

Abstract
Purpose of review
To describe a recently characterized autoimmune, inflammatory central nervous system (CNS) disorder known as autoimmune glial fibrillary acidic protein (GFAP) astrocytopathy.
Recent findings
Affected patients present with symptoms of one or more of meningitis (headache and neck ache), encephalitis (delirium, tremor, seizures, or psychiatric symptoms), and myelitis (sensory symptoms and weakness). Optic disc papillitis (blurred vision) is common. CNS inflammation is evident in characteristic T1 postgadolinium enhancement of GFAP-enriched CNS regions, and lymphocytic cerebrospinal fluid (CSF) white cell count elevation. CSF is more reliable than serum for GFAP-immunoglobulin G (IgG) testing. Ovarian teratoma commonly coexists, particularly among patients with accompanying N-methyl-D-aspartate receptor or aquaporin-4 autoimmunity. Parainfectious autoimmunity is suspected in some other patients, though the culprit organism is rarely verified. Pathophysiologic relevance of T cells is underscored by neuropathology and cases of dysregulated T-cell function (HIV or checkpoint inhibitor cancer therapy). Corticosteroid-responsiveness is a hallmark of the disease. Relapses occur in approximately 20% of patients, necessitating transition to a steroid-sparing drug. Reported outcomes vary, though in the authors’ experience, early and sustained intervention usually portends recovery.
Summary
Autoimmune GFAP astrocytopathy is a treatable autoimmune CNS disease diagnosable by GFAP-IgG testing in CSF. This disease presents opportunities to explore novel mechanisms of CNS autoimmunity and inflammation.
Keywords: encephalitis, glial fibrillary acidic protein, meningitis, myelitis, steroids
INTRODUCTION
Autoimmune glial fibrillary acidic protein (GFAP) astrocytopathy is an inflammatory central nervous system (CNS) disorder. It may affect any anatomic region, rostrocaudally, from optic nerve to spinal cord, though meningoencephalitis is predominant. The disorder is largely defined by detection and confirmation in CSF of immunoglobulin G (IgG) reactive with GFAP, an intracellular astrocytic intermediate filament [1▪▪]. The disorder is typically corticosteroid-responsive, though a relapsing course necessitating long-term immunosuppression occurs in a sizeable minority. Although a paraneoplastic cause is discernible in some patients (usually related to teratoma), the underlying cause (or causes) remain to be elucidated in the majority. Herein, we describe what is known and what remains to be discovered.
Box 1.
no caption available
DIAGNOSTIC ANTIBODY
Four patients with autoimmune encephalitis occurring in the context of ovarian teratoma were initially encountered as part of the Mayo Clinic Neuroimmunology Laboratory's extramural diagnostic serological practice. CSF from each produced identical novel IgG staining patterns of adult mouse cerebrum, but not cerebellum, in the course of routine screening by indirect immunofluorescence assay (IFA). This IgG staining pattern was filamentous appearing, and predominated in the following regions: subependymal and periventricular midbrain, pial and subpial midbrain and cortical surfaces, white matter, perivascular grey matter, and myenteric plexus (Fig. 1) [1▪▪]. A more classical GFAP staining pattern, involving also cerebellar Bergmann glia (identical to that produced by commercial pan-GFAP or GFAP mature [α] isoform-specific antibodies) is produced by patient GFAP-IgG when rat brain (rather than from mouse) is utilized as substrate [2▪]. Concurrently, a chronic, relapsing steroid-dependent meningoencephalitis was described at Mayo Clinic on a clinicopathological basis [3]. This disorder, usually nonparaneoplastic, was named chronic microglial encephalomyelitis, because of chronic inflammation with an abundance of microglia in biopsied brain tissue. Evaluation of CSF revealed the same immunohistochemical pattern of IgG staining as the teratoma patient group. Ultimately, patients from both groups were determined to harbor GFAP-IgG in CSF [4▪▪].
FIGURE 1.
