

Abstract
PABA/NO is a diazeniumdiolate of structure Me2NN(O)=NOAr (where Ar is a 5-substituted-2,4-dinitrophenyl ring whose 5-substituent is N-methyl-p-aminobenzoic acid). It has shown activity against human ovarian cancer xenografts in mice rivaling that of cisplatin, but it is poorly soluble and relatively unstable in water. Here we report structure-based optimization efforts resulting in three analogues with improved solubility and stability in aqueous solution. We sought to explain PABA/NO’s physicochemical uniqueness among these four compounds, whose aminobenzoic acid precursors differ structurally only in the presence or absence of the N-methyl group and/or the position of the carboxyl moiety (meta or para). Studies revealed that PABA/NO’s N-methyl-p-aminobenzoic acid substituent is bound to the dinitrobenzene ring via its carboxyl oxygen while the other three are linked through the aniline nitrogen. This constitutes a revision of the previously published PABA/NO structure. All four analogues reacted with GSH to produce bioactive nitric oxide (NO), but PABA/NO was the most reactive. Consistent with PABA/NO’s potent suppression of A2780 human ovarian cancer xenograft growth in mice, it was the most potent of the four in the OVCAR-3 cell line.
Introduction
PABA/NO is an O2-arylated diazeniumdiolate that has shown tumoristatic activity against A2780 human ovarian cancer xenografts in female SCID mice with a potency comparable to that of cisplatin.1 It was designed using a structure-based modeling approach to generate potentially cytolytic nitric oxide (NO) through mediation of π-class glutathione S-transferase (GST), which is overexpressed in many tumors. Consistent with the structure-based design considerations, PABA/NO was metabolized to NO more efficiently by GSTπ than by GSTα,1 which is the predominant isoform in the liver. Its cytotoxicity was shown to increase with increases in the intracellular expression levels of GSTπ. Conversely, elevated levels of multidrug resistance-associated protein 1 (MRP1), a key component of the cellular efflux pump system, conveyed enhanced resistance to the drug. PABA/NO also increased levels of phosphorylated and thus activated forms of the stress-activated protein kinases c-Jun NH2-terminal kinase (JNK) and p38, events that have been linked to drug-induced apoptosis.1
On the basis of its in vivo activity and the relevant mechanistic findings summarized above, we consider PABA/NO to be a promising anticancer lead deserving further preclinical characterization. In the present work, we compare it with three close structural analogues with regard to structure, solubility, hydrolytic stability, reactivity with GSH, and in vitro anticancer activity.
Results
Synthesis and Solubility.
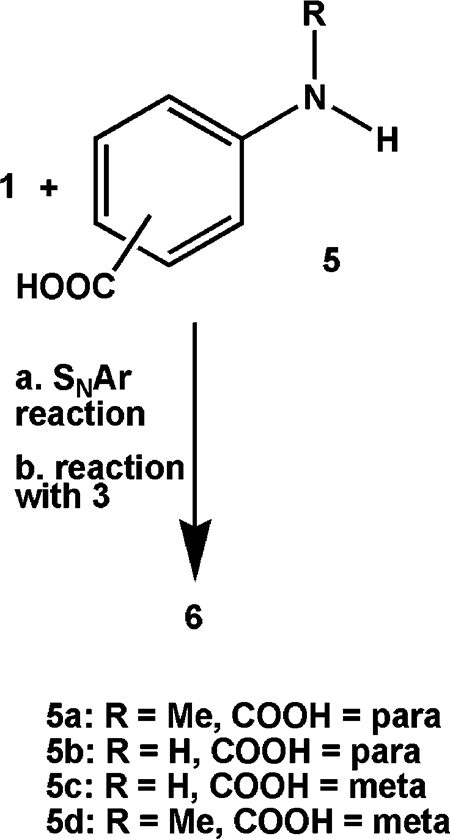
A milestone in our structure-based drug design effort was the synthesis of 4, which satisfied all the criteria for a good fit in the active site of GSTπ but was predicted to be poorly accommodated in that of GSTα. It was synthesized as in Scheme 1 and confirmed by X-ray crystallography to have the structure shown,2 but it proved too insoluble to test. Reasoning that a carboxyl group should overcome that barrier, we substituted N-methylated PABA (p-aminobenzoic acid) 5a for N-methylaniline in the reaction of Scheme 1 (see also eq 1) and isolated PABA/NO (6a). Encouraged by the promising initial testing results described above but concerned about its low solubility (<2 μM), we repeated the reaction of Scheme 1 using 5a analogues 5b, 5c, and 5d in place of N-methylated PABA and isolated three new compounds, 6b-6d. All were 3 orders of magnitude more soluble in pH 7.4 buffer (Table 1).
 |
(1) |
Scheme 1.

