

Abstract
Limited proteolysis of Gasdermin D (GSDMD) generates an N-terminal pore-forming fragment that controls pyroptosis in macrophages. GSDMD is processed via inflammasome-activated caspase-1 or −11. It is currently unknown if macrophage GSDMD can be processed by other mechanisms. Here we describe an additional pathway controlling GSDMD processing. The inhibition of TGF-β activated kinase-1 (TAK1) or IKK kinases by the Yersinia effector protein YopJ elicits RIPK1-and caspase-8-dependent cleavage of GSDMD which subsequently results in cell death. GSDMD processing also contributes to the NLRP3 inflammasome-dependent release of IL-1β. Thus, caspase-8 acts as a regulator of GSDMD-driven cell death, and our study establishes the importance of TAK1 and IKK activity in the control of GSDMD cleavage and cytotoxicity.
One-Sentence Summary:
TAK1 blockade by Yersinia bacteria unleashes caspase-8 dependent cleavage of GSDMD, cell death and IL-1β release.
The robust and rapid induction of innate immune signaling is a hallmark of the host response to microbial infection. Successful pathogens subvert, thwart, or dismantle these defensive measures. There is growing evidence that the host recognizes these disruptive efforts eliciting effective backup measures. Cell death processes including apoptosis and pyroptosis are integral components of the host response to infection. Multiprotein inflammasome complexes sense the presence of pathogens and activate inflammatory caspases, typically caspase-1 or caspase-11, leading to pyroptotic cell death and maturation of the inflammatory cytokines IL-1β and IL-18. Pyroptosis is an inflammasome-driven cytotoxic process, which occurs in macrophages following limited proteolysis of Gasdermin D (GSDMD). The generation of an N-terminal cleaved fragment then creates large oligomeric membrane pores and causes lytic cell death (1–7). At present, caspase-1 and caspase-11 are the only known regulators of GSDMD in macrophages (5, 7), although neutrophil elastase controls GSDMD cleavage in neutrophils (8).
Caspase-8 is an upstream activator of caspase-3 and controls apoptotic cell death. In addition, Caspase-8 prevents RIPK3–MLKL dependent necroptosis. Increasing evidence indicates important roles for caspase-8 in inflammatory responses in macrophages infected with diverse pathogens. Caspase-8 activation can trigger NLRP3 inflammasomes (9) and may also serve as a back-up measure when the caspase-1 pathway is blocked or deleted (10, 11). Pathogenic bacteria of the genus Yersinia include Y. pseudotuberculosis and Y. enterocolitica, which cause gastrointestinal disease, and Y. pestis, the etiologic agent of plague. Yersinia bacteria, through the action of their type III secretion systems, release effectors that manipulate host cells. One of these, YopJ, is a strong activator of caspase-8 via RIPK1 (12, 13). YopJ is an acetyl transferase that blocks phosphorylation and activation of kinases TAK1, IKKβ, and MAPKK (14–17). The inhibition of TAK1 is associated with cell death and inflammation (18–20), and is not unique to Yersinia. Several additional pathogens including Pseudomonas, Vibrio and enteroviruses also target TAK1 (21–23).
Yersinia bacteria induce cell death, caspase-1 cleavage and IL-1β release, whereas mutants lacking YopJ do not (12, 13) (Fig. S1A). By comparing these responses in wild-type macrophages to those lacking RIPK3 or RIPK3–Caspase-8, we previously found that caspase-8 is important for all of these effects (12). To investigate the pathways involved, we examined the inhibition of TAK1 kinase activity with 5z-7-oxozeaneol (5z7, TAK1-i), a specific small-molecule inhibitor, as genetic deletion of TAK1 in macrophages leads to spontaneous death (20). TAK1-i induced cell death and IL-1β release in LPS-stimulated macrophages (Fig. 1A–C, Fig. S1B–S1D). Similar findings were made with a second inhibitor of TAK1 (Fig. S1C). TAK1-i also restored caspase-8-dependent death and IL-1β release in cells infected with YopJ-mutant Yersinia (Fig. 1A, B). We found that TNF together with TAK1-i could induce similar responses (Fig. 1D, E). RIPK1 can control caspase-8 activation, and is necessary for cell death induced by Yersinia and TAK1 inhibition (12) (Fig. S1E–I). One function of TAK1 is to activate inhibitor of ĸB kinase (IKK), which also controls RIPK1 activity, hence the blockade of IKK could effectively trigger a similar pathway as TAK1 inhibition (Fig. 1F, Fig. S1I). Thus, TAK1 effects on IKK are likely key early events in this pathway, rather than effects on MAPKK and MAPK such as p38 (Fig S1J). These responses may serve as a host mechanism to detect the pathological disturbance of TAK1 kinase activity.