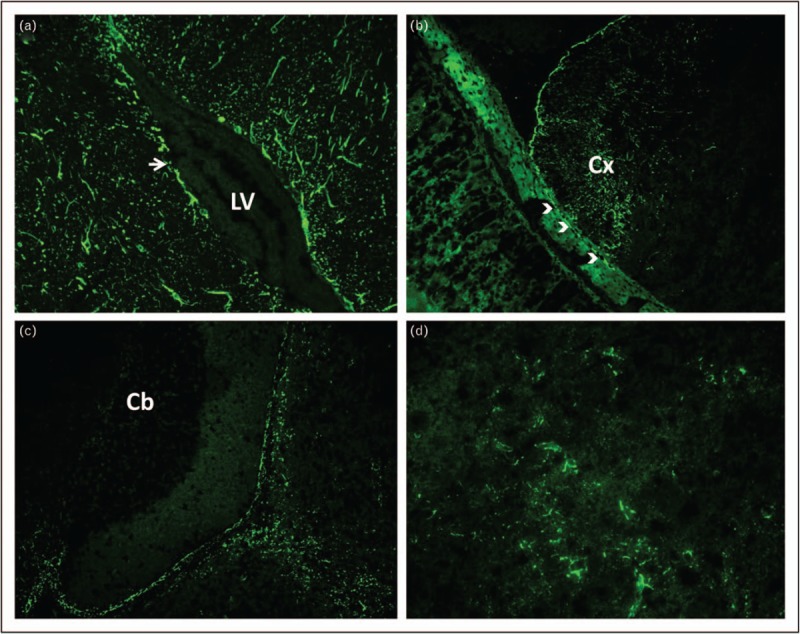
Indirect immunofluorescence assay: staining pattern of glial elements within a composite of adult mouse brain and gastric tissues produced by patient GFAP-IgG in CSF. Patient IgG produces filamentous staining of: (a) brain parenchyma adjacent to the lateral ventricle (LV, 20×), most prominently in the subependymal zone (arrow); (b) pia and subpial region of cerebral cortex (Cx, 20×) and adjacent myenteric plexus glial cells (arrow heads); (c) pial and subpial midbrain, but sparing cerebellum (Cb, 20×). (d) 40× magnification of midbrain demonstrates filamentous staining around small blood vessels. CSF, cerebrospinal fluid; GFAP, glial fibrillary acidic protein; IgG, immunoglobulin G.
Filamentous staining of the sub (lateral) ventricular zone was an early clue to an intermediate filament being the antigen of interest. Other clues included GFAP-IgG as a biomarker of autoimmune encephalitis in pug dogs, and an adoptive GFAP-specific T-cell animal model of CNS inflammation in mice [5,6].
Identification of GFAP as autoantigen was undertaken using Western blot to detect a 50 kDa protein band common to the studied patients, and mass spectrometry of that product [1▪▪]. The IFA pattern was replicated by applying patient IgG eluted from the 50 kDa nitrocellulose band to mouse tissue. Antigen specificity was further confirmed by GFAP-transfected HEK293 cell-based assay. Because the GFAP-IgG IFA staining pattern spared the cerebellum, and several patients developed meningoencephalitis in the context of cancer, an immature GFAP isoform (such as δ/ε or κ) was hypothesized to be most pertinent (GFAP isoforms have identical N-terminal head, and coiled-coil rod domains, but divergent C-terminal tails) [7]. Despite this, all patients had GFAP-IgGs reactive with the mature (α) GFAP isoform, which is predominant in adult astrocytes. Eighty percent had one or both of GFAP δ/ε or κ IgGs detected additionally (and never in isolation) [4▪▪]. The profile of GFAP isoform IgGs was neither predictive of cancer nor neurological phenotype [4▪▪].
