

Abstract
Protodepalladation is the redox-neutral conversion of a C-Pd(II) bond to a C-H bond promoted by a Brønsted acid. It can be viewed as the microscopic reserves of Pd(II)-mediated C-H cleavage. In the context of catalytic reaction development, protodepalladation offers a means of converting organopalladium(II) intermediates to organic products without a change in oxidation state at the metal center. Hence, when integrated into catalytic cycles, it can be a uniquely enabling elementary step. The goal of this Review is to provide an overview of protodepalladation, including exploration of different reactions types, discussion of literature examples, and analysis of mechanistic features. Our hope is that this review will stimulate other researchers in the field to pursue new applications of this underexploited step in catalysis.
Keywords: palladium, palladacycles, alkenes, alkynes, cyclization, transition metals, protonation, metalation
Graphical Abstract
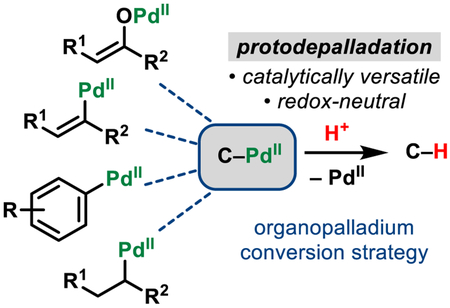
Introduction
The interchange of C-H and C-M bonds has been a prominent topic of study throughout the history of organometallic chemistry.2 C-H activation by transition metals, i.e. the conversion of a C-H bond to a C-M bond, is known to take place by a variety of mechanisms, and by the principle of microscopic reversibility, there exists an equally diverse set of mechanisms for transforming C-M bonds into C-H bonds (Scheme 1). While reactions to convert C-M into C-H bonds are intriguing from a mechanistic organometallic perspective, this mode of reactivity also offers exciting possibilities in the context of organic synthesis, given that organometallic (C-M) intermediates can be accessed from various classes of organic precursors during the course of catalysis. Protodemetalation, the redox-neutral conversion of a C-M bond to a C-H bond via reaction with a Brønsted acid (HX), is an elementary step that is of special interest, both mechanistically and synthetically (Scheme 1). In the literature, it has also been referred to as protiodemetalation, protonolysis, protic cleavage or simply protonation.
Scheme 1.

Microscopic reversibility of C-H activation.
Mechanistically, it can be viewed as the microscopic reverse of a metal-mediated C-H activation process proceeding via electrophilic activation or concerted metalation/deprotonation (CMD). Protodemetalation is perhaps best precedented in the context of copper- and gold-catalyzed reactions. For example, copper-catalyzed 1,4-addition reactions to α,β-unsaturated carbonyl compounds involve protodecupration of a copper(I) enolate intermediate (Scheme 2A). Gold-catalyzed alkyne and alkene hydrofunctionalization reactions similarly involve protodeauration of vinyl- and alkylgold(I) intermediates, respectively (Scheme 2B). Palladium is among the most catalytically versatile transition metals, and organopalladium(II) intermediates can be accessed in a variety of ways, such as oxidative addition to aryl halides and activated carbonheteroatom bonds or nucleometalation across C-C π-bonds. Protodepalladation, however, remains comparatively underexploited, despite its unique possibilities for bond construction. This review focuses on recent advances in protodepalladation as a productive and synthetically enabling step in catalysis that has intriguing potential for continued development.
Scheme 2.

Examples of protodemetalation in Cu(I)- and Au(I)-catalyzed reactions.
In the field of C-H activation, proto/deuterodepalladation has routinely been studied to elucidate reaction mechanisms, as H/D scrambling at the reactive center gives valuable information about how a substrate interacts with the metal catalyst, specifically pertaining to the reversibility of the C-H activation step.3 With the rise in current interest of medicinal chemistry in “heavy” drugs,4 which can benefit from greater metabolic and shelf stability than their protonated equivalents, there has also emerged a renewed drive toward the development of novel deuteration methodology. Traditional strategies rely on acid or base mediated exchange reactions. Homogeneous palladium(II)-catalyzed C-H activation/deuteration—using deuterodepalladation as the crucial step—offers a mild, highly selective alternative5 that can complement other mechanistically distinct approaches involving Ru,6 Rh,7 Ir,8 Pd,9 Pt,10 and Fe catalysts.11 As the number of catalytic reactions involving deuterodepalladation is somewhat extensive, this body of literature is outside of the current review. Instead we focus our attention on palladium-catalyzed C-C and C-heteroatom bond breaking or forming reactions involving a protodepalladation step. As such, this review will cover four main types of reactivity: (1) the functionalization of alkynes via alkenylpalladium intermediates, arene functionalization, (3) conjugate addition to generate palladium-enolates, and (4) the reactivity of alkylpalladium species (Scheme 3).
Scheme 3.

Pd-catalyzed reactions showcasing protodepalladation as an enabling transformation.
Scheme 4 shows a general catalytic cycle for the hydrofunctionalization of unsaturated substrates using palladium(II) catalysis with a protodepalladation termination step: palladium(II) acts as a π-Lewis acid, coordinating to the C-C π-bond and activating the substrate towards nucleopalladation—either with an external nucleophile (NuH) or one previously installed on the palladium catalyst (R1). The challenge in designing such a system is that protodepalladation of the resulting palladium(II) complex 1 is often kinetically unfavorable due to competing side-reactions. If 1 is an alkylpalladium species, β-hydride elimination must be suppressed (vide infra), while in alkenylpalladium complexes oligomerization may dominate.12 We hope that this review will give the reader useful strategies to favor protodepalladation over these competing elementary steps, making it a more accessible strategy for the synthetic community.
Scheme 4.

General catalytic cycle for PdII-catalyzed hydrofunctionalization of unsaturated substrates.
The reader will note that most of the catalytic protodepalladation examples are redox neutral and use Pd(II) as the active catalyst (Scheme 5i). Similar overall transformations can be achieved through a reductive elimination rather than a redox neutral protodepalladation with addition of formic acid, which leads to the formation of a palladium hydride intermediate via a decarboxylation event (Scheme 5ii) (e.g. Cacchi hydroarylations, see Scheme 6).13 Since these reactions are not generally believed to involve a protodepalladation step, they will not be covered in this review except when illustrative of certain principles. Another interesting and relevant mode of reactivity that will not be covered in this review, since it is unclear in most cases whether it involves a protodepalladation step, is aryl-aryl, aryl-vinyl or aryl-benzyl palladium 1,4-migration (and the less common 1,5-migration) (Scheme 7).14
Scheme 5.

Conversion of C-Pd(II) to C-H via (i) protodepalladation (this review) and (ii) palladium hydride formation (not covered).
Scheme 6.

Cacchi syn-hydroarylation of alkynes via palladium-hydride intermediates.
Scheme 7.

Aryl-vinyl palladium 1,4-migration.
Examples of Synthetically Enabling Protodepalladation Reactions
(1). Alkenylpalladium Species
Protodepalladation of alkenylpalladium(II) species offers a powerful platform for the synthesis of substituted alkenes, particularly in the context of ring-closing reactions to prepare heterocycles. Alkenylpalladium(II) intermediates are commonly access from alkynes, which can be activated toward nucleophilic attack upon coordination of palladium(II), which acts as a π-Lewis acid.
Alkyne Cyclization for the Synthesis of Carbo- and Heterocycles
Intramolecular nucleopalladation of these activated alkynes by pendant nucleophiles forms the kinetically favored five-membered rings which undergo protodepalladation to furnish the final product (Scheme 8A). This approach offers a useful synthetic route toward different carbo-15 and heterocycles, including furans,16 lactones,17 coumarins,18 isochromans,19 indoles and pyrroles,20 alkylidene indanones,21 benzazepines22 and multifused N-heterocycles23 (Scheme 8B).
Scheme 8.

Synthesis of various carbo- and heterocycles via PdII-catalyzed alkyne cyclization.
Alkynediol cycloisomerization allows access to interesting spirocycles (Scheme 9).24 The addition of chiral ligands enables the enantioselective synthesis of heterocycles.25 Polyfunctionalized quinolines can be synthesized by dimerization of N-aryl propargylamines (Scheme 10).26 The first step in this reaction is thought to be intramolecular palladium(II) catalyzed cyclization of the aromatic ring onto the alkyne, followed by hydroarylation of a second substrate molecule with the resulting quinolinylpalladium species. Intermolecular variants for the synthesis of oxindoles,27 benzoxepines,28 and thiadiazafluorenones29 have been successful as well. An interesting variation of this chemistry was developed by Aurrecoechea and co-workers, who accessed trien-1-ols 3 by reduction of 4,5-epoxyalk-2-ynyl esters 2 with samarium iodide. In the presence of a palladium(II) catalyst and a proton source, these compounds underwent facile cycloisomerization to afford substituted furans (Scheme 11).30
Scheme 9.