Fig. 1. Inhibition of TAK1 by Yersinia YopJ or small molecules triggers cell death, inflammasome activation and GSDMD cleavage.

BMDMs from C57BL/6, Ripk3−/−, and Ripk3−/−Caspase8−/− mice were challenged with indicated stimuli in the presence or absence of TAK1 inhibitor 5z7 (TAK1-i) or IKK inhibitor (IKK-i) (A, B, D-F). Cell death was measured by LDH release after 4 h (A, D and F) and IL-1β release by ELISA after 5h (B and E). (C) C57BL/6 BMDMs were challenged with indicated stimuli for 5 h, and cell lysates plus supernatants were analyzed by immunoblot for caspase-1 and IL-1β cleavage. (G-J) Oligomerization of ASC in the inflammasome-enriched and cross-linked lysates was detected in C57BL/6 (G, J) or the indicated KO BMDMs infected or stimulated for 3 hrs with LPS + TAK1- i, with or without 1 h pretreatment with inhibitors of pan-caspase (zVAD-fmk, 25 μM), RIP1 (1 μM) or RIP3 (3 μM). (K) C57BL/6 BMDMs were treated with indicated stimuli for 3 h and cell lysates were analyzed by immunoblot for GSDMD cleavage. Illustration of GSDMD, predicted cleavage sites and fragment sizes are indicated. Data are presented as mean ± SD of triplicate wells from three or more independent experiments. n.d.: not detected. For comparisons we used t-test. *p<0.05, **p<0.01, ***p<0.001.
TAK1 inhibition by Yersinia or by TAK1-i was associated with ASC oligomerization (Fig. 1G, H). This oligomerization was dependent on RIPK1, caspase-8 and NLRP3, but not caspase-1/11 (Fig. 1H–J, Fig. S1K). Notably, pan-caspase-inhibition blocked ASC oligomerization and IL-1β release, but not cytotoxicity. This was likely due to RIPK3-dependent necroptosis triggered by caspase-8 inhibition (Fig 1J, Fig S1L–N). Macrophage death was independent of the NLRP3 inflammasome and IL-1β release was partially dependent on NLRP3 (24) (Fig. S2A–D). Loss of TLR4, TRIF or TNFR1 showed a significant reduction in cell death after bacterial infection or LPS+TAK1-i (Fig. S3A–C). This suggests activation of the RIPK1-caspase-8 death pathway downstream of TLR4–TRIF activation, perhaps with other contributing bacterial ligands, with concurrent TNF production and TNFR1 autocrine signaling (25). Indeed, TAK1-i combined with additional stimulations, such as TLR2 and TLR7/8 ligands could also trigger TNFR1, MyD88 and caspase-8 dependent cell death and IL-1β release at later time points (Fig. S3D–H). Cell death induced by Yersinia has been proposed to be caspase-8–dependent apoptosis (12). Although caspase-3 and caspase-7 are activated by Yersinia (12, 13) (Fig. S4A), deficiency of these caspases did not impact cell death (13) (Fig. S4B, Table S1), and the combined action of multiple components may be necessary. Many Yersinia-infected cells display features of apoptosis including membrane blebs, nuclear condensation, and DNA laddering (12). However, we observed significant cleavage of GSDMD after infection, indicating the simultaneous presence of pyroptotic processes (Fig. 1K). The detection antibody we utilized recognizes the N-terminal 30-kDa pore-forming pyroptotic fragment of GSDMD. Yersinia also induced a smaller 20-kDa fragment and a larger p45 fragment, suggesting further cleavage of full-length protein (26) or p30 by caspase-3 at GSDMD D88 (Fig. S4C). Importantly, we found that GSDMD was a central mediator of both cell death and release of IL-1β and IL-18 induced by Yersinia infection and LPS or TNF combined with TAK1-i/IKK inhibition, events controlled by RIPK1–caspase-8 (Fig. 2A–G, Fig. S1E–I, Fig. S4D, E). GSDMD also influenced YopJ/TAK1-i induced caspase-1 p20 cleavage and ASC oligomerization (Fig. 2D, H, I. Table S2). Excess extracellular potassium completely blocked ASC oligomerization and partially inhibited IL-1β release, but did not affect cell death (Fig. 2J, K, L, Table S2). Thus, RIPK1–caspase-8 appears to exert full control of membrane damage that triggers potassium efflux. GSDMD displays partial control—possibly via GSDMD pore formation—over potassium efflux, and these signals direct NLRP3 activation, ASC oligomerization and ultimately IL-1β release. However, NLRP3 and caspase-1/11 do not contribute in a major way to cell death in this case (Fig. S2A, B).
