

SUMMARY
In response to retinal damage, the Müller glial cells (MGs) of the zebrafish retina have the ability to undergo a cellular reprogramming event in which they enter the cell cycle and divide asymmetrically, thereby producing multipotent retinal progenitors capable of regenerating lost retinal neurons. However, mammalian MGs do not exhibit such a proliferative and regenerative ability. Here, we identify Hippo pathway-mediated repression of the transcription cofactor YAP as a core regulatory mechanism that normally blocks mammalian MG proliferation and cellular reprogramming. MG-specific deletion of Hippo pathway components Lats1 and Lats2, as well as transgenic expression of a Hippo non-responsive form of YAP (YAP5SA), resulted in dramatic Cyclin D1 upregulation, loss of adult MG identity, and attainment of a highly proliferative, progenitor-like cellular state. Our results reveal that mammalian MGs may have latent regenerative capacity that can be stimulated by repressing Hippo signaling.
In Brief
Rueda et al. identify the Hippo pathway as an endogenous molecular mechanism normally preventing mammalian Müller glial reprogramming to a proliferative, progenitor-like state.
Graphical Abstract
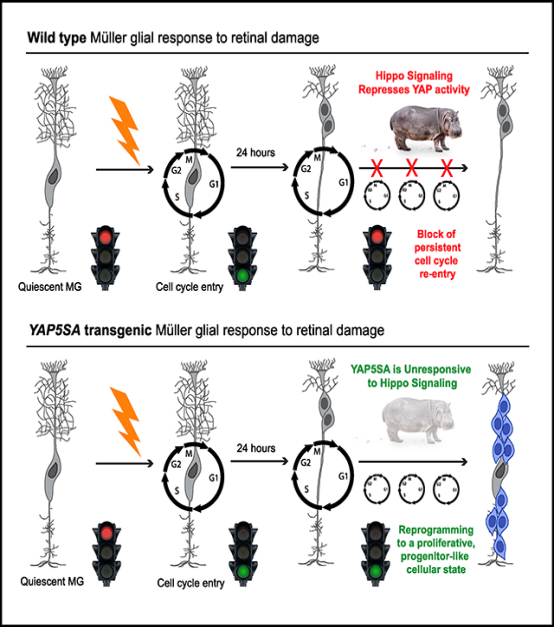
INTRODUCTION
The adult retinae of several non-mammalian vertebrate species, such as the zebrafish, are capable of a robust regenerative response that leads to the production of new photoreceptors and vision restoration. This feat comes from the ability of resident Müller glial cells (MGs) to reenter the cell cycle and produce proliferative, multipotent progenitor cells (Bernardos et al., 2007; Fausett and Goldman, 2006; Fimbel et al., 2007; Goldman, 2014; Qin et al., 2009; Ramachandran et al., 2010; Thummel et al., 2008). Unfortunately, mammalian MGs lack this regenerative response. Past studies have indicated that mouse MGs have some inherent ability to reenter the cell cycle in response to retinal damage, but this response is short lived and does not result in regeneration (Close et al., 2006; Dyer and Cepko, 2000; Karl et al., 2008; Ooto et al., 2004). In a landmark study, quiescent adult mouse MGs were shown to simultaneously express the cell-cycle G1 and/or S phase-promoting CYCLIN D3 protein and the cyclin kinase inhibitor p27KIP1 (Dyer and Cepko, 2000). p27KIP1 is known to inhibit the CYCLIN D/cyclin dependent kinase (CDK) complex, thereby preventing the G1- to S-phase transition (Dyer and Cepko, 2001a, 2001b). Coexpression of these proteins suggests that quiescent mouse MGs are primed for cell-cycle entry upon retinal damage. Indeed, 24 h after drug-induced retinal neuron death, a small subset of MGs entered S phase coincident with loss of P27KIP1 expression while CYCLIN D3 expression persisted, presumably in a derepressed state (Dyer and Cepko, 2000). Over an additional 24 h, through an unknown mechanism, CYCLIN D3 expression was turned off and proliferation stopped (Dyer and Cepko, 2000). These data indicate that the proliferative and regenerative machinery that drives retinal self-repair in the zebrafish may be present in mammals, but it is actively maintained in a dormant state.
Studies have shown that forced expression of transcription factors, as well as treatment with growth factors and drugs targeting chromatin-modifying enzymes, have the ability to push MGs to enter a proliferative state and transdifferentiate into retinal neurons. For example, ectopic expression of the proneural transcription factor Ascl1 in adult mouse MGs, along with intravitreal injection of the histone deacetylase inhibitor trichostatin-A, resulted in direct transdifferentiation of adult MGs to new retinal neurons, albeit limited to bipolar and amacrine-like identities (Jorstad et al., 2017). However, in contrast to zebrafish retinal regeneration, these adult MGs did not initially reprogram to a proliferative, progenitor state. This finding indicates that a full regenerative response, including the clonal expansion of a multipotent cell population, requires additional, unidentified molecular players. In another set of studies, adeno-associated virus (AAV)-driven expression of β-catenin in adult mouse MGs promoted spontaneous cell-cycle reentry in uninjured retinae (Yao et al., 2016). When these cells were subsequently exposed to AAV-driven rod-specifying factors Otx2, Crx, and Nrl, they were reported to differentiate into rods capable of vison restoration in a mouse model of congenital blindness (Yao et al., 2018). While these studies establish the transdifferentiation and regenerative potential of mouse MGs, they used primarily gain-of-function approaches. Therefore, the precise cellular reprogramming mechanism driving the regenerative response and the endogenous blocking mechanism normally preventing its sustained activity remain unknown. Here, we report the identification of the Hippo pathway as an endogenous blocking mechanism normally preventing MGs from adopting a proliferative, progenitor-like state.
Originally identified in Drosophila, the Hippo pathway is a conserved kinase cascade that regulates organ growth during development (Harvey et al., 2003; Heallen et al., 2011; Udan et al., 2003; Wu et al., 2003). Within the core mammalian Hippo pathway, the MST½ (Hippo) kinases phosphorylate the scaffold proteins SALV and MOB1A/B, which assist in the recruitment and phosphorylation of the LATS½ kinases. Phospho-LATS½ in turn phosphorylates YAP and TAZ, leading to phospho-YAP/TAZ binding by 14–3-3, which causes cytoplasmic retention and eventually proteasomal degradation. The end result is that YAP and TAZ are unable to translocate to the nucleus to function as TEAD cofactors. This repression of YAP/TAZ-TEAD complex formation results in loss of expression of target genes typically involved in processes such as the cell cycle, cell migration, and cellular potency (Moya and Halder, 2019). Beyond its role in developmental growth control, the Hippo pathway is known to regulate cellular renewal in tissues such as the liver, lung, skin, and intestine (Moya and Halder, 2019). In the most striking case, studies have shown that disruption of Hippo signaling, or activation of its effector YAP, promotes adult mouse cardiomyocyte regeneration in response to myocardial infarction (Leach et al., 2017; Lin et al., 2014).
We show that in response to retinal damage, Hippo signaling prevents sustained YAP activity in reactive MGs, leading to transcriptional downregulation of YAP target genes that normally promote cell-cycle reentry. Genetic loss or bypass of Hippo signaling within adult MGs causes spontaneous MG proliferation, along with the loss of MG identity and the acquisition of a cellular state more reminiscent of a progenitor cell. These data provide an essential molecular entry point from which to devise strategies to promote the MG-mediated regeneration of the mammalian retina.
RESULTS
YAP Expression in Adult Mouse MGs
To investigate whether Hippo pathway activity is present within adult mammalian MGs, we first performed YAP immunofluorescence and confocal microscopy on post-natal day 21 (P21) mouse retinal cryosections. Consistent with a previous report (Hamon et al., 2017), nuclear-localized, active YAP was detected within the center of the inner nuclear layer (INL) and colocalized with nuclear SOX9 expression, confirming MG identity (Figure 1A). In addition, YAP was observed in the MG cytoplasm, suggesting negative regulation by the Hippo pathway (Figure 1A, arrowheads). This finding was supported by immunofluorescence with an antibody that specifically recognizes phosphorylated YAP (pYAP). While no nuclear expression of pYAP was observed, colabeling with glutamine synthetase (GS) (Figure 1B, arrowheads) and total YAP antibodies (Figures S1A–S1C) confirmed expression specifically in MG cytoplasm. We also determined that the transcription factor TEAD1 is specifically expressed in adult mouse MG nuclei and coimmunoprecipitated with YAP in retinal lysates (Figures S1D and S1E). These data indicate that YAP and TEAD1 form a transcription factor complex in quiescent mouse MGs and that a steady state of Hippo regulation is present.
Figure 1. Negative Regulation of YAP in Response to Retinal Damage.

(A) P21 retinal immunofluorescence with an antibody against total YAP and Müller glial cell (MG) nuclear marker SOX9.
(B) Labeling with an antibody specific to phosphorylated YAP (pYAP) and MG marker glutamine synthetase (GS).
(C and D) Immunofluorescence for MG markers SOX2 (magenta) and GFAP (green) 48 h after intraocular injection of PBS or N-methyl-D-aspartate (NMDA).
(E) SOX2/PH3 immunofluorescence 48 h after NMDA injection.
(F and G) Time course western blots (F) and quantification (G; normalized to vinculin) of pYAP and YAP after NMDA injection.
(H and I) YAP (magenta)/SOX9 (cyan) and pYAP immunofluorescence (green) (H) and quantification (I) 24 h post-NMDA.
(J and K) Cell fraction time course western blots (J) and quantification (K) of YAP and pYAP 24 and 48 h post-NMDA.
For western blots, levels are given as a.u. ± SEM relative to PBS or no-treatment (NT) controls harvested at the 6-h time point (set to 1) (n = 3 independent pooled samples per group; Student’s t test). For pixel intensity measurements, levels are given as mean ± SEM (n = 3 per group; Wilcoxon rank-sum test). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
The Hippo Pathway and Cyclin D1 Expression Are Responsive to Retinal Damage
Because YAP is expressed in quiescent MGs, we next asked whether YAP expression or activity is affected in reactive MGs responding to retinal damage. We performed intravitreal injections of the excitotoxin NMDA (N-methyl-D-aspartate). By 48 h post-injection, the NMDA-damaged retinae were thinner than the PBS controls, suggesting loss of amacrine and ganglion cells as previously described (Ueki et al., 2015; Yao et al., 2016). We also confirmed that MGs upregulated GFAP expression, indicating reactive gliosis (Figures 1C and 1D) (Karl and Reh, 2012). We occasionally observed SOX2+ MGs that labeled with antibodies against phospho-histone H3 (PH3), consistent with previous reports of rare and transient cell-cycle entry of reactive MGs (Figure 1E) (Close et al., 2006; Dyer and Cepko, 2000; Karl et al., 2008; Ooto et al., 2004). Western blot analysis of whole retinal lysates from NMDA-damaged retinae showed a progressive increase in YAP phosphorylation beginning as early as 6 h post-damage (Figures 1F and 1G). Labeling of cryosections at 24 h post-NMDA confirmed that YAP nuclear localization (indicated by SOX9 colabeling) is reduced specifically within MGs while cytoplasmic localization of YAP and pYAP signal intensity is increased (Figures 1H and 1I). In addition, cellular fractions from retinal lysates showed the expected increase in pYAP in the cytoplasm (Figures 1J and 1K). In total, these data indicate that within 24 h of retinal damage, YAP activity in reactive MGs is repressed by Hippo signaling. This finding also raised the possibility that sustained YAP activity may be required for persistent MG proliferation.