Spirocycle synthesis by PdII-catalyzed cycloisomerization.
Scheme 10.

PdII-catalyzed dimerization of N-aryl propargylamines for the synthesis of quinolines.
Scheme 11.

Synthesis of furans via PdII-catalyzed allene cyclization.
Intermolecular Hydrofunctionalization of Alkynes
Aside from the palladium-mediated intramolecular cyclization of alkynes, another highly useful class of transformations is the reaction of alkenylpalladium intermediates that are formed upon outer-sphere attack by an external nucleophile. Oh et al. discovered that arylboronic acids undergo cross-coupling reactions with activated alkynes to afford alkenylpalladium species that lead to trisubstituted alkenes upon protodepalladation; however the regioselectivities observed in this reaction were low.31 Pincer ligands were successful in directing hydroarylation and -vinylation to the more electron-rich carbon with regioselectivities of >9:1 (Scheme 12A).32 The Engle group demonstrated that very high regio- and stereoselectivity can be achieved when a removable bidentate directing group is employed that coordinates strongly but reversibly to the catalyst (Scheme 12B), allowing for highly selective alkyne syn-hydroarylation33 and anti-hydrochlorination.34
Scheme 12.

Regioselective syn-hydroarylation of alkynes.
Fujiwara and co-workers discovered that electron-rich arenes undergo C-H activation in the presence of triflic acid and a Pd(II) or Pt(II) catalyst, affording trans-hydroarylation of C-C π-bonds (Scheme 13A).13a,35 Arenes, arylboronic acids31,36 and aryl sulfonates37 have been successfully used as coupling partners in the Fujiwara hydroarylation reaction. The intramolecular variant furnishes coumarins, quinolinones38 and fluorenes15i in good yields (Scheme 13B). Mechanistically, Fujiwara and co-workers proposed trans-carbopalladation of an arylpalladium species across the alkyne (Scheme 13C). KIE studies by Tunge and Forsee, however, showed an inverse KIE inconsistent with known C-H activation mechanisms with electrophilic palladium.39 This suggests the alkenylpalladium species may instead be formed by electrophilic aromatic substitution, in which case the highly electrophilic cationic [Pd(OCOCF3)nLm]+ active catalyst formed in situ acts as a π-acid, increasing the electrophilicity of the alkyne. These findings were further supported by extensive1H NMR and mass spectrometry experiments carried out recently by the Eberlin and Correira groups.13a
Scheme 13.

Fujiwara trans-hydroarylation of alkynes and alkenes.
Aside from hydroarylation, hydroamidation has been shown to selectively afford Z-enamides from terminal alkynes.40 The Zhang and Yoshikai groups developed interesting three-component condensation approaches for the synthesis of highly substituted furans (Scheme 14).41 Another elegant example of an intermolecular ring-synthesis involving deprotonation of an alkenylpalladium intermediated was demonstrated by Jarvo and co-workers who showed that allenylboronic esters can be used to access propargylpalladium species 5 which undergoes conjugate addition to alkenes bearing ester or nitrile groups (Scheme 15). The resulting palladium enolate or imine intermediates cyclize under the reaction conditions to diastereoselectively afford cyclopentenes.42
Scheme 14.

PdII-catalyzed three-component condensation for the synthesis of furans.
Scheme 15.

PdII-catalyzed diastereoselective cyclopentene synthesis.
In brief, the reaction of alkenes and alkynes with intra- or intermolecular nucleophiles under palladium(II) catalysis offers a useful strategy for the synthesis of carbo- and heterocycles as well as highly substituted alkenes. Rational design of substrates or ligands allows for high levels of regio- and stereoselectivity.
(2). Arylpalladium Species
Transformations involving protodepalladation of arylpalladium(II) species have been less extensively pursued than other categories of reactions described in this review. This is likely due at least in part to the fact that the common pathways for accessing arylpalladium(II) intermediates—oxidative addition (aryl halides), transmetalation (aryl boronates) and C-H activation (arenes)—involve starting materials that are more highly functionalized than the putative products (or equivalently functionalized starting materials in the case of C-H activation), which means protodepalladation is typically not a synthetically enabling step. Nevertheless, in recent years, creative applications of arylpalladium(II) protodepalladation have demonstrated the potential of this step in enabling unique transformations.
Aromatic Protodecarboxylation
Inspired by transition-metal catalyzed decarboxylative cross-coupling reactions, Kozlowski et al. hypothesized that it would be possible to intercept the arylpalladium species formed during these transformations with a proton source to afford mild aromatic protodecarboxylation. Indeed, they found that bis-ortfto-methoxy substituted aromatic substrates successfully underwent protodecarboxylation in the presence of a palladium(II) catalyst and trifluoroacetic acid (Scheme 16).43 Detailed mechanistic studies found that the reaction proceeds by decarboxylative palladation via a 4-membered transition state, followed by protodepalladation via an electrophilic aromatic substitution mechanism.44 This discovery paved the way for the use of carboxylic acids as traceless directing groups in palladium-catalyzed C-H activation that are easily removed following the functionalization of interest (Scheme 17).45
Scheme 16.

Palladium-catalyzed aromatic protodecarboxylation.
Scheme 17.

Carboxylic acids as traceless directing groups in PdII-catalyzed C-H activation.
Norbornene-Mediated Arene-Functionalization
Directed functionalization of C-H bonds immediately adjacent to suitable directing groups is a well-known transformation in transition-metal catalysis.46 Remote functionalization of unactivated C-H bonds is more challenging and has become the focus of several research groups.46g,47 Since the discovery by Catellani and co-workers in 1997 that norbornene (6) reversibly inserts into Pd-C(sp2) bonds under mild conditions and subsequently acts as a powerful transient ortho-directing auxiliary,48 this strategy has been used for a diverse range of ipso-, ortho- and meta-functionalization reactions of arenes.46f,49 Mechanistically, these methods rely on a relay of oxidative addition and reductive elimination steps, which are usually terminated by a Heck-, Sonogashira-, or Suzuki-Miyaura cross-coupling reaction (Scheme 18). The Yu group realized that a Catellani-type catalytic cycle could be entered through directed ortho-C-H activation and terminated by a protodepalladation step. Addition of norbornene to amide-directed arene C-H alkylation and arylation reactions thus switched the selectivity from ortho- to meta-functionalization, enabling very selective remote C-H functionalization (Scheme 19i).50 A similar approach was concomitantly developed by Dong et al. using amine directing groups (Scheme 19ii).51
Scheme 18.

Mechanism of Catellani-type reactions.
Scheme 19.

Directed, Catellani-type meta-C-H functionalization of arenes.
(3). Palladium-Enolates
Among all types of organopalladium species covered in this review, palladium(II)-enolates are perhaps the best precedented intermediates in the context of protodepalladation reactivity. Palladium(II)-enolates are mostly commonly generated via 1,4-conjugate addition of organopalladium species to α,β-unsaturated carbonyl compounds or functional group equivalents, a process that can take place via either in an inter- or intramolecular fashion.
Conjugate addition to α,β-unsaturated carbonyls is a powerful reaction enabling C-C bond formation, and the use of palladium catalysis significantly expands the range of compatible carbon nucleophiles. The first palladium-catalyzed conjugate addition was reported by Yamamura in 1978, who showed that benzene undergoes conjugate addition with bulky, electron-poor α,β-unsaturated ketones in the presence of stoichiometric amounts of Pd(OAc)2 to afford predominantly protodepalladated alkane products, with small amounts of alkene by-products arising from β-hydride elimination (Scheme 20A).52 Catalytic variants were developed soon thereafter by Cacchi and Lu for the conjugate addition of arylmagnesium, aryltin and arylboronic acid coupling partners to α,β-unsaturated aldehydes and ketones (Scheme 20B).53 Nucleopalladation of the alkene occurs selectively to deliver the aryl group to the β-carbon, with formation of a stabilized palladium enolate intermediate (7). This intermediate can undergo either β-hydride elimination, or protodepalladation to afford the desired products (Scheme 20C). Steric bulk on the β-carbon as well as conformationally rigid cyclic structures were found to favor protodepalladation, presumably by restricting rotation around the C-C bond necessary to obtain the correct conformation for syn-β-hydride elimination.13c Bidentate nitrogen-containing ligands such as 2,2’-bipyridine and excess halide anions were also found to suppress β-hydride elimination, while simultaneously stabilizing the palladium enolate intermediate 7 and favoring protodepalladation.53d,54
Scheme 20.