Fig. 2. GSDMD regulates cell death and inflammasome activation in response to TAK1 inhibition.

(A-G) The indicated BMDMs were challenged with Y. pestis or a mutant lacking YopJ, or S. Typhimurium, or stimulated with indicated ligands, TAK1-i, or ligands + TAK1-i. Cell death was measured by LDH release after 3 h (A and B), and IL-1β release after 5 h (C, E-G), or caspase-1 cleavage after 5h (D). (H-J) Oligomerization of ASC in the inflammasome-enriched and cross-linked lysates was detected in BMDMs after Y. pestis or LPS + TAK1-I challenge after the indicated time points (I) or 3 h. (J-L) BMDMs treated with Y. pestis, S. Typhimurium, or nigericin + LPS in the presence or absence of 50 mM (J-L) or 25 mM (K) KCl, and oligomerization of ASC was detected by immunoblot, cell death was measured by LDH release, or IL-1β by ELISA. Data are presented as mean ± SD of triplicate wells from three or more independent experiments. n.d.: not detected. For comparisons between two groups we used t-test, for more than two groups we used ANOVA. *p<0.05, **p<0.01, ***p<0.001.
Cleavage of GSDMD has previously been associated with caspase-1 and caspase-11–activity (2, 4, 5, 7). In contrast, we found that Yersinia and TAK1-i induced GSDMD p30 was entirely dependent upon RIPK1–caspase-8 with minor contributions from caspase-1/11 and RIPK3 (Fig 3A–E, Fig S5A–D). We also observed cleavage of caspase-8 and GSDMD before the appearance of any detectable cleaved caspase-1 (Fig. 3F, Fig S5E). Thus we have identified an additional pathway leading to processing of GSDMD. In addition, immunoprecipitated cleaved caspase-8 (p18) from TAK1-i/LPS treated GSDMD-deficient cells was capable of cleaving purified mouse GSDMD (Fig. 3G, Fig. S5F–H) and generate the p30 pore-forming fragment. Recombinant caspase-8 may not process GSDMD as strongly as caspase-1 (5, 26), however, increased concentrations of mouse and human caspase-8 cleaved GSDMD (Fig 3H, Fig S5I). In addition, a mixture of both caspase-8 and caspase-3 reproduced the cleavage pattern induced by Yersinia or TAK1/IKK inhibition (Fig 3H). Loss of GSDMD had no effect on caspase-8 activation, consistent with the hypothesis that GSDMD acts downstream of caspase-8 (Fig. 3I and Fig. S5J). We observed YopJ-dependent caspase-8 cleavage, GSDMD processing, and cell death with Y. pseudotuberculosis, Y. enterocolitica and Y. pestis, suggesting that the triggering of this pathway is conserved among these Yersinia species (Fig. 3J, Fig. S1A, Fig. S5J). Salmonella, normally a strong activator of caspase-1 and GSDMD, induces processing of caspase-8 when GSDMD is absent (Fig. 3I), as others have suggested (10, 11). We note that Yersinia has several effectors inhibiting caspase-1 cleavage (27, 28). This could contribute to the activation of caspase-8 upon infection by this pathogen. Treatment of macrophages from a GSDMD D276A knock-in mouse (29), containing a mutation in the caspase-1/11 cleavage motif of GSDMD 273LLSD276, with an LPS–TAK1-i combination, revealed that the caspase-8-directed cleavage of GSDMD depended upon the GSDMD D276 cleavage site (Fig. 4A–C). These observations expand upon the requirement of caspase-1/11 to proteolytically process GSDMD at D276 for cytotoxicity (5, 7). Cell death time-course experiments showed that the absence of GSDMD, but not of caspase-8, could be overcome by prolonged incubations with bacteria or TAK1-i/LPS (Fig. 4C–G, Fig. S6A–H). Thus, GSDMD is not the only mediator of cell death downstream of caspase-8 in the TAK1–IKK regulated pathway. Additional as yet to be defined components also participate in this process.
