendritic Spine Structural Plasticity and Nuclear Signaling. Cell Rep. 19, 321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, and Fields RD (2001). Mitogen-activated protein kinase/extracellular signal-regulated kinase activation in somatodendritic compartments: roles of action potentials, frequency, and mode of calcium entry. J. Neurosci 21, RC122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, and Fields RD (2002). Somatic action potentials are sufficient for late-phase LTP-related cell signaling. Proc. Natl. Acad. Sci. USA 99, 3962–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey U, Frey S, Schollmeier F, and Krug M (1996). Influence of actinomycin D, a RNA synthesis inhibitor, on long-term potentiation in rat hippocampal neurons in vivo and in vitro. J. Physiol 490, 703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii H, Inoue M, Okuno H, Sano Y, Takemoto-Kimura S, Kitamura K, Kano M, and Bito H (2013). Nonlinear decoding and asymmetric representation of neuronal input information by CaMKIIα and calcineurin. Cell Rep. 3, 978–987. [DOI] [PubMed] [Google Scholar]
- Goedhart J, von Stetten D, Noirclerc-Savoye M, Lelimousin M, Joosen L, Hink MA, van Weeren L, Gadella TWJ Jr., and Royant A (2012). Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat. Commun 3, 751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding NL, Jung HY, Mickus T, and Spruston N (1999). Dendritic calcium spike initiation and repolarization are controlled by distinct potassium channel subtypes in CA1 pyramidal neurons. J. Neurosci 19, 8789–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, and Crabtree GR (1999). L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature 401, 703–708. [DOI] [PubMed] [Google Scholar]
- Greer PL, and Greenberg ME (2008). From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron 59, 846–860. [DOI] [PubMed] [Google Scholar]
- Grienberger C, Chen X, and Konnerth A (2014). NMDA receptor-dependent multidendrite Ca(2+) spikes required for hippocampal burst firing in vivo. Neuron 81, 1274–1281. [DOI] [PubMed] [Google Scholar]
- Hagenston AM, and Bading H (2011). Calcium signaling in synapse-to-nucleus communication. Cold Spring Harb. Perspect. Biol 3, a004564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, and Catterall WA (1993). Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J. Cell Biol 123, 949–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst WA, and Martin KC (2017). Regulated transport of signaling proteins from synapse to nucleus. Curr. Opin. Neurobiol 45, 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoey T, Sun YL, Williamson K, and Xu X (1995). Isolation of two new members of the NF-AT gene family and functional characterization of the NF-AT proteins. Immunity 2, 461–472. [DOI] [PubMed] [Google Scholar]
- Hoshi N, Langeberg LK, and Scott JD (2005). Distinct enzyme combinations in AKAP signalling complexes permit functional diversity. Nat. Cell Biol 7, 1066–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova A, Mikhaylova M, Bera S, Bär J, Reddy PP, Behnisch T, Rankovic V, Spilker C, Bethge P, Sahin J, et al. (2013). Encoding and transducing the synaptic or extrasynaptic origin of NMDA receptor signals to the nucleus. Cell 152, 1119–1133. [DOI] [PubMed] [Google Scholar]
- Kipanyula MJ, Kimaro WH, and Seke Etet PF (2016). The Emerging Roles of the Calcineurin-Nuclear Factor of Activated T-Lymphocytes Pathway in Nervous System Functions and Diseases. J. Aging Res 2016, 5081021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremers GJ, Goedhart J, van den Heuvel DJ, Gerritsen HC, and Gadella TWJ Jr. (2007). Improved green and blue fluorescent proteins for expression in bacteria and mammalian cells. Biochemistry 46, 3775–3783. [DOI] [PubMed] [Google Scholar]
- Lai K-O, Zhao Y, Ch’ng TH, and Martin KC (2008). Importin-mediated retrograde transport of CREB2 from distal processes to the nucleus in neurons. Proc. Natl. Acad. Sci. USA 105, 17175–17180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, and Zhu JJ (2002). Signaling of layer 1 and whisker-evoked Ca2+ and Na+ action potentials in distal and terminal dendrites of rat neocortical pyramidal neurons in vitro and in vivo. J. Neurosci 22, 6991–7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PR, Cohen JE, Iacobas DA, Iacobas S, and Fields RD (2017). Gene networks activated by specific patterns of action potentials in dorsal root ganglia neurons. Sci. Rep 7, 43765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Pink MD, Murphy JG, Stein A, Dell’Acqua ML, and Hogan PG (2012). Balanced interactions of calcineurin with AKAP79 regulate Ca2+-calcineurin-NFAT signaling. Nat. Struct. Mol. Biol 19, 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Sun W, Kung F, Dell’Acqua ML, and Hoffman DA (2011). AKAP79/150 impacts intrinsic excitability of hippocampal neurons through phosphoregulation of A-type K+ channel trafficking. J. Neurosci 31, 1323–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Kidd PB, Dobosiewicz M, and Bargmann CI (2018). C. elegans AWA Olfactory Neurons Fire Calcium-Mediated All-or-None Action Potentials. Cell 175, 57–70.e17. [DOI] [PubMed] [Google Scholar]