In a previous study, quiescent adult mouse MGs were shown to simultaneously express the cell-cycle G1 and/or S phase-promoting CYCLIN D3 protein and the cyclin kinase inhibitor p27KIP1 (Dyer and Cepko, 2000). p27KIP1 normally inhibits the CYCLIN D/CDK complex to prevent G1-to S-phase transition (Dyer and Cepko, 2001a, 2001b). 24 h after retinal damage, P27Kip1 expression was shown to turn off while CYCLIN D3 expression persisted and was presumed to drive a brief period of S-phase entry before being silenced (Dyer and Cepko, 2000). These data indicated that mouse MGs have proliferative ability but that within 24 h, it is suppressed by an unknown mechanism. Surprisingly, when we independently analyzed CYCLIN D3 in response to NMDA damage in vivo, we did not observe significant changes in either protein or mRNA (Figures 2A–2C). However, between 12 and 24 h, we observed a dramatic burst of CYCLIN D1 expression that was followed by a sharp decline by 48 h post-damage (Figures 2A, 2D, and 2E). Immunofluorescence confirmed that the CYCLIN D1 expression changes occur specifically in GS+/SOX2+ MGs (Figure 2F; Figures S1F and S1G).
Figure 2. CYCLIN D3 and Cyclin D1 Expression in Response to Retinal Injury and CKO of Yap and Taz.

(A) CYCLIN D1 and CYCLIN D3 western blot time course of NMDA-damaged retinae.
(B and C) Quantification of CYCLIN D3 western blots (B) and qRT-PCR (C) analysis of mRNA.
(D and E) Quantification of CYCLIN D1 western blots (D) and qRT-PCR (E) analysis of mRNA.
(F) CYCLIN D1 immunofluorescence 12, 24, and 48 h post-injury.
(G and H) YAP (magenta) and SOX9 (cyan) immunofluorescence on control (G) and CKO MGs (H) (labeled with GFP from the ROSE26R-mTmG Cre reporter).
(I and J) CYCLIN D3 (magenta) and SOX9 (cyan) immunofluorescence on control (I) and CKO MGs (J).
(K and L) CYCLIN D1 (magenta) and SOX2 (cyan) immunofluorescence on control (K) and CKO MGs (L).
For western blots, levels are given as a.u. ± SEM relative to the 6-h post-injection PBS control (set to 1) (n = 3 independent pooled samples per group; Student’s t test). For qRT-PCR, mRNA levels are relative to the no-treatment (NT) control (set to 1) and depicted as fold change ± SEM (n = 3 independent pooled samples per group; Student’s t test). *p ≤ 0.05, **p ≤ 0.01.
Yap and Taz Are Required for MG Expression of Cyclin D1 and CYCLIN D3
Because the observed transient increase Cyclin D1 expression post-NMDA damage coincided with a progressive increase in YAP phosphorylation (Figures 1F–1K), we hypothesized that Hippo pathway negative regulation of YAP activity is the means by which Cyclin D1 expression is repressed. Therefore, we next sought to determine whether Yap and Taz are required for Cyclin D1 expression in MGs. We generated adult Glast-CreERT2+/tg; Yapflox/flox, Tazflox/flox; ROSA26R-mTmG+/tg (Yap/Taz conditional knockouts [CKOs]) mice. Glast-CreERT2 is a bacterial artificial chromosome (BAC) transgenic line that expresses CreERT2 under the control of the Glast promoter (Nathans, 2010). Retinal expression of Glast-CreERT2 has been reported to be specific to adult MGs (de Melo et al., 2012; Rattner et al., 2014), and we have independently verified this finding (Figure S2).
To induce Cre activity in MGs, mice were given three injections of tamoxifen per week for 3 weeks. After an additional week, qRTPCR analysis of whole CKO retinal lysates confirmed significant reduction of Yap and Taz transcripts (Figure S3A). We next performed immunofluorescence for YAP, CYCLIN D3, CYCLIN D1, and the MG nuclear marker SOX9 or SOX2 (Poché et al., 2008; Taranova et al., 2006). Cre activity was visualized with GFP fluorescence from the ROSA26R-mTmG reporter (Muzumdar et al., 2007). Retinal sections showed clear loss of YAP expression in SOX9+ MGs, suggesting that Yap and Taz are not required to maintain MG identity or survival (Figures 2G and 2H, arrowheads). However, when we stained for CYCLIN D3 and CYCLIN D1, we observed a cell-autonomous loss in Yap/Taz CKO MGs (Figures 2I–2L, arrowheads) that also coincided with reduction in Cyclin D1 and CYCLIN D3 mRNA levels (Figure S3A). Broad CKO of Yap and Taz during retinal development using Chx10-Cre (Rowan and Cepko, 2004) resulted in the same MG-specific loss of CYCLIN D1 and CYCLIN D3 without loss of SOX9+ MGs (Figures S3B and S3C). In total, these data suggested that YAP/TAZ-TEAD may be required to maintain CYCLIN D3 and Cyclin D1 gene expression in quiescent MGs. To investigate this possibility, we performed YAP chromatin immunoprecipitation (ChIP) from the ImM10 MG-like cell line (Otteson and Phillips, 2010), followed by qPCR analysis of a putative TEAD binding sites approximately 3.7 kb upstream of the Cyclin D1 start codon and another site in intron 3 (Figure S3D). We first confirmed that ImM10s express YAP, CYCLIN D1, TEAD1, and SOX9 and that YAP and TEAD1 coimmunoprecipitate (Figures S3E and S3F). ChIP-qPCR showed clear YAP enrichment at the upstream TEAD motif, while the motif in intron 3 did not have such enrichment (Figure S3G). Therefore, YAP/TEAD1 is likely a direct transcriptional regulator of Cyclin D1 in MGs.
The Hippo Pathway Suppresses Spontaneous MG Proliferation
To determine whether Hippo regulation of YAP activity is an endogenous mechanism normally restraining persistent MG proliferation, we first generated Glast-CreERT2+/tg; Lats1flox/flox; Lats2flox/flox (Lats1/Lats2 CKOs) mice followed by assessment of CKO efficiency (Figure S4A). We observed residual Lats1 and Lats2 expression in the CKOs. This finding is likely due to mosaic Cre expression, inefficiencies in Cre-mediated recombination of all four floxed loci, and Lats1 and Lats2 expression in retinal endothelial cells (Neto et al., 2018; Sakabe et al., 2017), which do not express Glast-CreERT2 (de Melo et al., 2012; Rattner et al., 2014). Despite the inefficiency in globally deleting Lats1 and Lats2 in MGs, immunofluorescence detected discrete MGs with increased CYCLIN D1 levels, which was confirmed by qRT-PCR (Figures S4A–S4E). We determined that these MGs with upregulated CYCLIN D1 also had a more intense YAP nuclear immunofluorescent signal, which indicates loss of the Hippo pathway’s negative regulation of YAP (Figures S4F–S4I).
To determine whether the increased CYCLIN D1 expression causes spontaneous cell-cycle entry, we performed 5-ethynyl-2′-deoxyuridine (EdU) labeling and found that uninjured, tamoxifen-treated Lats1/Lats2 CKO retinae contained EdU+ cells, which were never observed in the corn oil controls (Figures 3A–3G). In addition to being localized to the INL (Figure 3B, arrowheads), we observed EdU+ cells within the outer nuclear layer (ONL) (Figure 3C, arrowhead), and some cells appeared to be undergoing cell division (Figure 3D, arrowhead and inset). Labeling with MG marker SOX2 or SOX9 and cell-cycle marker KI67 confirmed that the EdU+ cells in the Lats1/Lats2 CKOs are spontaneously proliferating MGs (Figure 3E, arrowhead, and 3F; Figures S4J and S4K). In total, these data suggest that sustained YAP activity in MGs responding to damage is required for persistent Cyclin D1 expression and successive rounds of MG cell division. However, this response is normally blocked by Hippo signaling. If this is the case, bypassing Hippo regulation at the level of YAP phosphorylation and degradation should drive MGs into a proliferative state.
Figure 3. Genetic Loss or Bypass of Hippo Signaling Results in Spontaneous MG Proliferation.

(A–D) EdU labeling (magenta) of Lats1/Lats2 on control (A) and CKO (B, C, and D) retinae.
(E and F) EdU labeling (magenta) and immunofluorescence for SOX2 (green) and KI67 (cyan) in CKO (E and F). (F) shows the same nuclei as in (E) (arrowhead).
(G) Quantification of EdU+ cells in tamoxifen-injected mice versus corn oil vehicle controls.
(H) Map of the Yap5SA transgene.
(I and J) EdU labeling (magenta) and SOX9 (green) immunofluorescence on control (I) and YAP5SA-expressing (J) retinae (the boxed nucleus in J is shown in the insets).
(K) EdU labeling of radially oriented nuclear clusters.
(L) EdU labeling of YAP5SA+ retinae that were also exposed to NMDA damage.
(M and N) Flow cytometry (M) and quantification (N) of EdU-labeled, tdTomato+ cells from untreated and NMDA-damaged retinae expressing YAP5SA.
(O) CYCLIN D1 immunofluorescence and EdU labeling of YAP5SA+ retinae.
(P) Cyclin D1 qRT-PCR from three independent pools of tamoxifen-induced Glast-CreERT2+/tg; Yap5SA+/tg retinae (green bars) compared to tamoxifen-induced Glast-CreERT2+/tg retinae (black bar).