Fig. 3. Caspase-8 dependent GSDMD processing with little contribution by caspase-1 and -11.
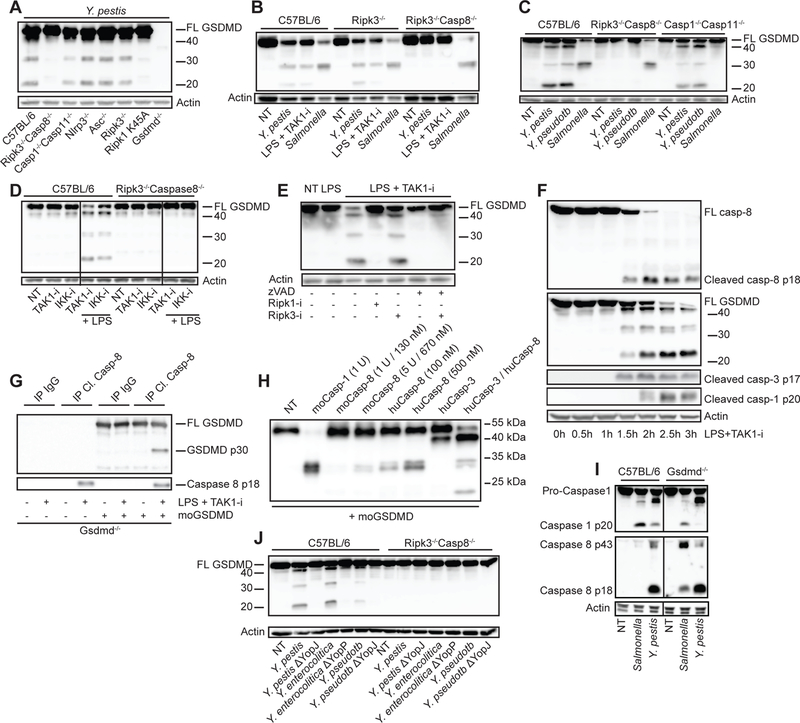
BMDMs from the indicated mouse strains were challenged with the bacteria Y. pestis, Y. pseudotuberculosis, Y. enterocolitica, their YopJ (YopP in Y.e.) mutants, or S. Typhimurium, or treated with LPS + TAK1-i. Cell lysates were harvested after 3–4 h unless otherwise stated and analyzed by immunoblots for GSDMD cleavage (A-F, and J) or caspase-1, caspase-3 and caspase-8 cleavage (F, I). (D) BMDMs from C57BL/6 or Ripk3−/−Casp8−/− mice were treated with IKK-i and TAK1-i in the absence or presence of LPS. (E) BMDMs were pretreated with pan-caspase inhibitor zVAD-fmk, RIPK1-i, or RIPK3-i for 1 h before being treated with LPS+TAK1-i. (G) Gsdmd−/− BMDMs were treated with LPS+TAK1-i for 3 h and then immunoprecipitated with cleaved caspase-8 antibody. Immunoprecipitates (G) or recombinant caspases (H) were incubated with recombinant mouse GSDMD at 37°C for 1 h in a protein cleavage buffer, before being analyzed by immunoblots. Figures are representative of three or more independent experiments.
Fig. 4. The GSDMD Asp276 residue is critical for GSDMD processing and for regulation of cell death downstream of TAK1 inhibition.
