

Abstract
The emergence of highly sensitive molecular diagnostic approaches, such as droplet digital PCR, has allowed the accurate identification of low-frequency variant alleles in clinical specimens; however, the multiplex capabilities of droplet digital PCR for variant detection are inadequate. The incorporation of molecular barcodes or unique IDs into next-generation sequencing libraries through PCR has enabled the detection of low-frequency variant alleles across multiple genomic regions. However, rational library preparation and sequencing data analytic strategies that integrate molecular barcodes have rarely been applied to clinical settings. In this study, we evaluated the parameters that are crucial in the use of molecular barcodes in next-generation sequencing for genotyping clinical specimens from patients with hematologic malignancies. The uniform incorporation of molecular barcodes into DNA templates through PCR was found to be crucial, and the extent of uniformity was governed by multiple interdependent variables. An error elimination strategy was developed for removing sequencing background errors by using molecular barcode sequence information as an alternative to the conventional error correction approach. This approach was successfully used to identify mutations with frequencies as low as 0.15%, and the clonal heterogeneity of hematologic malignancies was revealed. These findings have implications for elucidating heterogeneity and temporal and spatial clonal evolution, evaluating response to therapy, and monitoring relapse in patients with hematologic malignancies.
CME Accreditation Statement: This activity (“JMD 2019 CME Program in Molecular Diagnostics”) has been planned and implemented in accordance with the accreditation requirements and policies of the Accreditation Council for Continuing Medical Education (ACCME) through the joint providership of the American Society for Clinical Pathology (ASCP) and the American Society for Investigative Pathology (ASIP). ASCP is accredited by the ACCME to provide continuing medical education for physicians.
The ASCP designates this journal-based CME activity (“JMD 2019 CME Program in Molecular Diagnostics”) for a maximum of 18.0 AMA PRA Category 1 Credit(s)™. Physicians should claim only credit commensurate with the extent of their participation in the activity.
CME Disclosures: The authors of this article and the planning committee members and staff have no relevant financial relationships with commercial interests to disclose.
In recent years, next-generation sequencing (NGS) technologies have revolutionized the field of clinical genomics.1, 2, 3, 4 These technologies have allowed massive parallel sequencing of hundreds to thousands of genes in a single tube reaction.5 However, there are several challenges to using NGS-based assays for cancer management.6 They are relatively expensive for frequent use7 and have a high background error rate.8, 9, 10, 11, 12, 13
The mainstay targeted NGS technologies use either PCR amplification or hybridization capture-based strategies to enrich the target sequences during the preparation of sequencing-ready libraries.14 Both of these strategies have merits and limitations.15 An amplification-based enrichment strategy can be used to sequence tens to hundreds of genes. However, with increases in the complexity of primer sequences and in the number of primers per reaction, these assays require superior technical validation. In contrast, hybridization capture-based enrichment approaches can be used to sequence hundreds to thousands of genes, with less cumbersome technical optimizations.16, 17, 18 However, hybridization capture-based approaches have also been associated with incumbent costs higher than those of amplification-based NGS assays.
The identification of low-frequency genetic variants has implications in cancer management.19 Low-frequency variants provide valuable insight into tumor heterogeneity, the temporal and spatial clonal evolution of tumors, tumor responses to therapy, and disease relapse.20 In previous studies, therapy-resistant clones or mutations were shown to exist as early as the therapy was initiated.21, 22, 23, 24 In other studies, these mutations were found to be acquired de novo, after therapy had begun.21 In either scenario, the early identification of low-frequency mutations will reshape the therapeutic measures needed to prevent disease relapse.21, 25, 26
The detection of authentic low-frequency genetic variants is challenging since sequencing background errors contribute to false-positive variant alleles below a frequency of 5% in conventional NGS experiments.27 The development of highly sensitive approaches such as droplet digital–PCR (ddPCR) has revolutionized low-frequency variant detection and pushed the lower limits of detection to 0.001%.28 However, ddPCR requires prior knowledge of the variant that is being interrogated, and its multiplex capabilities are very limited. The incorporation of unique molecular barcode sequences during sequencing library preparation has enabled the re-derivation of sequences of original DNA templates, provided the opportunity to eliminate background errors associated with sequencing, and improved specificity and lower limits of detection.9, 29, 30 In contrast to ddPCR, molecular barcoded NGS could also facilitate the identification of de novo variants across multiple regions.
Sequencing read duplicates that share molecular barcode tag information are essential for removing the errors accrued during library preparation or sequencing. However, we lack library-preparation approaches that yield a uniform sequencing-read duplication, as well as an error removal strategy that can be performed on a minimum number of duplicate reads. Here, we developed a simplified library-preparation approach that would yield uniform duplicates from each targeted region. Furthermore, we developed an efficient error elimination approach that uses a minimum number of sequencing-read duplicates and aids in the identification of true variants that occur at low frequencies. As a proof of concept, we identified low-frequency variants in bone marrow samples obtained from patients with hematologic malignancy at various stages of disease follow-up. Mutation frequencies as low as 0.15% were accurately detected in these patients with this approach.
Materials and Methods
Genomic DNA
High–molecular weight genomic DNA was extracted from A375, Raji, NCI-1355 cells (obtained from ATCC, Manassas, VA) and OCI-AML3 cells [kindly provided by Dr. Michael Andreeff (The University of Texas MD Anderson Cancer Center, Houston, TX)]. In brief, the cell pellets were resuspended in lysis buffer (10 mmol/L Tris-Cl, pH 8.0; 5 mmol/L EDTA; 200 mmol/L NaCl; and 0.2% SDS) supplemented with 10 μg/mL proteinase K (Sigma-Aldrich, St. Louis, MO) and incubated overnight at 56°C. After proteinase K digestion, cell lysates were extracted with equal volumes of phenol, chloroform, and isoamyl alcohol (25:24:1; Sigma-Aldrich). DNA was precipitated with double volumes of absolute ethanol. Precipitated DNA pellets were washed once with n-butanol and twice with 70% alcohol before being resuspended in 10 mmol/L Tris-Cl (pH 8.0). Bone marrow samples from patients with hematologic malignancies (chronic myelomonocytic leukemia grade 1, 3; myelodysplastic syndrome, 3; plasma cell myeloma, 3; acute myeloid leukemia, 2; chronic myelomonocytic leukemia grade 0, 2; therapy-related acute myeloid leukemia, 2; B-cell acute lymphoblastic leukemia, subtype B-lymphoblastic leukemia/lymphoma, 2; acute myelomonocytic leukemia, 1; myelofibrosis grade 3, 1; and therapy-related myelodysplastic syndrome, 1) were obtained from the molecular diagnostics laboratory at MD Anderson. These samples were collected at varying stages of disease follow-up. Genomic DNA was extracted using ReliaPrep Large Volume HT gDNA, following the manufacturer's guidelines (Promega, Madison, WI). DNA concentrations were measured using a Qubit 2.0 fluorometer (Thermo Fisher Scientific, Waltham, MA). The study protocol was approved by the institutional review board at MD Anderson Cancer Center, and the study was conducted in accordance with the Declaration of Helsinki.
Sequencing Panel Design
Primer pairs that can yield 130- to 211-bp amplification products were designed using the PrimerQuest Tool (Integrated DNA Technologies, Coralville, IA) and synthesized from Integrated DNA Technologies. Each primer pair was evaluated separately to determine whether it could generate specific products in real-time quantitative PCR (qPCR) using 1× SYBR Green master mix (Thermo Fisher Scientific), 50 ng of OCI-AML3 genomic DNA, and 0.5 μmol/L primer mix. Primer pairs that produced a difference from the median Ct value of >3 on qPCR analysis were redesigned. The specificity of the amplified products from the qPCR reaction was verified by matching the observed and expected melting temperatures of the amplified product. A difference of >2°C between the observed and expected melting temperatures was considered nonspecific, and the primer pairs were redesigned. Primer pairs were also evaluated independently with a PCR amplification reaction using 1× HotStarTaq Plus master mix (Qiagen, Valencia, CA), 50 ng of OCI-AML3 genomic DNA, and 0.5 μmol/L primer mix. The amplification products were purified with 1× volume of solid-phase reversible immobilization (SPRI) beads (Beckman Coulter, Brea, CA) and analyzed with the Agilent D1000 DNA bioanalyzer kit (Agilent Technologies, Santa Clara, CA); a single band of an expected size was considered a specific product. These prescreened primer pairs were appended with adaptor sequences, custom sequencing primer sequences, and a molecular barcode sequence and synthesized from Integrated DNA Technologies. Target-specific forward primers were incorporated with P5 adaptor (5′ adaptor), custom sequencing primer 1 binding sequence, and 12-bp random nucleotides as a molecular barcode sequence in the hairpin structures described previously.29, 31 Target-specific reverse primers were incorporated with a P7 adaptor (3′ adaptor) and custom sequencing primer 2 binding sequence. Molecular barcode–containing primer pairs were reevaluated using qPCR and the Agilent D1000 bioanalyzer, as mentioned earlier in this paragraph. To create a sequencing-ready, 21-plex panel, all molecular barcode–containing, target-specific forward and reverse primers were pooled to a final concentration of 10 μmol/L. The list of primer sequences is provided in Table 1.