Synthesis of 42
Table 1.
Physicochemical Parameters for PABA/NO and Three Analogues
| compound | solubilitya (mM) | khydrolysisb (s−1) | kGSHc (M−1 s−1) | νC=O (cm−1) |
|---|---|---|---|---|
| 6a (PABA/NO) | <0.002 | (4.9 ± 2.4) × 10−4 d | 0.31 ± 0.02d | 1730 |
| 6b | >3 | ~0 | 0.043 ± 0.003 | 1685 |
| 6c | >2 | ~0 | 0.056 ± 0.003 | 1684 |
| 6d | 3 | ~0 | 0.13 ± 0.004 | 1685 |
Maximum solubility in 0.1 M phosphate buffer, 37 °C.
First-order rate constants for hydrolysis at pH 7.4 and, except as noted, 37 °C.
Secondorder rate constants for reaction with glutathione at pH 7.4 and, except as noted, 37 °C.
Rate data for PABA/NO were obtained at room temperature.
Structure Assignments, Including Revisions.
The significant difference in solubility between PABA/NO and its three analogues suggested that PABA/NO is fundamentally different from the other three, despite the fact that all four compounds were prepared by the same two-step (difluoride 1 plus substituted aniline 5, then product plus diazeniumdiolate ion 3) reaction sequence as 4, whose structure was confirmed as shown in Scheme 1 by X-ray crystallography.
PABA/NO differed from its three analogues not only in its solubility at physiological pH but also in the fact that it was the only one of the four that was not soluble in sodium bicarbonate solution. Moreover, its infrared carbonyl stretching frequency was the only one to appear at 1730 cm−1, consistent with an ester linkage, with the other three in the free carboxylate range, 1684–1685 cm−1. These physical data strongly suggest that PABA/NO is the only one of the four without a free carboxyl group, the PABA moiety being bound to the remainder of the molecule through an ester linkage rather than via the anilino nitrogen. This was confirmed by X-ray crystallography, which provided an unambiguous view of the connectivity (Figure 1).
Figure 1.

Structure of 6a showing the labeling of the non-hydrogen atoms. Thermal ellipsoids are shown at the 50% probability level.
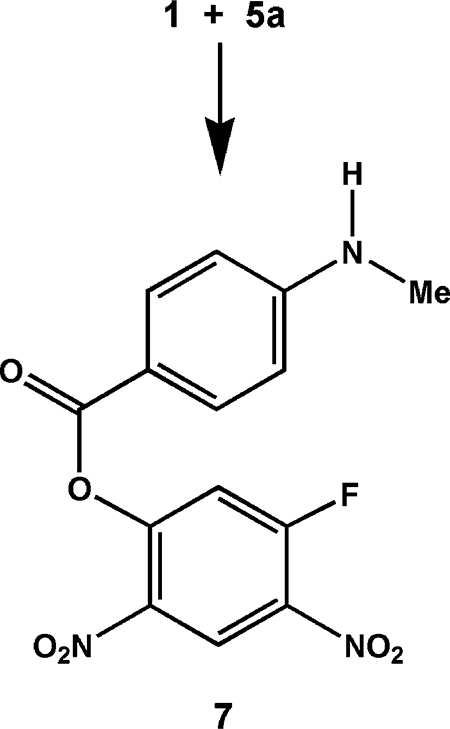
On the basis of these physicochemical data, summarized in Table 1 and the Supporting Information, we conclude that the structures of these compounds are as shown in Scheme 2. Thus, PABA/NO has structure 6a, a revision of the one reported previously.1 This implies that the structure of its immediate precursor, i.e., the one formed by reaction of 1 with 5a as shown in eq 1, was also incorrect as originally assigned1 and must be revised. The correct connectivity is as shown in eq 2, structure 7.
 |
(2) |
Scheme 2.

Structures of PABA/NO and Three Analogues Investigated Here
While the infrared and acid–base behavior of 6b, 6c, and 6d strongly support the structural assignments listed in Scheme 2, we sought confirmation through further crystallographic studies. Thus far, only 6c has yielded a suitable crystal (Figure 2), which crystallized with four molecules in the asymmetric unit, three of which were disordered. In these three molecules, the N3 atom and the two attached carbons of each dimethylamino group were disordered over two positions, with ratios of 48:52, 69:31, and 48:52, respectively. This contributed to the poor scattering exhibited by the crystal and resulted in slightly higher R1 and wR2 values than typically expected. Structurally, the four unique molecules varied significantly only in the orientation of the benzoate ring (Figure S1 of Supporting Information). The torsion angles about the N7–C8 bonds of molecules A and D were −35.5° and −37.0°, respectively, while molecules B and C have torsion angles of 44.5° and 36.2°, respectively.