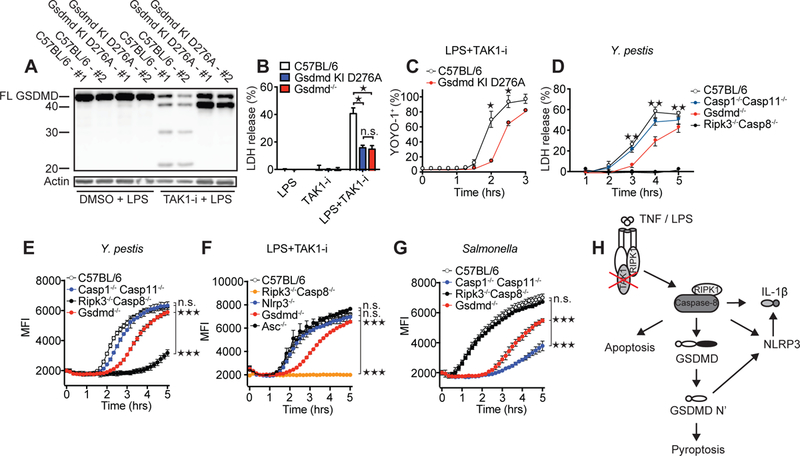
(A) Immunoblot of GSDMD cleavage in C57BL/6 or Gsdmd D276A mutant BMDMs after treatment with LPS and LPS + TAK1-i. Cell death as measured by LDH release (B and D) or by % YOYO-1+ cells (C) after challenge of indicated genotypes with LPS + TAK1-i or Y. pestis. (E-G) Cell death as measured by entry of EthD-1 into the cell. Samples were read every 10 min at 530 nm excitation, 645 nm emission wavelength. BMDMs from the indicated genotypes were treated with Y. pestis (E), LPS + TAK1-i (F), or S. Typhimurium (G). Infections in E, G are without gentamicin. For (E-G) statistics were performed on the area under the curve (AUC) for the whole time course. (H) Proposed model of TAK1-inhibition-induced caspase-8 activation and cell death. Data are presented as mean ± SD of triplicate wells from two (A-C) or three or more (D-G) independent experiments. n.d. not detected. For comparisons between two groups we used t-test, for more than two groups we used ANOVA. *p<0.05, **p<0.01, ***p<0.001.
The ability of cells to mobilize countermeasures to detect pathogenic inhibition of key signaling pathways is becoming increasingly apparent. One recent example is the Pyrin inflammasome pathway, where disturbances in RhoA GTPase by bacterial toxins trigger Pyrin-driven caspase-1 activation (27, 28, 30). Based on the studies herein, we propose that pathogen blockade of TAK1–IKK triggers a caspase-8 controlled cell death and inflammatory pathway involving GSDMD. GSDMD-mediated cell death has been considered a defining feature of pyroptosis (4, 5, 31). Our observations suggest that blockade of TAK1–IKK pathways leads to cytotoxicity with features of both apoptosis and pyroptosis (Fig 4H, Fig S7). Thus, conditions that impact many cellular processes, such as when bacteria and their secretion system components concurrently impact different signaling proteins (27, 28), can lead to cell death involving multiple pathways. Inhibition of TAK1 represents a strategy that several pathogens may use to their advantage. However, in the arms race between pathogens and their hosts, the host can sense these disturbances as pathogenic and counter these efforts with cytotoxicity and inflammation. Our study broadens the understanding of pathways leading to GSDMD activation and underscores the importance of GSDMD as a key driver of cell death and inflammation induced by many different pathogens.
Supplementary Material
Acknowledgments:
We thank G. Germain for help with mice; V. Dixit, N. Kayagaki and K. Newton (Genentech, Inc.) for providing GSDMD and RIP3 KO mice; W. Kaiser, E. Mocarski, D. R. Green and C. Dillon for sending caspase-8 RIP3 dKO mice; M. O’Riordan for providing S. Typhimurium; I. Brodsky and J. Bliska for providing Y. pseudotuberculosis; R. Adkins and G.Cornelis for providing Y. enterocolitica; G. Salvesen and S. Snipas for providing recombinant caspase-8 and caspase-3; GlaxoSmithKline and J. Bertin for providing RIPK1 and RIPK3 inhibitors, as well as RIPK1 K45A mutant mice. We also thank A. Poltorak lab for discussions and sharing data from their unpublished studies, and N. Silverman and T. Espevik for critically reading the manuscript.
Funding: The work was supported by National Institutes of Health Grants AI07538 and AI129527 (to EL), AI067497 and AI083713 (to KAF), AI095213 (to DR), AI139914 (to HW), AI075118 (to MAK), HL092610 (to AB), the Norwegian Cancer Society grants 7699133 (to KS) and B05035/001 (to EL), the UMass Medical School Summer Undergraduate Research Experience Program funding to ZB, and the Research Council of Norway Center of Excellence Funding Scheme, Project 223255/F50 (to PO, EL, KAF and KS).
