

Abstract
In this essay, I propose that DNA-binding anti-cancer drugs work more via chromatin disruption than DNA damage. Success of long-awaited drugs targeting cancer-specific drivers is limited by the heterogeneity of tumors. Therefore, chemotherapy acting via universal targets (e.g., DNA) is still the mainstream treatment for cancer. Nevertheless, the problem with targeting DNA is insufficient efficacy due to high toxicity. I propose that this problem stems from the presumption that DNA damage is critical for the anti-cancer activity of these drugs. DNA in cells exists as chromatin, and many DNA-targeting drugs alter chromatin structure by destabilizing nucleosomes and inducing histone eviction from chromatin. This effect has been largely ignored because DNA damage is seen as the major reason for anti-cancer activity. I discuss how DNA-binding molecules destabilize chromatin, why this effect is more toxic to tumoral than normal cells, and why cells die as a result of chromatin destabilization.
Keywords: chromatin damage, DNA damage, anti-cancer treatment, DNA-binding small molecules
1. Introduction
Cancer therapy targeting tumor-specific oncogenic drivers faces the problem of tumor heterogeneity determined by the genetic variability and epigenetic plasticity of tumor cells. Moreover, recent achievements in deciphering the genomic and proteomic content of thousands of tumors have led to the realization that “perfect” anti-cancer targets -- molecules or processes that are universally abnormal in tumor cells while being completely dispensable for normal cells -- likely do not exist [1,2]. Because of this phenomenon, conventional chemotherapy targeting DNA is still the most widely and successfully used anti-cancer treatment [3]. However, the efficacy of DNA-targeting therapy is limited by the inability to deliver doses high enough to kill all tumor cells without causing serious and long-lasting side effects to normal tissues.
The induction of DNA damage (Fig.1) has long been accepted as the major mechanism by which DNA-targeting anti-cancer therapies work, and thus, a key driver of their efficacy [4]. At the same time, DNA damage is the reason for the inefficiency and toxicity of current DNA-targeting therapies, because it irreversibly damages normal tissues, causes long-term side effects, and further increases the genomic heterogeneity of tumors.
Figure 1.
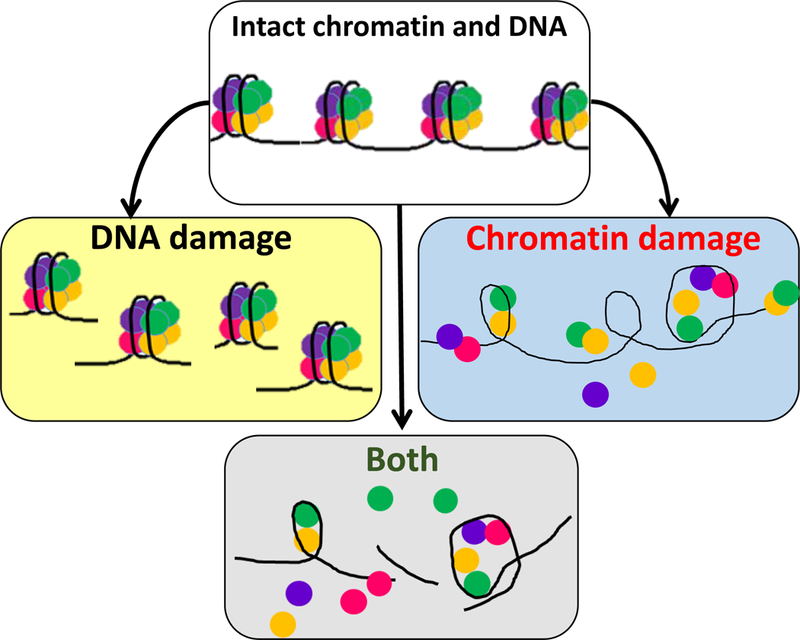
Schematic representation of DNA and chromatin damage as individual and combined events. DNA damage is any chemical modification of DNA (e.g., double- and single-strand breaks, base modifications). Chromatin damage is the loss of histones from nucleosomes in the absence of chemical modifications of DNA. In many cases, both types of damage are observed together because DNA damage may destabilize nucleosomes, and loss of histones makes DNA more sensitive to chemical modifications. In contrast to DNA damage, the causes and consequences of chromatin damage are poorly studied.
Interestingly, while it is well known that DNA exists as part of a nucleoprotein complex (chromatin) and chromatin alterations are observed in cancer [5], the effects of DNA-targeting drugs on chromatin have not been widely explored, and targeting of chromatin per se (Fig.1) has not been explicitly proposed as an anti-cancer approach. While it has been noted that some DNA-targeting drugs alter chromatin [6–8], the presumption that DNA damage is the primary source of their anti-cancer activity has placed their effect on chromatin outside the scope of major anti-cancer research.
In this essay, I review the published data from our and other groups demonstrating the destabilizing effects of DNA-binding small molecules on chromatin. Furthermore, I speculate as to why this effect is more toxic for tumoral than normal cells. Building on the accumulated data describing chromatin alterations in cancer and established anti-cancer activity of chromatin-destabilizing drugs, I hypothesize that destabilized chromatin -- i.e., chromatin in which nucleosomes are less stable and therefore DNA is easier to access -- is a specific feature of malignant (aggressive) tumor cells. This chromatin conformation provides such cells with selective advantages in the form of phenotypic plasticity. Yet, complete loss of chromatin is highly toxic for all cells for reasons not fully understood. Here, I propose some of these reasons based on our group’s experimental findings. The combination of these two facets of tumor genomes (the presence of destabilized chromatin and the toxicity of further reduction in chromatin stability) suggests the potential of exploring chromatin-damaging therapy as a novel anti-cancer approach.
2. DNA-binding compounds may kill tumor cells without inducing DNA damage
Our group questioned the role of DNA damage in the anti-cancer activity of small molecules when we ran a phenotypic screen for novel p53-activating compounds using tumor cells with wild-type, but not fully functional, p53, which did not respond to DNA damage [9]. Indeed, the identified p53-activating compounds did not cause DNA damage. The most active carbazole-based compounds were named curaxins. Curaxins were more toxic to tumoral than normal cells, making them promising anti-cancer agents [10]. However, they were also toxic to p53-deficient cells, suggesting that they did not act solely via p53 [10]. Even more puzzling, the curaxins bound DNA, and DNA binding was obligatory for their anti-cancer activity.
Further investigation revealed that although curaxins do not cause any chemical DNA modifications (i.e., breaks or nucleotide alterations), they do change the physical properties of DNA [11]. Intercalation of the carbazole planar “body” between the DNA bases increases the inter-base pair distance such that the DNA becomes longer and thinner. The carbazole side chains protrude into the major and minor grooves, anchoring the molecule to the DNA and providing positively charged nitrogen, respectively. These effects make the helix less flexible and less negatively charged. While the DNA itself can tolerate these changes without undergoing any disintegration, the basic unit of chromatin, the nucleosome, cannot.
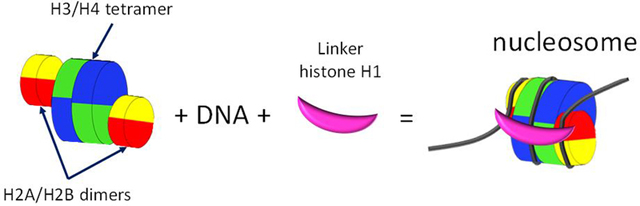
The nucleosome (Box 1) consists of two polar components: the positively-charged histone octamer and negatively-charged DNA wrapped around the octamer [12]. The octamer does not exist as a standalone structure at physiological conditions due to the high positive charge of the histones. However, the nucleosome is very stable due to electrostatic attraction between the histones and DNA, binding of several histone amino acids to DNA, and the perfect fit between the flexibility of the DNA helix and the diameter of an octamer [12]. Curaxin binding to DNA disturbs each of the components required for nucleosome stability: (i) it reduces the negative charge of DNA; (ii) shifts the positions of base pairs relative to the amino acids due to the lengthening of DNA; (iii) occupies the sites used by the histone amino acids in the DNA; (iv) makes DNA rigid such that it cannot wrap tightly around the octamer. As a result, nucleosomes are destabilized and disassembled in curaxin-treated cells, leading to the loss of histones from chromatin [11].