Figure 2.

Structure of 6c showing the labeling of the non-hydrogen atoms. Thermal ellipsoids are shown at the 30% probability level. Only one of the four molecules in the asymmetric unit is shown. Molecules 2–4 as well as disordered positions for N3, C3′, and C3″ are omitted for clarity.
Hydrolysis and Reaction with GSH.
PABA/NO also distinguished itself from 6b-6d in its reactivity. Its reaction rates could not be followed easily by the standard ultraviolet method used for the other three compounds, partly because of the complexity of the reaction and partly because the products’ spectra were closely similar to that of PABA/NO. Consequently, the reactivity of 6a was characterized by plotting the decrease in its HPLC peak area as a function of time. Because its hydrolysis and its reaction with glutathione were too fast to follow reliably by this method at 37 °C, its rate constants were determined at room temperature. The results are given in Table 1. Despite the lower temperature (by approximately 15 °C), PABA/NO’s kGSH was 2–7 times as great as those of its analogues. Chemiluminescence traces confirming the generation of NO in the reactions of 6a–6d with GSH are shown in Figures S2 and S3 of Supporting Information.
Hydrolysis of PABA/NO (6a) occurred at both the ester and diazeniumdiolate groups (Scheme 3), so the observed khydrolysis rate reflects a combination of two parallel processes. Integrating the HPLC peaks for products 8 and 9 showed that hydrolysis of the ester was at least an order of magnitude faster than cleavage of the diazeniumdiolate group. Once hydrolysis occurred at one position, though, further hydrolysis became extremely slow. No significant hydrolysis of analogues 6b–6d was observed under these conditions.
Scheme 3.

PABA/NO Hydrolysis Products Determined by HPLC, LC/MS, and Chemiluminescence Analysis
To further probe PABA/NO’s reactivity with GSH, we conducted the reaction in the presence of GSTμ: to catalyze the initial glutathionylation. GSH adduct 10 was formed very rapidly, dominating the HPLC–UV trace within 35 min of mixing (Figure 3); NO was seen as a byproduct by chemiluminescence. Cleavage of the ester function of 6a also occurred, giving rise to 8. On further incubation at 37 °C, the ester group of 10 was cleaved to generate hydrolysis products 5a and 11. The products formed on prolonged incubation under these conditions are shown in Scheme 4.
Figure 3.

Chromatograms of reaction mixtures containing 5 μM PABA/NO (6a), 0.1 mM GSH, and 2.9 nM GSTμ (molecular weight 25.7 kDa) in 0.1 M phosphate at pH 7.4 and 37 °C at (A) 2 min and (B) 35 min after mixing.
Scheme 4.

Products Observed in the Reaction of PABA/NO (6a) with Glutathione (GSH) under Catalysis by Glutathione S-Transferase μ (GSTμ) in 0.1 M Phosphate Buffer (pH 7.4)a
a *Organic product identities were confirmed by mass spectrometry or HPLC, with the exception** of 3 and dimethylamine. Production of NO was confirmed by chemiluminescence.
Biological Results.
Initial in vitro screening was performed with HL-60 human leukemia cell cultures. Results were disappointing, giving IC50 values that were at least an order of magnitude higher than that of JS-K, an O2-arylated diazeniumdiolate that has consistently shown sub-micromolar potency in this screen and has proven active against human leukemia and prostate cancer xenografts in NOD-SCID mice.3 Similar results were seen with NIH3T3 mouse skin fibroblasts, a cell line used in the mechanistic studies with PABA/NO described earlier.1 IC50 values for compounds 6a–6d in these two cell lines are summarized in the Supporting Information.
The compounds were next subjected to the NCI 51-cell-line screen. The dose-response curves revealed that the overall inhibitory potency of PABA/NO and its three analogues ranked in the order 6a > 6b > 6c > 6d. PABA/NO exhibited the greatest growth inhibition against all the tested cell lines with a mean GI50 value of 9.8 μM, a mean TGI value of 33 μM, and a mean LC50 value of 69 μM. Analogue 6b showed the next greatest potency in inhibiting the 51 cell lines with a mean GI50 value of 23 μM, a mean TGI value of 52 μM, and mean LC50 value of 85 μM. Compound 6c inhibited the 51 cell lines with a mean GI50 value of 68 μM, a mean TGI value of 96 μM, and a mean LC50 value of more than 100 μM. Compound 6d showed the least inhibitory potency with mean GI50, TGI, and LC50 values of more than 100 μM.