Further verification studies determined that the highest specificity for inflammatory CNS disease is assured by a two-step diagnostic process utilizing CSF: GFAP-IgG meeting all diagnostic criteria by IFA (or other tissue-based immunohistochemical technique), Fig. 1; specificity confirmation by GFP-tagged GFAPα cell-based IFA [4▪▪]. Utilizing one assay alone may yield nonspecific results. Some patients have GFAP-IgG detected in serum and CSF, whereas others have it detected in CSF only: almost all have an inflammatory meningoencephalomyelitis phenotype. Others are GFAP-IgG positive in serum only (in which CSF has been tested and is negative), and have diverse neurological phenotypes that may or may not have an autoimmune cause.
PATIENT DEMOGRAPHICS
There are four large clinical series published to date, two from Mayo Clinic (one retrospective, and one follow-up confirmatory prospective 1-year experience), one from Italy, and one from China [2▪,4▪▪,8▪,9▪] Shorter studies from the Chinese group focused on coexisting antibodies, pathology, and treatment [10–12]. Autoimmune GFAP astrocytopathy may have onset at any age (median 44–50 years). Men and women are affected equally, despite female predominance among paraneoplastic teratoma-accompanied cases. Children account for approximately 10% of cases. No racial predilection is known. The prevalence of GFAP astrocytopathy was calculated at 0.6/100 000 in Olmsted County, Minnesota [13].
NEUROLOGICAL PHENOTYPIC FEATURES
In the largest series (102 patients), 94% with GFAP-IgG positivity in CSF had one or more of meningitis, encephalitis, or myelitis. Meningoencephalitis was the predominant phenotype, and isolated myelitis was rare [4▪▪]. Preceding flu-like symptoms were very common (40–66%). The most common clinical features included encephalopathy, seizures (antiepileptic drug resistant), psychiatric symptoms, tremor, and meningeal symptoms (including headache in up to two-thirds), Table 1.
Table 1.
Clinical features of autoimmune glial fibrillary acidic protein meningoencephalomyelitis cases
| Early symptoms (40%) |
| ≥1 rhinorrhea, sore throat, fever, and cough |
| Syndromes |
| Meningoencephalitis (55%) |
| Meningoencephalomyelitis (40%) |
| Myelitis (5%) |
| Meningoencephalopathic symptoms |
| Delirium (60%) |
| Seizures (20%) |
| Psychiatric symptoms (30%, depression, anxiety, psychosis, insomnia, vivid dreams) |
| Headache, neck stiffness, vomiting (60%) |
| Blurred vision (30%, with optic disc edema) |
| Tremor (40%) |
| Myelopathic symptoms |
| Numbness, paresthesias, weakness (25%, signs include brisk reflexes, extensor plantars) |
| Accompaniments of meningoencephalomyelitis |
| Ataxia (40%) |
| Autonomic dysfunction (20%; orthostasis, GI motility disorders, erectile dysfunction, bladder dysfunction) |
| Peripheral neuropathy (<5%) |
GI, gastrointestinal.
Except where aquaporin-4 (AQP4)-IgG-positive-neuromyelitis optica spectrum disorders coexist, the opticospinal manifestations of autoimmune GFAP astrocytopathy are distinct from optic neuritis or myelitis. Rather than plegia from myelitis, patients experience myelopathic symptoms (sensory and mild motor). Rather than painful vision loss from optic neuritis, patients experience blurred vision because of optic disk edema [14]. This mimics papilledema, but patients have normal or mildly elevated CSF opening pressure. Visual acuity is normal, and visual field testing demonstrates mild arcuate defects only. Slit lamp examination reveals mild vitritis. Ocular coherence tomography demonstrates retinal nerve fiber layer thickening. Fluorescein-based retinal angiography is normal. Rarer neurological accompaniments of GFAP-IgG in CSF include movement disorders (chorea, myoclonus, and ataxia), neuropathy, and dysautonomia [2▪,4▪▪,8▪,9▪]. Adults and children have similar findings [8▪].