Table 1.
List of Primers Used for Preparing Molecular Barcode–Containing Libraries and for Sequencing
| Primer | Chromosome: start-end | Forward primer | Reverse primer |
|---|---|---|---|
| Universal sequence | 5′-GGACACTCTTTCCCTACACGACG-CTCTTCCGATCTCTGNNNNNNNNNN-NNATGGGAAAGAGTGTCC-3′ | 5′-GTGACTGGAGTTCAGACGTGTGCT-CTTCCGATCTGAC-3′ | |
| TP53Exon 2_1 | chr17:7579811-7579985 | 5′-GGGTTGGAAGTGTCTCA-3′ | 5′-GGCCTGCCCTTCCAATG-3′ |
| TP53Exon 3_1 | chr17:7579562-7579747 | 5′-ACTGACTTTCTGCTCTTGT-3′ | 5′-TCATCCATTGCTTGGGACG-3′ |
| TP53Exon 4_1 | chr17:7579491-7579623 | 5′-GGTCCTCTGACTGCTCTTT-3′ | 5′-TTCTGGGAGCTTCATCTGG-3′ |
| TP53Exon 4_2 | chr17:7579369-7579569 | 5′-ATGGATGATTTGATGCTGTCC-3′ | 5′-GCTGCCCTGGTAGGTTT-3′ |
| TP53Exon 4_3 | chr17:7579269-7579411 | 5′-CCTGTCATCTTCTGTCCCTT-3′ | 5′-GGCATTGAAGTCTCATGGAAG-3′ |
| TP53Exon 5_1 | chr17:7578428-7578581 | 5′-AACTCTGTCTCCTTCCTCTTC-3′ | 5′-GCTGTGACTGCTTGTAGATG-3′ |
| TP53Exon 5_2 | chr17:7578337-7578547 | 5′-CTGCCCTCAACAAGATGTTT-3′ | 5′-CAGCCCTGTCGTCTCTC-3′ |
| TP53Exon 6_1 | chr17:7578123-7578321 | 5′-CAGGCCTCTGATTCCTC-3′ | 5′-CACTGACAACCACCCTTA-3′ |
| TP53Exon 7_1 | chr17:7577435-7577645 | 5′-CGCACTGGCCTCATCTT-3′ | 5′-GTCAGAGGCAAGCAGAG-3′ |
| TP53Exon 8_1 | chr17:7576990-7577190 | 5′-ACTGCCTCTTGCTTCTCTT-3′ | 5′-CTCCACCGCTTCTTGTC-3′ |
| TP53Exon 9_1 | chr17:7576822-7576950 | 5′-CACCTTTCCTTGCCTCTTTC-3′ | 5′-CCACTTGATAAGAGGTCCCA-3′ |
| TP53Exon 10_1 | chr17:7573863-7574068 | 5′-ATATACTTACTTCTCCCCCTC-3′ | 5′-TCCTATGGCTTTCCAACCTA-3′ |
| TP53Exon 11_1 | chr17:7572889-7573061 | 5′-GACCCTCTCACTCATGTGAT-3′ | 5′-GTGGGAGGCTGTCAGTG-3′ |
| BRAFExon 11_1 | chr7:140481354-140481532 | 5′-TGTTTGGCTTGACTTGACTT-3′ | 5′-TGTCACCACATTACATACTTACC-3′ |
| BRAFExon 15_1 | chr7:140453058-140453256 | 5′-TCATAATGCTTGCTCTGATAGG-3′ | 5′-ATAGCCTCAATTCTTACCATCC-3′ |
| KRASExon 2_1 | chr12:25398169-25398337 | 5′-ATTATAAGGCCTGCTGAAA-3′ | 5′-GTCCTGCACCAGTAATATG-3′ |
| KRASExon 3_1 | chr12:25380240-25380369 | 5′-AGACTGTGTTTCTCCCTTCT-3′ | 5′-CTCATGTACTGGTCCCTCAT-3′ |
| KRASExon 4_1 | chr12:25378535-25378687 | 5′-AGGACTCTGAAGATGTACCTATG-3′ | 5′-CAGTGTTACTTACCTGTCTTGTC-3′ |
| NRASExon 2_1 | chr1:115258669-115258832 | 5′-TCGCCAATTAACCCTGATTAC-3′ | 5′-ACCTCTATGGTGGGATCATATT-3′ |
| NRASExon 3_1 | chr1:115256459-115256621 | 5′-CCCTTACCCTCCACACC-3′ | 5′-GATGGCAAATACACAGAGGAA-3′ |
| NRASExon 4_1 | chr1:115252120-115252258 | 5′-AAACAAGCCCACGAACTG-3′ | 5′-GGATCACATCTCTACCAGAGT-3′ |
| Custom sequencing primer | 5′-ACACTCTTTCCCTACACGACGC-TCTTCCGATCTCTG-3′ | 5′-GTGACTGGAGTTCAGACGTGTGCTCT-TCCGATCTGAC-3′ |
Note that universal forward and reverse sequences are appended to the 5′ end of each target-specific forward and reverse primer sequence, respectively, before synthesizing the primers.
Sequencing-Ready Library Preparation
PCR Cycles
The library was prepared in a two-stage PCR setup. The first stage of PCR was performed in 20 μL volume using 1× TaqMan genotyping master mix (Thermo Fisher Scientific), HotStarTaq Plus master mix or NEBNext Ultra II Q5 master mix (New England BioLabs, Ipswich, MA), 50 or 100 ng of genomic DNA, and 0.5 μmol/L pooled primer mix. The first-stage PCR started with denaturation at 94°C for 5 minutes; followed by 3 cycles of 94°C for 30 seconds, 55°C for 10 minutes, and 72°C for 30 seconds; and a final incubation at 74°C for 2 minutes. The second stage of PCR was performed in 40 μL volume using 1× NEBNext Ultra II Q5 master mix, HotStarTaq Plus master mix, or 1× TaqMan genotyping master mix; 17 μL of purified product from the first-stage PCR; and 0.5 μmol/L Illumina index primers (San Diego, CA). The second stage started with denaturation at 98°C for 30 seconds; followed by 10 cycles at 98°C for 10 seconds, 85°C for 1 second, 68°C for 6 minutes, and 74°C for 30 seconds; 9 to 17 cycles at 98°C for 10 seconds, 85°C for 1 second, 68°C for 30 seconds, and 74°C for 30 seconds; and a final incubation at 74°C for 5 minutes. During the second-stage PCR cycling, ramping at a rate of 0.2°C/second was applied at temperature transitions from 85°C to 68°C and from 68°C to 74°C.
Removal of Molecular Barcode Primers
After first-stage PCR, 1 μL of exonuclease I (20 U/μL; New England BioLabs) was added to the reactions and incubated at 37°C for 30 minutes.
SPRI Bead Cleanup
After the completion of the first-stage PCR or exonuclease I treatment, PCR products were purified with 1× volume of SPRI beads and eluted in 20 μL of 10 mmol/L Tris-HCl (pH 8.0). The second-stage PCR products were purified with 0.8× volume of SPRI beads and eluted in 50 μL of 10 mmol/L Tris-Cl (pH 8.0).
Size Selection
Double size selection was performed on the purified libraries obtained after second-stage PCR. In brief, fragments of ≥600 bp were removed with 0.56× SPRI beads; the desired fragments, ranging from 250 to 600 bp, were selected with 0.85× SPRI beads and purified. Size-selected libraries were eluted in 20 μL of 10 mmol/L Tris-HCl (pH 8.0).
Library Quantification
Libraries were quantified using a Kapa Library Quantification Kit for Illumina platforms (Roche, Basel, Switzerland). The presence of primer dimers and the size of the fragments in the library were verified by a bioanalysis using the Agilent High-Sensitivity DNA Kit (Agilent Technologies). Typically, library concentrations were within the range of 1 to 10 nmol/L. In the libraries prepared from positive reference samples, the expected allelic frequencies of the BRAF V600E mutation were verified with ddPCR (Bio-Rad Laboratories, Hercules, CA).
Library Pooling and Sequencing
Each library was uniquely indexed in the second stage of PCR. These indexed libraries were pooled to a final concentration of 0.5 to 2 nmol/L, denatured with 0.2 N NaOH, neutralized with 200 mmol/L Tris-Cl (pH 7.0), and diluted in hybridization buffer, according to the manufacturer's instructions (Illumina). To impart diversity into the 21-plex libraries prepared in this study, a uniquely indexed control library with 548 amplicons was included to 20% fraction of the total library pool. The denatured libraries were loaded at a 6.5 pmol/L concentration onto a MiSeq Nano flow cell for sequencing with 500-cycle v2 chemistry, or at an 8 pmol/L concentration onto a MiSeq v3 flow cell with 600-cycle v3 chemistry. Sequencing was performed with custom sequencing primers 1 and 2; the sequences are provided in Table 1. Typically, 2 libraries were sequenced on a Nano flow cell, and 11 libraries were sequenced on a v3 flow cell.