For qRT-PCR, mRNA levels are relative to the control (set to 1) and depicted as fold change ± SEM (n = 3 independent pooled samples per group; Student’s t test). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Genetic Bypass of Endogenous Hippo Signaling Results in Robust MG Proliferation
To investigate the role of Hippo-dependent phosphorylation of YAP in blocking sustained MG proliferation, we used the Yap5SA transgenic mouse (Monroe et al., 2019). YAP5SA is a phosphodeficient form of YAP in which all serines targeted by LATS½ kinases have been mutated to alanines, thereby rendering YAP5SA non-responsive to Hippo signaling (Figure S5A) (Zhao et al., 2007). The Yap5SA transgene is randomly inserted into the genome and is driven by the ubiquitous CAG promoter. It encodes a GFP-stop cassette flanked by loxP sites followed by an N-terminal FLAG-tagged Yap5SA cDNA and an internal ribosome entry site (IRES)-LacZ. Upon Cre-mediated recombination, the GFP-stop is deleted, leading to CAG-driven Yap5SA-IRES-LacZ (Figure 3H). Before using this line, we confirmed that the uninduced transgene, indicated by GFP fluorescence, is mosaically expressed in adult SOX2+ MGs (Figure S5B). This expression pattern allowed for a side-by-side comparison of YAP5SA-negative and YAP5SA+ cells in the same retinal sections. In addition, we generated Glast-CreERT2+/tg; Yap5SA+/tg mice and verified that a 5-day tamoxifen treatment results in expression of YAP5SA and LacZ in the same subset of MGs (Figure S5C). Retinae from Glast-CreERT2+/tg; Yap5SA+/tg mice injected with tamoxifen showed a striking increase of EdU+/SOX9+ cells within the INL that was never observed in the corn oiltreated controls (Figures 3I and 3J, arrowheads and insets from boxed region). This finding indicated that YAP5SA is capable of spontaneously driving MGs into the cell cycle. We also observed radial clusters of EdU+ cells that looked similar to clonally expanding MG-derived progenitor cells in the zebrafish retina (Figure 3K, boxed region) (Powell et al., 2016; Raymond et al., 2006; Wan et al., 2012). Upon NMDA damage, we found that Glast-CreERT2+/tg; Yap5SA+/tg retinae had more EdU+ cells versus PBS-injected controls (Figures 3K and 3L). To quantify this difference, we generated Glast-CreERT2+/tg; Yap5SA+/tg mice that also carried the ROSA26R-tdTomato reporter (Madisen et al., 2010). TdTomato+/EdU+ MGs from NMDA-damaged and undamaged retinae were counted using flow cytometry. We found that the average percentage of EdU+ tdTomato+ cells versus total tdTomato+ cells was significantly greater in the NMDA-damaged retinae (16.98% ± 2.50%) versus the no-treatment controls (7.29% ± 0.97%) (Figures 3M and 3N). This result suggests that the YAP5SA+ MG proliferation is not completely deregulated and that the cells are still capable of responding to damage cues. We also confirmed that the EdU+ cells exhibited the same upregulation of CYCLIN D1 protein expression as in the Lats1/Lats2 CKOs (Figure 3O, arrowheads) and that this occurred at the mRNA level (Figure 3P). From these data, we conclude that forced expression of YAP5SA results in MGs that bypass regulation by the Hippo pathway, thereby allowing persistent expression of Cyclin D1 and successive rounds of spontaneous cell-cycle reentry.
YAP5SA Activity Drives MGs to a Proliferative, Progenitor-like State
To track the fate of YAP5SA+ MGs, we used the LacZ cassette that, upon Cre recombination, is expressed from the Yap5SA transgene (Figure 3H). Consistent with our data suggesting Cyclin D1 is regulated by YAP, immunofluorescence showed LacZ+ MGs with a dramatic, cell-autonomous increase in CYCLIN D1 that was well above the level of endogenous expression in the neighboring Yap5SA-negative MGs (Figure S6A). Analysis of NMDA-damaged Glast-CreERT2+/tg; Yap5SA+/tg retinae showed dramatic expansion of the CYCLIN D1+/LacZ+ population (Figure S6B). The in vivo role of Cyclin D1 was originally described in retinal progenitor cells (RPCs), which were found to have one of the highest Cyclin D1 expression levels in the developing embryo (Sicinski et al., 1995). More recently published data indicate that in addition to promoting RPC G1- to S-phase transition, CYCLIN D1 functions as an RPC transcription factor (Bienvenu et al., 2010). Therefore, we reasoned that the high level of prolonged CYCLIN D1 experienced by YAP5SA+ MGs might be a sign of reprogramming to an RPC-like state. Consistent with this idea, when we analyzed tamoxifen-induced and non-induced Glast-CreERT2+/tg; Yap5SA+/tg retinae for GFAP upregulation in response to NMDA damage, we found that the YAP5SA+ MGs failed to express GFAP but the adjacent non-transgenic cells did express GFAP (Figures S6C–S6E). This result indicated the possibility that YAP5SA+ cells lost MG identity.
To more precisely define the identity of the YAP5SA+ cells, we next performed unbiased single-cell mRNA sequencing (scRNA-seq) transcriptome analysis. Adult Glast-CreERT2+/tg; Yap5SA+/tg; ROSA26R-tdTomato+/tg mice were induced with tamoxifen followed by NMDA injection. After 2 days, tdTomato+ cells were isolated by fluorescence-activated cell sorting (FACS) followed by scRNA-seq using the 10X Genomics platform (Figure 4A). Because the Yap5SA transgene is mosaically expressed within the MG population, the sorted cells contain both tdTomato+/YAP5SA+ cells and tdTomato+-only cells, which serve as an internal negative control. After computational processing (see STAR Methods), we performed graph-based clustering on all significant principal components, which allowed us to identify 11 MG cell clusters. The data were visualized through a non-linear dimensional reduction algorithm, t-Distributed Stochastic Neighbor Embedding (t-SNE) (Figure 4B). Differential expression analysis supported the considerable heterogeneity present in the injured mutant tissue. Moreover, we were able to clearly distinguish between the YAP5SA+ and the YAP5SA-negative clusters, because the YAP5SA+ clusters expressed many canonical YAP target genes, like Amotl2, Birc5, and Vgll3 (Figures S7A and S7B). The YAP5SA-negative control MGs displayed a high degree of heterogeneity, with 7 unique clusters identified (MGC-1 to MGC-7). The most obvious source of heterogeneity was derived from the immune response being sensed by clusters MGC-5, MGC-6, and MGC-7, suggesting different levels of immune activation and the interferon response (Figure S7A). These data imply that reactive MGs respond to wounding by eliciting a robust transcriptional immune response, although they do so in a heterogeneous manner.
Figure 4. Single-Cell mRNA Sequencing of YAP5SA+ Cells.

(A) Approach for unbiased single-cell mRNA sequencing (scRNA-seq) transcriptome analysis of YAP5SA+ MGs.
(B) Two-dimensional colored t-SNE representation of 13,511 single-cell transcriptomes.
(C) Cell-cycle phase analysis of single tdTomato+ cells. The approximate cell-cycle phase was calculated by scoring individual cells on their expression for S-,G1-, and G2- and/or M-phase transition genes as defined by Kowalczyk et al. (2015).
(D–F) Feature plots showing the expression of D-type cyclin genes (D) and known MG (E) and RPC (F) genes projected across t-SNE. Red indicates high gene expression. (G) Dot plot showing significant enrichment of Lhx2 expression within the YAP5SA+ cell cluster compared with the control. The color represents the average expression level, and the size represents the percentage of cells expressing Lhx2.
(H) GO-term enrichment of the YAP5SA-4 cluster over the MCG-1 to MGC-3 clusters.
(I and J) Feature plots (I) and dot plots (J) showing the presence of a rare population of YAP5SA+ cells that express the neuronal transcripts Elavl2 and Elavl4.
(K) HuC/D immunofluorescence confirming the presence of the Elavl2/Elavl4+ cell population (arrowhead and arrows).
The differential expression analysis revealed that much of the heterogeneity observed in the YAP5SA+ clusters (YAP5SA1–YAP5SA14) could be attributed to cell-cycle genes (Table S1). Thus, we next scored each tdTomato+ cell for S-, G1-, and G2- and/or M-phase transition genes (Kowalczyk et al., 2015) and found enrichment of such genes in the YAP5SA+ cluster, but not in the YAP5SA-negative cluster (Figure 4C). These data indicated that only the YAP5SA+ cells were actively dividing. When comparing the expression of known cell-type-specific markers, we determined that the YAP5SA+ clusters displayed the expected upregulation of Cyclin D1 expression (Figure 4D; Figures S7C–S7E). Cyclin D2 and CYCLIN D3 were also increased relative to the YAP5SA-negative population (Figure 4D). Analysis of known MG transcripts Gfap, Kcnj10, and Glul indicated a dramatic reduction within the YAP5SA+ cluster (Figure 4E; Figures S7F and S7G). Comparison of markers that are expressed in both MGs and RPCs showed that the YAP5SA+ cluster had a statistically significant increase in Lhx2 expression; other RPC markers, such as Pax6 and Six3, were also expressed. However, we did not observe enrichment for other transcripts, such as Vsx2, Rx, and Notch1 (Figures 4F and 4G). Altogether, these data indicate that YAP5SA+ cells lose MG identity and acquire an identity more reminiscent of proliferative, progenitor-like cells.
We next performed Gene Ontology (GO) analysis between YAP5SA+ and YAP5SA-negative cells (Table S2). To highlight gene categories outside of the canonical cell-cycle pathway (S, G2, and/or M phase) and inflammatory response, we focused our analysis on the YAP5SA-4 (G1 phase) cluster and the control MGC-1 to MGC-3 (non-inflamed) clusters (Figure 4H). The most represented GO term was MYC target genes. In addition to being a transcriptional target of YAP, MYC is known to cooperate with YAP to regulate genes required for cell-cycle entry and organ growth (Cai et al., 2018; Croci et al., 2017). MTORC1 signaling was also enriched and is known to crosstalk with the Hippo pathway (Tumaneng et al., 2012). The phosphatidylinositol 3-kinase (PI3K)-Akt-mTORC1 pathway has been discovered to promote mouse RPC proliferation by regulating the synthesis and degradation of CYCLIN proteins with the interesting exception of CYCLIN D1 (Choi et al., 2018). MTOR was also shown to be required for chick MG proliferation in response to damage (Zelinka et al., 2016). These data suggest that both MYC and mTORC1 activity may function downstream of YAP to promote robust and sustained MG-cycle reentry.
Previous studies of the Ascl1-GFP transgenic mouse retina have suggested that in response to NMDA damage, a small subset of GFP+ MGs give rise to a population of HuC/D+ amacrine and ganglion cells (Jorstad et al., 2017; Ueki et al., 2015). The HuC/D antibody used in these studies (Invitrogen A-21271) recognizes the gene products of Elavl2, Elavl3, and Elavl4, which are neuronal RNA binding proteins. In our scRNA-seq analysis, we noticed a small percentage of YAP5SA+ cells that expressed Elavl2 and Elavl4, suggesting that a similar population of cells emerges from YAP5SA+ MGs (Figures 4I and 4J). Using the HuC/D antibody, we occasionally observed HuC/D+, EdU+ cells within injured Glast-CreERT2+/tg; Yap5SA+/tg retinae (Figure 4K, arrowhead and arrows). These data indicate that a small subset of proliferative YAP5SA+ MGs may differentiate into neurons.