Box 1. Nucleosome structure and stability.
Nucleosomes are the structural units of chromatin, which are located every 200+/40 base pairs throughout all eukaryotic genomes. Eighty percent of the human DNA is wrapped into nucleosomes. Nucleosome-depleted regions are found at the promoter, enhancer, transcription start and end sites. Whether these regions have fewer nucleosomes or the nucleosomes are more sensitive to the nuclease digestion used to detect nucleosomes is still a matter of debate.
Nucleosomes consist of 145–147 base pairs of DNA wrapped 1.65 times around histone octamers – disc-like structures consisting of four pairs of core histones. The H3/H4 tetramer is located in the center of the nucleosome relative to the DNA dyad point (base pair located 72 or 73 base pairs from both ends of the nucleosomal DNA), and H2A and H2B form dimers located on both sides of the tetramer and are wrapped with DNA at the nucleosome entry and exit points.
Nucleosomes with completely wrapped DNA are known as folded or closed nucleosomes. Closed nucleosomes may be additionally locked by linker histone H1, which binds three threads of DNA (entering, exiting, and internal), and the core histones. Nucleosomes without a linker histone and with different degrees of DNA unwrapping are known as opened nucleosomes.
Histones are among the most positively charged proteins in eukaryotic cells. Due to this charge, histone octamers do not exist as stand-alone structures under physiological conditions. The only stable components of the nucleosome are the H2A/H2B and H3/H4 dimers. The H3/H4 tetramer is less stable than the dimers but can exist at physiological salt concentration.
Exposure of free histones to DNA at physiological salt concentration results in precipitation. Thus, nucleosomes cannot be self-assembled. The positive charge of histones needs to be buffered, which can be achieved in vitro with the help of high salt (e.g., 2 M NaCl), the presence of other nucleic acids (e.g., RNA), or histone chaperones, which also serve this function in cells.
The stability of assembled nucleosomes depends on the wrapping of negatively charged DNA around the positively charged core, the fitness between the DNA flexibility and diameter of the octamer, and several spatially-oriented contacts of the histone amino acids with DNA (e.g., the arginine side chain entering the minor groove when the minor groove faces octamer).
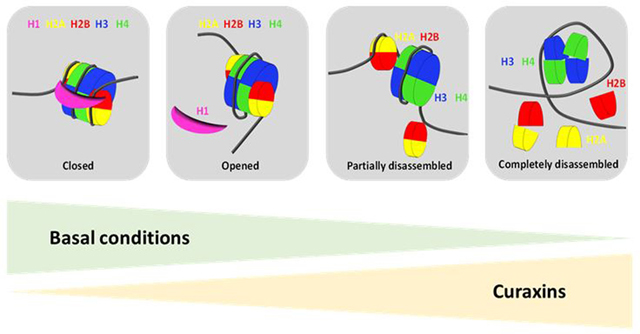
Because curaxins cause dose-dependent chromatin disassembly (Box 2) without inducing any detectable DNA damage, we named them “chromatin-damaging” agents. The anti-cancer activity of the lead curaxin CBL0137, which has now been shown in different tumor models by several groups [10,13–16], strongly suggests that chromatin-damaging agents can kill tumor cells without inducing DNA damage.
Box 2. Dose-dependent effect of curaxins on chromatin in cells.
Under basal conditions, nucleosomes in cells can be closed (e.g., locked with linker histone H1) or opened, and partially or completely disassembled (e.g., during transcription or replication). In the absence of treatment, their distribution is significantly skewed to the left (i.e., most nucleosomes are closed [heterochromatin is prevailing in most mammalian cells], some are opened [transcribed regions], and a few are disassembled [enhancer and promoter regions]).
Curaxins cause a dose-dependent shift in the distribution of nucleosomes to the right, which is accompanied by different phenotypic changes that are roughly divided into three phases:
-
I –
Transition from a state when most nucleosomes are closed to a state when most nucleosomes are opened. Marker: the occurrence of chromatin-free histone H1. Consequences: transcription of constitutive heterochromatin, activation of type I interferon response, activation of p53, and inhibition of several transcriptional factors (e.g., NF-kappaB, MYC, HSF1). Toxicity: mostly to tumor cells; short incubation with the drug is fully reversible, and prolonged incubation (> 24 h) causes the death of up to 50% of tumor cells (death occurs at approximately 72 h), but normal cells may only be growth arrested and resume proliferation upon drug removal.
-
II –
Partial disassembly of a significant proportion of nucleosomes. Marker: the appearance of free outer histones (H2A and H2B) in the nucleoplasm. Consequences: Redistribution of all FACT from the nucleoplasm to chromatin, loss of long-range chromatin interactions, and inhibition of transcription. Toxicity: partially reversible depending on the time of incubation. Tumor cells are more sensitive than normal cells. Incubation of tumor cells for > 1 h results in the death of 80 to 100% of cells in approximately 48 h. Normal cells undergo growth arrest and resume proliferation when the drug is removed.
-
III –
Complete disassembly of a significant proportion of nucleosomes. Markers: the appearance of chromatin-free inner histones (H3 and H4) in the nucleoplasm, and the transition of B-DNA into left-handed Z-DNA at regions of nucleosome loss, which is the point of no return for all cells. All cells eventually die if the drug is present when the inner histones start being lost from the chromatin (at least 30 min). Death occurs in approximately 24 h.
The introduction of this paradigm raises multiple questions. Is there anything specific in the curaxins’ structure that makes them capable of damaging chromatin, but not DNA? Is chromatin damage truly responsible for the toxicity of curaxins to tumor cells? How does chromatin damage kill cells? Why would chromatin damage be more toxic to tumoral than normal cells?
3. Which small molecules cause chromatin damage?
In principle, any small molecule that binds to DNA should alter the DNA shape, charge, or flexibility. If the reason for chromatin destabilization is that the small molecule alters these DNA properties, then theoretically all DNA-binding molecules should destabilize chromatin, which may occur independently of whether they can or cannot react chemically with DNA (i.e., cause DNA damage). In support of this hypothesis, there are reported effects of DNA-binding small molecules on chromatin [6–8,17]. However, what structural features of a small molecule result in DNA or chromatin damage is not completely clear.
The effect of a small molecule on chromatin likely depends on the mode of DNA binding by that molecule (Box 3). Theoretically, insertion of intercalators between DNA base pairs leading to an increased inter-base pair distance, untwisting, or reducing DNA flexibility should destabilize nucleosomes. Major and minor groove binders filling the corresponding grooves of DNA have little effect on DNA length and twist but may change DNA flexibility and charge. Minor groove binders may affect nucleosome stability by interfering with DNA/histone binding, Indeed, the tightest contact between a histone octamer and DNA occurs via the minor groove [12], [18].
Box 3. DNA-targeting small molecules and their potential effects on nucleosomes.
DNA-targeting compounds can be divided into several groups. First, there are compounds that directly bind DNA and those that do not bind DNA themselves or bind very weakly, but their binding is stabilized by DNA-binding proteins, such as the topoisomerases (e.g., etoposide or camptothecin).
DNA-binding drugs can bind DNA covalently (e.g., platinum compounds) or non-covalently (e.g., anthracyclines). Covalent binders cause chemical DNA modifications (i.e., DNA damage). There is a limited number of covalent DNA binders used in medicine. Because DNA consists of the same elements as all other organic molecules, the ability of a compound to react chemically with DNA means that it can react with many other molecules in cells and, therefore, is highly toxic.