POTENTIAL ETIOLOGIC CLUES: COEXISTING AUTOIMMUNITY, CANCERS, AND INFECTIONS
There may be no additional historical clues to an autoimmune GFAP astrocytopathy diagnosis. Coexisting autoimmune diseases are apparent in approximately 20% [4▪▪]. These include type 1 diabetes mellitus, autoimmune thyroid disease, and rheumatoid arthritis. Approximately 25% of patients have a neoplasm detected prospectively after autoimmune GFAP astrocytopathy onset. In one series, 22 of 102 patients had a neoplasm detected [4▪▪]. The only common oncological association was ovarian teratoma, which occurred in 75% of paraneoplastic cases (usually mature teratoma). Other cancers are rare and diverse: adenocarcinomas (of breast, lung, ovary, endometrium, esophagus, and kidney), head and neck squamous cell carcinoma, thymoma, pleomorphic parotid adenoma, gliomas, multiple myeloma, small-cell carcinoma, and carcinoid [2▪,4▪▪,8▪,9▪,15]. It was also of interest that a patient ultimately determined to have temporal lobe astrocytoma (initially suspected to be encephalitis) had GFAP-IgG detected in CSF [4▪▪].
The most common neural IgGs coexisting with GFAP-IgG are generally encountered in patients with teratoma: N-methyl-D-aspartate (NMDA)-receptor (R)–IgG, detected in CSF followed by AQP4-IgG (detected in serum or CSF). Myelin oligodendrocyte glycoprotein (MOG) autoimmunity has been reported in one case [4▪▪,12]. In the Chinese series of 30 patients, one-third of patients had at least one of those antibodies coexisting [2▪]. In the large Mayo Clinic retrospective series, teratoma was detected in 71% of patients with both NMDA-R–IgG and AQP4-IgG coexisting, 53% of patients with NMDA-R–IgG alone coexisting and two of three patients with AQP4-IgG coexisting alone [4▪▪]. Neurological phenotypes of patients with coexisting AQP4-IgG or MOG-IgG seem largely indistinguishable from those positive for GFAP-IgG alone, though there are occasional neuromyelitis optica spectrum disorder cases reported [4▪▪,12]. In contrast, patients with coexisting NMDA-R autoimmunity, have a clinical course more resembling NMDA-R encephalitis. Other coexisting antibodies encountered [specific for one or more of GAD65, striational, ganglionic acetylcholine receptors, and voltage gated ion channels (calcium or potassium)] occur at rates at or just slightly above what is encountered in autoimmune neurological diseases in general [4▪▪]. Nonneural antibodies (such as antinuclear antibody, Sjogren syndrome antigen A and double stranded DNA antibodies) were encountered in 75% of the Chinese cohort [2▪].
Other reported cases of autoimmune GFAP astrocytopathy have occurred in the context of dysregulated T-cell biology, with or without cancer. One patient treated with dual T-cell regulatory (‘checkpoint’) inhibitor therapy (ipilimumab and nivolumab) for cancer developed acute inflammatory demyelinating polyneuropathy (Guillain–Barre syndrome) [8▪]. Two others developed more characteristic encephalitis, one after ipilimumab therapy for melanoma, and the other accompanying HIV infection [4▪▪]. One further patient developed encephalitis while treated with the anti-CD25 therapy daclizumab for multiple sclerosis (targeting T-regulatory cells and natural killer cells) [16].
Many patients (30–40%) have symptoms suggestive of systemic infection before the onset of CNS symptoms, with rhinorrhea, sore throat, and cough being particularly common [4▪▪]. Others developed the disorder after well-defined herpes virus infections (Herpes Simplex type 1, or Varicella zoster) [8▪,17].
PARACLINICAL FINDINGS
Completely normal neuraxis MRI is rare in autoimmune GFAP astrocytopathy. Approximately half of patients have abnormalities on T2-weighted sequences, though these are generally limited in size. One patient with advanced disease from our experience, diagnosed 1 year after symptom onset, and some patients from the Chinese series, had extensive T2 abnormalities, somewhat resembling a leukodystrophy [2▪,4▪▪].