Among the 51 cell lines, colon cancer HCT-15, melanoma LOXIMVI, ovarian OVCAR-3, and renal CAKI-1 showed the greatest sensitivity to the compounds. Their proliferation was most inhibited by PABA/NO and next by 6c (Table 2). The low GI50 along with low LC50 values indicate that PABA/NO has both strong cytostatic and cytotoxic effects on these four cell lines, while 6c has cytotoxic effects less potent than PABA/ NO’s.
Table 2.
Cytotoxic Concentrations (μM) of 6a and 6c against the Most Sensitive Human Cancer Cell Lines in the NCI 51-Cell-Line Screena
| PABA/NO (6a) |
analogue 6c |
|||||
|---|---|---|---|---|---|---|
| cell line | GI50 | TGI | LC50 | GI50 | TGI | LC50 |
| colon HCT-15 | 2.0 | 5.3 | 19 | 20 | 36 | 68 |
| melanoma LOXIMVI | 1.9 | 4.4 | 10 | 19 | 3.4 | 60 |
| ovarian OVCAR-3 | 2.6 | 6.9 | 2.6 | 3.9 | 30 | 58 |
| renal CAKI-1 | 1.8 | 4.0 | 8.9 | 2.2 | 51 | >100 |
Compound 6b inhibited the ovarian OVCAR-3 cells with GI50 = 5.5 μM, TGI = 9 μM, and LC50 = 100 μM, and the other three cell lines with GI50, TGI, and LC50 > 100 μM. Analogue 6d was inactive at a concentration of 100 μM.
Discussion
This work was initiated in an effort to improve on structure 12 (JS-K, Scheme 5) as an anticancer lead. JS-K was active against both HL-60 human leukemia and PPC-1 human prostate cancer xenografts in NOD-SCID mice, but it was metabolized to the putative cytostatic/cytotoxic intermediate nitric oxide almost 100-fold more efficiently by the α isoform of GST than by the enzyme’s π isoform;3 π-susceptibility should allow for greater concentration of this active metabolite in tumor tissue, given that GSTπ expression is upregulated in many malignant cell types. Also, given the importance of the α isoform to cellular well-being, we sought to decrease the α-susceptibility so as to spare normal, α-expressing tissue from potentially deleterious collateral exposure to NO.
Scheme 5.

Results of Molecular Modeling Experiments1 Leading to the Design of PABA/NO (6a) as a Potential GSTπ-Selective Improvement over JS-K [12, O2-(2,4-Dinitrophenyl) 1-[(4-Ethoxycarbonyl)piperazin-l-yl]diazen-l-ium-1,2-diolate]
To these ends, an earlier structure-based drug design exercise3 involving transition state modeling of O2-arylated diazeniumdiolate structures in the active sites of three GST isoforms was conducted. The results (Scheme 5) suggested that decreasing the bulk on the amine side of the N2O2 group should make the substrate more π-compatible, while adding a sterically demanding substituent at the 5-position of the aryl ring should decrease its accommodation in the α active site. These considerations led to the synthesis of PABA/NO (6a), which was indeed metabolized more efficiently by GSTπ than GSTα, if only slightly so, and showed promising in vivo activity against a human ovarian cancer xenograft in SCID mice.1 As may have been predicted, in vitro experiments implicated proteins involved in clearing electrophilic species from the cell (GSTπ, MRP1, and γCS) in PABA/NO’s mechanism of action. The drug was also shown to activate the MAPK suite of protein kinases (ERK, JNK, and p38). Unfortunately, PABA/NO was not very soluble, and its spontaneous hydrolysis and its uncatalyzed reaction with glutathione were more rapid than might be desired, given the goal of systemically administering it for selective metabolism only in GSTπ-overexpressing cells without collaterally exposing other NO-sensitive compartments.
In view of PABA/NO’s promising activity against human ovarian cancer xenografts in mice, however, we began a lead optimization effort by preparing analogues 6b–6d and compared their chemistry with that of PABA/NO. All were orders of magnitude more soluble than PABA/NO. They were also significantly less reactive with GSH in the absence of GST. Unfortunately, their in vitro anticancer activity was not improved over that of PABA/NO.
With the view that a full physicochemical characterization of a medicinally promising agent is a crucial prerequisite to systematic lead optimization, we report here on the fundamental properties of PABA/NO. Not only have we provided new details concerning its reactivity, but a comparison with its analogues has revealed the need for a revision in its structure. PABA/NO is an ester, as diagrammed in Schemes 2 and 4, rather than the diarylamine derivative that was originally1 reported.