Non-covalent binders do not cause chemical modifications to DNA themselves. However, many of them damage DNA via one of two mechanisms. (i) They have reactive substituents that cause DNA damage (e.g., quinacrine mustard). The DNA-binding moiety of quinacrine mustard, 9-aminoacridine, directs the whole molecule to DNA and the nitrogen mustard causes a chemical modification to almost any organic molecule nearby. (ii) They cause a change in the helical shape of DNA, making it a poor substrate for enzymes that work with DNA. Cellular enzymes typically work with B-DNA. The right-handed helix consists of 10.5 base pairs per turn of 33.2 Å with a diameter of 20 Å. A deviation from these parameters makes DNA a poor substrate for these enzymes, and they make mistakes. These mistakes lead to DNA damage. The best-known examples are the topoisomerases, which either cannot cleave or religate DNA bound to the small molecules. In the latter case, topoisomerase activity leads to DNA breaks (i.e., DNA damage). This DNA damage is provoked by, but not directly caused by, the small molecule. However, in some cases, topoisomerase cannot cleave DNA bound to the small molecules. In this case, although the enzyme is inhibited, there are no breaks or DNA damage.
Non-covalent direct DNA binders are divided into three major types: (i) intercalators (e.g., curaxins, anthracyclines), (ii) major groove binders (MaGB, e,g., methyl green), and (iii) minor groove binders (MiGB, e.g., DAPI). Many compounds bind DNA via several types of binding simultaneously.
Intercalators are inserted between base pairs forming hydrogen bonds with neighboring nucleotides, which leads to an increase in the inter-base pair distance, untwisting of DNA, and a reduction in its flexibility, similar to the effects of curaxins.
MaGB and MiGB fill the corresponding grooves of DNA due to electrostatic interactions. They have little or no effect on DNA length and twist but change DNA flexibility and charge.
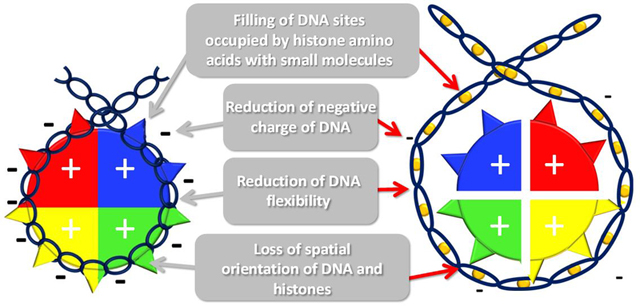
An important consideration in understanding how small molecules affect chromatin is whether the molecule can compete with histones for binding DNA. Molecules may be able to bind free DNA but not DNA that is wrapped around histones, because nucleosomal DNA has less freedom to adapt to structural changes, and binding interfaces may be hidden (e.g., minor groove). Another consideration is that even when a small molecule cannot bind a folded nucleosome, the dynamic nature of nucleosomes may provide an opportunity for the binding of naked DNA when the nucleosome breathes or is remodeled, which would make this DNA unsuitable for the wrapping of an octamer.
To test some of these assumptions, our group selected a set of compounds representing different types of DNA - small molecule interactions and tested their ability to induce disassembly of nucleosomes in living cells and cell-free conditions. We found that all of the tested agents that bound DNA directly, but not those that interacted with DNA indirectly (e.g., with the help of topoisomerase), reduced nucleosome stability under cell-free conditions. Intercalators had a much stronger effect than minor groove binders (no major groove binders were tested) [19]. The activity of these small molecules in cells was dependent on whether the compounds could access genomic DNA. For example, some DNA intercalators cannot enter living cells (propidium iodide) [20] or accumulate almost exclusively in mitochondria (ethidium bromide) [21]. Some compounds may bind primarily to the linker DNA (fragment of DNA between nucleosomes). It is not known how this binding affects nucleosome organization in cells.
Most importantly, when our group tested the ability of different DNA-binding compounds to destabilize chromatin or cause DNA damage using several tumor cell lines, a strong positive correlation was observed between the cytotoxicity of a compound and its ability to induce chromatin damage whereas a weak negative correlation was observed between cytotoxicity and DNA damage [19]. The negative correlation may be explained by the fact that DNA breaks reduce the accumulation of negative super-helicity due to nucleosome disassembly; and this negative super-helicity or DNA under-twisting amplifies the destabilization of chromatin. In addition, several molecules were toxic to tumor cells without causing any DNA damage but were potent inducers of chromatin damage [19]. Overall, these data suggest that at least some of the small molecules that directly bind to DNA are toxic to cells not because they damage DNA but because they destabilize chromatin. This proposition does not imply that DNA damage does not have an anti-cancer effect. For multiple small molecules with well-established anti-cancer activity and gamma-irradiation, DNA damage is one of the critical components of their anti-cancer activity. However, for some molecules (e.g., curaxins and some anthracyclines) that cause little or no DNA damage, chromatin damage may be the major critical component of their activity, worthy of consideration and investigation.
Well-established therapeutic efficacy of many “chromatin-damaging” molecules in animal models and human patients (e.g. [22]) infers that chromatin destabilization is more toxic to tumoral than normal cells. Although the therapeutic window (i.e., the difference between effective and toxic concentrations) for chromatin-damaging drugs, similarly to DNA damaging therapy, is quite narrow, especially for cells growing in vitro, a therapeutic effect still occurs in the absence of significant toxicity. For example, curaxin can cause tumors to shrink and disappear in some animals in several models without causing evident morbidity even in young mice, which have many proliferating cells in different tissues [10,14,15,23].
4. Why may chromatin damage work as an anti-cancer approach?
There are several possible explanations for how destabilization of chromatin might affect normal and tumor cells differently. First, normal cells may be more resistant to chromatin destabilization due to pre-existing differences in chromatin structure. Chromatin in normal cells may be more compact and thus accumulate fewer drug molecules, or it may withstand higher drug concentrations without losing histones. Normal cells may also have better buffering mechanisms. Finally, normal cells may have checkpoint mechanisms that block cell division in response to chromatin damage, which are disrupted in tumor cells. These scenarios imply that (i) chromatin organization is different in tumoral and normal cells; (ii) cells have mechanisms to control chromatin stability and detect chromatin destabilization.
What evidence is there that chromatin structure or function is different between normal and tumor cells? Altered chromatin structures in cancer cells were first noticed by Theodor Boveri in 1902 [24] and confirmed in multiple other studies (Table 1, rev in [25] ). However, the underlying causes of these alterations or their benefits to tumor cells remain unclear. Currently, the prevailing view on chromatin alterations is that they cause changes in the expression (up or down) of individual cancer drivers (rev, in [26]). An alternative view is that the basic function of chromatin is changed in cancer [27,28].
Table 1.