DISCUSSION
Our data have identified Hippo signaling as an essential regulatory mechanism normally blocking mammalian MGs from adopting a proliferative, progenitor-like state. In our proposed model, upon retinal damage, a YAP-dependent increase in Cyclin D1 transcription drives an initial burst of MG proliferation. If this response were sustained over time, as in the case of the GlastCreERT2+/tg; Yap5SA+/tg mice, proliferative MGs would clonally expand into a progenitor-like cell population. However, Hippo signaling normally represses YAP activity, thereby blocking persistent Cyclin D1 expression to halt subsequent cell-cycle reentries (Figure S8).
With forced YAP5SA expression, we were able to drive MG proliferation spontaneously in the absence of retinal damage. This finding is significant because typically, MG-cycle entry depends on retinal damage cues (Close et al., 2006; Dyer and Cepko, 2000; Karl et al., 2008; Ooto et al., 2004). Therefore, most experimental approaches to date have had to incorporate extensive retinal damage, leading to catastrophic neuron loss, into strategies to promote MG proliferation. Such methods would not be useful as a therapeutic approach to reverse retinal degeneration. Previously, in an attempt to bypass the requirement for damage to induce MG proliferation, Wnt signaling was investigated as a potential target (Yao et al., 2016). The rationale for this idea came from previous experiments showing that injury of the adult retina promotes Wnt signaling and that Wnt activation is required for damage-induced MG proliferation (Das et al., 2006; Liu et al., 2013). Others have suggested that Wnt signaling potentiates hematopoietic stem or progenitor cell fusion-mediated reprogramming of both MG and retinal neurons within damaged retinae (Sanges et al., 2013, 2016). When AAV was used to drive β-catenin, a transcriptional effector of canonical Wnt signaling, in adult mouse MGs, the cells underwent spontaneous cell-cycle reentry in uninjured retinae. Surprisingly, when these cells were subsequently exposed to AAV-driven Otx2, Crx, and Nrl, the β-catenin-expressing cells redifferentiated into rods that were reported to restore vision in a mouse model of congenital blindness (Yao et al., 2018). While this study suggests that mammalian MGs may have regenerative potential, it did not establish the endogenous molecular mechanism normally keeping that potential at bay.
Although we propose that the Hippo pathway blocks mammalian MG proliferation, it is unclear why this occurs in mammals while the zebrafish can sustain a proliferative response leading to regeneration. One possible explanation may stem from species differences in CYCLIN D1 activity or expression. In adult mouse MGs, CYCLIN D1 protein levels are normally maintained at a low level relative to CYCLIN D3. In contrast, during embryonic development, Cyclin D1 is highly expressed in RPCs, where it is required to maintain proliferation, whereas CYCLIN D3 is not (Das et al., 2009, 2012; Fantl et al., 1995; Geng et al., 1999; Sicinski et al., 1995). Furthermore, in this study, we showed that Cyclin D1 gene expression is highly, but transiently, upregulated in response to retinal damage. At this time, expansion of multipotential progenitor cells would be beneficial; however, CYCLIN D3 levels did not change in response to retinal damage. This dichotomy in Cyclin D1 and CYCLIN D3 expression and activities may be due to one of the emerging non-canonical roles for Cyclin D1. Specifically, in RPCs, CYCLIN D1 was shown to function as a transcriptional regulator and to directly influence the expression of genes such as Notch1 (Bienvenu et al., 2010). Therefore, it is possible that CYCLIN D1 has dual roles. In addition to driving cell-cycle entry, CYCLIN D1 may have the ability to broadly influence the MG transcriptome and directly promote a progenitorlike identity. Transgenic expression of Cyclin D1 in post-mitotic mouse photoreceptor precursors has been reported to induce proliferation and prevent photoreceptor differentiation, eventually leading to death (Skapek et al., 2001). While no RPC marker analyses were performed in that study, it is possible that ectopic CYCLIN D1 activity forced photoreceptors toward a persistent progenitor-like state. If this is the case, the mammalian retina would need to suppress prolonged CYCLIN D1 activity postretinal damage. Otherwise, large numbers of MGs would eventually undergo transcriptome changes to reprogram into progenitor-like cells and the retinal neurons would lose essential MG support roles (Reichenbach and Bringmann, 2013; Vecino et al., 2016). Future studies should be aimed at precisely defining the role of Cyclin D1 in reactive MGs and whether its continued expression is necessary and sufficient for MG reprogramming to a progenitor state.
Based on our findings, and that of the accompanying report by Hamon et al. (2019) in this issue of Cell Reports, we propose that MGs have reprogramming potential that can be awakened by targeting endogenous Hippo signaling. In the future, it will be important to elucidate whether there is crosstalk between the Hippo and the Wnt pathways in MG-mediated retinal regeneration as has been described for other systems (Azzolin et al., 2012, 2014; Heallen et al., 2011; Li et al., 2013; Meng et al., 2016; Varelas et al., 2010; Wang and Martin, 2017). In addition, the requirement of transcriptional and signaling mechanisms, such as MYC and MTORC1 downstream of YAP, will need to be determined (Cai et al., 2018; Croci et al., 2017; Tumaneng et al., 2012). Finally, it will be important to devise methods to transiently bypass the Hippo pathway in reactive MGs, followed by the introduction of differentiation factors coupled to lineage tracing, to determine whether bona fide retinal regeneration occurs.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ross Poché (poche@bcm.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Strains and Genotyping
Lats1flox/flox and Lats2flox/flox (Heallen et al., 2011), Yapflox/flox (Xin et al., 2011), Tazflox/flox (Xin et al., 2013), ROSA26R-mTmG+/tg (Gantz et al., 2012; Muzumdar et al., 2007), ROSA26R-tdTomato+/tg (Madisen et al., 2010), and Glast-CreERT+/tg (Nathans, 2010) mice were PCR genotyped using published protocols. Additionally, the Yap5SA+/tg mice (Monroe et al., 2019), ROSA26R-mTmG+/tg, and the ROSA26R-nTnG+/tg (Prigge et al., 2013; Schmidt, 2013) fluorescent reporter mice were genotyped by visualizing fluorescence from tail snips. All animal research was conducted according to protocols approved by the Institutional Animal Care and Use Committee (IACUC) of Baylor College of Medicine. All animals used were between post-natal day 21 (P21) and P120. Since there were no age- or sex-dependent differences in our observations, male and female data were combined for presentation herein. All genotyping primer sequences and references are included in Table S3 within the Supplemental Information.
Injections
To induce Glast-CreERT2, intraperitoneal (IP) injections of tamoxifen (Sigma, T5648) (75 mg/kg body weight) were given for five consecutive days or every other day for two weeks. To label cells in S phase, 5-ethynyl-2′-deoxyuridine (EdU) (Sigma 900584 or Santa Cruz sc-284628A) was injected IP at a concentration of 50 mg/kg body weight and mice were euthanized 2 or 24 hours later.
For intravitreal NMDA injections, mice were anesthetized using Isoflurane. One drop of Proparacaine Hydrochloride (0.5%) was placed on to the eye to be injected. The anesthetized mouse was positioned on its side under a dissecting microscope while on a warming plate and a sterile 28G hypodermic needle in inserted in the post-limbus region, with the bevel of the needle facing upward to avoid the lens. The puncture needle was then carefully removed, and a small amount of vitreous fluid was wicked away from the puncture site using a twisted Kimwipe. Then, using a Hamilton syringe, a sterile blunt needle (33G) was gently inserted into the hole, pass the lens, and into the vitreous chamber where 2 μl of either PBS or NMDA (100 mM) (Sigma: M3262) was injected. Following injection, veterinary ophthalmic antibacterial ointment (Bacitracin Zinc and Polymyxin B) was placed on the eye and the animals were monitored continuously until recovered from anesthesia (maintain sternal recumbency).
ImM10 Cells
ImM10 cells were cultured and maintain as recommended (Otteson and Phillips, 2010). Cells were thawed and incubated in neurobasal media containing 2% heat-inactivated FBS, 1x B27 neuronal supplement with retinoic acid, 2mM L-Glutamine, and 50U/mL mouse recombinant interferon gamma. For immunofluorescence, cells were washed in 0.1M PBS and fixed in 2% paraformaldehyde for 15 min at room temperature. Fixed cells were permeabilized for 30 min at room temperature in 0.5% Titron-X100 and then washed in 2% BSA before immunostaining.
METHOD DETAILS
Immunofluorescence, EdU labeling, and Confocal Microscopy
Enucleated eyes were immersed in 4% paraformaldehyde (PFA) in 0.1M Phosphate Buffered Saline (PBS) for 30 min at room temperature, corneas and lenses were removed. After fixation, eyecups were washed in PBS 3 times for 10 minutes on ice and then cryoprotected by immersing in 30% sucrose until the tissue sank to the bottom of the tubes. Then, tissue was embedded in OCT for 15–20 min and then flash-frozen in liquid nitrogen and stored at −80°C before sectioning. Retinal sections from controls and mutant mice were immunolabeled in parallel to insure identical processing. Cryosections were cut at 20 μm on a cryostat and mounted on Superfrost Plus slides (VWR Brand, Westchester, PA). Cryosections were thawed and post-fixed in 4% PFA for 15 min and then washed 3 times in PBS. For EdU, sections were permeabilized for 30 in 0.5% Titron-X100 diluted in 0.1M PBS and then washed using 3% BSA diluted in 0.1M PBS. The Click-iT EdU Alexa 488, 555, or 647 (Thermo Fisher) was used following the manufacturer’s instructions. For immunofluorescence, sections were rinsed with PBS and incubated for 2 hours at 4°C with 5% normal goat serum, 2% bovine serum albumin, and 0.3% Triton X-100 diluted in 0.1M PBS (pH 7.4). Slides were incubated for 24 hours in primary antibodies in a humid chamber. The following antibodies and concentrations were used: mouse-YAP (1:100, Santa Cruz, #101199,(Szymaniak et al., 2017; Zanconato et al., 2015)), rabbit-pYAP (1:500, Cell Signaling, #4911, (Heallen et al., 2013)), rabbit-SOX9 (1:500, Millipore, AB5535,(Poché et al., 2008)), mouse-TEAD1 (1:1000, BD transduction, 610923, (Cao et al., 2008)), mouse-SOX2 (1:500, Santa Cruz, 365823, (Xiao et al., 2018a)), rabbit-GFAP (1:1000, Dako, Z0334, (Poché et al., 2016)), rabbit-PH3 (1:500, Millipore, 06–570) (Barrasso et al., 2018), rabbit-Ki67 (1:500, Abcam 15580, (Poché et al., 2016)), rabbit-Cyclin D1 (1:1000, Thermo Fisher, 9104, (Poché et al., 2016)), mouse-CYCLIN D3 (1:200, Abcam, 28283, (Wang et al., 2017)), rabbit-GFP (1:500, Rockland, 600–401-215, (Kovalchuk et al., 2015)), mouse-Flag (1:250, Sigma, F1804, (Zanconato et al., 2015)), chick-beta-Gal (1:500, Abcam, 936, (Kim et al., 2018)), mouse-GS (1:500, Chemicon, MAB302, (Rueda et al., 2016)), mouse-HuC/D (1:100, Invitrogen, A-21271, (Jorstad et al., 2017; Ueki et al., 2015)).