PdII-catalyzed conjugate addition of arenes to α,β-unsaturated ketones and aldehydes.
Minaard et al. demonstrated that the use of a chiral DuPHOS ligand afforded highly chemo-, regio-, and entantioselective conjugate addition of arylboronic acids to cyclic alkenes under palladium(II) catalysis (Scheme 21i).55 Selectivities for linear substrates, however, were low. Similar results were obtained with BINAM-based NHC ligands56 and pre-formed palladacycles bearing chiral naphylphosphine ligands.57 Chiral pyridinooxazoline (PyOX) (Scheme 21ii)58 and bisoxazoline (BOX)59 ligands were effectively used to construct chiral quaternary centres. Miyaura and co-workers found that addition of a silver salt greatly enhanced the enantioselectivity in linear substrates, presumably by accelerating the transmetalation step of the arylboronic acid onto palladium (Scheme 21iii).60 Alternatively, a chiral amine co-catalyst can be used to activate the carbonyl and impart enantioselectivity.61 Finally, addition of 4-tert-butyloxazolidin-2-one to the carbonyl as a chiral auxiliary also afforded enantioselective conjugate addition of arylboronic acids to linear alkenes with moderate to high ee’s.62 Recently, Yang and Zhang demonstrated the use of nitrostyrene and dienes derived from Meldrum’s acid as viable Michael acceptors for the asymmetric conjugate addition of arylboronic acids under Pd(II) catalysis using Pyox-derived ligands (Scheme 21iv and v).63 Alkoxylation, acetoxylation and amidation of alkenes have been developed with alcohols, acetic acid and Cbz-protected amines as external nucleophiles, respectively.64 Panda and Yadav recently adapted an aza-Wacker reaction developed by Lloyd-Jones and Booker-Milburn65 for the synthesis of benzoxazolidines by forming a stabilized palladium(II) enolate intermediate which could be trapped by protodepalladation to afford the desired products (Scheme 22).66 In a similar vein, Wang and Shi developed an elegant synthesis of chiral [3.3.1]-bicyclic ketals and spiroketals via a Pd(II) catalyzed cascade of conjugate addition reactions in the presence of a chiral BINAP ligand (Scheme 23).
Scheme 21.

Enantioselective PdII-catalyzed conjugate addition of arylboronic acids.
Scheme 22.

Benzoxazolidine synthesis via palladium enolate intermediates.
Scheme 23.

Pd(II) cascade for the synthesis of chiral [3.3.1]-bicyclic ketals.
Widenhoefer demonstrated that 1,3-diketones intramolecularly add to unactivated alkenes in the presence of catalytic PdCl2(MeCN)2, giving alkane rather than alkene products. Subsequent mechanistic studies were consistent with chain-walking of the palladium to afford a stabilized palladium(II) enolate which undergoes protodepalladation rather than beta-hydride elimination (Scheme 24).67
Scheme 24.

Intramolecular hydrocarbofunctionalization of unactivated alkenes.
Furthermore, the alkenylpalladium species introduced in the previous section can be used as nucleophiles, opening up an interesting range of tandem reactions for the synthesis of beta-alkylated ketones and heterocycles (Scheme 25).68 An impressive asymmetric oxa-Diels-Alder/nucleophilic substitution cascade under cooperative PdII and chiral phosphoric acid catalysis was reported by Yao et al. (Scheme 26).69 Ligand exchange of the chiral phosphoric acid onto palladium after intramolecular nucleopalladation of the alkyne allows for enantioselective [4+2]-cyclization with the styrene coupling partner, forming three stereogenic centers with good to excellent ee’s. The resulting intermediate 9 undergoes cyclisation to form a stabilized palladium enolate which—upon protodepalladation—yields the desired products.
Scheme 25.

Tandem alkenylpalladium - palladium enolate chemistry.
Scheme 26.

Cooperative PdII/chiral phosphoric acid catalysis.
Palladium enolate chemistry has furthermore been exploited to achieve enantioselective protodecarboxylation protocols for cyclic ketones. Treatment of β-keto allyl esters with a palladium(II) catalyst in the presence of acid affords clean protodecarboxylation. The first example of this transformation was reported by Tsuji in 1985,70 followed by an asymmetric variant developed by Hénin and Muzart who used ephedrine as a chiral proton source to achieve moderate ee’s.71 Excellent enantioselectivities were attained by the Stoltz group with a chiral phosphinooxazoline (PHOX) ligand, which presumably leads to the formation of an asymmetric palladium enolate intermediate (10) that can be intercepted with acid before it undergoes β-allylation.72 Guiry and co-workers discovered that expedient choice of acid allows for enantiodivergent protodecarboxylation, the exact mechanism of which is still under investigation (Scheme 27).73
Scheme 27.

Enantioselective protodecarboxylation of cyclic ketones.
(4). Alkylpalladium Species
The aforementioned reactions all proceed through organopalladium intermediates (alkenyl, aryl, or enolate) that contain C(sp2)-Pd or O-Pd bonds. These species are comparatively stable owing both to the hybridization and polarity of the bonding atom and to the absence of accessible β-H atoms. In contrast, protodepalladation to alkylpalladium(II) systems is comparatively difficult for several reasons, including the typically rapid nature of competitive β-H elimination and the more sterically encumbered nature of because C(sp3)-Pd bonds.
Protodepalladation of π-allyl palladium(II) species
π Allyl/benzylpalladium(II) species are a special case owing to the presence of adjacent π-bond which enables adoption of a stabilizing η3-coordination mode and are not covered here in extensive detail. In an early representative example, Yoshida and co-workers demonstrated that stabilized π-allylpalladium complexes could also be accessed by treatment of conjugated dienes with stoichiometric amounts of PdCl2 and a suitable nucleophile (Scheme 28).74 Protodepalladation of these complexes with dimethylglyoxime in a protic solvent cleanly afforded sulfonylated Z-alkenes, presumably due to coordination of the sulfonyl group to palladium.
Protodepalladation of π-Allyl palladium(II) species
The stability and reactivity of σ-alkylpalladium intermediates is much harder to control, and therefore less well documented. One of the major challenges associated with this chemistry is the propensity for alkylpalladium species to undergo rapid beta-hydride elimination.75 For a long time, only substrates in which β-hydride elimination was unfavorable were implicated in alkylpalladium chemistry. Examples include the hydroarylation and hydrovinylation of substrates that do not contain β-hydrogens,76 as well as cyclic, strained alkenes—such as norbornene or porphyrins—in which syn-β-hydride elimination is geometrically impossible.77
The Anbarasan group exploited the first strategy in an elegant indoline synthesis: reaction of a palladium(II) catalyst with a diazonium salt is thought to form a palladium carbene which is trapped by a secondary amine to give the alpha-amino palladium enolate species 11. Attack on a pendant alkene closes the indoline ring, forming alkylpalladium species 12 adjacent to a quarternary carbon center which therefore cannot undergo beta-hydride elimination. Instead, protodepalladation yields the desired indoline products in good yields (Scheme 29).78
Scheme 29.

Indoline synthesis via palladium enolate chemistry.
β-Hydride elimination is initiated by the agostic interaction between a hydrogen atom’s sigma bond and an open coordination site on the metal catalyst. While in certain cases bidentate nitrogen ligands have been shown to sufficiently stabilize the alkylpalladium intermediate,44d,45 Michael and Chochran hypothesized that a tridentate pincer ligand would completely suppress undesired beta-hydride elimination by coordinatively saturating the metal.79 This approach allowed them to develop an intramolecular alkene hydroamination proceeding through a truly unstabilized alkylpalladium intermediate (Scheme 30). In the presence of a strong base such as 2,6-lutidine, complex (14+)(BF4−) was isolated and its structure determined by NMR and X-ray analysis, confirming the ability of the pincer ligand to prevent beta-hydride elimination. The Engle group was able to expand this reactivity to regioselective intermolecular hydroamination of unactivated alkenes by employing a removable bidentated 8-aminoquinoline directing group (Scheme 31).80 It is believed that the stability and conformational rigidity imparted by the directing group to palladacycle intermediate 14 prevents beta-hydride elimination and allows protodepalladation to occur instead.81 The concept was further expanded to hydrocarbofunctionalization of alkenes and dienes (Scheme 32).82 Chen et al. recently developed an asymmetric variant of this reaction using a chiral monodentate oxazoline ligand L5, achieving good to excellent ee’s (Scheme 32Aii).83 Engle and co-workers further developed an elegant formal C-X bond activation (where X = O, N, C, F, S) based on this transformation, by accessing alkene substrates in situ via β-X elimination (Scheme 33).84
Scheme 30.