Types of known chromatin alterations in cancer
| Type of chromatin organization |
Condition in normal cells |
Changes in cancer | Potential significance of changes in cancer |
|---|---|---|---|
| DNA methylation at CpG “islands” | Present at promoters of developmental genes, which are silenced in differentiated cells | Appears at promoters of tumor suppressor genes and genes involved in differentiation [86]* | Silencing of tumor suppressors and genes involved in differentiation |
| Widely spread methylation of cytosine at CpG dinucleotides (CpG “seas”) | Occurs throughout constitutive heterochromatin | Reduced or lost [87] | Desilencing of heterochromatin |
| Core histones | H2A, H2B, H3.1, H4 – wild type histones, basic constituents of chromatin | In frame point mutations [88] | Interferes with function of “readers” and “writers” – factors responsible for the introduction and recognition of histone PTMs; Make nucleosomes less stable [100] |
| Variant histones | Incorporated in certain defined regions depending on histone type, e.g. centromere, active enhancers and promoters | Mutations in histone variants; incorporation into wrong genomic regions [89] | Unclear |
| Linker histone H1 | Mostly locks nucleosomes present within heterochromatin | Reduced expression [90] | Desilencing of heterochromatin |
| Posttranslational modifications (PTMs**) of histones | Label different regions in chromatin | Multiple mutations in enzymes introducing and removing PTMs in cancer [91] | Gene expression changes |
| Non-histone chromatin proteins (HP1, HMGB family etc) | Condensate or decondensate chromatin | Overexpression of HMGB1 [92]; reduction in levels of HP1 family members [93] | Unclear, but most probably make chromatin in general more accessible |
| HATs/HDACs | Add or remove acetyl groups to/from histones | Abnormal expression and changes in substrate preferences [94,95] | Change of epigenetic code; Change of chromatin condensation |
| KMTs/KDMs | Add/remove methyl groups to/from histones | Mutations; overexpression and changes in substrate preferences [96] | Change of epigenetic code |
| ATP dependent chromatin remodelers | Reorganize nucleosomes at promoter, enhancers and TSS | Mostly loss of function mutations, gain of function mutations in some cancers [97] | unclear |
| Histone chaperones | Assemble and disassemble nucleosomes; exchange histones | Mostly overexpressed [98] | unclear |
| CTCF/cohesin | Creates loops governing long-range enhancer promoter interactions | Mutations in binding sites, reduced expression, methylation of binding sites [99] | Altered enhancer/promoter interactions; changes in gene expression |
| Chromatin domains (TAD, LAD, NAD etc) | Long-range chromatin loops organized into spatially restricted domains differing in transcriptional activity | Change of long range interactions between promoters and enhancers [26] | Oncogene activation; Gene translocations; Changes in gene expression |
| Distribution of euchromatin and heterochromatin | Separation of transcriptionally active and inactive chromatin between different nuclear locations | Reduction of heterochromatin [46] | General decondensation of chromatin |
only references to the recent reviews are given;
abbreviations: PTM – post-translational modifications, HAT – histone acetyl transferase, HDAC – histone diacetyl transferase, KMT – histone (lysine) methyl transferase, KDM – histone (lysine), CTCF - CCCTC-binding factor, TAD – topologically associated domain, LAD – lamina associated domain, NAD – nucleoli associated domain.
What is the normal function of chromatin? The most popular answer is to pack ~2 meters of genomic DNA into a tiny nucleus with a diameter less than 10−5 meters [29]. However, simple calculations convincingly overthrew this assumption [30]. The other common explanation links the packaging of DNA into chromatin with the necessity to separate duplicated genomic DNA threads between two daughter cells. However, this seemingly simple answer disappears when one realizes how many nucleosomes are in a cell (~1.3 million per average chromosome), a number that is likely to make the process of separation even more difficult. In fact, it was recently shown that metaphase chromosomes could be assembled from naked DNA in histone-depleted Xenopus egg extract with the help of condensins [31]. Therefore, chromatin is not essential even for the packaging of DNA into chromosomes for cell division.
Another hypothesis is that chromatin protects genomic DNA from DNA damage and access of non-histone proteins. To my mind, the major function of chromatin is to prevent the “free” access of the transcriptional machinery to DNA. However, why do eukaryotic cells need to restrict the access of the transcription machinery to genomic DNA? Textbooks suggest that transcription is regulated via the binding of sequence-specific transcription factors to DNA. However, incredible growth of RNA diversity (i.e., types of RNAs transcribed in eukaryotic cells, including long and short non-coding RNAs, promoter [32] and enhancer RNAs [33], and products of divergent transcription [34,35], [36] was revealed during last decade. Some of these RNAs likely play regulatory roles. However, their production is also consistent with the possibility that RNA polymerase transcribes whatever is accessible (i.e., not covered with chromatin). Our experiments with the induction of chromatin disassembly in cells by small molecules and published data suggest that transcription occurs almost everywhere that chromatin packaging is lost or reduced [34–40]. In light of these data, a major function of chromatin may be to protect DNA from unnecessary, “illicit” or pervasive transcription. The emergence of pervasive transcription can be considered evidence of the failure or weakening of chromatin function.
There are several types of pervasive transcription. First, there is the expression of constitutive heterochromatin (e.g., centromeric and pericentromeric repeats), which theoretically should not be expressed under any circumstances. It is well documented that these repeat elements are overexpressed in cancer [41–43]. Endogenous retroviruses are another component of the heterochromatin, silencing of which is important for the safety of the eukaryotic genome. Again, there are multiple examples of significantly elevated expression of these elements in different types of cancer, which correlates with poor clinical prognosis [43,44]. The third type of pervasive transcription is intra- and intergenic transcription that does not correspond to known open reading frames and is initiated from cryptic promoters that are not associated with known functional transcripts. At least part of these cryptic promoters are remnants of ancient Long Terminal Repeats (LTR) that are kept inactive due to nucleosomes positioned at these sequences [37]. An accurate comparison of the level of cryptic transcription between normal and tumor cells has not been performed. However, the well-known phenomenon of intron retention in cancer cells [45] can be explained not only by problems with splicing but also by increased transcription initiation from intragenic sequences. Based on these examples, I propose that chromatin is in general destabilized in cancer, and therefore, drugs causing further chromatin destabilization may exceed the limits of cell tolerability at lower concentrations than is required to cause the same state in normal cells.
5. Why may chromatin be destabilized in tumor cells?
First, what do I mean by destabilization of chromatin? This process may include: (i) a reduction in the number of nucleosomes per unit of length of genomic DNA; (ii) an increase in the number of opened nucleosomes (i.e., lacking a linker histone) or nucleosomes with destabilizing histone modifications; (iii) enhanced chromatin dynamics (i.e., chromatin in tumor cells loses histones more frequently, but these losses are compensated by elevated activity of factors that build new nucleosomes, such as histone chaperones). Although the number of nucleosomes may not change in this case, the access to nucleosomal DNA is simplified due to the higher chance of encountering nucleosome-free DNA. Differences in some of these parameters (e.g., chromatin dynamics or amount of heterochromatin [i.e., regions with closed nucleosomes]) were already noticed between non-tumor and tumor cells (rev. in [46], [47]). However, an accurate comparison of nucleosome occupancy, frequency, positioning, and stability, and genome-wide chromatin accessibility and distribution of histone post-translational modifications between normal and tumor cells needs to be performed. These comparisons must take into account the complexity of the situation: (i) the origin of tumoral and normal cells (often not clearly defined); (ii) the necessity to adjust for the rate of proliferation (i.e., all these parameters change with cell cycle and populations of cells with different cell cycle distributions may appear different); (iii) the significant variability in these parameters within a cell depending on the genomic regions.
Chromatin destabilization may happen because there are more ongoing cycles of replication and transcription occurring in tumor cells compared to normal cells. However, there may be benefits of having less stable chromatin, such as the acquisition of epigenetic plasticity, a property of tumor cells that allows phenotypic transitions to occur (e.g., epithelial vs. mesenchymal, stem vs. non-stem, drug-sensitive vs. drug-resistant) to adapt to different conditions. The phenotypic plasticity of tumor cells resembles the state of the epigenome of embryonic stem cells with bivalent marks of facultative heterochromatin [48] that allow transcription to be turned on and off without significant chromatin remodeling. Theoretically, the presence of fewer nucleosomes or nucleosomes with less well marked or irregular histone modifications (e.g., at or around promoters and transcriptional start sites [TSS]) presents a weak barrier for transcription initiation. The “leakiness” of transcription in this region can be quickly turned up or down depending on the circumstances without recruitment of bulky and complicated chromatin remodeling complexes that are required under normal conditions. This unstable chromatin creates the possibility of easy and rapid changes in the transcriptional program of a cell in response to different circumstances. If this effect is true, then chromatin alterations in tumor cells result not in a change in the transcriptional program from one to another, but provide tumor cells with the ability to change transcription as many times and as strongly as needed to adapt and continue to grow under various environmental conditions. Thus, chromatin destabilization may be beneficial to tumor cells and, therefore, selected during tumor development.