Sequencing Data Analysis
The data were analyzed using a combination of a custom-developed bioinformatics pipeline and a commercially available variant caller (NextGENe version 4.1.2; SoftGenetics, State College, PA). The front-end bioinformatics pipeline was used to derive consensus read sequences as follows: the first 12 nucleotides in the forward sequencing reads indicate the molecular barcode sequence. On the basis of the molecular barcode sequence information, reads in the forward sequencing read file and the corresponding reads in the reverse sequencing read file were sorted independently. In this way, sequencing reads that shared the same molecular barcode information were grouped into a single family; the number of reads present in each family indicates the size of that family.
From each family of sequencing reads, a consensus read sequence was developed. At a given position, if the same nucleotide was present in all reads of the family, it was chosen as a consensus nucleotide. If ambiguity was encountered (ie, if more than one type of nucleotide was present at a particular position), the consensus nucleotide was denoted with N. Consensus reads derived from molecular barcode families with two or more sequencing reads were used to identify variants. Variants were called and annotated with NextGENe. During variant identification, nucleotide positions that were denoted by N were ignored by the variant-calling algorithms of NextGENe. The back-end bioinformatics pipeline was used to process the variant calls and identify the true variants. With this approach, the potential false-positive variants caused by sequencing errors were effectively eliminated up to 0.05% frequency. This error elimination approach was used to identify true variants that were present in samples from patients with hematologic malignancy, down to 0.15% frequency. A read–balance ratio of >0.5 was used as a cutoff criterion for processing variant data and eliminating the residual sequencing artefacts that contributed to false-positive variants; the minimum numbers of nonreference reads required to identify single-nucleotide variants and insertions and deletions were set to 10 and 15, respectively.
ddPCR
Following the manufacturer's guidelines, ddPCR reactions were assembled in 20 μL volume by mixing 10 μL of 2× Supermix, 1 μL of 20× target primers/probe mix, and 30 ng of genomic DNA. The droplets were generated, the genomic DNA targets were amplified within the droplets, and the fluorescence intensity of the droplets was measured to quantify wild-type and mutant allele copies, as per the vendor's recommendation (Bio-Rad).
Results
Crucial Requirement of Different Polymerase Mixes during First- and Second-Stage PCR
In previous studies, molecular barcode–containing hairpin structures were shown to improve the formation of target-specific amplification products.29, 31 A similar hairpin structure was used to incorporate the molecular barcode sequences, and an NGS gene panel was created to cover all exons of TP53 and hot spot regions of KRAS (exons 2, 3, and 4), NRAS (exons 2, 3, and 4), and BRAF (exons 11 and 15). To obtain amplicons with partial adaptor sequences present at both ends, three-cycle amplification was performed during first-stage PCR with molecular barcode–containing, target-specific primers. For second-stage PCR, 19 cycles amplification were performed with Illumina index primers (Figure 1).
Figure 1.
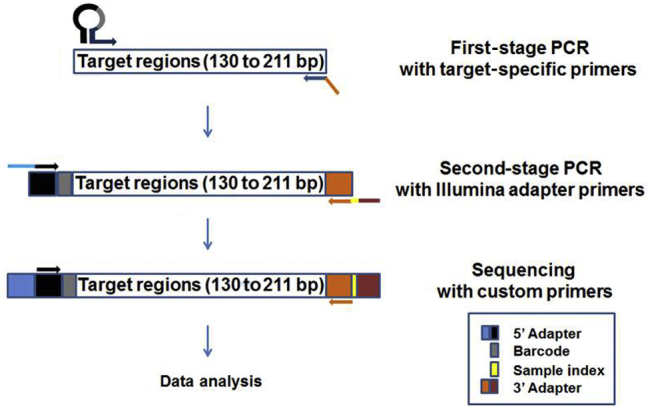
Next-generation sequencing (NGS)-ready library preparation workflow for the molecular barcode–containing 21-amplicon panel. The hairpin structure of the target-specific forward primers contains the partial P5 adaptor (5′ adaptor) sequence and a 12-bp random nucleotide sequence as a molecular barcode sequence. The target-specific reverse primers contain the partial P7 adaptor (3′ adaptor) sequence. The first stage of PCR was performed using target-specific forward and reverse primers, and a 130- to 211-bp region of interest was amplified. The second stage was performed with Illumina sample-indexing primers. The Illumina partial P5 and P7 adaptor sequences that were incorporated into the first-stage PCR product served as anchor sequences to the second-stage PCR primers; amplicons that contained full-length P5 and P7 adaptor sequences were generated by second-stage PCR. These amplicon libraries were subjected to sequencing on MiSeq with custom sequencing primers, and the data were analyzed with a custom bioinformatics pipeline and NextGENe NGS data–analysis software.
During the first stage, molecular barcodes and Illumina partial adaptor sequences were incorporated; during the second stage, full-length Illumina adaptor sequences were produced, along with sample indexes. Three different PCR mixes were evaluated during the first and second stages of PCR. Adding TaqMan genotyping master mix during first-stage PCR yielded highly specific amplification products (Figure 2A). When NEBNext Ultra II Q5 high-fidelity and HotStarTaq Plus polymerases were used in first-stage PCR, high–molecular weight amplification products were apparent (Figure 2A). In contrast, TaqMan genotyping master mix during second-stage PCR failed to yield optimal amplification products; however, NEBNext Ultra II Q5 high-fidelity polymerase mix and HotStarTaq Plus polymerase mix successfully generated abundant amplification products (Figure 2, B and C). These findings suggest that the right combinations of polymerase mixes are crucial for multiplex PCR amplification in the first stage and for singleplex PCR in the second stage.
Figure 2.
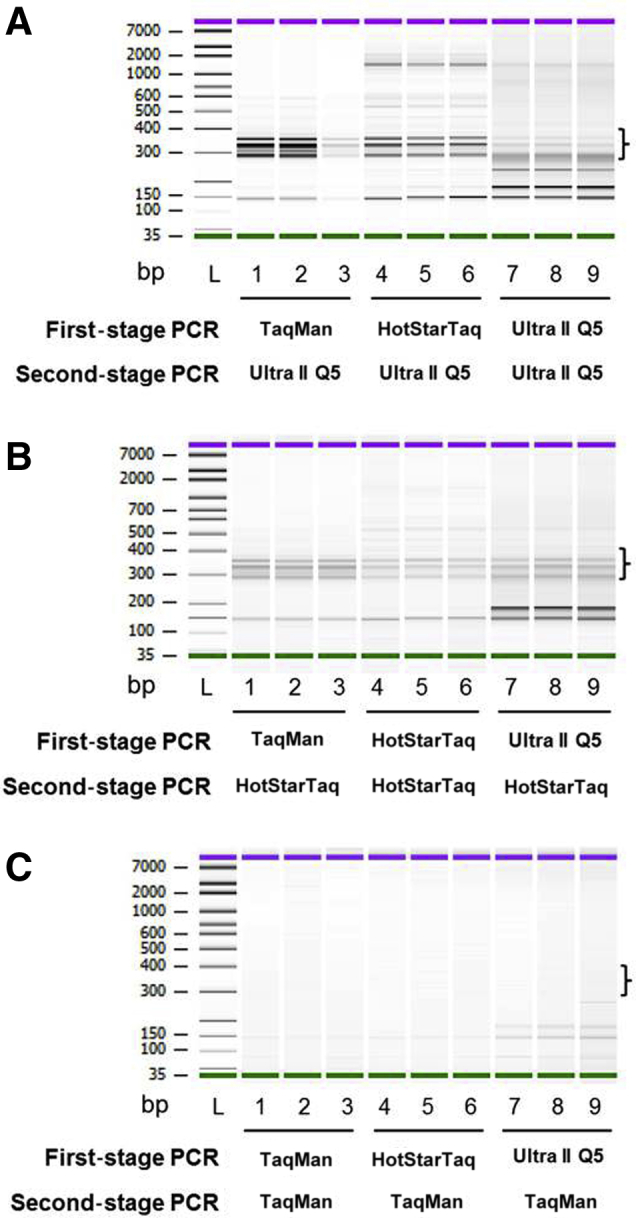
Evaluation of three different polymerase master mixes in the first and second stages of PCR, for sequencing library preparation. Amplification reaction mixes were assembled with TaqMan genotyping master mix, HotStarTaq Plus master mix, or NEBNext Ultra II Q5 mix during first-stage PCR. All of the first-stage PCR products were assembled in NEBNext Ultra II Q5 master mix (A), HotStarTaq Plus master mix (B), or TaqMan genotyping master mix (C) for second-stage PCR. Libraries were purified with solid-phase reversible immobilization beads and analyzed on an Agilent 2100 DNA bioanalyzer. A 300- to 400-bp target-specific library is indicated by a bracket. Note that the fragments of 100 to 200 bp predominantly contained primer dimers. Green and purple bars indicate lower and upper markers, respectively. All samples were evaluated in triplicate.