Footnotes
Competing interests: BL and NK are employees of Genentech. SBB, PJG and JB are employees and shareholders of GlaxoSmithKline.
Data and materials availability: All data needed to evaluate the conclusions in this paper are present either in the main text or the Supplementary Materials. Materials described in the paper are available upon completion and approval of MTAs with the respective parties.
References and Notes:
- 1.Sborgi L et al. , GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J 35, 1766–1778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu X et al. , Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kovacs SB, Miao EA, Gasdermins: Effectors of Pyroptosis. Trends Cell Biol 27, 673–684 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi J, Gao W, Shao F, Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci 42 (2017), pp. 245–254. [DOI] [PubMed] [Google Scholar]
- 5.Shi J et al. , Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Aglietti RA et al. , GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A 113, 7858–7863 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kayagaki N et al. , Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Kambara H et al. , Gasdermin D Exerts Anti-inflammatory Effects by Promoting Neutrophil Death. Cell Rep 22, 2809–2817 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vince JE, Silke J, The intersection of cell death and inflammasome activation. Cell. Mol. Life Sci 73, 2349–2367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rauch I et al. , NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and −8 Article NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Releas. Immunity 46, 649–659 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mascarenhas DPA et al. , Inhibition of caspase-1 or gasdermin-D enable caspase-8 activation in the Naip5 / NLRC4 / ASC inflammasome. PLoS Pathog 13, 1–28 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weng D et al. , Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc. Natl. Acad. Sci. U. S. A 111, 7391–7396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Philip NH et al. , Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-κB and MAPK signaling. Proc. Natl. Acad. Sci. U. S. A 111, 7385–7390 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meinzer U et al. , Yersinia pseudotuberculosis effector YopJ subverts the Nod2/RICK/TAK1 pathway and activates caspase-1 to induce intestinal barrier dysfunction. Cell Host Microbe 11, 337–351 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Mittal R, Peak-Chew S-Y, McMahon HT, Acetylation of MEK2 and I kappa B kinase (IKK) activation loop residues by YopJ inhibits signaling. Proc. Natl. Acad. Sci. U. S. A 103, 18574–18579 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mukherjee S et al. , Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312, 1211–1214 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Paquette N et al. , Serine / threonine acetylation of TGF β -activated kinase ( TAK1 ) by Yersinia pestis YopJ inhibits innate immune signaling. Proc. Natl. Acad. Sci 109, 12710–12715 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanlangenakker N et al. , cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ 18, 656–665 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menon MB et al. , p38 MAPK / MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat. Cell Biol 19, 1248–1259 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Malireddi RKS et al. , TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J. Exp. Med 215, 1023–1034 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lei X et al. , Enterovirus 71 3C Inhibits Cytokine Expression through Cleavage of the TAK1/TAB1/TAB2/TAB3 Complex. J. Virol 88, 9830–9841 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou X et al. , A Vibrio parahaemolyticus T3SS Effector Mediates Pathogenesis by Independently Enabling Intestinal Colonization and Inhibiting TAK1 Activation. Cell Rep 3, 1690–1702 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He C et al. , Bacterial nucleotidyl cyclase inhibits the host innate immune response by suppressing TAK1 activation. Infect. Immun 85 (2017), doi: 10.1128/IAI.00239-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng Y et al. , A yersinia effector with enhanced inhibitory activity on the NF-kappaB pathway activates the NLRP3/ASC/Caspase-1 inflammasome in macrophages. PLoS Pathog 7, 1–14 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peterson LW et al. , Cell-Extrinsic TNF Collaborates with TRIF Signaling To Promote Yersinia -Induced Apoptosis. J Immunol 197, 4110–4117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taabazuing CY, Okondo MC, Bachovchin DA, Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem. Biol 24, 507–514 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ratner D, Orning MPA, Lien E, Bacterial secretion systems and regulation of inflammasome activation. J. Leukoc. Biol 101, 165–181 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ratner D et al. , The Yersinia pestis Effector YopM Inhibits Pyrin Inflammasome Activation. PLOS Pathog 12, e1006035 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee BL et al. , Caspase-11 auto-proteolysis is crucial for non-canonical inflammasome activation. J. Exp. Med 215, 2279–2288 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu H et al. , Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513, 237–41 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Galluzzi L, Vitale I, Molecular mechanisms of cell death : recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25, 486–541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.