- Ma H, Groth RD, Cohen SM, Emery JF, Li B, Hoedt E, Zhang G, Neubert TA, and Tsien RW (2014). gCaMKII shuttles Ca2+/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell 159, 281–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin JS, Borghuis BG, Tian L, Cichon J, Harnett MT, Akerboom J, Gordus A, Renninger SL, Chen T-W, Bargmann CI, et al. (2013). An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat. Methods 10, 162–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojsa B, Mora S, Bossowski JP, Lassot I, and Desagher S (2015). Control of neuronal apoptosis by reciprocal regulation of NFATc3 and Trim17. Cell Death Differ. 22, 274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JG, Sanderson JL, Gorski JA, Scott JD, Catterall WA, Sather WA, and Dell’Acqua ML (2014). AKAP-anchored PKA maintains neuronal Ltype calcium channel activity and NFAT transcriptional signaling. Cell Rep. 7, 1577–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveria SF, Dell’Acqua ML, and Sather WA (2007). AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron 55, 261–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AE, Jin C, Reed JC, and Tsien RY (2004). Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc. Natl. Acad. Sci. USA 101, 17404–17409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panayotis N, Karpova A, Kreutz MR, and Fainzilber M (2015). Macromolecular transport in synapse to nucleus communication. Trends Neurosci. 38, 108–116. [DOI] [PubMed] [Google Scholar]
- Remy S, and Spruston N (2007). Dendritic spikes induce single-burst longterm potentiation. Proc. Natl. Acad. Sci. USA 104, 17192–17197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross WN (2012). Understanding calcium waves and sparks in central neurons. Nat. Rev. Neurosci 13, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield MEJ, and Dombeck DA (2015). Calcium transient prevalence across the dendritic arbour predicts place field properties. Nature 517, 200–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield MEJ, Adoff MD, and Dombeck DA (2017). Increased Prevalence of Calcium Transients across the Dendritic Arbor during Place Field Formation. Neuron 96, 490–504.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioda N, Han F, Moriguchi S, and Fukunaga K (2007). Constitutively active calcineurin mediates delayed neuronal death through Fas-ligand expression via activation of NFAT and FKHR transcriptional activities in mouse brain ischemia. J. Neurochem 102, 1506–1517. [DOI] [PubMed] [Google Scholar]
- Takahashi H, and Magee JC (2009). Pathway interactions and synaptic plasticity in the dendritic tuft regions of CA1 pyramidal neurons. Neuron 62, 102–111. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Dieterich DC, Ito HT, Kim SA, and Schuman EM (2010). Microfluidic local perfusion chambers for the visualization and manipulation of synapses. Neuron 66, 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson KR, Otis KO, Chen DY, Zhao Y, O’Dell TJ, and Martin KC (2004). Synapse to nucleus signaling during long-term synaptic plasticity; a role for the classical active nuclear import pathway. Neuron 44, 997–1009. [DOI] [PubMed] [Google Scholar]
- Tsay D, Dudman JT, and Siegelbaum SA (2007). HCN1 channels constrain synaptically evoked Ca2+ spikes in distal dendrites of CA1 pyramidal neurons. Neuron 56, 1076–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyssowski KM, DeStefino NR, Cho J-H, Dunn CJ, Poston RG, Carty CE, Jones RD, Chang SM, Romeo P, Wurzelmann MK, et al. (2018). Different Neuronal Activity Patterns Induce Different Gene Expression Programs. Neuron 98, 530–546.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich JD, Kim M-S, Houlihan PR, Shutov LP, Mohapatra DP, Strack S, and Usachev YM (2012). Distinct activation properties of the nuclear factor of activated T-cells (NFAT) isoforms NFATc3 and NFATc4 in neurons. J. Biol. Chem 287, 37594–37609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Marks CR, Perfitt TL, Nakagawa T, Lee A, Jacobson DA, and Colbran RJ (2017). A novel mechanism for Ca2+/calmodulin-dependent protein kinase II targeting to L-type Ca2+ channels that initiates long-range signaling to the nucleus. J. Biol. Chem 292, 17324–17336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DG, Barrett CF, Groth RD, Safa P, and Tsien RW (2008). CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation-transcription coupling. J. Cell Biol 183, 849–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P, and Tsien RW (2012). Ca(V)1 and Ca(V)2 channels engage distinct modes of Ca(2+) signaling to control CREB-dependent gene expression. Cell 149, 1112–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild AR, and Dell’Acqua ML (2018). Potential for therapeutic targeting of AKAP signaling complexes in nervous system disorders. Pharmacol. Ther 185, 99–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao JJ, Zhao QR, Liu DD, Chow CW, and Mei YA (2016). Neuritin Up-regulates Kv4.2 α-subunit of potassium channel expression and affects neuronal excitability by regulating the calcium-calcineurin-NFATc4 signaling pathway. J. Biol. Chem 291, 17369–17381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CE, and Yang CR (2004). Dopamine D1/D5 receptor modulates state-dependent switching of soma-dendritic Ca2+ potentials via differential protein kinase A and C activation in rat prefrontal cortical neurons. J. Neurosci 24, 8–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai S, Ark ED, Parra-Bueno P, and Yasuda R (2013). Long-distance integration of nuclear ERK signaling triggered by activation of a few dendritic spines. Science 342, 1107–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, and Shapiro MS (2012). Activity-dependent transcriptional regulation of M-Type (Kv7) K(+) channels by AKAP79/150-mediated NFAT actions. Neuron 76, 1133–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Chang H, Zhang Y, Yu J, Wu L, Ji W, Chen J, Liu B, Lu J, Liu Y, et al. (2012). Rational design of true monomeric and bright photoactivatable fluorescent proteins. Nat. Methods 9, 727–729. [DOI] [PubMed] [Google Scholar]
- Zhao M, Adams JP, and Dudek SM (2005). Pattern-dependent role of NMDA receptors in action potential generation: consequences on extracellular signal-regulated kinase activation. J. Neurosci 25, 7032–7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.