Intramolecular hydroamination of alkenes.
Scheme 31.

Intermolecular hydroamination of alkenes. (DG = directing group)
Scheme 32.

Hydrocarbofunctionalization of alkenes and conjugated dienes (AQ = 8-aminoquinoline).
Scheme 33.

PdII-catalyzed C-X functionalization via β-X elimination.
The reactivity and selectivity of these methodologies hinge on the kinetic and thermodynamic stability of 5-membered palladacycle intermediate 15 (Scheme 31). Remote functionalization—e.g. via the less stable 6-membered palladacycle intermediate—is more difficult, and usually only possible if the 5-membered intermediate is sterically inaccessible.85 Rational design of removable, pincer-like tridentate directing groups allowed Engle et al. to access the less stable 6-membered palladacycle 16 by suppressing beta-hydride elimination, further expanding the scope of their methodology to include highly regioselective remote alkene functionalization (Scheme 34).86
Scheme 34.

Tridentate directing groups for remote alkene hydrofunctionalization (PAQ = 2-pyridyl-8-aminoquinoline, PPA = 2,2’-bipyridylamide).
Mechanistic Considerations
Protodepalladation can occur via two possible mechanisms: (1) protonation of the PdII-C bond with external acid through a three-center-two-electron transition state (i), or via a retro-CMD mechanism (ii) (Scheme 33A). Or (2) oxidative addition of HX to the palladium(II) intermediate to form a PdIV hydride, followed by reductive elimination (Scheme 35B).
Scheme 35.

Possible mechanisms of protodepalladation.
One of the first mechanistic investigations into protodepalladation was carried out by Fryzuk and Bosnich, who developed an elegant stereospecific deuteration protocol for alkenes in 1979 using CF3CO2D as the deuterium source (Scheme 36):87 Reaction of vinyl bromide 17 with stoichiometric amounts of Pd(PPh3)4 afforded oxidative addition complex 18 which was identified by NMR and X-ray88 analysis. Treatment of this complex with CF3CO2D and trifluoroacetic anhydride afforded the deuterated alkene in 90% yield with retention of stereochemistry. This would suggest that the deuterated TFA is acting as an external proton source (mechanism A, Scheme 35).
Scheme 36.

Stoichiometric, Pd-mediated deuteration of vinyl bromide 17.
Michael and Cochran carried out variable NMR studies of their pincer-ligand assisted, palladium-catalyzed hydroamination of alkenes in order to investigate whether protodepalladation was happening via an external deprotonation mechanism (mechanism A, Scheme 35) or via a palladium(IV) hydride (mechanism B, Scheme 35) (Scheme 37).79b Screening temperatures ranging from −78 °C to room temperature, they were never able to observe an NMR signal in the spectral region indicative of a late metal hydride (0 to −60 ppm). While these results cannot fully rule out a palladium hydride intermediate, they strongly suggest that protodepalladation is occurring through protonation of the C-Pd bond, possibly through intramolecular assistance of the carbamate group. 1n the presence of 2,6-lutidine, complex (14+)(BF4−) was isolated and its structure confirmed by X-ray analysis. Treatment of the isolated alkylpalladium complex with different acids (TfOH, HBF4·Et2O, [Ph2NH2][BF4], [Ph2NH2][OTf]) resulted in complete conversion to product 20 and catalytically competent complex 19 in less than 15 minutes at room temperature.
Scheme 37.

Prododepalladation via (i) palladium hydride vs. (ii) protonation by external acid.
Computational studies on the protodepalladation mechanism of palladium enolates in methanol and acetic acid were carried out by the Houk (Scheme 38)89 and Wu (Scheme 39)90 groups, respectively. In both studies, the barrier toward intramolecular, solvent-assisted CMD-type protodepalladation was found to be lower than 15 kJ/mol. Furthermore, when comparing protodepalladation to competing beta-hydride elimination, Wu et al. found that the rate-determining step for the latter was isomerization to enable a Pd-H agostic interaction. While the energy of this transition state was relatively unaffected by ligand choice, the barrier for protodepalladation was significantly reduced by bidentate nitrogen or phosphine ligands on the palladium catalyst (compared to acetate). For example with a 2,2’-bipyridyl ligand, protodepalladation was found to be more favorable than beta-hydride elimination by 2.8 kcal/mol.90 This confirmed the experimental observation that bidentate ligands can be successful at suppressing unwanted beta-hydride elimination. Engle and Liu found that for the tridentate directing groups they developed (Scheme 34), beta-hydride elimination is disfavored by 7.1 kcal/mol compared to protodepalladation, since the former requires partial dissociation of the directing group from the palladium catalyst (Scheme 40).86 For similar reasons, intermolecular protodepalladation via a retro-CMD mechanism was strongly disfavored with these directing groups, since it would require association of HOAc to the palladium catalyst. Instead, intramolecular protodepalladation is thought to occur, with the nucleophile acting as a proton source (Scheme 40).
Scheme 38.

Protodepalladation of palladium enolate complexes in methanol.
Scheme 39.

Protodepalladation of palladium enolate complexes in acetic acid.
Scheme 40.

DFT calculations: protodepalladation with tridentate directing groups.
Based on the data presented in these studies, the authors of this review believe evidence points towards a retro-CMD-type protodepalladation mechanism with external acid for catalysts bearing mono- or bidentate ligands (Scheme 35A) and a novel, intramolecular mechanism with tridentate ligands (Scheme 40).
Conclusion
This review has described a series of catalytic transformations that involve protodepalladation of several different classes or organopalladium intermediates containing C(alkenyl)-, C(aryl)-, O(enolate)-, and C(alkyl)-Pd(II) bonds. In the context of synthetic methods development, this elementary step represents a unique means of converting organopalladium intermediates into organic products in a process that is redox-neutral at the metal center. The palladium(II) species that is thus generated can reenter the catalytic cycle in many cases without the need for oxidation or reduction.
We hope that the examples and mechanistic discussion covered in this review have demonstrated the synthetic utility of protodemetalation in palladium(II)-catalyzed C-C and C-heteroatom bond forming reactions, paving the way for further investigations into this field. By understanding how to favor this elementary step over competing side-reactions such as beta-hydride elimination or polymerization, we believe that this underappreciated mode of reactivity can be harnessed to enable invention of a plethora of interesting new transformations in the future.
Scheme 87.

Hydrosulfonylation of conjugated dienes.
Funding Information
We gratefully acknowledge The Scripps Research Institute (TSRI) and the National Institutes of Health (1R35GM125052) for funding and the DAAD for a postdoctoral fellowship (M.L.O.).
Biographies

Miriam L. O’Duill grew up in Ireland and Germany. In 2011 she obtained her MChem from the University of Oxford. She carried out graduate work at the same university under the supervision of Véronique Gouverneur, earning a PhD in Organic Chemistry in 2015. After a two-year stint as a DAAD Postdoctoral Fellow with Keary M. Engle at the Scripps Research Institute, Miriam joined the School of Chemistry at the National University of Ireland, Galway as a lecturer in January 2018.