6. Why is chromatin destabilization toxic?
If chromatin destabilization is in itself beneficial, one would expect a gradual degradation of chromatin until its eventual loss from cells in advanced tumors, which reach a state in which the cells of different tumors become just “Tumor Cells” – completely undifferentiated creatures that proliferate but lack any signs of the tissue of origin. However, this does not occur. Moreover, based on transcriptional profiling, tumor cells bear the well-preserved traits of their tissue of origin even when they lose any “geographical” connection with the original tissue (e.g., upon metastasizing or propagation in vitro [http://cancergenome.nih.gov/]). Because a transcriptional profile is defined by the chromatin organization, it means that, in general, chromatin organization is preserved in tumor cells. So why do HeLa cells with 82 chromosomes living in a dish for more than 60 years continue to be cervical epithelium? Why and how do tumor cells which easily lose multiple functions during tumor progression, continue to maintain altered but still well-organized and functional chromatin? I propose that neither loosening of chromatin nor its preservation are stochastic. There are pros and cons to having chromatin destabilized. However, tumor cells require a balance between the positive and negative consequences of chromatin destabilization.
The consequences of chromatin destabilization are poorly studied. The toxicity of curaxins towards tumor cells begins at concentrations where nucleosome opening but not disassembly, occurs [10,11] (Box 2). This observation indicates that cells suffer from the loss of proper chromatin organization before there is complete nucleosome loss. So why is nucleosome opening that is beyond what exists in tumor cells toxic? To answer this question, the accumulation of many more observations of what happens in cells in response to chromatin destabilization is needed. Below, I describe our observations in curaxin-treated cells and propose some mechanisms that likely contribute to the toxic effects of chromatin destabilization.
6.1. C-trapping of FACT
The first responder to chromatin destabilization in cells is the histone chaperone FACT(FAcilitates Chromatin Transcription) [11]. Histone chaperones allow the step-by-step assembly of nucleosomes from histones and DNA. The histone chaperones bind the histones and prevent their irregular, non-nucleosomal binding to DNA until proper nucleosomal interactions occur (rev. in [49]).
Our group became interested in FACT when we found that p53 is activated in curaxin-treated cells via its phosphorylation by casein kinase 2 (CK2) [10]. Based on the literature, CK2 phosphorylates p53 when it is in a complex with FACT [50,51]. FACT consists of two subunits, SSRP1 and SPT16. Both subunits are highly expressed in tumor cells, and their levels correlate with poor prognosis in several tumor types [15,16,52]. Moreover, most normal differentiated cells in mammals do not express FACT [53]. FACT’s function has been described in the literature as “facilitation” of transcription elongation through chromatin. This function was established based on the analysis of transcription of nucleosomal templates in a cell-free system [54,55]. However, most reported cell experiments failed to detect inhibition of transcription upon FACT inactivation (rev. in [56]). FACT binds almost all the components of the disassembled nucleosome, (H2A/H2B dimer, H3/H4 tetramer, and DNA) (rev. in [56]). However, it does not bind to the assembled, folded nucleosome [11,57,58] because the FACT-binding histone interfaces are hidden within the assembled nucleosome and binding to DNA is weakened by FACT post-translational modifications [59,60]. These data suggest that FACT only interacts with chromatin when it is destabilized.
Our group observed that under basal conditions in human tumor cells, most FACT is free in the nucleoplasm, and only a minor proportion is bound to chromatin at highly transcribed genomic regions [11,19]. Upon treatment with curaxins, DNA unwraps from the octamers to expose multiple FACT binding sites throughout the whole genome, which leads to the binding of all cellular FACT to chromatin, a phenomenon that we named chromatin trapping of FACT or c-trapping [11]. Surprisingly, among all histone chaperones tested so far, only FACT undergoes c-trapping in response to chromatin destabilization in cells.
After years of studying the effect of curaxins on FACT, manifested in the form of c-trapping [11], I propose that c-trapping is an extreme variation of basal FACT function or its performance in extreme conditions. This function is to prevent nucleosome loss under conditions of chromatin destabilization. FACT is a multi-domain module that binds all nucleosome components when they are available for binding (i.e., when nucleosomes are destabilized or disassembled) and keeps them together to prevent histone loss. Under normal conditions, this binding most likely occurs during transcription and replication, and the more intensive these processes are, the more FACT is needed. Curaxins are functional inhibitors of FACT that not only cause direct chromatin destabilization but also functionally exhaust FACT that normally prevents histone loss. FACT activity is critical for the viability of most tumor cells tested to date, but not most normal cells [10,15,53,61], which supports the proposition that keeping organized chromatin is more difficult for tumoral than normal cells. Overall, inhibition of FACT may partially explain the higher toxicity of curaxins to tumoral than normal cells, but inhibition of FACT cannot explain why chromatin destabilization is, in principle, toxic for cells.
6.2. TRAIN
Treatment of cells with curaxins at concentrations that cause nucleosome opening is accompanied by an increase in transcription of constitutive heterochromatin [38]. This response most likely reflects the loss of linker histone H1, which keeps the nucleosomes in a “closed” state. Because constitutive heterochromatin consists of multiple repeats, the transcripts originating from these repeats form double-stranded (ds) RNAs that activate the type I interferon (IFN) response, a phenomenon known as TRAIN (Transcription of Repeats Activates IFN) [39]. IFNs are known inducers of apoptosis (rev. in [62]). Moreover, type I IFNs are more toxic to transformed or tumor cells than to normal cells [63–67]. Therefore, it is possible that the induction of TRAIN could contribute to the higher toxicity of chromatin damage to tumoral than normal cells.
6.3. Disruption of long-range chromatin interactions
While curaxin treatment turns on transcription of heterochromatin, expression of a number of specific genes, including MYC, NF-kappaB, and HSF1 target genes, is shut down. The mechanism(s) responsible for reduced transcription of these genes was initially unclear because the same curaxin doses induced transcription of p53- and IFN-regulated genes, indicating that there was no general inhibition of transcription [10,38]. Our recent data demonstrated that suppression of gene expression by curaxins is determined at least in part by whether an enhancer element is involved in its expression. The activity of enhancers typically depends on long-range chromatin interactions and curaxin treatment disrupts these interactions in cells and cell-free conditions [68]. Therefore, I propose that proper nucleosome structure is needed for the long-range regulatory interactions controlling the expression of certain genes. However, this hypothesis has yet to be experimentally verified. Because super-enhancers play an important role in tumor cell viability [69], the effect of chromatin destabilization on long-range regulatory interactions could be an additional mechanism that contributes to the toxicity of curaxins in tumor cells.
6.4. Activation of p53
Activation of p53 by the curaxins suggests that p53 can respond to changes in chromatin structure or stability in the absence of DNA damage [10,11]. Despite extensive investigation, the mechanism(s) by which curaxins activate p53 are still not completely clear. When FACT is trapped in chromatin in curaxin-treated cells, FACT-associated CK2 phosphorylates p53 on serine 392, which results in p53 stabilization and full functional activation [10]. However, many details of this process, such as what exactly triggers CK2 towards p53 and how serine 392 phosphorylation makes p53 active, are still obscure. Another possible cause of p53 activation following curaxin treatment is the appearance of free histones that have been evicted from chromatin. Our group has found that histones accumulate in nucleoli if they are not accompanied by an appropriate histone chaperone or incorporated into chromatin [11,19]. In nucleoli, they can compete for binding to the histone chaperone nucleophosmin (B23) with ARF and several other nucleolar proteins that are known inhibitors of MDM2 [70–73]. Because MDM2 is a major negative regulator of p53, the release of ARF and other MDM2 inhibitors from the nucleoli caused by the accumulation of histones, could result in the activation of p53.