It appears that resonance interaction with the electron-withdrawing p-carboxyl group of 5a diminishes the nucleophilicity of the methylamino nitrogen, allowing a carboxyl oxygen to displace a fluoride ion of 1 by default, as shown in eq 2. It is understandable that this resonance effect would not be operative when the carboxyl group is meta to the amino nitrogen, leading to the diarylamine connectivity shown in Scheme 2 for 6c and 6d, but it is surprising that 5b did not displace a fluoride of 1 via its oxygen as 5a did. Apparently, the N-methyl group of 5a that distinguishes it from 5b, whose presence might have been expected to increase the inherent nucleophilicity of the nitrogen, offers enough steric interference at the nitrogen center to force attack on 1 by the oxygen of 5a.
We have been unable thus far to confirm the structural assignment of 6b by X-ray crystallography, but the structure shown in Scheme 2 is consistent with the data at hand and seems secure. Unlike PABA/NO (νC=O ) 1730 cm−1), but like 6c and 6d, 6b has a carbonyl stretching frequency (1685 cm−1) typical of a free carboxylic acid instead of an ester grouping (Table 1). The greater solubility of 6b as well as 6c and 6d (but not 6a) in mild alkali than in acid also supports the presence of a free carboxylate function in 6b–6d.
The significantly greater overall activity of 6a relative to its three analogues in the 51-cell-line screen appears to correlate with its greater reactivity and unique structure, possibly also with its greater lipophilicity (hence membrane permeability). Future work will be aimed at preparing differently substituted O2-arylated diazeniumdiolates patterned after the core PABA/NO structure in an effort to increase its solubility without lowering its cytostatic/cytotoxic activity against tumor cells, as well as to decrease its reactivity toward GSH in the absence of GST. With all three of the in vivo tests that have to our knowledge been conducted with O2-arylated diazeniumdiolates in anticancer screens thus far having been positive, we are hopeful that continued lead optimization work will generate further improvements still.
Experimental Section
NO was purchased from Matheson Gas Products (Montgomeryville, PA). Starting materials were purchased from Aldrich Chemical Co. (Milwaukee, WI) unless otherwise indicated. Proton NMR spectra were obtained in chloroform-d, acetone-d6, or dimethyl sulfoxide-d6. Chemical shifts (δ) are reported in parts per million (ppm) downfield from tetramethylsilane. Quantification of NO by chemiluminescence was done with a Thermal Energy Analyzer Model 502A instrument (Thermedics, Inc., Woburn, MA), as previously described.4 Elemental analyses were performed by Atlantic Microlab, Inc. (Norcross, GA) or Midwest Analytic (Indianapolis, IN). Compounds 6a and 7 were prepared as previously described1 (though see structure revisions reported above). The sodium salt of 3 was prepared as described earlier.2
4-[N-(2,4-Dinitro-5-fluorophenyl)amino]benzoic Acid.
A solution of 3.84 g (0.0187 mol) of 1,5-difluoro-2,4-dinitrobenzene in 50 mL of acetone was placed in a 500-mL flask equipped with a stirring bar and a pressure-equalizing addition funnel. To the stirring solution was added 2.56 g (0.0187 mol) of 4-aminobenzoic acid 5b in 50 mL of acetone. The resulting bright yellow solution was stirred at room temperature and the progress of the reaction was monitored by TLC on silica gel plates using 10:1 dichloromethane: methanol as the developer. After stirring for 24 h at room temperature, the reaction mixture was heated at reflux for 8 h. Filtration and evaporation left a residue that was recrystallized from ethanol:water to give 2.6 g of red-orange plates: mp 238–239 °C; 1H NMR (DMSO-d6) δ 7.10 (d, 2H, J = 14 Hz), 7.49–7.54 (m, 2H), 8.01–8.05 (m, 2H), 8.92 (d, 1H, J = 7.96 Hz), 10.24 (b, 1H), 12.99 (b, 1H). Anal. Calcd for C13H8N3O6F: C, 48.61; H, 2.51; N, 13.08. Found: C, 48.65; H, 2.60; N, 12.97.
O2-[2,4-Dinitro-5-(4-carboxylatophenyl)amino]phenyl 1-(N,N-Dimethylamino)diazen-1-ium-1,2-diolate (JS-38–12) (6b).
A solution of 731 mg (2.28 mmol) of 4-[N-(2,4-dinitro-5-fluorophenyl)-amino]benzoic acid in DMSO was added to a slurry of 508 mg (4 mmol) of 3 (sodium salt) in tetrahydrofuran and cooled to 0 °C under nitrogen. After gradually warming to room temperature and then stirring for another day, the reaction mixture was poured into ice-cold hydrochloric acid. The product was extracted with ethyl acetate, and the organic layer was extracted with 5% sodium bicarbonate. The aqueous layer was washed with dichloromethane, acidified, and extracted with ethyl acetate. The organic extract was dried and evaporated to give 414 mg of crude product. Purification of the product required two subsequent recrystallizations from ethanol: mp 163–165 °C; 1H NMR (DMSO-d6) δ 3.07 (s, 6H), 7.19 (s, 1H), 7.55 (d, 2H, J = 8.6 Hz), 8.02 (d, 2H, J = 8.4 Hz), 8.92 (s, 1H), 10.19 (s, 1H), 13.0 (b, 1H). Anal. Calcd for C15H14N6O8•0.5 ethanol: C, 44.76; H, 3.47; N, 19.57. Found: C, 44.90; H, 3.55; N 19.41.