Keary M. Engle grew up in Holland, MI, USA. In 2007 he obtained his BS from the University of Michigan, where he carried out research with Professor Adam J. Matzger. He spent the ensuing academic year at the MaxPlanck-Institut für Kohlenforschung as a Fulbright Scholar working with Professor Manfred T. Reetz. In 2013, Keary earned a Ph.D. in Chemistry from The Scripps Research Institute and a D.Phil in Biochemistry with Professors Jin-Quan Yu and Véronique Gouverneur, respectively. After a two-year appointment as a NIH Postdoctoral Fellow with Professor Robert H. Grubbs at the California Institute of Technology, Keary joined the Department of Chemistry at The Scripps Research Institute as an Assistant Professor in the summer of 2015.
References
- (2).For reviews, see:Arndtsen BA; Bergman RG; Mobley TA; Peterson TH Acc. Chem. Res 1995, 28, 154–162;Jia C; Kitamura T; Fujiwara Y Acc. Chem. Res 2001, 34, 633–639;Labinger JA; Bercaw JE Nature 2002, 417, 507–514;Crabtree RH J. Organomet. Chem 2004, 689, 4083–4091;Campeau L-C; Stuart DR; Fagnou K Aldrichimica Acta 2007, 40, 35–41;Chen X; Engle KM; Wang D-H; Yu J-Q Angew. Chem. Int. Ed 2009, 48, 5094–5115;Ackermann L Chem. Rev 2011, 111, 1315–1345;Wencel-Delord J; Dröge T; Liu F; Glorius F Chem. Soc. Rev 2011, 40, 4740–4761;Neufeldt SR; Sanford MS Acc. Chem. Res 2012, 45, 936–946;Daugulis O; Roane J; Tran LD Acc. Chem. Res 2015, 48, 1053–1064;Liu B; Hu F; Shi B-F ACS Catal. 2015, 5, 1863–1881;Hartwig JF J. Am. Chem. Soc 2016, 138, 2–24;Gensch T; Hopkinson MN; Glorius F; Wencel-Delord J Chem. Soc. Rev 2016, 45, 2900–2936;Hartwig JF J. Am. Chem. Soc 2016, 138, 2–24;Davies HML; Morton D J. Org. Chem 2016, 81, 343–350; for thematic issues on this topic, see:Chem. Soc. Rev 2011, 4;Beilstein J. Org. Chem 2016, 12;Chem. Rev 2017, 117.
- (3).For recent examples, see:Plata RE; Hill DE; Haines BE; Musaev DG; Chu L; Hickey DP; Sigman MS; Yu J-Q; Blackmond DG J. Am. Chem. Soc 2017, 139, 9238–9245;Deb A; Hazra A; Peng Q; Paton RS; Maiti D J. Am. Chem. Soc 2017. 139, 763–775;Mai BK; Kim Y Inorg. Chem 2016, 55, 9822–9829;Kefalidis CE; Davi M; Holstein PM; Clot E; Baudoin O J. Org. Chem 2014, 79, 11903–11910; for a tutorial review on how to interpret KIE data in C-H activation, see:Simmons EM; Hartwig JF Angew. Chem. Int. Ed 2012, 51, 3066–3072.
- (4).(a) Timmins GS Expert Opin. Ther. Pat 2017, 27, 1353–1361; [DOI] [PubMed] [Google Scholar]; (b) Atzrodt J; Derdau V; Kerr WJ; Reid M Angew. Chem. Int. Ed 2018, 57, 1758–1784; [DOI] [PubMed] [Google Scholar]; (c) Tung R Innovations in Pharmaceutical Technology, March 2010, 24–28. [Google Scholar]
- (5).Atzrodt J; Derdau V; Kerr WJ; Reid M Angew. Chem. Int. Ed 2018. 57, 3022–3047. [DOI] [PubMed] [Google Scholar]
- (6).(a) Prechtl MHG; Hölscher M; Ben-David Y; Theyssen N; Loschen R; Milstein D; Leitner W Angew. Chem. Int. Ed 2007, 46, 2269–2272; [DOI] [PubMed] [Google Scholar]; (b) Erdogan G; Grotjahn DB J. Am. Chem. Soc 2009, 131, 10354–10355; [DOI] [PubMed] [Google Scholar]; (c) Lockley WJS; Hesk D J. Label. Compd. Radiopharm 2010, 53, 704–715; [Google Scholar]; (d) Prades A; Poyatos M; Peris E Adv. Synth. Catal 2010, 352, 1155–1162; [Google Scholar]; (e) Neubert L; Michalik D; Bähn S; Imm S; Neumann H; Atzrodt J; Derdau V; Holla W; Beller M J. Am. Chem. Soc 2012, 134, 12239–12244; [DOI] [PubMed] [Google Scholar]; (f) Gröll B; Schnürch M; Mihovilovic MD J. Org. Chem 2012, 77, 4432–4437; [DOI] [PubMed] [Google Scholar]; (g) Lee SH; Gorelsky SI; Nikonov GI Organometallics 2013, 32, 6599–6604; [Google Scholar]; (h) Piola L; Fernández-Salas JA; Manzini S; Nolan SP Org. Biomol. Chem 2014, 12, 8683–8688; [DOI] [PubMed] [Google Scholar]; (i) Bai W; Lee KH; Tse SKS; Chan KW; Lin Z; Jia G Organometallics 2015, 34, 3686–3698; [Google Scholar]; (j) Chatterjee B; Krishnakumar V; Gunanathan C Org. Lett 2016, 18, 5892–5895; [DOI] [PubMed] [Google Scholar]; (k) Hale LVA; Szymczak NK J. Am. Chem. Soc 2016, 138, 13489–13492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Blake M; Garnett J; Gregor I J. Chem. Soc. Chem. Commun 1975, 930–932; [Google Scholar]; (b) Lockley WJS Tetrahedron Lett. 1982, 23, 3819–3822; [Google Scholar]; (c) Hesk D; Jones JR; Lockley WJS J. Label. Compd. Radiopharm 1990, 28, 1427–1436; [Google Scholar]; (d) Lenges CP; White PS; Brookhart M J. Am. Chem. Soc 1999, 121, 4385–4396; [Google Scholar]; (e) Chen S; Song G; Li X Tet. Lett 2008, 49, 6929–6932; [Google Scholar]; (f) Campos J; Esqueda AC; López-Serrano J; Sánchez L; Cossio FP; de Cozar A; Alvarez E; Maya C; Carmona E J. Am. Chem. Soc 2010, 132, 16765–16767 [DOI] [PubMed] [Google Scholar]; (g) Di Giuseppe A; Castarlenas R; Pérez-Torrente JJ; Lahoz FJ; Polo V; Oro LA Angew. Chem. Int. Ed 2011, 50, 3938–3942; [DOI] [PubMed] [Google Scholar]; (h) Rhinehart JL; Manbeck KA; Buzak SK; Lippa GM; Brennessel WW; Goldberg KI; Jones WD Organometallics 2012, 31, 1943–1952; [Google Scholar]; (i) Campos J; Rubio M; Esqueda AC; Carmona E J. Label. Compd. Radiopharm 2012, 55, 29–38; [Google Scholar]; (j) Di Giuseppe A; Castarlenas R; Pérez-Torrente JJ; Lahoz FJ; Oro LA Chem. Eur. J 2014, 20, 8391–8403; [DOI] [PubMed] [Google Scholar]; (k) Webster-Gardiner MS; Fu R; Fortman GC; Nielsen RJ; Gunnoe TB; Goddard III WA Catal. Sci. Technol 2015, 5, 96–100. [Google Scholar]
- (8).(a) Shu A; Chen W; Heys J J. Organomet. Chem 1996, 524, 87–93; [Google Scholar]; (b) Ellames G, Gibson J, Herbert J, McNeill A, Tetrahedron 2001, 57, 9487–9497; [Google Scholar]; (c) Heys JR J. Label. Compd. Radiopharm 2007. 50, 770–778; [Google Scholar]; (d) Zhou J; Hartwig JF Angew. Chem. Int. Ed 2008, 47, 5783–5787; [DOI] [PubMed] [Google Scholar]; (e) Salter R J. Label. Compd. Radiopharm 2010, 53, 645–657; [Google Scholar]; (f) Herbert JM J. Label. Compd. Radiopharm 2010, 53, 658–661; [Google Scholar]; (g) Nilsson GN; Kerr WJ J. Label. Compd. Radiopharm 2010, 53, 662–667; [Google Scholar]; (h) Lockley WJS J. Label. Compd. Radiopharm 2010, 53, 668–673; [Google Scholar]; (i) Cochrane AR; Irvine S; Kerr WJ; Reid M; Andersson S; Nilsson GN J. Label. Compd. Radiopharm 2013, 56, 451–454; [DOI] [PubMed] [Google Scholar]; (j) Parmentier M, Hartung T, Pfaltz A, Muri D, Chem. Eur. J 2014, 20, 11496–11504; [DOI] [PubMed] [Google Scholar]; (k) Kerr WJ; Reid M; Tuttle T ACS Catal. 2015, 5, 402–410; [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Hatano M, Nishimura T, Yorimitsu H, Org. Lett 2016, 18, 3674–3677. [DOI] [PubMed] [Google Scholar]