6.5. Histone “overdose”
It was previously shown that overexpression of histones (presumably accompanied by an increase in the pool of free histones) is highly toxic to cells [74]. This toxicity cannot be solely explained by the proposed mechanism by which free histones cause p53 activation through the release of MDM2 inhibitors because it has been observed in p53-deficient human cells and yeast lacking p53 [74]. The mechanism of histone toxicity in mammalian cells has not been adequately studied. Several scenarios can be proposed. First, histones are generally “sticky” to all nucleic acids and, therefore, may interfere with multiple DNA- and RNA-related processes. Alternatively, accumulation of “unattended histones” in nucleoli may be a mechanism to preserve cell viability until histone chaperones are recruited to incorporate them into chromatin, but this process can also cause nucleolar stress, a poorly understood phenomenon of nucleolar disintegration that is observed in cells treated with some DNA-damaging compounds [75]. Most, if not all, of these compounds cause chromatin damage in addition to DNA damage and, thus, could potentially drive histone accumulation in nucleoli. At the same time, significant histone loss from chromatin is observed at relatively high concentrations of chromatin-damaging drugs whereas toxicity occurs at doses that just cause chromatin opening.
6.6. Dysregulation of transcription
A final and potentially major mechanism underlying the toxicity of genome-wide chromatin destabilization is dysregulation of transcription. This hypothesis is based on the idea (which is suggested, but not well-elaborated on in the current literature) that nucleosomes present a much more serious barrier for transcription initiation than for elongation. The available data suggest that formation of the pre-initiating complex and initiation of transcription require nucleosome disassembly (rev. in [76], [77]). This process is supported by the occurrence of nucleosome-depleted regions at transcription start sites of transcribed genes (rev. in [77]). However, transcription elongation can occur in the presence of nucleosomes, as evidenced by the lack of any correlation between nucleosome occupancy in the coding regions of genes and the level of their transcription [78,79]. Moreover, nucleosomes are generally better organized (i.e., have higher occupancy and more accurate positioning) in transcribed regions of the genome (rev. in [80]). It is currently not completely clear what makes nucleosomes in cells “transparent” for polymerase passage. Co-factors, histone modifications, non-ideal nucleotide sequences (“ideal” nucleosome positioning sequences used in cell-free experiments do present a barrier for transcription elongation) or a combination of these may be involved in allowing elongation to proceed through the nucleosomes.
Because the presence of nucleosomes prevents initiation, but not elongation, of transcription, the major purpose of nucleosomes at the coding regions of transcribed genes may be to prevent cryptic initiation. It is well-known that elongating polymerase can degrade nucleosomes unless they are constantly restored by histone chaperones. Thus, if for some reason the polymerase elongates through the promoter region of an expressed gene, histone chaperones (e.g., FACT) add nucleosomes to the promoter to block initiation. In fact, the spt phenotype (Suppressor of Ty) from which the SPT16 subunit of FACT got its name is the loss of the inhibitory effect of expression of the Ty transposon on nearby genes, which is essential for yeast survival under special conditions. This effect may be achieved via inhibition of Ty expression or FACT nucleosome building activity. Thus, transcription of a non-coding RNA through the promoter of a nearby gene may lead to the inhibition of transcription of that gene. There are examples of this in yeast [81–85], and it likely occurs in higher eukaryotes, which means that pervasive transcription not only generates useless transcripts and activates endogenous viruses, but also leads to the loss or alterations of expression of necessary transcripts encoding proteins. It is likely that genome-wide opening of chromatin promotes multiple instances of cryptic transcription initiation which will, in turn, disturb expression of nearby genes. Whether these effects depend on the features of the genome landscape, such as the presence of cryptic initiation sites (e.g., LTRs), or the strength and frequency of the usage of nearby promoter elements is currently unclear. Moreover, the sequencing method, which is based on the analysis of short reads, does not allow accurate distinction between functional and pervasive transcripts. Therefore, cis-regulation of gene transcription within the context of all nearby coding and non-coding genomic elements (i.e., within the actual species-specific genomic landscape) is still awaiting careful investigation.
7. Conclusions and outlook
Chromatin damage represents the disassembly of nucleosomes with loss of histones from chromatin induced by small DNA-binding molecules independently of DNA damage. It may also occur following the loss of factors normally maintaining chromatin stability. The higher toxicity of chromatin-damaging drugs to tumoral than normal cells implies that tumor cells are more sensitive to chromatin destabilization. This hypothesis is likely the case because chromatin in tumor cells is already destabilized to some extent. Indeed, chromatin destabilization supports the epigenetic plasticity that enables cancer cells to thrive. Such destabilization may arise spontaneously due to the elevated chromatin dynamics in tumor cells, or because oncogenic mutations lead to transformation only when they occur in cells with destabilized chromatin (e.g., in cells transiting between epigenetic states). Exposure of tumor cells to chromatin-damaging drugs further destabilizes chromatin and results in cell death. The precise mechanisms that cause chromatin damage-induced cell death remain to be defined, but this presents a potentially new strategy for cancer treatment. The benefits of chromatin-damaging anti-cancer drugs include (i) broad efficacy due to the universality of DNA and nucleosome structure in all mammalian cells, and (ii) the low risk of development of drug resistance because mutations would have to occur simultaneously in several dozen histone genes. One limitation for this type of drug is that like DNA-damaging drugs, chromatin-damaging agents have a narrow therapeutic index and are likely to be toxic to at least some normal cells (e.g., stem cells). However, unlike the long-lasting toxicity associated with DNA damaging therapy, the toxicity from chromatin damage is likely temporary and should resolve after the end of drug administration. Multiple questions are still awaiting our experimental investigation, including the mechanisms controlling chromatin stability in cells, the existence of stress response pathways to chromatin destabilization, and the relationship between chromatin and DNA damage.
Acknowledgments:
I would like to thank Andrei Gudkov and members of my lab and students for critical reading and discussion of the manuscript; Incuron, Inc, Roswell Park Comprehensive Cancer Center, and the NIH (RO1CA197967) and Incuron, Inc for funding of our research. I am co-author of patent application “Carbazole compoundsand therapeutic use of the compounds”. I am consultant and recipient of research grants from Incuron, Inc.