3-[N-(2,4-Dinitro-5-fluorophenyl)amino]benzoic Acid.
A solution of 6.42 g (0.0313 mol) of 1,5-difluoro-2,4-dinitrobenzene in 100 mL of acetone was placed in a 500-mL flask equipped with a stirring bar and a pressure-equalizing addition funnel. To the stirred solution was added 4.29 g (0.0313 mol) of 3-aminobenzoic acid 5c in 100 mL of acetone. The resulting bright yellow solution was stirred at room temperature and the progress of the reaction was monitored by TLC on silica gel plates using 10:1 dichloromethane: methanol as the eluant. After 24 h the reaction mixture was filtered to remove any acetone-insoluble particles, and the yellow filtrate was evaporated under vacuum to give a yellow solid. The product was triturated with acetone:ether, collected by filtration, and dried under vacuum to give 7.12 g (71%) of product: mp 239–240 °C; UV (methanol) λmax 342 nm (ϵ = 8.9 mM−1 cm−1); 1H NMR (CDCl3) δ 6.86 (d, 1H, J = 14 Hz), 7.18–7.32 (m, 1H), 7.63–7.66 (m, 2H), 7.92 (s, 2H), 8.93 (d, 1H, J ) 8.0 Hz), 10.27 (s, 1H). Anal. Calcd for C13H8N3O6F: C, 48.61; H, 2.51; N, 13.08. Found: C, 48.21; H, 2.64; N, 12.73.
O2-[2,4-Dinitro-5-(3-carboxylatophenyl)amino]phenyl 1-(N,N-Dimethylamino)diazen-1-ium-1,2-diolate (JS-36–120) (6c).
A slurry of 800 mg (6.3 mmol) of 3 (sodium salt) in 25 mL of tetrahydrofuran was cooled to 0 °C under nitrogen. A solution of 836 mg (2.6 mmol) of 3-[N-(2,4-dinitro-5-fluorophenyl)amino]-benzoic acid in 15 mL of dimethyl sulfoxide (DMSO) was introduced slowly through a syringe and gradually allowed to warm to room temperature. After 24 h the solution was poured over ice–water containing hydrochloric acid and the product was extracted with ethyl acetate. The ethyl acetate solution was extracted with 5% sodium bicarbonate solution. The aqueous layer was separated and washed with dichloromethane to remove the DMSO and then acidified with dilute hydrochloric acid, and the product was extracted with ethyl acetate. The organic layer was dried over sodium sulfate, filtered through magnesium sulfate, and concentrated on a rotary evaporator to give 501 mg of product that was recrystallized from acetone: mp 175–176 °C; UV (in 5% sodium bicarbonate solution) λmax 354 nm (ϵ = 20.5 mM−1 cm−1); 1H NMR (CDCl3) δ 3.02 (s, 6H), 6.98 (s, 1H), 7.63–7.67 (m, 2H), 7.88–7.98 (m, 2H), 8.93 (s, 1H), 10.23 (s, 1H); 13C NMR (CDCl3) δ 100.36 ppm, 126.03, 126.59, 126.96, 127.19, 127.39, 129.97, 132.16, 138.10, 146.67, 154.14, 166.52. Anal. Calcd for C15H14N6O8•0.2 acetone: C, 44.83; H, 3.67; N, 20.11. Found: C, 44.83; H, 3.55; N, 19.82.
3-Methylaminobenzoic Acid (5d).
3-Aminobenzoic acid (42.5 g; 0.31 mol) was dissolved in 600 mL of water containing 25 g (0.62 mol) of sodium hydroxide. To the basic solution was added 29 mL (0.31 mol) of dimethyl sulfate, and the resulting mixture was stirred at room temperature overnight. The aqueous solution was washed with dichloromethane to remove any neutral organic material, acidified with 10% hydrochloric acid, and extracted with ethyl acetate. The solution was dried over sodium sulfate and filtered through magnesium sulfate. The solvent was removed on a rotary evaporator to give 13 g of a solid that contained the desired product along with starting material and 3-(N,N-dimethylamino)benzoic acid. The mixture was adsorbed on 20 g of silica gel and deposited on a 1.1- × 15-cm precolumn reservoir connected to a prepacked silica gel column (2.7 × 29 cm) that had been conditioned with 5:1 dichloromethane:acetonitrile and eluted with the same solvent system to give fractions containing 3.03 g of 3-methylaminobenzoic acid (5d): mp 115–116 °C (lit.5 mp 127 °C); 1H NMR (acetone-d6) δ 2.82 (s, 2H), 6.81–6.86 (m, 1H), 7.25–7.27 (m, 3H). Anal. Calcd for C8H9NO2: C, 63.56; H, 6.00; N, 9.27. Found: C, 63.54; H, 5.94; N 9.19.