Exonuclease I Treatment after First-Stage PCR Is Crucial
It is essential to remove the molecular barcode–containing, target-specific primers after the first stage of PCR to prevent the production of new barcode-containing amplicons during the second stage of PCR. The first-stage PCR product was treated with exonuclease I, which digests unincorporated single-strand primers from their 3′ end. Exonuclease I–treated libraries gave rise to a much higher yield and fewer primer dimers than did untreated libraries (Figure 3A).
Figure 3.
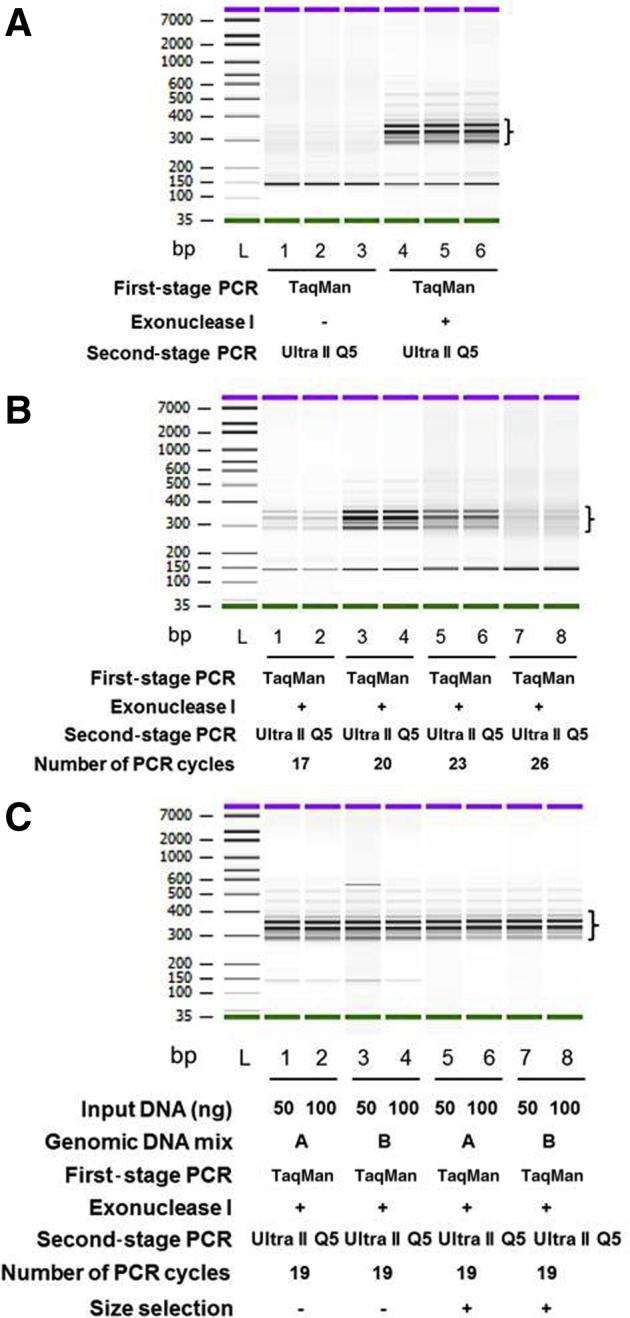
Identification of parameters crucial for improving the quality of molecular barcode–containing next-generation sequencing libraries. A: Exonuclease I treatment reduces the primer dimer concentration and improves the yield of sequencing libraries. First-stage PCR products were incubated with 1 μL of 10 mmol/L Tris-Cl (pH 8.0) or exonuclease I (20 U/μL) at 37°C for 30 minutes. B: Identification of an optimal number of second-stage PCR cycles for library preparation. The first-stage PCR amplification was performed in TaqMan genotyping master mix. The products were then digested with exonuclease I. The second-stage PCR amplification with Ultra II Q5 mix was performed for 17, 20, 23, or 26 cycles. The second-stage PCR products were purified with solid-phase reversible immobilization beads and run on the Agilent 2100 DNA bioanalyzer. C: Size selection efficiently eliminated primer dimers. Genomic DNA mixes A (1% A375, 0.5% Raji, 0.1% NCI-1355, and 98.4% OCI-AML3 DNA; lanes 1, 2, 5, and 6, respectively) and B (1% NCI-1355, 0.5% Raji, 0.1% A375, and 98.4% OCI-AML3 DNA; lanes 3, 4, 7, and 8, respectively) were created and subjected to first-stage PCR amplification, exonuclease I treatment, and second-stage PCR amplification. The purified second-stage PCR products were used for double-size selection with 056×/0.85× volumes of solid-phase reversible immobilization beads, and the size-selected libraries were analyzed on the Agilent 2100 DNA bioanalyzer. Note that a 300- to 400-bp target-specific library is indicated by brackets. Green and purple bars indicate lower and upper markers, respectively. All samples were evaluated in duplicate (B and C) or in triplicate (A).
PCR Yield and Barcode Family Size Is Dictated by the Number of PCR Cycles in Second-Stage PCR
During the first stage of PCR, each amplicon was tagged with a unique molecular barcode. The amplicons from the first stage of PCR served as templates in the second stage and yielded barcode families of varying sizes. High variation in the barcode family sizes of each template would decrease the quality of the library. To identify the crucial factor that governs the highly uniform size of barcode families, varying cycles of amplification were performed during the second stage of PCR (Figure 3B). Seventeen to 20 cycles of amplification yielded a library quantity (1 to 10 nmol/L) that was sufficient for sequencing. However, when the number of cycles was increased to 23 or 26, the yield of the libraries was reduced, accompanied by imbalanced PCR amplification, in which some barcode families were amplified at a higher rate than were others (data not shown). These findings suggest that an optimal number of PCR cycles is required to obtain a uniform barcode family size.
Size Selection Efficiently Improves the Quality of a Molecular Barcode–Containing NGS Library
In multiplex PCR, primer dimers form very frequently.32 The formation of primer dimers is essentially dictated by sequence complexity, primer length, and the number of primers contained in the multiplex PCR. The presence of primer dimers in the sequencing library results in poor-quality sequencing data. The sequencing library prepared from 21-plex primer pairs contained a significant proportion of primer dimers (Figure 3C). To eliminate 150-bp dimers, 0.56×/0.85× double size selection was performed after the second, cleanup PCR step. By incorporating this additional step, these dimers were effectively removed and 300- to 400-bp fragments selected (Figure 3C). To support these observations, the sequencing data also indicated an almost complete absence of dimers in the size-selected library.
Analytical Approach Improves the Detection Sensitivity of Low-Frequency Mutation
The sequencing-ready libraries were prepared from a cell line–derived DNA mix that contained 98.4% (p.Q61L) NRAS, 1% (p.E285K) TP53, 0.5% (p.R213Q; p.Y234H) TP53, 0.5% (p.G13C) KRAS, and 0.1% (p.V600E) BRAF mutations, and the relative representation of each amplicon in a 21-plex amplicon library was measured by qPCR. Each of the 21-amplicon Ct values were within the range of 19.4 to 22.6 (median, 21.2; mean, 21.0; SD, 0.79) and 17.8 to 20.6 (median, 19; mean, 18.9; SD, 0.73) for 50 and 100 ng of input DNA, respectively. All amplicons were amplified with a difference in Ct values of 3, suggesting that each of the 21 amplicons in the library was represented uniformly (Figure 4A).
Figure 4.

Evaluation of molecular barcode–containing libraries by real-time quantitative PCR (qPCR) and next-generation sequencing (NGS). A: qPCR evaluation of size-selected libraries indicate uniform representation of 21 amplicons in the libraries. Note that all amplicons were present within a 2- to 3-Ct value difference from the median. B: NGS indicates relatively uniform representation of the 21 amplicons in sequencing libraries. Libraries were sequenced in two independent runs, and the absolute read counts for each amplicon are depicted. C: An NGS data analysis without using molecular barcode information indicates the presence of abundant false-positive mutations at low frequencies. Libraries prepared from the reference DNA mix containing 98.4% (p.Q61L) NRAS, 1% (p.E285K) TP53, 0.5% (p.R213Q; p.Y234H) TP53, 0.5% (p.G13C) KRAS, and 0.1% (p.V600E) BRAF mutations were sequenced independently twice; the results of one sequencing run are shown. Note that the expected mutations above 0.3% allelic frequency were clearly apparent, although false-positive mutations appeared in this range within the amplicons covering KRAS exon 4 and NRAS exon 3. More false-positive mutations are observed between 0.05% and 0.3% allelic frequencies, and the true BRAF (V600E) mutation within this range is obscured by the false positives.