Next, slides were rinsed three times in PBS and blocked for 30 minutes in 5% normal goat serum, 2% bovine serum albumin, and 0.3% Triton X-100 diluted in 0.1M PBS (pH 7.4) before incubation in secondary antibodies. 1:400 dilutions of Alexa Fluor 405, 488, 555, and/or 647-conjugated secondary antibodies (Life Technologies) were applied and incubated for one hour in the dark at room temperature. DAPI was included in the secondary antibody solution at a 1:600 concentration. After incubation with secondary antibodies, slides were rinsed 3 times in PBS. Slides were coverslipped with Fluoromount-G (SouthernBiotech). Confocal images were acquired using a Zeiss LSM 780 20X and 40X objectives. For standardization between samples, all images were acquired with the same laser power, detector gain, scan speed, and pinhole size. Z stack images with an identical step-size and thickness were acquired for each retinal section and a maximal intensity projection were obtained.
RNA extraction and quantitative rtPCR
Retinae from control and mutant mice were dissected and total RNA was purified using the RNeasy Mini Kit (QIAGEN). The purified RNA was reverse transcribed using the SuperScript III first strand synthesis kit with Oligo (dT)20 and random hexamer priming (Invitrogen). The Taqman gene expression assay from Applied Biosystems was used for qrtPCR and the probes used to detect gene expression were CYCLIN D3 (Mm01612362), Cyclin D1 (Mm00432359), Yap (Mm01143263), Taz (Mm01289583), Lats1 (Mm01191886), and Lats2 (Mm00497217). Gapdh (Mm4352932) and Beta-actin (Mm4352933) were used for normalization. The conditions to perform qrtPCR using a StepOnePlus Real-Time PCR System (Life Technologies) were: 50°C for 2 minutes, 95°C for 10 minutes, 40 cycles of 95°C for 15 s and 60C for 1 minute.
Western Blot Analyses
Retinae were dissected in PBS and briefly washed in PBS containing protease inhibitor (Thermo Scientific 1862209) and phosphatase inhibitor (Thermo Scientific 78420). The PBS was removed, and the retinae were flash-frozen and kept at −80°C until used. Samples were thawed on ice for 10 minutes in ice cold lysis buffer (Thermo Scientific 1861603) containing protease and phosphatase inhibitors. Retinae were homogenized and centrifuged at 4°C (2,000 RPM, 10 minutes) and the supernatant was transfer to a new tube. Clarified protein lysates were quantified by the Bradford assay. 20 to 25 μg of protein was loaded onto a 4%–15% Tris/Glycine precast gels (BioRad, 5671084) for electrophoresis and subsequently transferred (30V, overnight) onto Immobilon-P PVDF membranes using the Criterion™ System BioRad. Membranes were washed in TBS-T pH 7.4 (20 mM Tris, 137 mM NaCl, 0.1% Tween 20) and blocked for 1 hour at RT with 5% non-fat milk solution to reduce non-specific antibody binding. At least 3 independent control and treated samples were probed with the following primary antibodies (in 5% milk, overnight at 4°C): YAP (1:2000, Santa Cruz, 101199) and pYAP (1:1000, Cell signaling, 4911). Vinculin (1:3000, Cell Signaling, 13901) was used as loading control. Following primary antibody incubation, membranes were washed in TBS-T, incubated with HRP-conjugated secondary antibodies (1-hour, room temperature), detected with Clarity™ Western ECL Substrate (Bio-Rad #170–5060) and imaged with the Chemidoc Touch Imager (Bio-Rad).
For western blots of nuclear or cytoplasmic fractions, sequential lysing was performed using the Cell Fractionation Kit (Cell Signaling #9038). Two to four retinae per sample were gently dissociated using a Wheaton tissue grinder and resuspended in lysis buffer as recommended by manufacturer. Dissociated retinae were centrifuged at 800 g for 5 minutes at 4°C to extract nuclear fractions. For cytoplasmic isolation, the supernatant was centrifuged at 48,000 RPM for 1 hour at 4°C using a Beckman TLA 100.3 ultracentrifuge rotor. 20 μg of nuclear or cytoplasmic fractions were loaded for YAP and pYAP western blot analysis as described above. Histone H3 (1:3000, Abcam, 1791) and GAPDH (1:3000, Millipore, MAB374) were utilized as loading and fractions validation controls. The blots were washed with TBS-T and incubated with HRP-conjugated secondary antibodies (1-hour, room temperature) from the appropriate species, detected with Clarity™ Western ECL Substrate (Bio-Rad #170–5060) and imaged with the Chemidoc Touch Imager (Bio-Rad).
Co-immunoprecipitation (Co-IP)
Retinae were homogenized in lysis buffer (20mM Tris-HCl [pH 7.5], 150mM NaCl, 1mM EDTA, 1mM EGTA, 1% Triton-X, 2.5mM sodium pyrophosphate, 1mM βglycerophosphate, 1mM Na3VO4). Aliquots of clarified protein lysate (500mg per sample) were diluted in 500μL lysis buffer supplemented with HaltTM Protease Inhibitor Cocktail (ThermoFisher, #78430) and incubated with 1μg of the following antibodies overnight at 4°C: mouse-YAP (Santa Cruz, 101199 (Zanconato et al., 2015)) or mouse-TEAD1 (BD Transduction, 610923, (Zanconato et al., 2015)), Rabbit IgG (Millipore, #12–370), Mouse IgG (Millipore, #12–371) overnight at 4°C (Poché et al., 2016). The protein/antibody complexes were captured using either Protein A/G Magnetic Beads (ThermoFisher, #88802) or TrueBlot anti-Rabbit Ig IP Beads (Rockland, #00–8800-25) by incubating for 1 hour at room temperature. The beads were then washed, and protein eluted with SDS-PAGE reducing sample buffer (50μL 1X PBS plus 10μL 6X sample buffer) by boiling for 10 minutes. Next, 2% of input protein and 15μL of IP reactions were loaded onto a 4%–15% Tris/Glycine precast gels (BioRad, 5671084) and western blots performed as described. The following secondary antibodies were used: True Blot HRP-conjugated anti-mouse IgG (1:1000, Rockland, #18–8817-33) and True Blot HRP-conjugated anti-rabbit IgG (1:1000, Rockland, #18–8816-33).
Chromatin Immunoprecipitation (ChIP)-qPCR
The ChIP-IT High-Sensitivity Kit (Active Motif, No. 53040) was used. Briefly, ImM10 cells grown to 50% confluency were cross-linked in Complete Cell Fixation Solution for 15 minutes. Stop Solution was added to stop the crosslinking, then cells were washed in icecold PBS Wash Buffer and centrifuged at 1,250 × g for 3 minutes at 4°C. Cell pellets were then resuspended and incubated in Chromatin Prep Buffer for 10 minutes. Pellets were then transferred to a chilled dounce homogenizer and using the Type A pestle, pellets were homogenized with 150 strokes. Homogenized samples were then centrifuged for 3 minutes at 1,250 × g at 4°C. Pellets were resuspended in ChIP Buffer supplemented with Protease inhibitor cocktail (PIC)/PMSF and then transferred to a microcentrifuge tube, incubated for 10 minutes on ice. This was followed by sonication of the resuspended pellets (25% amplitude pulse for 30 s on and 30 s off) for a total sonication on-time of 5 minutes and centrifugation at 14,800 rpm for 2 minutes at 4°C. The supernatant was then transferred to a new tube for immunoprecipitation. A rabbit, anti-YAP1 (Novus Biologicals, NB110–58358, (Lin et al., 2017)) or rabbit, anti-IgG antibody (Millipore, #12–370) was incubated with 10–20 μg chromatin per ChIP reaction. ChIP reactions components were added according to the Kit protocol and then incubated on a rotator overnight at 4°C. The next day the reactions were spun at 1250 × g for 1 minute. Protein G agarose beads were washed using TE buffer-pH 8.0 and then added to the ChIP reactions. The reactions were then incubated on a rotator at 4°C for 3 hours. After incubation, 600 μl of ChIP Buffer were added to each reaction and then transferred to the labeled ChIP Filtration Column. Columns were washed with Wash Buffer AM1 and then transferred to microcentrifuge tubes and spun at 1250 × g for 3 minutes at room temperature. Pre-warmed Elution Buffer AM4 was added to each column and, after 5 min incubation, the samples were centrifuged at 1250 × g for 3 minutes at room temperature. To reverse the crosslinking, each eluted ChIP DNA sample was then transferred to 250μl PCR tubes and 2μl of proteinase K was added. Samples were then heated in a thermocycler at 55°C for 30 minutes and then at 80°C for 2 hours. After incubation, DNA Purification Binding Buffer was added to each ChIP DNA sample and the pH was adjusted using Sodium Acetate. Samples were then poured into the DNA purification columns and spun at 14,000 rpm for 1 minute. DNA Purification Wash Buffer was run through the columns, then columns were spun at 14,000 rpm for 2 minutes and then transferred to microcentrifuge tubes. Prewarmed DNA Purification Elution Buffer was added to the columns, allowed to incubate for 1 minute at room temperature and then spun at 14,000 rpm for 1 minute. Purified DNA was then analyzed using qPCR.
We used the following primer pairs: TEAD motif #1 F: 5′-TCCGGGGTCACGGCTT-3′ and TEAD motif #1R: 5′-CGT CGCGTCCAAGGTTTA-3′, and TEAD motif #2F: 5′-CCCAACCCTAGCTAGCACATT-3′ and TEAD motif #2R 5′-AAGGCCAGGA GATCTGTCGT-3′. QPCR was performed using SYBR Green Master Mix (ThermoFisher, #4309155) and the ViiA 7 Real-Time PCR system (Applied Biosystems) with the following cycle conditions: 50°C for 2 minutes, 95°C for 10 minutes, 40 cycles of [95°C for 15 s, 60°C for 1 minute], 95°C for 15 s, 60°C for 1 minute, and 95°C 15 s.