3-[N-(2,4-Dinitro-5-fluoro)-N-methylamino]benzoic Acid.
A solution of 1.10 g (0.0073 mol) of 3-methylaminobenzoic acid (5d) in 50 mL of 5% sodium bicarbonate solution was cooled in an ice-bath. A solution of 1.50 g (0.0073 mol) of 1,5-difluoro-2,4-dinitrobenzene in 15 mL of tetrahydrofuran and 25 mL of tert-butyl alcohol was added. The resulting orange reaction mixture was stirred for 4 h, concentrated under vacuum, extracted with water, and washed with ethyl acetate. The aqueous solution was acidified with 10% hydrochloric acid, extracted with ethyl acetate, dried over sodium sulfate, filtered, and concentrated under vacuum to give 538 mg of an orange glass. The crude product was flash chromatographed on silica gel using a glass column and eluted with 5:1 dichloromethane:ethyl acetate. The fractions containing the desired product were pooled and evaporated to give 342 mg of a yellow powder: mp 95–96 °C; 1H NMR (CDCl3) δ 3.50 (s, 3H), 7.00 (d, 1H, J = 12.9 Hz), 7.45–7.60 (m, 2H), 7.74–7.97 (m, 2H), 8.62 (d, 1H, J = 7.8 Hz). Anal. Calcd for C14H10N3O6FC•1/3H2O: C, 49.27; H, 3.15; N, 12.31. Found: C, 49.10; H, 3.21; N, 12.11.
O2-{2,4-Dinitro-5-[N-(3-carboxylatophenyl)-N-methyl]amino}phenyl 1-(N,N-Dimethylamino)diazen-1-ium-1,2-diolate (JS-39–47) (6d).
A slurry of 239 mg (1.88 mmol) of 3 (as the sodium salt) in 2.5 mL of tetrahydrofuran under nitrogen was cooled to 0 °C. A solution of 324 mg (0.91 mmol) of 3-[N-(2,4-dinitro-5-fluoro)-N-methylamino]-benzoic acid in 2.5 mL of tetrahydrofuran was added to the cold slurry, which was gradually allowed to warm to room temperature with stirring overnight. Ethyl acetate was added and the mixture was poured over dilute hydrochloric acid. The aqueous portion was discarded and the organic layer was extracted with 5% sodium bicarbonate. The aqueous solution was separated, acidified with hydrochloric acid, and extracted with ethyl acetate. The solution was dried over sodium sulfate, filtered, and concentrated under vacuum to give 196 mg of a brown glass. Flash chromatography of the material was performed on silica gel using a 1.3- × 13.5-cm glass column and elution with 9:1 dichloromethane:methanol. A second round of chromatography was necessary for better purification, giving 12 mg of product: mp 150–153 °C; 1H NMR (DMSO-d6) δ 3.21 (s, 6H), 3.49 (s, 3H), 7.21–7.34 (m, 2H), 7.37 (s, 1H), 7.57–7.69 (m, 2H), 8.56 (s, 1H). Anal. Calcd for C16H16N6O8•0.5H2O: C, 44.76; H, 3.99; N, 19.57. Found: C, 44.58; H, 3.86; N, 19.34.
Characterization of GSH Adduct 10 Isolated from the GSTμ-Catalyzed Reaction of PABA/NO with GSH.
A solution of PABA/NO (50 μM final concentration), GSH (2 mM final concentration), and GSTμ (3 fμ/mL final concentration) in 100 mM phosphate containing 1% DMSO, pH 7.4, was incubated at 37 °C for 5 min, whereupon the entire 1-mL volume was injected into a 1-mL loop for isolation by HPLC. The mobile phase consisted of 25:75 acetonitrile:water with 0.1% formic acid for 5 min followed by a linear gradient to 70:30 acetonitrile:water over the next 10 min at a flow rate of 1 mL/min. The separation was performed on a Phenomenex 5-μm C18 Luna (2) column (250 × 4.6 mm) with 315-nm detection.