The sequencing data further confirmed the uniform coverage of 21 amplicons in the library (Figure 4B). In the 21-amplicon library, a median coverage of 144,825 was observed, and most of the amplicons were within twofold of the median coverage, suggesting highly uniform sequencing coverage for all of the amplicons in the 21-plex library. The sequencing data were initially analyzed to identify the expected variants without integration of the molecular barcodes. The analysis indicated that the expected mutations above 0.3% were clearly distinguishable from false-positive mutations, although false-positive mutations were seen in some amplicons. The false-positive mutations became abundant when the allele frequency was below 0.3%, and the true mutations between 0.05% and 0.3% were completely obscured by the false-positive mutations (Figure 4C).
The molecular barcodes were used to eliminate errors that had accrued in the sequencing data. Sequencing reads that contained the same molecular barcode tags were grouped into individual barcode families, and then a consensus read sequence was derived from reads in each family. To derive consensus reads, molecular barcode families that contained two or more sequencing reads were chosen. In deriving the consensus read sequence from a group of reads that was present in each molecular barcode family, a 100%-match criterion was used: For any given nucleotide position, when all reads in the barcode family contained the same nucleotide (100% match), that nucleotide was chosen as the consensus nucleotide (Supplemental Figure S1A). If any mismatches were encountered, the consensus nucleotide assigned for that position was N (Supplemental Figure S1A). The nucleotide positions where N was assigned in the consensus reads were categorically ignored when allelic variants were determined (Supplemental Figure S1B). This error elimination approach was developed instead of using the error correction strategy described in earlier studies.29, 31
Error Elimination Approach Effectively Removes False-Positive Variants and Identifies True Variants at Low Frequencies
A positive reference sample containing 98.4% (p.Q61L) NRAS, 1% (p.E285K) TP53, 0.5% (p.R213Q; p.Y234H) TP53, 0.5% (p.G13C) KRAS, and 0.1% (p.V600E) BRAF mutations was sequenced, and the error elimination approach was used to remove false-positive variants occurring at a low frequency. The sequencing analysis indicated that approximately 87% consensus reads were derived from molecular barcode families that contained at least two sequencing reads (Supplemental Figure S2), and 50% consensus reads were derived from families that contained at least four sequencing reads (Figure 5A), suggesting that in a larger fraction of consensus reads, the error was eliminated effectively. Before error elimination, in some amplicons, the error was null; in others, it occurred at a range of 1 to 32 nucleotide positions per amplicon. After application of the error elimination strategy, the error was completely removed in all 21 amplicons to allele frequencies as low as 0.05% (Supplemental Figure S3).
Figure 5.
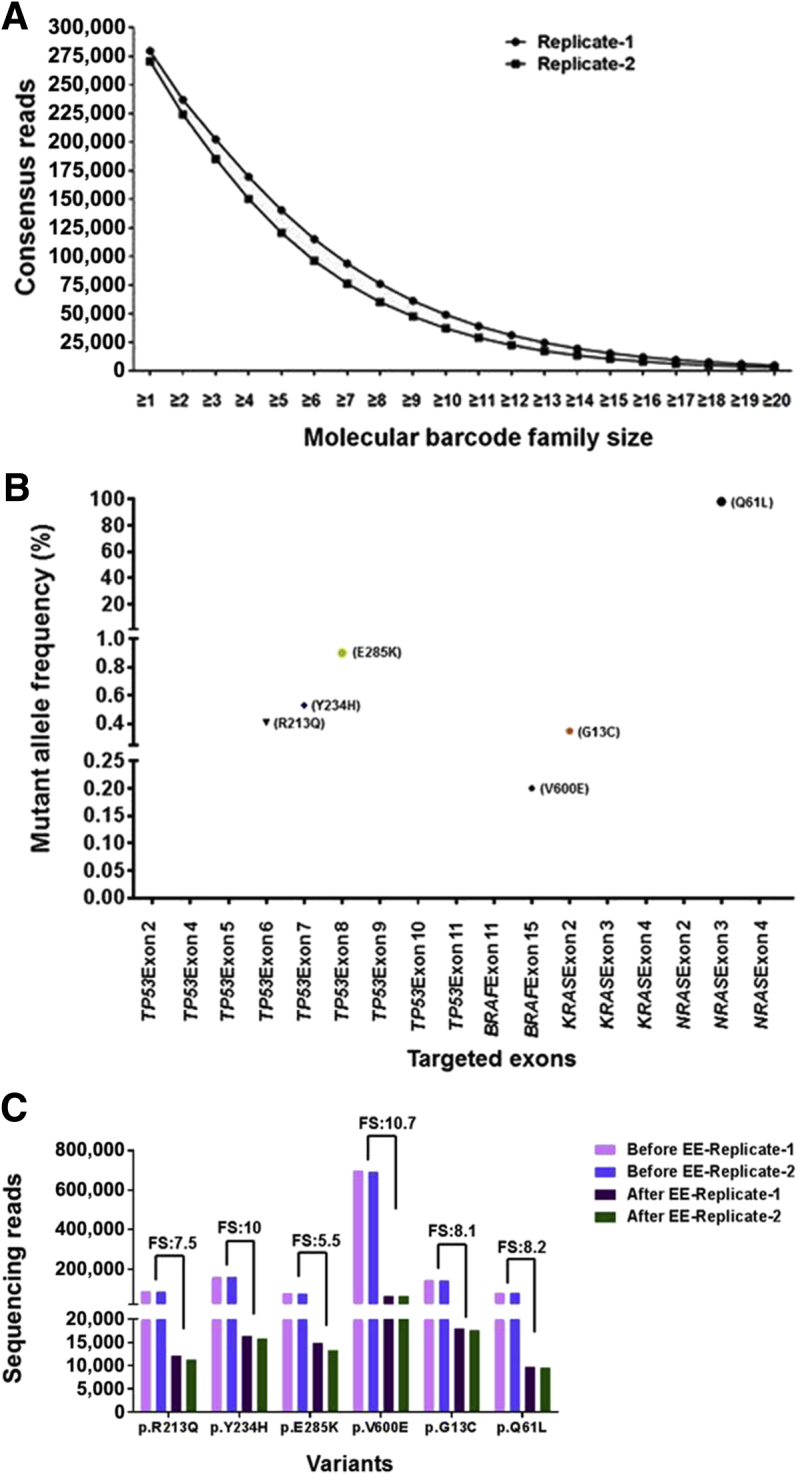
The error elimination (EE) approach efficiently removes false-positive variants that occur at low frequencies. Libraries prepared from genomic DNA mix containing 98.4% (p.Q61L) NRAS, 1% (p.E285K) TP53, 0.5% (p.R213Q; p.Y234H) TP53, 0.5% (p.G13C) KRAS, and 0.1% (p.V600E) BRAF mutations were sequenced in two independent runs. A: Distribution pattern of consensus reads. Note that 50% of the consensus reads were derived from molecular barcode families with at least four sequencing reads. B: Identification of all expected variants in positive reference DNA without yielding any false-positive variants down to 0.05%. Note that consensus reads that had been derived from molecular barcode families with two or more sequencing reads were used for variant identification. Results from an individual sequencing run are shown. C: Determination of calculated family size (FS) that yields each consensus read at selected variant positions. Note that the calculated FS varied between 5.5 and 10.7, indicating that each consensus read is derived from a molecular barcode family that contains 5.5 to 10.7 sequencing reads. Calculated FSs were determined by dividing the number of raw reads covering a variant position (without consensus read derivation) by the number of reads covering the same variant position after deriving the consensus reads.
All of the expected mutations in the reference samples were identified without yielding false-positive mutations, suggesting that the error elimination strategy is effective in detecting low-frequency variants (Figure 5B). The mean size of a molecular barcode family that produced a consensus read at the variant positions identified in the positive reference samples was calculated. Each consensus read was found to be derived from molecular barcode families with 5.5 to 10.7 reads, suggesting that the errors were corrected more effectively, as an optimal number of reads could be obtained in each family for deriving the consensus sequence (Figure 5C).
After the establishment of the library preparation and bioinformatics pipeline for the custom molecular barcode–containing NGS panel, 20 samples from patients with hematologic malignancies were genotyped using the custom panel, and the results were compared with an 81-gene end-leukemia panel developed in the Clinical Laboratory Improvement Amendments–certified molecular diagnostics laboratory at MD Anderson. A mean coverage of 17,085 in the targeted regions and a minimum coverage of over 30 in the mutant alleles were found (Supplemental Figure S4). The end-leukemia panel reported variants occurring at allele frequencies above 1%; mutant allele frequencies above 1% were highly concordant between two panels (Figure 6A).
Figure 6.