Tissue Dissociation and Cell Sorting
Retinae were dissected in ice-cold PBS and dissociated in 10 units Papain/60 units DNase (500μL/30μL respectively for 3–6 retinae) for 25–30 min in a shaking water bath at 37°C. Dissociated retinae were triturated 10 times with a polished pipette and EBSS-Ovomucoid-DNase was added to stop digestion. The cell suspension was filtered using a 40μm strainer and centrifuged at 900 RPM for 8–10 min. For single cell analysis, cell pellets were resuspended and kept in DNase/Ovomucoid/Neurobasal (1:1:10 respectively) during cell sorting. Cell sorting was performed using the Aria III cell sorter (BD Biosciences). Tomato positive and negative cells were sorted using an 80-micron nozzle, captured in chilled tubes and resuspended in 0.04% ultrapure BSA in 0.1M PBS. Fresh cell suspensions were immediately subjected to the scRNA-seq protocol.
For flow cytometry and quantification of EdU positive cells after retinal dissociation, cell pellets were washed in 0.1M PBS and resuspended in 2% paraformaldehyde for 30 min at room temperature. Fixed cells were permeabilized for 30 min at room temperature in 0.5% Titron-X100 and then washed in 2% BSA before EdU detection. Cells were incubated for 30 min in the Click-iT reaction cocktail using the Plus EdU Alexa Fluor 647 kit (ThermoFisher), washed in 2% BSA, and resuspended on 0.1M PBS. Tomato+ cells and EdU+ cells were quantified using the Attune NxT cell cytometer (Life Technologies).
Single-cell mRNA sequencing (scRNA-seq)
FACS sorted TdTomato+ cells were counted and diluted in 1X PBS with 0.04% BSA (Sigma Cat# SRE0036) before being loaded onto the 10X Genomics Chromium instrument. The scRNA-seq libraries were generated using the 10X Chromium Single Cell 3′ v2 reagent kit according to the manufacturer’s instructions and were sequenced on an Illumina Nextseq500.
QUANTIFICATION AND STATISTICAL ANALYSIS
For quantification of nuclear YAP immunofluorescence (Figure 1I), MATLAB was used to quantify YAP pixel intensities when coincident with SOX9+ pixels (to denote nuclear localization). Cytosolic YAP pixel intensities were quantified by non-zero intensities not coinciding with SOX9+ pixels (to denote non-nuclear/cytosolic localization). For EdU quantification of confocal images (Figure 3G), EdU+ cells were manually counted in each image and retinal area was measured in MATLAB to determine EdU+ cells per mm2. To measure pixel co-localization across MGs within a given retina (Figures S1C, S4I, and S6E) MATLAB was used to plot pixel intensities along a line drawn across the image and Pearson’s Correlation Coefficient (R) was calculated. For quantification of relative pixel intensity per μm2 (Figure S4E), total pixel intensity was measured in MATLAB and divided by the total area of each image. For all figure panels, images were imported into Adobe Photoshop CS software for identical and minimal processing. For each control, mutant, or treated experimental group, three to six independent retinae from different mice were examined and the results shown are representative images from at least three separate experiments. A Student’s t test was used to compare measurements between controls and mutant mice. P values < 0.05 were considered significant.
For quantification of western blots (Figures 1F, 1G, 1J, 1K, 2A, 2B, and 2D), images were taken with the Chemidoc Touch Imager (Bio-Rad Laboratories Inc, Hercules CA) and scanned at high-resolution for protein band size and signal intensity. Densitometry measurements were obtained using the Image Lab™ software (Bio-Rad). All bands were normalized to their corresponding loading control. The results shown are from 3 biological replicates. The Student’s t test was used to determine differences between mutants and controls. P values < 0.05 were considered significant.
For quantification of mRNA by qrtPCR (Figures 2C, 2E, and 3P; Figures S3A and S4A), each qrtPCR reaction was performed in triplicate for each independent control (N = 3), CKO (N = 3), or treatment group (N = 3) cDNA samples. The mean ΔCt values were normalized against the housekeeping genes Gapdh or Beta-actin and corresponding ΔΔCt values were log2-transformed to obtain fold change values. For data analysis, the Pfaffl method was used to determine relative gene expression ratios and a p value of < 0.05 was considered significant (Pfaffl, 2001).
For quantification of sorted cells (Figure 3N), td-Tomato positive cells and EdU positive cells were quantified using the Attune NxT cell cytometer (Life Technologies). The Student’s t test was used to determine differences between cells from treated and untreated tissue. A p value of < 0.05 was considered significant.
For single cell mRNA sequencing analysis (Figure 4; Figure S7), the scRNA data was analyzed as previously described with minor modifications (Li et al., 2018; Xiao et al., 2018b). Briefly, raw sequencing data were handled using the 10X Genomics Cellranger software (https://www.10xgenomics.com/). Fastq files were mapped to the mm10 genome, and gene counts were quantified using Cellranger count function. Subsequently, expression matrices from each experiment were merged and were then imported into Seurat (version 2.3.4) where log normalization was performed (Butler et al., 2018). We corrected for batch effects by regressing out the number of molecules per cell and the batch identity with the Seurat implementation of regression (ScaleData function). Next, we performed a principle components analysis (PCA), and significant principle components (PCs) were used as input for graph-based clustering. We used tSNE for 2-dimensional visualization of the multi-dimensional dataset. Differential expression of the individual clusters was achieved by using the Wilcoxon rank sum test (FindMarkers, default). To avoid over-clustering, we merged clusters that were not transcriptionally distinct into a single cluster. Clusters composed of doublets (2 different cell types within a single droplet) were removed from the dataset. And clusters that were not MGCs were also removed, including rod cells, cone cells, and astrocytes. Gene ontology analysis was performed using Metascape (http://metascape.org/gp/index.html). The approximate cell cycle phase of each cell was calculated using Seurat by scoring individual cells on their expression for S-phase, G1, and G2M genes. (Kowalczyk et al., 2015).
DATA AND SOFTWARE AVAILABILITY
The accession number for the scRNA-seq data reported in this paper is GEO: GSE121707 and is accessible through the NCBI’s Gene Expression Omnibus (GEO) (Edgar et al., 2002).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-YAP | Santa Cruz | Cat# sc-101199; RRID: AB_1131430 |
| Rabbit anti-YAP | Novus Bio | Cat# NB110-58358; RRID: AB_922796 |
| Rabbit anti-pYAP (Ser 127) | Cell Signaling | Cat# 4911; RRID: AB_2218913 |
| Rabbit anti-SOX9 | Millipore | Cat# AB5535; RRID: AB_2239761 |
| Mouse anti-TEAD1 | BD Biosciences | Cat# 610923; RRID: AB_398238 |
| Mouse anti-SOX2 | Santa Cruz | Cat# sc-365823; RRID: AB_10842165 |
| Rabbit anti-GFAP | Dako/Aligent | Cat# Z0334; RRID: AB_10013382 |
| Rabbit anti-PH3 | Millipore | Cat# 06-570; RRID: AB_310177 |
| Rabbit anti-CYCLIN D1 | Thermo Fisher | Cat# 9104; RRID: AB_149911 |
| Mouse anti-CYCLIN D3 | Abcam | Cat# Ab28283; RRID: AB_2070798 |
| Rabbit anti-Vinculin | Cell Signaling | Cat# 13901; RRID: AB_2728768 |
| Rabbit anti-GFP | Rockland | Cat# 600-401-215; RRID: AB_828167 |
| Rabbit anti-Ki67 | Abcam | Cat# Ab16667; RRID: AB_302459 |
| Mouse anti-FLAG | Sigma | Cat# F1804; RRID: AB_262044 |
| Chicken anti-B-GAL | Abcam | Cat# Ab9361; RRID: AB_307210 |
| Mouse anti-Glutamine Synthetase (GS) | Chemicon | Cat# MAB302; RRID: AB_2110656 |
| Rabbit anti-HISTONE 3 | Abcam | Cat# Ab1791; RRID: AB_302613 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tamoxifen | Sigma | Cat# T5648 |
| 5-ethynyl-2′-deoxyuridine (EdU) | Sigma | Cat# 900584 |
| N-methyl-D-aspartate (NMDA) | Sigma | Cat# M3262 |
| Critical Commercial Assays | ||
| Click-iT EdU Alexa Fluor® 488;555; and 647 Imaging Kit |
Thermo Fisher Scientific | Cat# C10337; C10338; C10340 |
| ChIP-IT-Sensitivity Kit | Active Motif | Cat# 53040 |
| Papain Dissociation System Kit | Worthington | Cat# LK003150 |
| Single Cell 3′ v2 Kit | 10X Genomics | REF: PN-120267 |
| Single Cell A Chip v2 Kit | 10X Genomics | REF: PN-120236 |
| TG NextSeq 500/550 High Output Kit v2 | Illumina | Cat# FC-404-2005 |
| Deposited Data | ||
| Single cell mRNA sequencing | NCBI’s Gene Expression Omnibus | GEO: GSE121707 |
| Experimental Models: Cell Lines | ||
| ImM10 Cells | Otteson and Phillips, 2010 | N/A |
| Experimental Models: Organisms/Strains | ||
| R26R-mTmG+/tg | The Jackson Laboratory (Muzumdar et al., 2007) |
Cat# JAX:007676, RRID:IMSR_JAX:007676 |
| R26R-nTnG+/tg | The Jackson Laboratory (Prigge et al., 2013; Schmidt, 2013) |
Cat# JAX:023035; RRID:IMSR_JAX:023035 |
| R26R-tdTomato+/tg | The Jackson Laboratory (Madisen et al., 2010) |
Cat# JAX:007909, RRID:IMSR_JAX:007909 |
| Glast-CreERT2+/tg | The Jackson Laboratory (Nathans, 2010) |
Cat#JAX:012586, RRID:IMSR JAX:012586 |
| Chx10-Cre+/tg | The Jackson Laboratory (Rowan and Cepko, 2004) |
Cat# JAX:005105, RRID:IMSR_JAX:005105 |
| Yapflox/flox | (Xin et al., 2011) | N/A |
| Tazflox/flox | (Xin et al., 2013) | N/A |
| Lats1flox/flox | The Jackson Laboratory (Heallen et al., 2011) |
Cat# JAX:024941, RRID:IMSR_JAX:024941 |
| Lats2flox/flox | The Jackson Laboratory (Heallen et al., 2011) |
Cat# JAX:025428, RRID:IMSR_JAX:025428 |
| Yap5SA+/tg | (Monroe et al., 2019) | N/A |
| Oligonucleotides | ||
| CYCLIN D3 -Taqman | Thermo Fisher, Applied Systems | Mm01612362 |
| Cyclin D1 -Taqman | Thermo Fisher, Applied Systems | Mm00432359 |
| Yap -Taqman | Thermo Fisher, Applied Systems | Mm01143263 |
| Taz -Taqman | Thermo Fisher, Applied Systems | Mm01289583 |
| Lats1 -Taqman | Thermo Fisher, Applied Systems | Mm01191886 |
| Lats2 -Taqman | Thermo Fisher, Applied Systems | Mm00497217 |
| Beta-actin -Taqman | Thermo Fisher, Applied Systems | Mm4352933 |
| Gapdh -Taqman | Thermo Fisher, Applied Systems | Mm4352932 |
| Software and Algorithms | ||
| 10X Cellranger 2.0.0 | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/overview/welcome |
| Seurat (version 2.3.4) | Butler et al., 2018 | https://github.com/satijalab/seurat/tree/develop |
Highlights.