- (9).(a) Korsager PS; Taaning RH; Lindhardt AT; Skrydstrup T J. Org. Chem 2013, 78, 6112–6120; [DOI] [PubMed] [Google Scholar]; (b) Ma S; Villa G; Thuy-Boun PS; Homs A; Yu J-Q Angew. Chem. Int. Ed 2014, 53, 734–737; [DOI] [PubMed] [Google Scholar]; (c) Zhao D; Luo H; Chen B; Chen W; Zhang G; Yu Y J. Org. Chem 2018, DOI: 10.1021/acs.joc.8b00734 [DOI] [PubMed] [Google Scholar]
- (10).(a) Garnett J; Hodges R J. Am. Chem. Soc 1967, 89, 4546–4547; [Google Scholar]; (b) Emmert MH; Gary JB; Villalobos JM; Sanford MS Angew. Chem. Int. Ed 2010, 49, 5884–5886; [DOI] [PubMed] [Google Scholar]; (c) Villalobos JM; Hickman AJ; Sanford MS Organometallics 2010, 29, 257–262; [Google Scholar]; (d) Hickman AJ; Cismesia MA; Sanford MS Organometallics 2012, 31, 1761–1766. [Google Scholar]
- (11).Yu RP; Hesk D; Rivera N; Pelczer I; Chirik PJ Nature 2016, 529, 195–199. [DOI] [PubMed] [Google Scholar]
- (12).Miyaki N; Tomita I; Endo T Polymer Bulletin 1997, 39, 677–684. [Google Scholar]
- (13).For a good review on this topic, see: for a good review on the topic, see:Godoi MN; de Azambuja F; Martinez PDG; Morgon NH; Santos VG; Regiani T; Lesage D; Dossmann H; Cole RB; Eberlin MN; Correia CRD Eur. J. Org. Chem 2017, 1794–1803; for Cacchi hydroarylation, see:Cacchi S; Felici M; Pietroni B Tet. Lett 1984, 25, 3137–3140;Cacchi S Pure & Appl. Chem 1990, 62, 713–722;Cacchi S; Fabrizi G; Moro L; Pace P Synlett 1997, 1367–1370; for coordination-controlled regioselectivity in these types of reactions, see:Kim N; Kim KS; Gupta AK; Oh CH Chem. Commun 2004, 618–619.
- (14).For a review on palladium 1,4-migration, see:Ma S; Gu Z Angew. Chem. Int. Ed 2005, 44, 7512–7517; for selected examples of palladium 1,4-migration, see:Campo MA; Larock RC J. Am. Chem. Soc 2002, 124, 14326–14327;Campo MA; Huang Q; Yao T; Tian Q; Larock RC J. Am. Chem. Soc 2003, 125, 11506–11507;Kesharwani T; Larock RC Tetrahedron 2008. 64, 6090–6102;Bhunia SK; Polley A; Natarajan R; Jana R Chem. Eur. J 2015, 21, 16786–16791;Hu T-J; Zhang G; Chen Y-H; Feng C-G; Lin G-Q J. Am. Chem. Soc 2016, 138, 2897–2900; for selected examples of palladium 1,5-migration, see:Bour C; Suffert J Org. Lett 2005, 7, 653–656;Mota AJ; Dedieu A; Bour C; Suffert J J. Am. Chem. Soc 2005, 127, 7171–7182.
- (15).(a) Burns B; Grigg R; Sridharan V; Worakun T Tet. Lett 1988, 29, 4325–4328; [Google Scholar]; (b) Burns B; Grigg R; Ratananukul P; Sridharan V; Stevenson P; Sukirthalingam S; Worakun T Tet. Lett 1988, 29, 5565–5568; [Google Scholar]; (c) Burns B; Grigg R; Sridharan V; Stevenson P Tet. Lett 1989, 30, 1135–1138; [Google Scholar]; (d) Zhang Y; Negishi E J. Am. Chem. Soc 1989, 111, 3454–3456. Monteiro N; [Google Scholar]; (e) Gore J; Balme G Tetrahedron 1992, 48, 10103–10114; [Google Scholar]; (f) Marat X; Monteiro N; Balme G Synlett 1997, 845–847; [Google Scholar]; (g) Tsukada N; Yamamoto Y Angew. Chem. Int. Ed 1997, 36, 2477–2480; [Google Scholar]; (h) Nakamura I; Bajracharya GB; Wu H; Oishi K; Mizushima Y; Gridnev ID; Yamamoto Y J. Am. Chem. Soc 2004, 126, 15423–15430; [DOI] [PubMed] [Google Scholar]; (i) Chernyak N; Gevorgyan V J. Am. Chem Soc 2008, 130, 5636–5637; [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Chernyak N; Gevorgyan V Adv. Synth. Catal 2009, 351, 1101–1114; [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Hwang JH; Jung YH; Hong YY; Jeon SL; Jeong IH J. Fl. Chem 2011, 132, 1227–1231; [Google Scholar]; (l) Aziz J; Frison G; Le Menez P; Brion J-D; Hamze A; Alami M Adv. Synth. Catal 2013, 355, 3425–3436; [Google Scholar]; (m) Kumar S; Saunthwal RK; Mujahid M; Aggarwal T; Verma AK J. Org. Chem 2016, 81, 9912–9923. [DOI] [PubMed] [Google Scholar]
- (16).(a) Wakabayashi Y; Fukuda Y; Shiragami H; Utimoto K; Nozaki H Tetrahedron 1985, 41, 3655–3661; [Google Scholar]; (b) Fukuda Y; Shiragami H; Utimota K; Nozaki H J. Org. Chem 1991, 56, 5816–5819; [Google Scholar]; (c) Schabbert S; Schaumann E Eur. J. Org. Chem 1998, 1873–1878; [Google Scholar]; (d) Minami Y; Sakai M; Sakamaki T; Hiyama T Chem. Asian J 2017, 12, 2399–2403. [DOI] [PubMed] [Google Scholar]
- (17).(a) Lambert C; Utimoto K; Nozaki H Tet. Lett 1984, 25, 5323–5326; [Google Scholar]; (b) Mandai T; Ohta K; Baba N; Kawada M; Tsuji J Synlett 1992, 671–672; [Google Scholar]; (c) Monot J; Brunel P; Kefalidis CE; Espinosa-Jalapa NA; Maron L; Martin-Vaca B; Bourissou D Chem. Sci 2016, 7, 2179–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).(a) Kitamura T; Otsubo K Heterocycles 2012, 86, 759–766; [Google Scholar]; (b) Kitamura T; Otsubo K J. Org. Chem 2012, 77, 2978–2982. [DOI] [PubMed] [Google Scholar]
- (19).Nandakumar A; Balakrishnan K; Perumal PT Synlett 2011, 18, 2733–2739. [Google Scholar]
- (20).(a) Iritani K; Matsubara S; Utimoto K Tet. Lett 1988, 29, 1799–1802; [Google Scholar]; (b) Cacchi S J. Organomet. Chem 1999, 576, 42–64 and references therein; [Google Scholar]; (c) Müller TE; Grosche M; Herdtweck E; Pleier A-K; Walter E; Yan Y-K Organometallics 2000, 19, 170–183; [Google Scholar]; (d) Ren L; Shi Z; Jiao N Tetrahedron 2013, 69, 4408–4414; [Google Scholar]; (e) Fan X; Yan M; Wang Y; Zhang X J. Org. Chem 2015, 80, 10536–10547; [DOI] [PubMed] [Google Scholar]; (f) Ngo TN; Janert F; Ehlers P; Hoang DH; Dang TT; Villinger A; Lochbrunner S; Langer P Org. Biomol. Chem 2016, 14, 1293–1301. [DOI] [PubMed] [Google Scholar]
- (21).Chernyak N; Gorelsky SI; Gevorgyan V Angew. Chem. Int. Ed 2011,123, 2390–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Peshkov VA; Pereshivko OP; Donets PA; Mehta VP; van der Eycken EV Eur. J. Org. Chem 2010, 4861–4867. [Google Scholar]