Two-thirds of patients have abnormalities on T1-weighted, postgadolinium images. These findings are not pathognomonic but aid diagnosis considerably [4▪▪]. Over half of affected patients have a characteristic linear, radial perivascular pattern of enhancement, through the cerebral white matter, emanating from GFAP-enriched peri-lateral ventricular regions (Fig. 2a). This same pattern of enhancement had been previously reported (likely erroneously) as being characteristic of ‘angiogram-negative microvasculitis’ [18]. Indeed, in cases of autoimmune GFAP astrocytopathy reported to date, no angiographic abnormalities have been encountered. Other cerebral hemispheric patterns of enhancement reported include leptomeningeal, punctate, serpentine, and ependymal (Fig. 2b–d). In occasional cases a similar pattern of radial enhancement is encountered in the cerebellum, emanating from the peri-IVth ventricular region. PET imaging of brain may reveal hypermetabolism corresponding to areas of abnormality on MRI. Diffusion-weighted imaging is usually normal.
FIGURE 2.
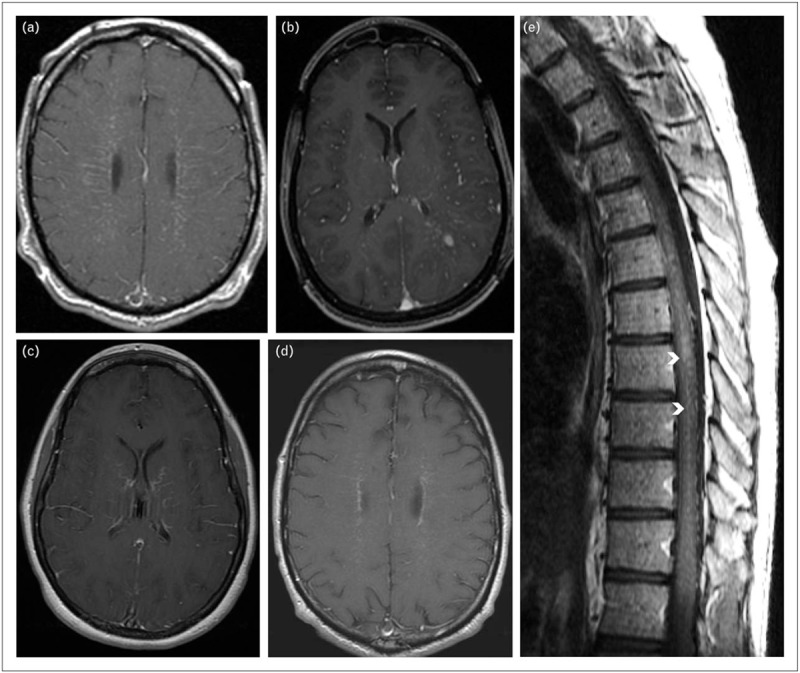
Characteristic T1 postgadolinium MR images of autoimmune GFAP astrocytopathy (axial brain, a–d; sagittal spine, e). Patterns of brain enhancement include: (a) radial periventricular; (b) leptomeningeal and punctate; (c) serpiginous; and (d), periependymal. Spinal cord enhancement, e, is characteristically central, often adjacent to the canal (arrow heads). GFAP, glial fibrillary acidic protein; MR, magnetic resonance.
In the spinal cord, longitudinally extensive T2 signal change may be encountered, though this tends to be more subtle and hazy than reported for AQP4-IgG or MOG-IgG-related transverse myelitis [4▪▪]. Sometimes a central predominant postgadolinium enhancement can be appreciated on T1 sagittal images (Fig. 2e) in the GFAP-enriched region adjacent to the central spinal canal. Patients with GFAP mutations (Alexander disease) may also have central spinal cord T2 hyperintensity [19].
CSF demonstrates marked inflammatory changes in almost all patients. Ninety percent have a lymphocyte-predominant elevation in white blood cells (average 80/μl), 80% have elevated protein, and half have CSF-exclusive oligoclonal bands [4▪▪].