Hippo represses YAP activity in Müller glial cells of the damaged mammalian retina
Genetic bypass of Hippo signaling causes spontaneous Müller glial proliferation
Single-cell transcriptomic analysis of reprogrammed Müller glia
Hippo prevents Müller glial reprogramming to a proliferative, progenitor-like state
ACKNOWLEDGMENTS
We thank members of the Poché and Martin labs for critical reading of this manuscript. We thank Dr. Shang Wang for assistance in image analyses and Dr. Chang Seok Lee for guidance with cell fractionation. We thank Joel Sederstrom, Christine Beeton, and the BCM Cytometry and Cell Sorting Core for assistance in flow cytometry (NIH P30 AI036211, P30 CA125123, and S10 RR024574). We thank Jason Kirk, Mary Dickinson, and the Optical Imaging and Vital Microscopy Core for assistance with confocal imaging (NIH U54 HG006348). This work was supported by grants from the NIH (R01 EY024906 to R.A.P.; R01 DE023177, R01 HL127717, R01 HL118761, and R01 HL130804 to J.F.M.; and F31 HL136065 to M.C.H.), the Vivian L. Smith Foundation and MacDonald Research Fund award (16RDM001 to J.F.M.), and the Bright Focus Foundation Macular Degeneration Research Grant (to R.A.P.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.04.047.
REFERENCES
- Azzolin L, Zanconato F, Bresolin S, Forcato M, Basso G, Bicciato S, Cordenonsi M, and Piccolo S (2012). Role of TAZ as mediator of Wnt signaling. Cell 151, 1443–1456. [DOI] [PubMed] [Google Scholar]
- Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S, Bresolin S, Frasson C, Basso G, Guzzardo V, et al. (2014). YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell 158, 157–170. [DOI] [PubMed] [Google Scholar]
- Barrasso AP, Wang S, Tong X, Christiansen AE, Larina IV, and Poché RA (2018). Live imaging of developing mouse retinal slices. Neural Dev. 13, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardos RL, Barthel LK, Meyers JR, and Raymond PA (2007). Late-stage neuronal progenitors in the retina are radial Müller glia that function as retinal stem cells. J. Neurosci 27, 7028–7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienvenu F, Jirawatnotai S, Elias JE, Meyer CA, Mizeracka K, Marson A, Frampton GM, Cole MF, Odom DT, Odajima J, et al. (2010). Transcriptional role of Cyclin D1 in development revealed by a genetic-proteomic screen. Nature 463, 374–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Song X, Wang W, Watnick T, Pei Y, Qian F, and Pan D (2018). A RhoA-YAP-c-Myc signaling axis promotes the development of polycystic kidney disease. Genes Dev. 32, 781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Pfaff SL, and Gage FH (2008). YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev. 22, 3320–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Jo HS, Lim S, Kim HT, Lee KW, Moon KH, Ha T, Kwak SS, Kim Y, Lee EJ, et al. (2018). mTORC1 accelerates retinal development via the immunoproteasome. Nat. Commun. 9, 2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Close JL, Liu J, Gumuscu B, and Reh TA (2006). Epidermal growth factor receptor expression regulates proliferation in the postnatal rat retina. Glia 54, 94–104. [DOI] [PubMed] [Google Scholar]
- Croci O, De Fazio S, Biagioni F, Donato E, Caganova M, Curti L, Doni M, Sberna S, Aldeghi D, Biancotto C, et al. (2017). Transcriptional integration of mitogenic and mechanical signals by Myc and YAP. Genes Dev. 31, 2017–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das AV, Mallya KB, Zhao X, Ahmad F, Bhattacharya S, Thoreson WB, Hegde GV, and Ahmad I (2006). Neural stem cell properties of Müller glia in the mammalian retina: regulation by Notch and Wnt signaling. Dev. Biol 299, 283–302. [DOI] [PubMed] [Google Scholar]
- Das G, Choi Y, Sicinski P, and Levine EM (2009). Cyclin D1 fine-tunes the neurogenic output of embryonic retinal progenitor cells. Neural Dev. 4, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das G, Clark AM, and Levine EM (2012). Cyclin D1 inactivation extends proliferation and alters histogenesis in the postnatal mouse retina. Dev. Dyn 241, 941–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Melo J, Miki K, Rattner A, Smallwood P, Zibetti C, Hirokawa K, Monuki ES, Campochiaro PA, and Blackshaw S (2012). Injury-independent induction of reactive gliosis in retina by loss of function of the LIM homeodomain transcription factor Lhx2. Proc. Natl. Acad. Sci. USA 109, 4657–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer MA, and Cepko CL (2000). Control of Müller glial cell proliferation and activation following retinal injury. Nat. Neurosci 3, 873–880. [DOI] [PubMed] [Google Scholar]
- Dyer MA, and Cepko CL (2001a). p27Kip1 and p57Kip2 regulate proliferation in distinct retinal progenitor cell populations. J. Neurosci 21, 4259–4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer MA, and Cepko CL (2001b). Regulating proliferation during retinal development. Nat. Rev. Neurosci 2, 333–342. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantl V, Stamp G, Andrews A, Rosewell I, and Dickson C (1995). Mice lacking Cyclin D1 are small and show defects in eye and mammary gland development. Genes Dev. 9, 2364–2372. [DOI] [PubMed] [Google Scholar]
- Fausett BV, and Goldman D (2006). A role for alpha1 tubulin-expressing Müller glia in regeneration of the injured zebrafish retina. J. Neurosci 26, 6303–6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimbel SM, Montgomery JE, Burket CT, and Hyde DR (2007). Regeneration of inner retinal neurons after intravitreal injection of ouabain in zebrafish. J. Neurosci 27, 1712–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantz JA, Palpant NJ, Welikson RE, Hauschka SD, Murry CE, and Laflamme MA (2012). Targeted genomic integration of a selectable floxed dual fluorescence reporter in human embryonic stem cells. PLoS ONE 7, e46971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Whoriskey W, Park MY, Bronson RT, Medema RH, Li T, Weinberg RA, and Sicinski P (1999). Rescue of Cyclin D1 deficiency by knockin cyclin E. Cell 97, 767–777. [DOI] [PubMed] [Google Scholar]
- Goldman D (2014). Müller glial cell reprogramming and retina regeneration. Nat. Rev. Neurosci 15, 431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamon A, Masson C, Bitard J, Gieser L, Roger JE, and Perron M (2017). Retinal Degeneration Triggers the Activation of YAP/TEAD in Reactive Müller Cells. Invest. Ophthalmol. Vis. Sci 58, 1941–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamon A, García-García D, Ail D, Bitard J, Chesneau A, Dalkara D, Locker M, Roger JE, and Perron M (2019). Linking YAP to Müller glia quiescence exit in the degenerative retina. Cell Rep 27, this issue, 1712–1725. [DOI] [PubMed] [Google Scholar]
- Harvey KF, Pfleger CM, and Hariharan IK (2003). The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 114, 457–467. [DOI] [PubMed] [Google Scholar]
- Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, and Martin JF (2011). Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 332, 458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heallen T, Morikawa Y, Leach J, Tao G, Willerson JT, Johnson RL, and Martin JF (2013). Hippo signaling impedes adult heart regeneration. Development 140, 4683–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorstad NL, Wilken MS, Grimes WN, Wohl SG, VandenBosch LS, Yoshimatsu T, Wong RO, Rieke F, and Reh TA (2017). Stimulation of functional neuronal regeneration from Müller glia in adult mice. Nature 548, 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karl MO, and Reh TA (2012). Studying the generation of regenerated retinal neuron from Müller glia in the mouse eye. Methods Mol. Biol 884, 213–227. [DOI] [PubMed] [Google Scholar]
- Karl MO, Hayes S, Nelson BR, Tan K, Buckingham B, and Reh TA (2008). Stimulation of neural regeneration in the mouse retina. Proc. Natl. Acad. Sci. USA 105, 19508–19513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Osborn DP, Lee JY, Araki M, Araki K, Mohun T, Känsäkoski J, Brandstack N, Kim HT, Miralles F, et al. (2018). WDR11-mediated Hedgehog signalling defects underlie a new ciliopathy related to Kallmann syndrome. EMBO Rep. 19, 269–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalchuk Y, Homma R, Liang Y, Maslyukov A, Hermes M, Thestrup T, Griesbeck O, Ninkovic J, Cohen LB, and Garaschuk O (2015). In vivo odourant response properties of migrating adult-born neurons in the mouse olfactory bulb. Nat. Commun 6, 6349. [DOI] [PubMed] [Google Scholar]
- Kowalczyk MS, Tirosh I, Heckl D, Rao TN, Dixit A, Haas BJ, Schneider RK, Wagers AJ, Ebert BL, and Regev A (2015). Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 25, 1860–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach JP, Heallen T, Zhang M, Rahmani M, Morikawa Y, Hill MC, Segura A, Willerson JT, and Martin JF (2017). Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 550, 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Chen X, Ding X, Cheng Y, Zhao B, Lai ZC, Al Hezaimi K, Hakem R, Guan KL, and Wang CY (2013). LATS2 suppresses oncogenic Wnt signaling by disrupting β-catenin/BCL9 interaction. Cell Rep. 5, 1650–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Tao G, Hill MC, Zhang M, Morikawa Y, and Martin JF (2018). Pitx2 maintains mitochondrial function during regeneration to prevent myocardial fat deposition. Development 145, dev168609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, von Gise A, Zhou P, Gu F, Ma Q, Jiang J, Yau AL, Buck JN, Gouin KA, van Gorp PR, et al. (2014). Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ. Res 115, 354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Yao E, Zhang K, Jiang X, Croll S, Thompson-Peer K, and Chuang PT (2017). YAP is essential for mechanical force production and epithelial cell proliferation during lung branching morphogenesis. eLife 6, e21130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Hunter DJ, Rooker S, Chan A, Paulus YM, Leucht P, Nusse Y, Nomoto H, and Helms JA (2013). Wnt signaling promotes Müller cell proliferation and survival after injury. Invest. Ophthalmol. Vis. Sci 54, 444–453. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Moroishi T, and Guan KL (2016). Mechanisms of Hippo pathway regulation. Genes Dev. 30, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe TO, Hill MC, Morikawa Y, Leach JP, Heallen T, Cao S, Krijger PHL, de Laat W, Wehrens XHT, Rodney GG, and Martin JF (2019). YAP Partially Reprograms Chromatin Accessibility to Directly Induce Adult Cardiogenesis In vivo. Dev. Cell 48, 765–779.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya IM, and Halder G (2019). Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Biol 20, 211–226. [DOI] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, and Luo L (2007). A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605. [DOI] [PubMed] [Google Scholar]
- Nathans J (2010). Generation of an inducible Slc1a3-cre/ERT transgenic allele (MGI Direct Data Submission).