References
- 1.Iranzo J, Martincorena I, Koonin EV, Proc. Natl. Acad. Sci. U S A 2018, 115, E6010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGranahan N, Swanton C, Cell, 2017, 168, 613. [DOI] [PubMed] [Google Scholar]
- 3.Dilruba S, Kalayda GV, Cancer Chemother. Pharmacol. 2016, 77, 1103. [DOI] [PubMed] [Google Scholar]
- 4.Cheung-Ong K, Giaever G, Nislow C. Chem. Biol. 2013, 20, 648. [DOI] [PubMed] [Google Scholar]
- 5.Ferraro A, Cell. Oncol. (Dordr), 2016, 39, 195. [DOI] [PubMed] [Google Scholar]
- 6.Fitzgerald DJ, Anderson JN, J. Biol. Chem. 1999, 274, 27128. [DOI] [PubMed] [Google Scholar]
- 7.Pang B, Qiao X, Janssen L, Velds A, Groothuis T, Kerkhoven R, Nieuwland M, Ovaa H, Rottenberg S, van Tellingen O, Janssen J, Huijgens P, Zwart W, Neefjes J, Nat. Commun. 2013, 4, 1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang F, Kemp CJ, Henikoff S, Curr. Biol. 2013, 23, 782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gurova KV, Hill JE, Guo C, Prokvolit A, Burdelya LG, Samoylova E, Khodyakova AV, Ganapathi R, Ganapathi M, Tararova ND, Bosykh D, Lvovskiy D, Webb TR, Stark GR, Gudkov AV, Proc. Natl. Acad. Sci. U S A, 2005, 102, 17448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gasparian AV, Burkhart CA, Purmal AA, Brodsky L, Pal M, Saranadasa M, Bosykh DA, Commane M, Guryanova OA, Pal S, Safina A, Sviridov S, Koman IE, Veith J, Komar AA, Gudkov AV, Gurova KV, Sci. Transl. Med. 2011, 3, 95–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Safina A, Cheney P, Pal M, Brodsky L, Ivanov A, Kirsanov K, Lesovaya E, Naberezhnov D, Nesher E, Koman I, Wang D, Wang J, Yakubovskaya M, Winkler D, Gurova K, Nucleic Acids Res. 2017, 45, 1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ, Nature, 1997, 389, 251. [DOI] [PubMed] [Google Scholar]
- 13.Barone TA, Burkhart CA, Safina A, Haderski G, Gurova KV, Purmal AA, Gudkov AV, Plunkett RJ, Neuro. Oncol. 2017, 19, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burkhart C, Fleyshman D, Kohrn R, Commane M, Garrigan J, Kurbatov V, Toshkov I, Ramachandran R, Martello L, Gurova KV, Oncotarget, 2014, 5, 11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carter DR, Murray J, Cheung BB, Gamble L, Koach J, Tsang J, Sutton S, Kalla H, Syed S, Gifford AJ, Issaeva N, Biktasova A, Atmadibrata B, Sun Y, Sokolowski N, Ling D, Kim PY, Webber H, Clark A, Ruhle M, Liu B, Oberthuer A, Fischer M, Byrne J, Saletta F, Thwe l. M., Purmal A, Haderski G, Burkhart C, Speleman F, De Preter K, Beckers A, Ziegler DS, Liu T, Gurova KV, Gudkov AV, Norris MD, Haber M, Marshall GM, Sci. Transl. Med. 2015, 7, 312–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dermawan JK, Hitomi M, Silver DJ, Wu Q, Sandlesh P, Sloan AE, Purmal AA, Gurova KV, Rich JN, Lathia JD, Stark GR, Venere M, Cancer Res. 2016, 76, 2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rabbani A, Iskandar M, Ausio J, J. Biol. Chem. 1999, 274, 18401. [DOI] [PubMed] [Google Scholar]
- 18.West SM, Rohs R, Mann RS, Honig B, J. Biomol. Struct. Dyn. 2010, 27, 861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nesher E, Safina A, Aljahdali I, Portwood S, Wang ES, Koman I, Wang J, Gurova KV, Cancer Res, 2018, 78, 1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wlodkowic D, Faley S, Darzynkiewicz Z, Cooper JM, Methods Mol. Biol. 2011, 731, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayakawa T, Noda M, Yasuda K, Yorifuji H, Taniguchi S, Miwa I, Sakura H, Terauchi Y, Hayashi J, Sharp GW, Kanazawa Y, Akanuma Y, Yazaki Y, Kadowaki Y, J. Biol. Chem. 1998, 273, 20300. [DOI] [PubMed] [Google Scholar]
- 22.Warrell RP Jr, Drugs Exp. Clin. Res. 1986, 12, 275. [PubMed] [Google Scholar]
- 23.Koman IE, Commane M, Paszkiewicz G, Hoonjan B, Pal S, Safina A, Toshkov I, Purmal AA, Wang D, Liu S, Morrison C, Gudkov AV, Gurova KV, Cancer Prev. Res. (Phila), 2012, 5, 1025. [DOI] [PubMed] [Google Scholar]
- 24.Boveri T, J. Cell Sci. 2008, 121 Suppl 1, 1. [DOI] [PubMed] [Google Scholar]
- 25.Zink D, Fischer AH, Nickerson JA, Nat. Rev. Cancer, 2004, 4, 677. [DOI] [PubMed] [Google Scholar]
- 26.Lupianez DG, Spielmann M, Mundlos S, Trends Genet. 2016, 32, 225. [DOI] [PubMed] [Google Scholar]
- 27.Flavahan WA, Gaskell E, Bernstein BE, Science, 2017, 357, 1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wainwright EN, Scaffidi P, Trends Cancer, 2017, 3, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parker SP, 1997. McGraw-Hill Encyclopedia of Science & Technology. In Parker SP, ed: McGraw-Hill; p 5000. [Google Scholar]
- 30. https://luysii.wordpress.com/2010/03/22/the-cell-nucleus-and-its-dna-on-a-human-scale-i/.
- 31.Shintomi K, Inoue F, Watanabe H, Ohsumi K, Ohsugi M, Hirano T, Science, 2017, 356, 1284. [DOI] [PubMed] [Google Scholar]
- 32.Matsui M, Chu Y, Zhang H, Gagnon KT, Shaikh S, Kuchimanchi S, Manoharan M, Corey DR, Janowski BA, Nucleic Acids Res. 2013, 41, 10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, Markenscoff-Papadimitriou E, Kuhl D, Bito H, Worley PF, Kreiman G, Greenberg ME, Nature 2010, 465, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neil H, Malabat C, d’Aubenton-Carafa Y, Xu Z, Steinmetz LM, Jacquier A, Nature, 2009, 457, 1038. [DOI] [PubMed] [Google Scholar]
- 35.Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Münster S, Camblong J, Guffanti E, Stutz F, Huber W, Steinmetz LM, Nature, 2009, 457, 1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cech TR, Steitz JA, Cell, 2014, 157, 77. [DOI] [PubMed] [Google Scholar]
- 37.Brocks D, Schmidt CR, Daskalakis M, Jang HS, Shah NM, Li D, Li J, Zhang B, Hou Y, Laudato S, Lipka DB, Schott J, Bierhoff H, Assenov Y, Helf M, Ressnerova A, Islam MS, Lindroth AM, Haas S, Essers M, Imbusch CD, Brors B, Oehme I, Witt O, Lübbert M, Mallm JP, Rippe K, Will R, Weichenhan D, Stoecklin G, Gerhäuser C, Oakes CC, Wang T, Plass C, 2017, Nat. Genet. 49, 1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leonova K, Safina A, Nesher E, Sandlesh P, Pratt R, Burkhart C, Lipchick B, Gitlin I, Frangou C, Koman I, Wang J, Kirsanov K, Yakubovskaya MG, Gudkov AV, Gurova K, Elife, 2018, 7, e30842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leonova KI, Brodsky L, Lipchick B, Pal M, Novototskaya L, Chenchik AA, Sen GC, Komarova EA, Gudkov AV, Proc. Natl. Acad. Sci. U S A, 2013, 110, E89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neri F, Rapelli S, Krepelova A Incarnato D, Parlato C, Basile G, Maldotti M, Anselmi F, Oliviero S, Nature 2017, 543, 72. [DOI] [PubMed] [Google Scholar]
- 41.Bersani F, Lee E, Kharchenko PV, Xu AW,Liu M, Xega K, MacKenzie OC, Brannigan BW, Wittner BS, Jung H, Ramaswamy S, Park PJ, Maheswaran S, Ting DT, Haber DA, Proc. Natl. Acad. Sci. U. S .A. 2015, 15148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dejardin J, Trends Genet. 2015, 31, 661–72. [DOI] [PubMed] [Google Scholar]
- 43.Ting DT, Lipson D, Paul S, Brannigan BW, Wittner BS, Jung H, Ramaswamy S, Park PJ, Maheswaran S, Ting DT, Haber DA, Science 2011, 331, 593.21233348 [Google Scholar]
- 44.Ferreira D, Meles S, Escudeiro A, Mendes-da-Silva A, Adega F, Chaves R, Chromosome Res. 2015, 23: 479. [DOI] [PubMed] [Google Scholar]