- (23).Alam K; Hong SW; Oh KH; Park JK Angew. Chem. Int. Ed 2017. 56, 13387–13391. [DOI] [PubMed] [Google Scholar]
- (24).Ramana CV; Suryawanshi SB; Gonnade RG J. Org. Chem 2009, 74, 2842–2845. [DOI] [PubMed] [Google Scholar]
- (25).(a) Song J; Shen Q; Xu F; Lu X Org. Lett 2007, 9, 2947–2950; [DOI] [PubMed] [Google Scholar]; (b) Shibuya T; Shibata Y; Noguchi K; Tanaka K Angew. Chem. Int. Ed 2011, 50, 3963–3967. [DOI] [PubMed] [Google Scholar]
- (26).Li X-F; Zhang X-G; Hu B-L; Zhang X-H Org. Biomol. Chem 2018. 6, 1736–1744. [DOI] [PubMed] [Google Scholar]
- (27).Kamijo S; Sasaki Y; Kanazawa C; Schüßeler T; Yamamoto Y Angew. Chem. Int. Ed 2005, 44, 7718–7721. [DOI] [PubMed] [Google Scholar]
- (28).Liu G Lu X Adv. Synth. Catal 2007, 349, 2247–2252. [Google Scholar]
- (29).Veltri L; Paladino V; Plastina P; Gabriele P J. Org. Chem 2016, 81, 6106–6111. [DOI] [PubMed] [Google Scholar]
- (30).Aurrecoechea JM; Pérez E; Solay M J. Org. Chem 2001, 66, 564–569. [DOI] [PubMed] [Google Scholar]
- (31).Oh CH; Jung HH; Kim KS; Kim N Angew. Chem. Int. Ed 2003, 42, 805–808. [DOI] [PubMed] [Google Scholar]
- (32).Gupta AK; Kim KS; Oh CH Synlett 2005, 3, 457–460. [Google Scholar]
- (33).Liu Z; Derosa J; Engle KM J. Am. Chem. Soc 2016, 138, 13076–13081. [DOI] [PubMed] [Google Scholar]
- (34).Derosa J; Cantu AL; Boulous MN; O’Duill ML; Turnbull JL; Liu Z; De La Torre DM; Engle KM J. Am. Chem. Soc 2017, 139, 5183–5193. [DOI] [PubMed] [Google Scholar]
- (35).(a) Jia C; Piao D; Oyamada J; Lu W; Kitamura T; Fujiwara Y Science 2000, 287, 1992–1995; [DOI] [PubMed] [Google Scholar]; (b) Jia C; Lu W; Oyamada J; Kitamura T; Matsuda K; Irie M; Fujiwara Y J. Am. Chem. Soc 2000, 122, 7252–7263; [Google Scholar]; (c) Jia C; Kitamura T; Fujiwara Y J. Synth. Org. Chem., Jpn 2001, 59, 1052–1061. [Google Scholar]
- (36).Cacchi S; Fabrizi G; Goggiamani A; Persiani D Tetrahedron 2010, 66, 2433–2438. [Google Scholar]
- (37).Liu S; Bai Y; Cao X; Xiao F; Deng G-J Chem. Commun 2013, 49, 7501–7503. [DOI] [PubMed] [Google Scholar]
- (38).Jia C; Piao D; Kitamura T; Fujiwara Y J. Org. Chem 2000, 65, 7516–7522. [DOI] [PubMed] [Google Scholar]
- (39).Tunge JA; Foresee LN Organometallics 2005, 24, 6440–6444. [Google Scholar]
- (40).Panda N; Mothkuri R J. Org. Chem 2012, 77, 9407–9412. [DOI] [PubMed] [Google Scholar]
- (41).(a) Liu R; Zhang J Chem. Eur. J 2009, 15, 9303–9306; [DOI] [PubMed] [Google Scholar]; (b) Wu J; Yoshikai N Angew. Chem. Int. Ed 2015, 54, 11107–11111. [DOI] [PubMed] [Google Scholar]
- (42).Kohn BL; Jarvo ER Org. Lett 2011, 13, 4858–4861. [DOI] [PubMed] [Google Scholar]
- (43).Dickstein JS; Mulrooney CA; O’Brien EM; Morgan BJ; Kozlowski MC Org. Lett 2007, 9, 2441–2444. [DOI] [PubMed] [Google Scholar]
- (44).(a) Xue L; Su W; Lin Z Dalton Trans. 2010, 39, 9815–9822; [DOI] [PubMed] [Google Scholar]; (b) Dickstein JS; Curto JM; Gutierrez O; Mulrooney CA; Kozlowski MC J. Org. Chem 2013, 78, 4744–4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).For a good review on the use of carboxylic acids as traceless directing groups, see:Font M; Quibell JM; Perry GJP; Larrosa I Chem. Commun 2017, 53, 5584–5597.
- (46).For recent reviews on directed C-H activation with palladium, see:Lyons TW; Sanford MS Chem Rev. 2010, 110, 1147–1169;Davies HML; Du Bois J; Yu J-Q Chem. Soc. Rev 2011, 40, 1855–1856;Zhang M; Zhang Y; Jie X; Zhao H; Li G; Su W Org. Chem. Front 2014, 1, 843–895;Shi G; Zhang Y Adv. Synth. Catal 2014, 356, 1419–1442;Rit RK; Yadav MR; Ghosh K; Sahoo AK Tetrahedron 2015, 71, 4450–4459;Mehta VP; Garcia-Lopez J-A Chem. Cat. Chem 2017, 7, 1149–1156;He J; Wasa M; Chan KSL; Shao Q; Yu J-Q Chem. Rev 2017, 117, 8754–8786.
- (47).(a) Topczewski JT; Cabrera PJ; Saper NI; Sanford MS Nature 2016, 531, 220–224; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang F-L; Hong K; Li T-J; Park H; Yu J-Q Science 2016, 351, 252–256; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen G; Gong W; Zhuang Z; Andra MS; Chen Y-Q; Hong X; Yang Y-F; Liu T; Houk KN; Yu J-Q Science 2016, 353, 1023–1027; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liu Y; Ge H Nat. Chem 2017, 9, 26–32; [Google Scholar]; (e) Davis HJ; Phipps RJ Chem. Sci 2017, 8, 864–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Catellani M; Frignani F; Rangoni A Angew. Chem. Int. Ed 1997, 36, 119–122. [Google Scholar]
- (49).For recent reviews, see:Della Ca’ N; Fontana M; Motti E; Catellani M Acc. Chem. Res 2016, 49, 1389–1400.
- (50).Wang X-C; Gong W; Fang L-Z; Zhu R-Y; Li S; Engle KM; Yu J-Q Nature 2015, 519, 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Dong Z; Wang J; Dong G J. Am. Chem. Soc 2015, 137, 5887–5890. [DOI] [PubMed] [Google Scholar]
- (52).Yamamura K J. Org. Chem 1978, 43, 724–727. [Google Scholar]
- (53).(a) Cacchi S; La Torre F; Misiti D Tet. Lett 1979, 20, 4591–4594; [Google Scholar]; (b) Lu X; Lin S J. Org. Chem 2005, 70, 9651–9653. [DOI] [PubMed] [Google Scholar]
- (54).(a) Wang Z; Zhang Z; Lu X Organometallics 2000, 19, 775–780; [Google Scholar]; (b) Zhao L; Lu X Org. Lett 2002, 4, 3903–3906. [DOI] [PubMed] [Google Scholar]
- (55).Gini F; Hessen B; Minnaard AJ Org. Lett 2005, 7, 5309–5312. [DOI] [PubMed] [Google Scholar]
- (56).Zhang T; Shi M Chem. Eur. J 2008, 14, 3759–3764. [DOI] [PubMed] [Google Scholar]
- (57).Wong J; Gan K; Chen HJ; Pullarkat SA Adv. Synth Catal 2014, 356, 3391–3400. [Google Scholar]
- (58).(a) Kikushima K; Holder JC; Gatti M; Stoltz BM J. Am. Chem. Soc 2011, 133, 6902–6905; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Holder JC; Goodman ED; Kikushima K; Gatti M; Marziale AN; Stoltz BM Tetrahedron 2015, 71, 5781–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Gottumukkala AL; Matcha K; Lutz M; de Vries JG; Minnaard AJ Chem. Eur. J 2012, 18, 6907–6914. [DOI] [PubMed] [Google Scholar]
- (60).(a) Nishikata T; Yamamoto Y; Miyaura N Angew. Chem. Int. Ed 2003, 42, 2768–2770; [DOI] [PubMed] [Google Scholar]; (b) Nishikata T; Yamamoto Y; Gridnev ID; Miyaura N Organometallics 2005, 24, 5025–5032; [Google Scholar]; (c) Nishikata T; Yamamoto Y; Miyaura N Adv. Synth. Catal 2007, 349, 1759–1764. [Google Scholar]