- Neto F, Klaus-Bergmann A, Ong YT, Alt S, Vion AC, Szymborska A, Carvalho JR, Hollfinger I, Bartels-Klein E, Franco CA, et al. (2018). YAP and TAZ regulate adherens junction dynamics and endothelial cell distribution during vascular development. eLife 7, e31037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooto S, Akagi T, Kageyama R, Akita J, Mandai M, Honda Y, and Takahashi M (2004). Potential for neural regeneration after neurotoxic injury in the adult mammalian retina. Proc. Natl. Acad. Sci. USA 101, 13654–13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otteson DC, and Phillips MJ (2010). A conditional immortalized mouse Müller glial cell line expressing glial and retinal stem cell genes. Invest. Ophthalmol. Vis. Sci 51, 5991–6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poché RA, Furuta Y, Chaboissier MC, Schedl A, and Behringer RR (2008). Sox9 is expressed in mouse multipotent retinal progenitor cells and functions in Müller glial cell development. J. Comp. Neurol 510, 237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poché RA, Zhang M, Rueda EM, Tong X, McElwee ML, Wong L, Hsu CW, Dejosez M, Burns AR, Fox DA, et al. (2016). RONIN Is an Essential Transcriptional Regulator of Genes Required for Mitochondrial Function in the Developing Retina. Cell Rep. 14, 1684–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell C, Cornblath E, Elsaeidi F, Wan J, and Goldman D (2016). Zebrafish Müller glia-derived progenitors are multipotent, exhibit proliferative biases and regenerate excess neurons. Sci. Rep 6, 24851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigge JR, Wiley JA, Talago EA, Young EM, Johns LL, Kundert JA, Sonsteng KM, Halford WP, Capecchi MR, and Schmidt EE (2013). Nuclear double-fluorescent reporter for in vivo and ex vivo analyses of biological transitions in mouse nuclei. Mamm. Genome 24, 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z, Barthel LK, and Raymond PA (2009). Genetic evidence for shared mechanisms of epimorphic regeneration in zebrafish. Proc. Natl. Acad. Sci. USA 106, 9310–9315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Reifler A, Parent JM, and Goldman D (2010). Conditional gene expression and lineage tracing of tuba1a expressing cells during zebrafish development and retina regeneration. J. Comp. Neurol 518, 4196–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattner A, Wang Y, Zhou Y, Williams J, and Nathans J (2014). The role of the hypoxia response in shaping retinal vascular development in the absence of Norrin/Frizzled4 signaling. Invest. Ophthalmol. Vis. Sci 55, 8614–8625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond PA, Barthel LK, Bernardos RL, and Perkowski JJ (2006). Molecular characterization of retinal stem cells and their niches in adult zebrafish. BMC Dev. Biol 6, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenbach A, and Bringmann A (2013). New functions of Müller cells. Glia 61, 651–678. [DOI] [PubMed] [Google Scholar]
- Rowan S, and Cepko CL (2004). Genetic analysis of the homeodomain transcription factor Chx10 in the retina using a novel multifunctional BAC transgenic mouse reporter. Dev. Biol 271, 388–402. [DOI] [PubMed] [Google Scholar]
- Rueda EM, Johnson JE Jr., Giddabasappa A, Swaroop A, Brooks MJ, Sigel I, Chaney SY, and Fox DA (2016). The cellular and compartmental profile of mouse retinal glycolysis, tricarboxylic acid cycle, oxidative phosphorylation, and P transferring kinases. Mol. Vis 22, 847–885. [PMC free article] [PubMed] [Google Scholar]
- Sakabe M, Fan J, Odaka Y, Liu N, Hassan A, Duan X, Stump P, Byerly L, Donaldson M, Hao J, et al. (2017). YAP/TAZ-CDC42 signaling regulates vascular tip cell migration. Proc. Natl. Acad. Sci. USA 114, 10918–10923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanges D, Romo N, Simonte G, Di Vicino U, Tahoces AD, Fernández E, and Cosma MP (2013). Wnt/β-catenin signaling triggers neuron reprogramming and regeneration in the mouse retina. Cell Rep. 4, 271–286. [DOI] [PubMed] [Google Scholar]
- Sanges D, Simonte G, Di Vicino U, Romo N, Pinilla I, Nicolás M, and Cosma MP (2016). Reprogramming Müller glia via in vivo cell fusion regenerates murine photoreceptors. J. Clin. Invest 126, 3104–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EE (2013). Direct Data Submission Gt(ROSA)26Sor. MGI Direct Data Submission. [Google Scholar]
- Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, Haslam SZ, Bronson RT, Elledge SJ, and Weinberg RA (1995). Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell 82, 621–630. [DOI] [PubMed] [Google Scholar]
- Skapek SX, Lin SC, Jablonski MM, McKeller RN, Tan M, Hu N, and Lee EY (2001). Persistent expression of Cyclin D1 disrupts normal photoreceptor differentiation and retina development. Oncogene 20, 6742–6751. [DOI] [PubMed] [Google Scholar]
- Szymaniak AD, Mi R, McCarthy SE, Gower AC, Reynolds TL, Mingueneau M, Kukuruzinska M, and Varelas X (2017). The Hippo pathway effector YAP is an essential regulator of ductal progenitor patterning in the mouse submandibular gland. eLife 6, e23499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taranova OV, Magness ST, Fagan BM, Wu Y, Surzenko N, Hutton SR, and Pevny LH (2006). SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev. 20, 1187–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thummel R, Kassen SC, Montgomery JE, Enright JM, and Hyde DR (2008). Inhibition of Müller glial cell division blocks regeneration of the light-damaged zebrafish retina. Dev. Neurobiol 68, 392–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumaneng K, Schlegelmilch K, Russell RC, Yimlamai D, Basnet H, Mahadevan N, Fitamant J, Bardeesy N, Camargo FD, and Guan KL (2012). YAP mediates crosstalk between the Hippo and PI(3)K-TOR pathways by suppressing PTEN via miR-29. Nat. Cell Biol 14, 1322–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udan RS, Kango-Singh M, Nolo R, Tao C, and Halder G (2003). Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat. Cell Biol 5, 914–920. [DOI] [PubMed] [Google Scholar]
- Ueki Y, Wilken MS, Cox KE, Chipman L, Jorstad N, Sternhagen K, Simic M, Ullom K, Nakafuku M, and Reh TA (2015). Transgenic expression of the proneural transcription factor Ascl1 in Müller glia stimulates retinal regeneration in young mice. Proc. Natl. Acad. Sci. USA 112, 13717–13722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varelas X, Miller BW, Sopko R, Song S, Gregorieff A, Fellouse FA, Sakuma R, Pawson T, Hunziker W, McNeill H, et al. (2010). The Hippo pathway regulates Wnt/beta-catenin signaling. Dev. Cell 18, 579–591. [DOI] [PubMed] [Google Scholar]
- Vecino E, Rodriguez FD, Ruzafa N, Pereiro X, and Sharma SC (2016). Glia-neuron interactions in the mammalian retina. Prog. Retin. Eye Res 51, 1–40. [DOI] [PubMed] [Google Scholar]
- Wan J, Ramachandran R, and Goldman D (2012). HB-EGF is necessary and sufficient for Müller glia dedifferentiation and retina regeneration. Dev. Cell 22, 334–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, and Martin JF (2017). Hippo Pathway: An Emerging Regulator of Craniofacial and Dental Development. J. Dent. Res 96, 1229–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Nicolay BN, Chick JM, Gao X, Geng Y, Ren H, Gao H, Yang G, Williams JA, Suski JM, et al. (2017). The metabolic function of CYCLIN D3-CDK6 kinase in cancer cell survival. Nature 546, 426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Huang J, Dong J, and Pan D (2003). hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 114, 445–456. [DOI] [PubMed] [Google Scholar]
- Xiao D, Liu X, Zhang M, Zou M, Deng Q, Sun D, Bian X, Cai Y, Guo Y, Liu S, et al. (2018a). Direct reprogramming of fibroblasts into neural stem cells by single non-neural progenitor transcription factor Ptf1a. Nat. Commun 9, 2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Hill MC, Zhang M, Martin TJ, Morikawa Y, Wang S, Moise AR, Wythe JD, and Martin JF (2018b). Hippo signaling plays an essential role in cell state transitions during cardiac fibroblast development. Dev. Cell 45, 153–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M, Kim Y, Sutherland LB, Qi X, McAnally J, Schwartz RJ, Richardson JA, Bassel-Duby R, and Olson EN (2011). Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci. Signal 4, ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, et al. (2013). Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl. Acad. Sci. USA 110, 13839–13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao K, Qiu S, Tian L, Snider WD, Flannery JG, Schaffer DV, and Chen B (2016). Wnt regulates proliferation and neurogenic potential of Müller glial cells via a Lin28/let-7 miRNA-dependent pathway in adult mammalian retinas. Cell Rep. 17, 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao K, Qiu S, Wang YV, Park SJH, Mohns EJ, Mehta B, Liu X, Chang B, Zenisek D, Crair MC, et al. (2018). Restoration of vision after de novo genesis of rod photoreceptors in mammalian retinas. Nature 560, 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, and Piccolo S (2015). Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 17, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelinka CP, Volkov L, Goodman ZA, Todd L, Palazzo I, Bishop WA, and Fischer AJ (2016). mTor signaling is required for the formation of proliferating Müller glia-derived progenitor cells in the chick retina. Development 143, 1859–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. (2007). Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 21, 2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the scRNA-seq data reported in this paper is GEO: GSE121707 and is accessible through the NCBI’s Gene Expression Omnibus (GEO) (Edgar et al., 2002).