Electroencephalogram, in general, demonstrates nonspecific abnormalities, such as generalized slowing [4▪▪]. One patient with δ wave-diffuse slowing with superimposed β-range fast activity (extreme δ brush) has been reported. Unlike previous reports of this electroencephalogram finding, the patient had neither NMDA-R encephalitis coexisting nor teratoma [20].
NEUROPATHOLOGY
The Mayo Clinic series, published in abstract form, reported chronic inflammation, with microglia abundant, without evidence of vasculitis [3]. The Chinese series included more detailed neuropathological findings encountered in evaluation of biopsied brains of four patients [2▪]. All had similar neuropathological findings. Extensive inflammation (infiltration of lymphocytes, monocytes, and neutrophils) was encountered, particularly around microvessels, paralleling the radial inflammatory MRI changes. In addition, microglial activation was apparent. Immunohistochemical analysis demonstrated prominent perivascular B cells (CD20+), brain parenchymal T-cell infiltrates (CD3+), and abundant CD138+ plasma cells in the Virchow–Robin spaces. Stains for GFAP and AQP4 were diminished in the lesions of three patients, and absent in a patient with coexisting AQP4-IgG detected in CSF. An additional patient, reported by the same group, had serum and CSF testing revealing IgGs reactive with MOG, AQP4, and GFAP [12]. Immunopathology of a biopsied lesion from that patient revealed absent GFAP, and AQP4, but preserved MOG expression. In contrast, another report from the same group demonstrated a similar inflammatory infiltrate, but preserved GFAP, AQP4, and MOG expression [10]. Analysis of leptomeningeal tissue from one Italian patient revealed an inflammatory infiltrate with cytotoxic (CD8+) T lymphocytes, macrophages, and some multinucleated giant cells [9▪].
Ovarian teratoma, in one reported case of a teenage girl with NMDA receptor and GFAP autoimmunity coexisting, demonstrated extensive CD3+ T-cell infiltrate [15]. In another case, a patient's serological IgG profile matched her mature ovarian teratoma immunostaining (NMDA-receptor and GFAP detected, but not AQP4) [8▪].
PATHOPHYSIOLOGY
GFAP, the main intermediate filament protein in mature astrocytes and an important component of the cytoskeleton, is also involved in multiple astrocyte functions, which are important during regeneration, synaptic plasticity, and reactive gliosis [7,21]. The intracellular antigen location supports GFAP-specific cytotoxic T cells as central to the immune response, rather than GFAP-IgG. Given that neoplasms encountered in patients express GFAP, it is likely that the priming of T cells occurs in the periphery, in paraneoplastic cases. In an animal model of relapsing-remitting CNS autoimmunity GFAP-specific CD8, T cells could avoid tolerance mechanisms before entering into the CNS [6]. In the same study, different triggers lead to different clinical phenotypes of GFAP-dependent-autoimmunity. It is unclear at this stage if infection is a common trigger for GFAP autoimmunity in humans. Other components of the immune system (microglia, macrophages, cytokines, and chemokines) could contribute to pathogenesis. Immune-mediated astrocyte dysfunction could lead to chemokine release that further recruits inflammatory cells, and enables spread of the immune attack to healthy nervous system tissue. There may also be yet undiscovered plasma membrane protein-directed IgGs that initiate a primary autoimmune event, disrupting astrocytic function, with GFAP autoimmunity occurring as a secondary phenomenon.