- (61).Ibrahem I; Ma G; Afewerki S; Córdova A Angew. Chem. Int. Ed 2013, 52, 878–882. [DOI] [PubMed] [Google Scholar]
- (62).Zhi W; Li J; Zou D; Wu Y; Wu Y J. Org. Chem 2017, 82, 12286–12293. [DOI] [PubMed] [Google Scholar]
- (63).(a) He Q; Xie F; Fu G; Quan M; Shen C; Yang G; Gridnev ID; Zhang W Org. Lett 2015, 17, 2250–2253; [DOI] [PubMed] [Google Scholar]; (b) Chen S; Wu L; Shao O; Yang G; Zhang W Chem. Commun 2018, 54, 2522–2525. [DOI] [PubMed] [Google Scholar]
- (64).(a) Hosokawa T; Shinohara T; Ooka Y; Murahashi S-I Chem. Lett 1989, 2001–2004; [Google Scholar]; (b) Gaunt MJ; Spencer JB Org. Lett 2001, 3, 25–28. [DOI] [PubMed] [Google Scholar]
- (65).Elliott LD; Wrigglesworth JW; Cox B; Lloyd-Jones GC; Booker-Milburn KI Org. Lett 2011, 13, 728–731. [DOI] [PubMed] [Google Scholar]
- (66).Panda N; Yadav SA Tetrahedron 2018, 74, 1497–1504. [Google Scholar]
- (67).(a) Pei T; Widenhoefer RA J. Am. Chem. Soc 2001, 123, 11290–11291; [DOI] [PubMed] [Google Scholar]; (b) Qian H; Widenhoefer RA J. Am. Chem. Soc 2003, 125, 2056–2057. [DOI] [PubMed] [Google Scholar]
- (68).(a) Wang Z; Lu L J. Org. Chem 1996, 61, 2254–3355; [Google Scholar]; (b) Quan LG; Gevorgyan V; Yamamoto Y J. Am. Chem. Soc 1999, 121, 3545–3546; [Google Scholar]; (c) Lei A; Lu X Org. Lett 2000, 2, 2699–2702; [DOI] [PubMed] [Google Scholar]; (d) Liu G; Lu X Org. Lett 2001, 3, 3879–3882; [DOI] [PubMed] [Google Scholar]; (e) Shen Z; Lu X Tetrahedron 2006, 62, 10896–10899; [Google Scholar]; (f) Tello-Aburto R; Harned AM Org. Lett 2009, 11, 3998–4000. [DOI] [PubMed] [Google Scholar]
- (69).Yu S-Y; Zhang H; Gao Y; Mo L; Wang S; Yao Z-Y J. Am. Chem. Soc 2013, 135, 11402–11407. [DOI] [PubMed] [Google Scholar]
- (70).Tsuji J; Nisar M; Shimizu I J. Org. Chem 1985, 50, 3416–3417. [Google Scholar]
- (71).Héin F; Muzart J Tetrahedron: Asymm 1992, 3, 1161–1164. [Google Scholar]
- (72).(a) Mohr JT; Nishimata T; Behenna DC; Stoltz BM J. Am. Chem. Soc 2006, 128, 11348–11349; [DOI] [PubMed] [Google Scholar]; (b) Marinescu SC; Nishimata T; Mohr JT; Stoltz BM Org. Lett 2008, 10, 1039–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).(a) Doran R; Guiry PJ J. Org. Chem 2014, 79, 9112–9124; [DOI] [PubMed] [Google Scholar]; (b) Kingston C; Guiry PJ J. Org. Chem 2017, 82, 3806–3819. [DOI] [PubMed] [Google Scholar]
- (74).Tamaru Y; Yamada Y; Kagotani M; Ochiai H; Nakajo E; Suzuki R; Yoshida Z J. Org. Chem 1983, 48, 4669–4681. [Google Scholar]
- (75).Hegedus LS Angew. Chem., Int. Ed 1988, 27, 1113–1116. [Google Scholar]
- (76).Larock RC; Babu S Tet. Lett 1987, 28, 5291–5294. [Google Scholar]
- (77).(a) Smith KM; Langry KC; Minnetian OM J. Org. Chem 1984, 49, 4602–4609; [Google Scholar]; (b) Arcadi A; Marinelli F; Bernocchi E; Cacchi S; Ortar G J. Organomet. Chem 1989, 368, 249–256; [Google Scholar]; (c) Larock RC; Johnson PL J. Chem. Soc., Chem. Commun 1989, 1368–1370. [Google Scholar]
- (78).Reddy ACS; Choutipalli VSK; Ghorai J; Subramanian V; Anbarasan P ACS Catal. 2017, 7, 6283–6288. [Google Scholar]
- (79).(a) Michael FE; Cochran BM J. Am. Chem. Soc 2006, 128, 4246–4247; [DOI] [PubMed] [Google Scholar]; (b) Cochran BM; Michael FE J. Am. Chem. Soc 2008, 130, 2786–2792. [DOI] [PubMed] [Google Scholar]
- (80).(a) Gurak JA Jr.; Yang KS; Liu Z; Engle KM J. Am. Chem. Soc 2016, 138, 5805–5808; [DOI] [PubMed] [Google Scholar]; (b) Gurak JA Jr.; Tran VT; Sroda MM; Engle KM Tetrahedron 2017, 73, 3636–3642; [Google Scholar]; (c) Gurak JA Jr.; Engle KM Synlett 2017, 28, 2057–2065. [Google Scholar]
- (81).Shabashov D; Daugulis O J. Am. Chem. Soc 2010, 132, 3965–3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).(a) Yang K; Gurak JA Jr.; Liu Z; Engle KM J. Am. Chem. Soc 2016, 138, 14705–14712, [DOI] [PubMed] [Google Scholar]; (b) Matsuura R; Jankins T; Hill D; Yang K, Gallego G; Yang S; He M; Wang F; Marsters R; McAlpine I; Engle K 2018, manuscript submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Wang H; Bai Z; Jiao T; Deng Z; Tong H; He G; Peng Q; Chen G J. Am. Chem. Soc 2018, 140, 3542–3546. [DOI] [PubMed] [Google Scholar]
- (84).Tran VT; Gurak JA Jr.; Yang KS; Engle KM 2018, manuscript submitted. [Google Scholar]
- (85).(a) Reddy BVS; Reddy LR; Corey E J. Org. Lett 2006, 8, 3391–3394; [DOI] [PubMed] [Google Scholar]; (b) Ano Y; Tobisu M; Chatani N J. Am. Chem. Soc 2011, 133, 12984–12986; [DOI] [PubMed] [Google Scholar]; (c) He G; Zhao Y; Zhang S; Lu C; Chen G J. Am. Chem. Soc 2012, 134, 3–6; [DOI] [PubMed] [Google Scholar]; (d) Nadres ET; Daugulis O J. Am. Chem. Soc 2012, 134, 7–10; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhang S-Y; He G; Nack WA; Zhao Y; Li Q; Chen G J. Am. Chem. Soc 2013, 135, 2124–2127; [DOI] [PubMed] [Google Scholar]; (f) He G; Zhang S-Y; Nack WA; Li Q; Chen G Angew. Chem. Int. Ed 2013, 52, 11124–11128; [DOI] [PubMed] [Google Scholar]; (g) Cui W; Chen S; Wu J-Q; Zhao X; Hu W; Wang H Org. Lett 2014, 16, 4288–4291; [DOI] [PubMed] [Google Scholar]; (h) He G; Zhang S-Y; Nack WA; Pearson R; Rabb-Lynch J; Chen G Org. Lett 2014, 16, 6488–6491; [DOI] [PubMed] [Google Scholar]; (i) Li S; Zhu R-Y; Xiao K-J; Yu J-Q Angew. Chem. Int. Ed 2016, 55, 4317–4321; [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Xu J-W; Zhang Z-Z; Rao W-H; Shi B-F J. Am. Chem. Soc 2016, 138, 10750–10753. [DOI] [PubMed] [Google Scholar]
- (86).O’Duill ML; Matsuura R; Wang Y; Turnbull JL; Gurak JA Jr.; Gao D-W; Lu G; Liu P; Engle KM J. Am. Chem. Soc 2017, 139, 15576–15579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Fryzuk MD; Bosnich B J. Am. Chem. Soc 1979, 101, 3043–3049. [Google Scholar]
- (88).The crystal structure was not published, but alluded to in personal communications between the authors and N. C. Payne (see ref. 78).
- (89).Lan Y; Houk KN J. Org. Chem 2011, 76, 4905–4909. [DOI] [PubMed] [Google Scholar]
- (90).Peng Q; Yan H; Zhang X; Wu Y-D J. Org. Chem 2012, 77, 7487–7496 [DOI] [PubMed] [Google Scholar]