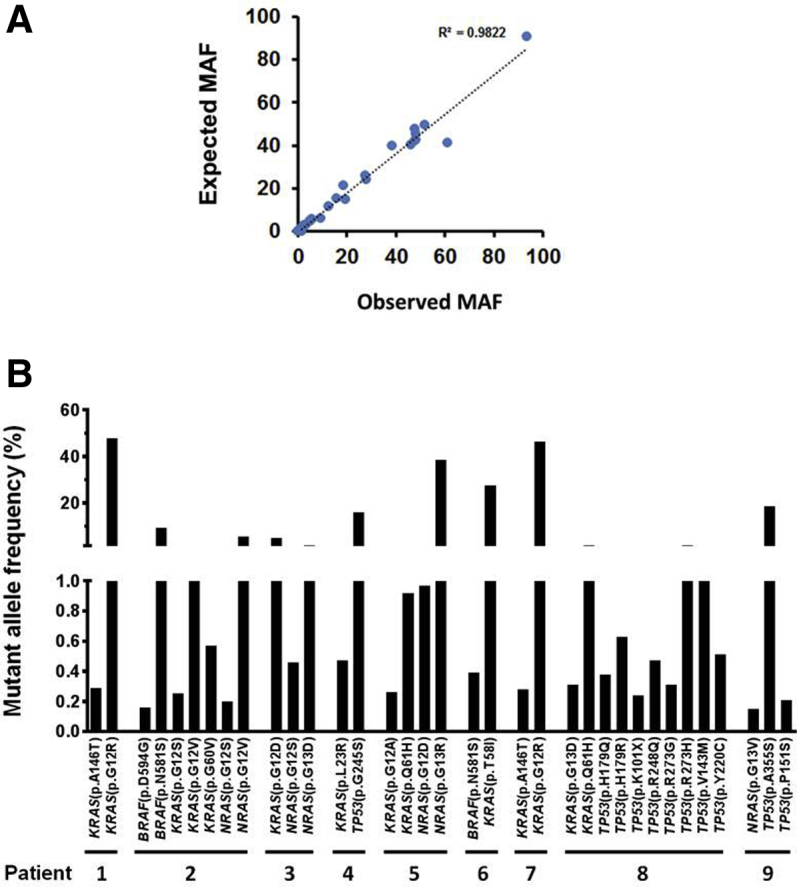
Identification of low-frequency mutations down to 0.15% in hematologic malignancy patients. A: Determination of expected and observed mutant allele frequency (MAF) concordance in samples. The expected mutant allele frequencies of 20 samples were retrieved from the sequencing results of an 81-gene end-leukemia panel developed in MD Anderson's molecular diagnostics CLIA laboratory. Note that the end-leukemia panel identifies mutations that were present at a frequency above 1%. B: Low-frequency mutational profiles identified by a molecular barcode–containing 21-amplicon next-generation sequencing panel. Of the 20 samples sequenced, nine yielded low-frequency mutations.
Of the 20 samples sequenced, 9 contained low-frequency mutations below 1%, in addition to high-frequency mutations above 1%. These low-frequency mutations were noticed in several hotspot mutation regions from KRAS, NRAS, BRAF, and TP53 (Figure 6B) and were verified using ddPCR as an orthogonal validation method. They were accurately identified by molecular barcode–incorporated NGS (Supplemental Table S1). Manual examination of closely located mutations in the NextGENe viewer or Integrative Genomics Viewer revealed that high- and low-frequency mutations in the same genes were present in mutually exclusive reads. The coexistence of these mutations in the same clinical specimens and their identification by distinct sets of sequencing reads indicate that the malignant cells were heterogeneous, which could be determined using molecular barcode–containing NGS with error elimination.
Discussion
In this study, we developed a well-balanced molecular barcode–containing NGS library preparation workflow and a custom bioinformatics pipeline to identify variants down to 0.15% in patients with hematologic malignancies. In this approach, each template is tagged with a random nucleotide sequence as a template index, which is often referred to as the molecular barcode or unique ID. Although the potential of molecular barcodes to significantly improve the detection limits of variants has been recognized, the widespread application of molecular barcode–containing NGS still requires tremendous technologic advancement: The incorporation of barcodes into DNA templates through PCR is not uniform, and there are only a limited number of rational analytical approaches that can effectively remove the sequencing errors.33, 34, 35
When a long stretch of 12 random nucleotides is used as a molecular barcode sequence, it can contribute to mispriming events from target-specific primer sequences and yield significant amounts of nonspecific product.29, 31 Therefore, the molecular barcode sequence was incorporated into the hairpin structure appended to target-specific forward primers. In a simplified protocol of simple, multiplexed, PCR-based barcoding of DNA for sensitive mutation detection using sequencing that also uses molecular barcode–containing hairpin structures, a combination of lower primer concentrations and extended annealing temperatures during the first-stage PCR cycle, and a dilution strategy for first-stage amplified products, were adopted to overcome the tedious purification steps.29, 31 A limitation of this approach is that the carryover of molecular barcode–containing primers into the second stage of PCR, which occurs at minute concentrations, might contribute to the occurrence of molecular barcode families of lesser size. To overcome this primer carryover issue, high-depth sequencing was performed in earlier studies, and only barcode families with 10 reads or more were used to correct the sequencing errors.29, 31
The optimal concentrations of input DNA and the number of PCR cycles with molecular barcode–containing, target-specific primers are reported to be crucial for incorporating molecular barcodes into a higher fraction of DNA templates through PCR.29, 35 Here, molecular barcodes were incorporated into the hairpin structures appended to target-specific forward primers, and the crucial parameters that contributed to the high degree of uniformity in molecular barcode–containing NGS were evaluated. The extent of uniformity was found to be influenced by highly interdependent variables, including target-specific primer sequences, the selection of DNA polymerases, PCR cycling conditions, the removal of molecular barcode primers carried over from the first stage of PCR, and the removal of primer dimers. Typically, 50 ng of input DNA, 3 cycles of first-stage PCR with 0.5 μmol/L molecular barcode–containing 21-plex target-specific primer pairs, and 19 cycles of second-stage PCR with 0.5 μmol/L Illumina indexing primers yielded 1 to 10 nmol/L sequencing-ready libraries and produced molecular barcode families with a uniform size distribution.
When the cutoff threshold for calling the variants from raw sequencing reads was reduced to 0.01%, > 50% of nucleotides in any given amplicon were found to yield low allele frequency variants (data not shown). Characteristically, A-to-G, G-to-A, C-to-T, and T-to-C nucleotide transitions were found in those identified variants. These variants likely originated from sequencing steps rather than from library-preparation stages. At any given position, in a very small fraction of reads, erroneous base calls are reported consistently during sequencing, and these sequencing artefacts predominantly contributed to the low allele frequency variants.8, 9, 10, 11, 12, 13 In the past few years, molecular barcodes have been used to correct these sequencing errors by incorporating an error correction strategy.9, 29, 30 Because a potential limitation of error correction approaches is that the error will remain uncorrected when families of smaller sizes are used, sequencing was performed at greater depths to obtain molecular barcode families of larger sizes. Typically, families with 10 reads or more were used for efficient error correction.29, 31 In this study, an error elimination approach was developed as an alternative to an error correction strategy, which allowed the use of molecular barcode families with two or more reads to remove sequencing errors. Technically, in those families that contain only single reads, errors cannot be eliminated; therefore, the molecular barcode families with single reads were omitted from the analysis. In this error elimination approach, error-accrued positions were waived from participation during variant identification, which allowed the detection of true variants down to 0.15%.
In this study, a simplified library-preparation approach was developed that allowed the uniform incorporation of molecular barcode tags through the targeted amplification of template DNA. In contrast to the error correction strategies reported in previous studies,31 an error elimination strategy was developed that allowed us to use molecular barcode families containing at least two sequencing reads to derive consensus sequence reads. This approach allowed us to not only remove the errors more effectively but also retain the maximal number of consensus reads for identifying the variants. As a proof of concept, a 21-amplicon panel was developed and its application demonstrated by the identification of low-frequency variants in hematologic malignancy patients who were at different stages of disease follow-up. The panel developed in this study will have direct applications in clinical settings for evaluating therapeutic responses and monitoring minimal residual disease. In addition, the approaches described in this study may lead to the development of larger sequencing panels that can be used with extended genomic regions that are pertinent to hematologic malignancies; such panels may help us to understand clonal heterogeneity.
Clonal heterogeneity plays a crucial role in cancer therapy resistance21 and is indirectly reflected by the existence of distinct subsets of mutant allele frequencies.36, 37 In our study, molecular barcodes facilitated the accurate identification of low-frequency variants down to 0.15% and aided the identification of subclonal diversity in patients with hematologic malignancies. These findings may help us to understand clonal evolution in hematologic malignancies and to better manage the disease.25, 38 As evidenced by this study, low-frequency mutations identified in BRAF, KRAS, and NRAS genes can be targeted with therapeutic inhibitors.39, 40 Early therapies directed toward subclonal populations, which contribute to low mutant allele frequencies, can be integrated into mainstay therapies to prevent relapses from these malignant subclones. However, further clinical studies to evaluate the benefits of these combinatorial therapeutic approaches are warranted.