TREATMENT AND PROGNOSIS
All treatment and outcome data are observational, and not acquired from prospective, controlled studies [2▪,4▪▪,8▪,9▪,11]. Early observations revealed rapid clinical and radiological improvements upon treatment with corticosteroids. For patients with monophasic courses, this may suffice. Approximately 20–50% of patients have relapsing courses, and require more prolonged therapy [2▪,4▪▪,8▪,9▪,11]. Treatments utilized to date for refractory or relapsing cases include mycophenolate mofetil, azathioprine, rituximab, and cyclophosphamide. For relapsing disease, the authors typically utilize a combination of oral therapies (prednisone and a steroid-sparing immune suppressant with broad moderate immune suppressive effect on lymphocyte populations). Prednisone dosing is typically 60 mg/day (or 1 mg/kg/day, not to exceed 100 mg/day) for 3 months, followed by 10 mg/month taper until at 10 mg, followed by 1 mg/month taper thereafter. Oral immune suppressants could include mycophenolate mofetil 1000 mg twice daily or azathioprine 2.5 mg/kg/day. Steroid side-effect prophylactic recommendations include: for osteoporosis, calcium 1500 mg/day, vitamin D 1000 U/day, and a bisphosphonate if osteopenic; for pneumocystis infection, trimethoprim-sulfamethoxazole, double strength 1 tablet three times per week, or daily atovaquone, if sulfa-allergic.
Prognosis varies, though overall seems good. A median modified Rankin score of 1 was reported among 38 patients, after a median of 20 months of follow-up at Mayo Clinic [4▪▪]. Another reported a mean modified Rankin score of 1.5 among 19 Italian patients, after follow-up of 8.5 months [9▪]. In contrast, long-term follow-up in seven Chinese patients revealed severe disability and poor outcomes as the norm [11]. It is possible that this latter group of patients was diagnosed late in the disease course (median symptom duration 12 months at diagnosis) or that more severe disease occurs in Chinese patients. In contrast to patients with GFAP-IgG positivity only, patients with coexisting NMDA-R autoimmunity usually have clinical courses similar to the reported literature for that disorder: prolonged encephalitic illnesses with multiple immune therapies used, but with later recovery in most [8▪].
CONCLUSION
Autoimmune GFAP astrocytopathy, though presumed to be cytotoxic T-cell-mediated, seems to respond better to treatment than other T-cell-mediated paraneoplastic CNS diseases [22]. The triggers and pathophysiological mechanisms of GFAP autoimmunity remain to be elucidated, as well as the best long-term treatment for patients with relapsing disease.
Acknowledgements
None.
Financial support and sponsorship
A.M. has received research funding from Alexion, Euroimmun, Grifols, and Medimmune.
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▪ of special interest
▪▪ of outstanding interest
REFERENCES
- 1▪▪.Fang B, McKeon A, Hinson SR, et al. Autoimmune glial fibrillary acidic protein astrocytopathy: a novel meningoencephalomyelitis. JAMA Neurol 2016; 73:1297–1307. [DOI] [PubMed] [Google Scholar]; The original study describing autoimmune GFAP astrocytopathy.
- 2▪.Long Y, Liang J, Xu H, et al. Autoimmune glial fibrillary acidic protein astrocytopathy in Chinese patients: a retrospective study. Eur J Neurol 2018; 25:477–483. [DOI] [PubMed] [Google Scholar]; This article contains a large experience and includes some pathological data.
- 3.Aksamit AJ, Parisi J. Chronic microglial encephalomyelitis. Ann Neurol 2012; 72:S110. [Google Scholar]
- 4▪▪.Flanagan EP, Hinson SR, Lennon VA, et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: analysis of 102 patients. Ann Neurol 2017; 81:298–309. [DOI] [PubMed] [Google Scholar]; This article is the largest and most detailed experience of autoimmune GFAP astrocytopathy published to date.