- 45.Dvinge H, Bradley RK, Genome Med. 2015, 7, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morgan MA, Shilatifard A, Genes Dev. 2015, 29, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carone DM, Lawrence JB, Semin. Cancer Biol. 2013, 23, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath L, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES, Cell 2006. 125: 315. [DOI] [PubMed] [Google Scholar]
- 49.Hammond CM, Stromme CB, Huang H, Patel DJ, Groth A, Nat. Rev. Mol. Cell Biol. 2017, 18, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keller DM, Lu H, J. Biol. Chem. 2002, 277, 50206. [DOI] [PubMed] [Google Scholar]
- 51.Keller DM, Zeng X, Wang Y, Zhang QH, Kapoor M, Shu H, Goodman R, Lozano G, Zhao Y, Lu H, Mol. Cell 2001, 7, 283. [DOI] [PubMed] [Google Scholar]
- 52.Garcia H, Miecznikowski JC, Safina A, Commane M, Ruusulehto A, Kilpinen S, Leach RW, Attwood K, Li Y, Degan S, Omilian AR, Guryanova O, Papantonopoulou O, Wang J, Buck M, Liu S, Morrison C, Gurova KV, Cell Rep. 2013, 4, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia H, Fleyshman D, Kolesnikova K, Safina A, Commane M, Paszkiewicz G, Omelian A, Morrison C, Gurova K, Oncotarget 2011, 2, 783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Orphanides G, LeRoy G, Chang CH, Luse DS, Reinberg D, Cell 1998, 92, 105. [DOI] [PubMed] [Google Scholar]
- 55.Orphanides G, Wu WH, Lane WS, Hampsey M, Reinberg D, Nature 1999, 400, 284. [DOI] [PubMed] [Google Scholar]
- 56.Gurova K, Studitskii VM, Biochimica et Biophysica Acta 2018. submitted. [Google Scholar]
- 57.Tsunaka Y, Fujiwara Y, Oyama T, Hirose S, Morikawa K, Genes Dev. 2016. 30, 673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang T, Liu Y, Edwards G, Krzizike D, Scherman H, Luger K, Life Sci. Alliance, 2018, 1, e201800107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krohn NM, Stemmer C, Fojan P, Grimm R, Grasser KD, J. Biol. Chem, 2003, 278, 12710. [DOI] [PubMed] [Google Scholar]
- 60.Li Y, Keller DM, Scott JD, Lu H, J. Biol. Chem. 2005, 280, 11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fleyshman D, Prendergast L, Safina A, Paszkiewicz G, Commane M, Morgan K, Attwood K,Gurova K, Oncotarget 2017, 8, 20525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chawla-Sarkar M, Lindner DJ, Liu YF, Williams BR, Sen GC, Silverman RH, Borden EC, Apoptosis 2003, 8, 237. [DOI] [PubMed] [Google Scholar]
- 63.Bekisz J, Baron S, Balinsky C, Morrow A, Pharmaceuticals K.C.Zoon (Basel), 2010, 3, 994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Otsuki T, Yamada O, Sakaguchi H, Tomokuni A, Wada H, Yawata Y, Ueki A, Br. J. Haematol. 1998, 103, 518. [DOI] [PubMed] [Google Scholar]
- 65.Parker BS, Rautela J, Hertzog PJ, Nat. Rev. Cancer 2016, 16, 131. [DOI] [PubMed] [Google Scholar]
- 66.Sanceau J, Hiscott J, Delattre O, Wietzerbin J, Oncogene 2000, 19, 3372. [DOI] [PubMed] [Google Scholar]
- 67.Thyrell L, Erickson S, Zhivotovsky B, Pokrovskaja K, Sangfelt O, Castro J, Einhorn S, Grandér D, Oncogene 2002, 21, 1251. [DOI] [PubMed] [Google Scholar]
- 68.Kantidze OL, Luzhin AV, Nizovtseva EV, Golov AK, Velichko AK, Lyubitelev AV, Feofanov AV, Gurova KV, Studitsky VM, Razin SV, Nature Communications 2018, sumitted. [Google Scholar]
- 69.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. Cell 2013, 155, 934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jin A, Itahana K, O’Keefe K, Zhang Y, Mol. Cell. Biol. 2004, 24, 7669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kurki S, Peltonen K,Latonen L, Kiviharju TM, Ojala PM, Meek D, Laiho M, Cancer Cell 2004, 5, 465. [DOI] [PubMed] [Google Scholar]
- 72.Lo D, Lu H, Cell Cycle 2010, 9, 3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D, Nat. Cell. Biol. 1999, 1, 20. [DOI] [PubMed] [Google Scholar]
- 74.Singh RK, Liang D, Gajjalaiahvari UR, Kabbaj MH, Paik J, Gunjan A, Cell Cycle 2010, 9, 4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.James A, Wang Y, Raje H, Rosby R, DiMario P, Nucleus 2014, 5, 402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang C, Pugh BF, Nat. Rev. Genet. 2009, 10, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Radman-Livaja M, Rando OJ, Dev. Biol. 2010, 339, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jonkers I, Kwak H, Lis JT, Elife 2014, 3, e02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Danko CG, Hah N, Luo X, Martins AL, Core L, Lis JT, Siepel A, Kraus WL, Mol. Cell. 2013, 50, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lai WKM, Pugh BF, Nat. Rev. Mol. Cell. Biol. 2017, 18, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Erkina TY, Erkine A, Cell Stress Chaperones 2015, 20, 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jamai A, Puglisi A, Strubin M, Mol. Cell. 2009, 35, 377. [DOI] [PubMed] [Google Scholar]
- 83.Morillo-Huesca M, Maya D, Munoz-Centeno MC, Singh RK, Oreal V, Reddy GU, Liang D, Géli V, Gunjan A, Chávez S, PLoS Genet. 2010, 6, e1000964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.True JD, Muldoon JJ, Carver MN, Poorey K, Shetty SJ, Bekiranov S, Auble DT, J. Biol. Chem. 2016, 291, 15307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Voth WP, Takahata S, Nishikawa JL, Metcalfe BM, Näär AM, Stillman DJ, PLoS One 2014, 9, e84092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Klutstein M, Nejman D, Greenfield R, Cedar H. Cancer Res. 2016, 76, 3446. [DOI] [PubMed] [Google Scholar]
- 87.Ehrlich M, Epigenomics 2009, 1, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Weinberg DN, Allis CD, Lu C, Cold Spring Harb. Perspect. Med, 2017, 7, a026443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vardabasso C, Hasson D, Ratnakumar K, Chung CY, Duarte LF, Bernstein E, Cell Mol. Life Sci. 2014, 71, 379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Scaffidi P, Biochim. Biophys. Acta. 2016, 1859, 533. [DOI] [PubMed] [Google Scholar]
- 91.Khan SA, Reddy D, Gupta S, World J Biol. Chem. 2015, 6, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hock R, Furusawa T, Ueda T, Bustin M, Trends Cell. Biol. 2007, 17, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dialynas GK, Vitalini MW, Wallrath LL, Mutat. Res. 2008, 647, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ceccacci E, Minucci S, Br. J. Cancer 2016, 114, 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen HP, Zhao YT, Zhao TC, Crit. Rev. Oncog. 2015, 20, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McCabe MT, Mohammad HP, Barbash O, Kruger RG, Cancer J 2017, 23, 292. [DOI] [PubMed] [Google Scholar]
- 97.Kadoch C, Crabtree GR, Sci. Adv. 2015, 1, e1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Burgess RJ, Zhang Z, Nat. Struct. Mol. Biol. 2013, 20, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Song SH, Kim TY, Genomics Inform. 2017, 15, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Arimura Y, Ikura M, Fujita R, Noda M, Kobayashi W, Horikoshi N, Sun J, Shi L, Kusakabe M, Harata M, Ohkawa Y, Tashiro S, Kimura H, Ikura T, Kurumizaka H. Nucleic Acids Res. 2018, gky661. [DOI] [PMC free article] [PubMed] [Google Scholar]