Acknowledgments
We thank Helene Pelicano, Lan Chieh, Justin P. Windham, and Yujin Kudo for helpful discussions; Ronald Abraham and Harsha R. Patel for help with sample procurement; and Ann Sutton for editing the manuscript. OCI-AML3 cells were a gift from Dr. Michael Andreeff, The University of Texas MD Anderson Cancer Center.
S.M., X.S., and R.L. planned the study, designed the experiments, analyzed the data, and wrote the manuscript; S.M. and D.Y.D. performed most of the experiments; S.Z. contributed to ddPCR optimization and sequencing library preparation; S.M. and M.A.H. developed the custom bioinformatics pipeline; M.M. and R.K.-S. analyzed the NGS data; and I.I.W., X.S., and R.L. designed and supervised the study.
Footnotes
Supported by NIH grant 1R21 AI103652-01A1 (X.S.), American Cancer Society Research Scholar grant RSG-10-131-01-DDC (X.S.), MD Anderson Institutional Research grants 114844 and 114845 (X.S.), and MD Anderson Leukemia SPORE grant CA100632 (X.S.).
Disclosures: None declared.
Supplemental material for this article can be found at http://doi.org/10.1016/j.jmoldx.2019.01.008.
Contributor Information
Xiaoping Sun, Email: xsun@mdanderson.org.
Rajyalakshmi Luthra, Email: rluthra@mdanderson.org.
Supplemental Data
Supplemental Figure S1.
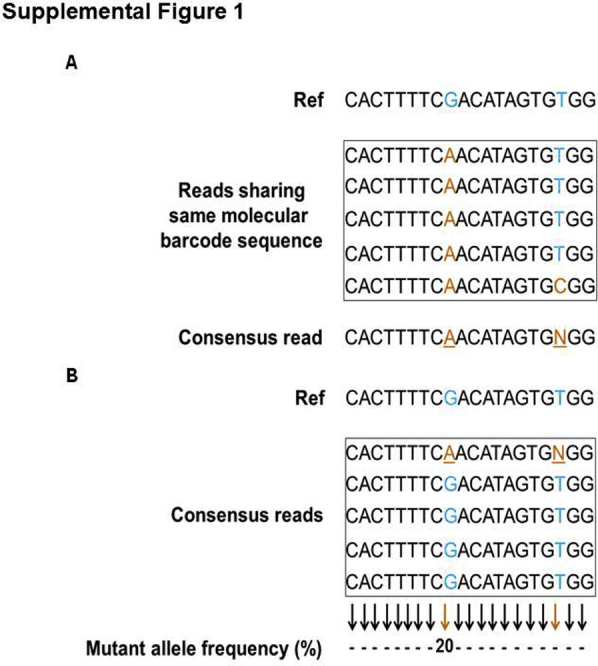
Error elimination approach to removing false-positive variant alleles. A: Derivation of the consensus read sequence. All sequencing reads are grouped into families on the basis of molecular barcode information. All reads within the family share the same molecular barcode sequence; a consensus read sequence is derived from these reads. For any chosen position, if the same nucleotide is present in all of the reads of the family, it is chosen as a consensus nucleotide for that position. If a fraction of reads contains a particular nucleotide and the remaining reads contain a different nucleotide, a consensus base for that position is assigned the letter N. Note that in this example, the underlined nucleotides A and N are the consensus nucleotides, derived from reads that shared the same molecular barcode sequence. N denotes the nucleotide ambiguity of the assigned position. B: Identification of true variant alleles. Variant alleles are determined from the derived consensus read sequences of each molecular barcode family. In this example, a fraction of reads contains the nonreference nucleotide A; it is a true variant allele and accounts for 20% allelic frequency. N was ambiguous and was eliminated during variant allele determination. The positions of the reference sequence, at which variants are shown in the piled sequencing reads, are denoted blue; the variant positions in the original sequencing reads or derived consensus reads are shown in orange, and the corresponding wild-type nucleotide positions are indicated with blue. The orange and black arrows denote the expected mutant allele frequencies at the positions where variants were and were not detected, respectively.
Supplemental Figure S2.

Sample-wise distribution metrics for total and usable consensus sequences. Each consensus sequence was derived from a group of sequencing reads that share the same unique molecular barcode. Note that the number of sequencing reads in each group indicates the size of each molecular barcode family. Total consensus sequences include all consensus sequences derived from molecular barcode families with a size of ≥1; however, usable consensus sequences include only those derived from families with a size of ≥2. On average, 87% of the total consensus sequences (181,670 of 206,914) were usable for identifying variants. PR, positive reference.
Supplemental Figure S3.
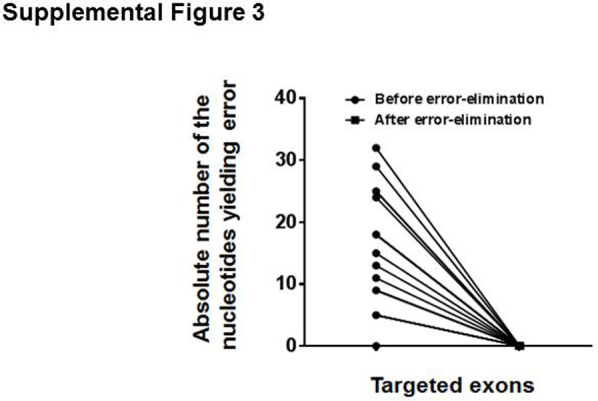
The error elimination approach effectively eliminates errors down to 0.05%. Note that errors were detected in 0 to 32 nucleotides per amplicon and that these errors were eliminated completely with the error elimination approach. Results from an individual sequencing run are shown.
Supplemental Figure S4.

Coverage analysis of variants detected in samples from patients with hematologic malignancy. Each node on the x axis indicates a single variant. The coverage directly indicates the number of unique molecular barcodes or consensus reads supporting either the wild-type (WT) and mutant (Mut) allele or the mutant allele only. Note that the observed mean coverage of 17,085 for each targeted region suggests that each targeted region is covered with a mean of 17,085 unique molecular barcodes. In the bioinformatics pipeline, to identify true variants, the minimum variant coverages required were set to 10 for single-nucleotide variants and to 15 for insertions and deletions. With this variant coverage threshold and the observed total coverage of 17,085, true single-nucleotide variants and insertions and deletions are expected to be identified down to frequencies of 0.058% and 0.087%, respectively.