- 5.Shibuya M, Matsuki N, Fujiwara K, et al. Autoantibodies against glial fibrillary acidic protein (GFAP) in cerebrospinal fluids from Pug dogs with necrotizing meningoencephalitis. J Vet Med Sci 2007; 69:241–245. [DOI] [PubMed] [Google Scholar]
- 6.Sasaki K, Bean A, Shah S, et al. Relapsing-remitting central nervous system autoimmunity mediated by GFAP-specific CD8 T cells. J Immunol 2014; 192:3029–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Middeldorp J, Hol EM. GFAP in health and disease. Prog Neurobiol 2011; 93:421–443. [DOI] [PubMed] [Google Scholar]
- 8▪.Dubey D, Hinson SR, Jolliffe EA, et al. Autoimmune GFAP astrocytopathy: prospective evaluation of 90 patients in 1year. J Neuroimmunol 2018; 321:157–163. [DOI] [PubMed] [Google Scholar]; This article provides some prospective data, utility of CSF versus serum testing, and ovarian teratoma disorder.
- 9▪.Iorio R, Damato V, Evoli A, et al. Clinical and immunological characteristics of the spectrum of GFAP autoimmunity: a case series of 22 patients. J Neurol Neurosurg Psychiatry 2018; 89:138–146. [DOI] [PubMed] [Google Scholar]; Contains a large experience with some pathological data.
- 10.Shu Y, Long Y, Chang Y, et al. Brain immunohistopathology in a patient with autoimmune glial fibrillary acidic protein astrocytopathy. Neuroimmunomodulation 2018; 25:1–6. [DOI] [PubMed] [Google Scholar]
- 11.Yang X, Liang J, Huang Q, et al. Treatment of autoimmune glial fibrillary acidic protein astrocytopathy: follow-up in 7 cases. Neuroimmunomodulation 2017; 24:113–119. [DOI] [PubMed] [Google Scholar]
- 12.Yang X, Xu H, Ding M, et al. Overlapping autoimmune syndromes in patients with glial fibrillary acidic protein antibodies. Front Neurol 2018; 9:251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dubey D, Pittock SJ, Kelly CR, et al. Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis. Ann Neurol 2018; 83:166–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen JJ, Aksamit AJ, McKeon A, et al. Optic disc edema in glial fibrillary acidic protein autoantibody-positive meningoencephalitis. J Neuroophthalmol 2018; 38:276–281. [DOI] [PubMed] [Google Scholar]
- 15.Martin AL, Jolliffe E, Hertweck SP. Ovarian teratoma associated with coexisting anti-N-methyl-D-aspartate receptor and glial fibrillary acidic protein autoimmune meningoencephalitis in an adolescent girl: a case report. J Pediatr Adolesc Gynecol 2018; 31:321–324. [DOI] [PubMed] [Google Scholar]
- 16.Luessi F, Engel S, Spreer A, et al. GFAPalpha IgG-associated encephalitis upon daclizumab treatment of MS. Neurol Neuroimmunol Neuroinflamm 2018; 5:e481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Xu Y, Ren H, et al. Autoimmune GFAP astrocytopathy after viral encephalitis: a case report. Mult Scler Relat Disord 2018; 21:84–87. [DOI] [PubMed] [Google Scholar]
- 18.Salvarani C, Brown RD, Jr, Calamia KT, et al. Angiography-negative primary central nervous system vasculitis: a syndrome involving small cerebral vessels. Medicine (Baltimore) 2008; 87:264–271. [DOI] [PubMed] [Google Scholar]
- 19.Farina L, Pareyson D, Minati L, et al. Can MR imaging diagnose adult-onset Alexander disease? AJNR Am J Neuroradiol 2008; 29:1190–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Theroux LM, Goodkin HP, Heinan KC, et al. Extreme delta brush and distinctive imaging in a pediatric patient with autoimmune GFAP astrocytopathy. Mult Scler Relat Disord 2018; 26:121–123. [DOI] [PubMed] [Google Scholar]
- 21.McKeon A, Benarroch EE. Glial fibrillary acid protein: functions and involvement in disease. Neurology 2018; 90:925–930. [DOI] [PubMed] [Google Scholar]
- 22.McKeon A, Tracy JA, Pittock SJ, et al. Purkinje cell cytoplasmic autoantibody type 1 accompaniments: the cerebellum and beyond. Arch Neurol 2011; 68:1282–1289. [DOI] [PubMed] [Google Scholar]