References
- 1.Koboldt D.C., Steinberg K.M., Larson D.E., Wilson R.K., Mardis E.R. The next-generation sequencing revolution and its impact on genomics. Cell. 2013;155:27–38. doi: 10.1016/j.cell.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Dijk E.L., Auger H., Jaszczyszyn Y., Thermes C. Ten years of next-generation sequencing technology. Trends Genet. 2014;30:418–426. doi: 10.1016/j.tig.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 3.Rehm H.L., Bale S.J., Bayrak-Toydemir P., Berg J.S., Brown K.K., Deignan J.L., Friez M.J., Funke B.H., Hegde M.R., Lyon E., Working Group of the American College of Medical Genetics and Genomics Laboratory Quality Assurance Committee ACMG clinical laboratory standards for next-generation sequencing. Genet Med. 2013;15:733–747. doi: 10.1038/gim.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vijay P., McIntyre A.B., Mason C.E., Greenfield J.P., Li S. Clinical genomics: challenges and opportunities. Crit Rev Eukaryot Gene Expr. 2016;26:97–113. doi: 10.1615/CritRevEukaryotGeneExpr.2016015724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.ten Bosch J.R., Grody W.W. Keeping up with the next generation: massively parallel sequencing in clinical diagnostics. J Mol Diagn. 2008;10:484–492. doi: 10.2353/jmoldx.2008.080027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cummings C.A., Peters E., Lacroix L., Andre F., Lackner M.R. The role of next-generation sequencing in enabling personalized oncology therapy. Clin Transl Sci. 2016;9:283–292. doi: 10.1111/cts.12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sboner A., Mu X.J., Greenbaum D., Auerbach R.K., Gerstein M.B. The real cost of sequencing: higher than you think! Genome Biol. 2011;12:125. doi: 10.1186/gb-2011-12-8-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fox E.J., Reid-Bayliss K.S., Emond M.J., Loeb L.A. Accuracy of next generation sequencing platforms. Next Gener Seq Appl. 2014;1 doi: 10.4172/jngsa.1000106. pii: 1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmitt M.W., Kennedy S.R., Salk J.J., Fox E.J., Hiatt J.B., Loeb L.A. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci U S A. 2012;109:14508–14513. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen P., Ma J., Pei D., Obert C., Cheng C., Geiger T.L. Identification of errors introduced during high throughput sequencing of the T cell receptor repertoire. BMC Genomics. 2011;12:106. doi: 10.1186/1471-2164-12-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meacham F., Boffelli D., Dhahbi J., Martin D.I., Singer M., Pachter L. Identification and correction of systematic error in high-throughput sequence data. BMC Bioinformatics. 2011;12:451. doi: 10.1186/1471-2105-12-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang T.H., Wu N.C., Sun R. A benchmark study on error-correction by read-pairing and tag-clustering in amplicon-based deep sequencing. BMC Genomics. 2016;17:108. doi: 10.1186/s12864-016-2388-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakamura K., Oshima T., Morimoto T., Ikeda S., Yoshikawa H., Shiwa Y., Ishikawa S., Linak M.C., Hirai A., Takahashi H., Altaf-Ul-Amin M., Ogasawara N., Kanaya S. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res. 2011;39:e90. doi: 10.1093/nar/gkr344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ballester L.Y., Luthra R., Kanagal-Shamanna R., Singh R.R. Advances in clinical next-generation sequencing: target enrichment and sequencing technologies. Expert Rev Mol Diagn. 2016;16:357–372. doi: 10.1586/14737159.2016.1133298. [DOI] [PubMed] [Google Scholar]
- 15.Samorodnitsky E., Jewell B.M., Hagopian R., Miya J., Wing M.R., Lyon E., Damodaran S., Bhatt D., Reeser J.W., Datta J., Roychowdhury S. Evaluation of hybridization capture versus amplicon-based methods for whole-exome sequencing. Hum Mutat. 2015;36:903–914. doi: 10.1002/humu.22825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia-Garcia G., Baux D., Faugere V., Moclyn M., Koenig M., Claustres M., Roux A.F. Assessment of the latest NGS enrichment capture methods in clinical context. Sci Rep. 2016;6:20948. doi: 10.1038/srep20948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rennert H., Eng K., Zhang T., Tan A., Xiang J., Romanel A., Kim R., Tam W., Liu Y.C., Bhinder B., Cyrta J., Beltran H., Robinson B., Mosquera J.M., Fernandes H., Demichelis F., Sboner A., Kluk M., Rubin M.A., Elemento O. Development and validation of a whole-exome sequencing test for simultaneous detection of point mutations, indels and copy-number alterations for precision cancer care. NPJ Genom Med. 2016;1 doi: 10.1038/npjgenmed.2016.19. pii: 16019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meienberg J., Zerjavic K., Keller I., Okoniewski M., Patrignani A., Ludin K., Xu Z., Steinmann B., Carrel T., Rothlisberger B., Schlapbach R., Bruggmann R., Matyas G. New insights into the performance of human whole-exome capture platforms. Nucleic Acids Res. 2015;43:e76. doi: 10.1093/nar/gkv216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young A.L., Wong T.N., Hughes A.E., Heath S.E., Ley T.J., Link D.C., Druley T.E. Quantifying ultra-rare pre-leukemic clones via targeted error-corrected sequencing. Leukemia. 2015;29:1608–1611. doi: 10.1038/leu.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diaz L.A., Jr., Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32:579–586. doi: 10.1200/JCO.2012.45.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmitt M.W., Loeb L.A., Salk J.J. The influence of subclonal resistance mutations on targeted cancer therapy. Nat Rev Clin Oncol. 2016;13:335–347. doi: 10.1038/nrclinonc.2015.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mullighan C.G., Phillips L.A., Su X., Ma J., Miller C.B., Shurtleff S.A., Downing J.R. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008;322:1377–1380. doi: 10.1126/science.1164266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong T.N., Ramsingh G., Young A.L., Miller C.A., Touma W., Welch J.S., Lamprecht T.L., Shen D., Hundal J., Fulton R.S., Heath S., Baty J.D., Klco J.M., Ding L., Mardis E.R., Westervelt P., DiPersio J.F., Walter M.J., Graubert T.A., Ley T.J., Druley T., Link D.C., Wilson R.K. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518:552–555. doi: 10.1038/nature13968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhang H.E., Ruddy D.A., Krishnamurthy Radhakrishna V., Caushi J.X., Zhao R., Hims M.M., Singh A.P., Kao I., Rakiec D., Shaw P., Balak M., Raza A., Ackley E., Keen N., Schlabach M.R., Palmer M., Leary R.J., Chiang D.Y., Sellers W.R., Michor F., Cooke V.G., Korn J.M., Stegmeier F. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat Med. 2015;21:440–448. doi: 10.1038/nm.3841. [DOI] [PubMed] [Google Scholar]
- 25.Landau D.A., Carter S.L., Getz G., Wu C.J. Clonal evolution in hematological malignancies and therapeutic implications. Leukemia. 2014;28:34–43. doi: 10.1038/leu.2013.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hiley C., de Bruin E.C., McGranahan N., Swanton C. Deciphering intratumor heterogeneity and temporal acquisition of driver events to refine precision medicine. Genome Biol. 2014;15:453. doi: 10.1186/s13059-014-0453-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fisher K.E., Zhang L., Wang J., Smith G.H., Newman S., Schneider T.M., Pillai R.N., Kudchadkar R.R., Owonikoko T.K., Ramalingam S.S., Lawson D.H., Delman K.A., El-Rayes B.F., Wilson M.M., Sullivan H.C., Morrison A.S., Balci S., Adsay N.V., Gal A.A., Sica G.L., Saxe D.F., Mann K.P., Hill C.E., Khuri F.R., Rossi M.R. Clinical validation and implementation of a targeted next-generation sequencing assay to detect somatic variants in non-small cell lung, melanoma, and gastrointestinal malignancies. J Mol Diagn. 2016;18:299–315. doi: 10.1016/j.jmoldx.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Milbury C.A., Zhong Q., Lin J., Williams M., Olson J., Link D.R., Hutchison B. Determining lower limits of detection of digital PCR assays for cancer-related gene mutations. Biomol Detect Quantif. 2014;1:8–22. doi: 10.1016/j.bdq.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stahlberg A., Krzyzanowski P.M., Jackson J.B., Egyud M., Stein L., Godfrey T.E. Simple, multiplexed, PCR-based barcoding of DNA enables sensitive mutation detection in liquid biopsies using sequencing. Nucleic Acids Res. 2016;44:e105. doi: 10.1093/nar/gkw224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kinde I., Wu J., Papadopoulos N., Kinzler K.W., Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108:9530–9535. doi: 10.1073/pnas.1105422108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stahlberg A., Krzyzanowski P.M., Egyud M., Filges S., Stein L., Godfrey T.E. Simple multiplexed PCR-based barcoding of DNA for ultrasensitive mutation detection by next-generation sequencing. Nat Protoc. 2017;12:664–682. doi: 10.1038/nprot.2017.006. [DOI] [PubMed] [Google Scholar]
- 32.Brownie J., Shawcross S., Theaker J., Whitcombe D., Ferrie R., Newton C., Little S. The elimination of primer-dimer accumulation in PCR. Nucleic Acids Res. 1997;25:3235–3241. doi: 10.1093/nar/25.16.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brodin J., Hedskog C., Heddini A., Benard E., Neher R.A., Mild M., Albert J. Challenges with using primer IDs to improve accuracy of next generation sequencing. PLoS One. 2015;10:e0119123. doi: 10.1371/journal.pone.0119123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kou R., Lam H., Duan H., Ye L., Jongkam N., Chen W., Zhang S., Li S. Benefits and challenges with applying unique molecular identifiers in next generation sequencing to detect low frequency mutations. PLoS One. 2016;11:e0146638. doi: 10.1371/journal.pone.0146638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng Q., Vijaya Satya R., Lewis M., Randad P., Wang Y. Reducing amplification artifacts in high multiplex amplicon sequencing by using molecular barcodes. BMC Genomics. 2015;16:589. doi: 10.1186/s12864-015-1806-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salk J.J., Fox E.J., Loeb L.A. Mutational heterogeneity in human cancers: origin and consequences. Annu Rev Pathol. 2010;5:51–75. doi: 10.1146/annurev-pathol-121808-102113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gawad C., Koh W., Quake S.R. Dissecting the clonal origins of childhood acute lymphoblastic leukemia by single-cell genomics. Proc Natl Acad Sci U S A. 2014;111:17947–17952. doi: 10.1073/pnas.1420822111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rossi D., Khiabanian H., Spina V., Ciardullo C., Bruscaggin A., Fama R., Rasi S., Monti S., Deambrogi C., De Paoli L., Wang J., Gattei V., Guarini A., Foa R., Rabadan R., Gaidano G. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood. 2014;123:2139–2147. doi: 10.1182/blood-2013-11-539726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ward A.F., Braun B.S., Shannon K.M. Targeting oncogenic Ras signaling in hematologic malignancies. Blood. 2012;120:3397–3406. doi: 10.1182/blood-2012-05-378596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Irving J., Matheson E., Minto L., Blair H., Case M., Halsey C., Swidenbank I., Ponthan F., Kirschner-Schwabe R., Groeneveld-Krentz S., Hof J., Allan J., Harrison C., Vormoor J., von Stackelberg A., Eckert C. Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood. 2014;124:3420–3430. doi: 10.1182/blood-2014-04-531871. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.