The product was isolated by collecting the fraction at 13.8 min, which had a λmax of 320 nm. The solution was frozen and lyophilized. The resulting off-white solid was stored in the refrigerator overnight. For LC–MS, the residue was dissolved in 100 mM, pH 7.4 sodium phosphate buffer and sampled periodically over 3 h. Initially, there was a very large peak in the total ion chromatogram with an (M + H)+ value of 623 m/z, corresponding to 10. On continued incubation, the intensity of this peak began to decrease, giving way to two other peaks having (M + H)+ values of 490 and 152 m/z, corresponding to 11 and 5a, respectively.
Hydrolysis of PABA/NO at Physiological pH.
PABA/NO (final concentration 5 μM) in 1 mL of 0.1 M, pH 7.4 phosphate buffer containing 0.1% DMSO was analyzed for hydrolytic decomposition by HPLC. The HPLC conditions utilized consisted of acetonitrile: water with 0.1% formic acid added, and the separation was performed using a Phenomenex 5-μm C18 Luna (2) column (250 × 4.6 mm) at a flow rate of 1 mL/min with 315-nm detection. The gradient consisted of 25% acetonitrile for 5 min, followed by a linear gradient to 70% acetonitrile over the next 10 min. Upon adding the PABA/NO to the buffer, the solution was immediately analyzed using the above HPLC conditions. A second analysis was performed after the solution had been incubated at 37 °C for 35 min.
Anticancer Screen of PABA/NO Analogues against the NCI 51 Human Cancer Cell Lines.
The anticancer screening test was conducted as previously described.6,7 Cells from nine cancer subpanels of 51 human lines were grown in RPMI 1640 culture medium supplemented with 5% fetal bovine serum and 2 mM L-glutamine for 24 h at 37 °C to allow stabilization prior to addition of the PABA/NO analogues. The nine human cancer subpanels are leukemia, non-small-cell lung, colon, melanoma, ovarian, renal, prostate, breast, and central nervous system cancer. Stock solutions of the analogues in DMSO were serially diluted with the RPMI 1640 medium and added immediately to the microtiter plates to produce five concentrations of the analogues, i.e., 10−4, 10−5, 10−6, 10−7, and 10−8 M. Each analogue was incubated with cells for 48 h. At the end of the incubation, the cells were fixed in situ by 10% trichloroacetic acid, washed five times with water, and dried. Sulforhodamine B (0.4% in 1% acetic acid) was added to each well and incubated for 10 min at room temperature. Unbound sulforhodamine B was removed by washing five times with acetic acid. Then the plates were air-dried. Bound stain was solubilized with Tris buffer, and the optical densities were read at 515 nm. The optical densities generated from sulforhodamine B staining are a function of cell mass and growth rate. The dose–response curve (not shown) was created by plotting the percent growth against the log of the corresponding PABA/NO analogue’s concentrations for each cell line and grouped by disease subpanels. The molar analogue concentrations that cause 50% growth inhibition (GI50), total growth inhibition (TGI), and 50% cell killing (LC50) of the 51 cell lines were determined, respectively.
Rate Constant Determinations.
The reaction of 5 μM PABA/NO with GSH was monitored by HPLC analysis of PABA/NO concentrations, obtained from peak areas with reference to a 5 μM PABA/NO standard as a function of time, in 0.10 M phosphate buffer at pH 7.4. The greater solubility and simpler decomposition behavior of the three PABA/NO analogues permitted their reactions to be monitored directly by ultraviolet spectrophotometry using a Hewlett-Packard 8452A diode-array spectrophotometer. The wavelengths followed were 341, 335, and 346 nm for 6b, 6c, and 6d, respectively, and their starting concentrations were 20, 10, and 50 μM, respectively. The decreases in substrate concentration displayed excellent first-order behavior over several half-lives. Pseudo-firstorder rate constants were obtained with GSH in large excess of substrate. Measured values showed a linear dependence on the concentration of GSH. The slopes and y-intercepts of the plots yielded rate constants for the reactions with GSH and water, respectively.
Single-Crystal X-ray Diffraction Studies.
For 6a, a clear, orange needle of dimensions 0.08 × 0.09 × 0.43 mm3 was mounted on glass fiber using a small amount of Cargille immersion oil. Data were collected on a Bruker four-circle diffractometer equipped with a SMART 1000 CCD detector. The crystals were irradiated using graphite-monochromated Mo Kα radiation (λ = 0.71073). An MSC X-Stream low-temperature device was used to keep the crystals at a constant −170 °C during data collection. Data collection was performed and the unit cell was initially refined using SMART (v5.625).8 The crystals were monoclinic, P21/n, with a = 14.153(4) Å, b = 7.881(2) Å, c = 18.163(5) Å, β = 112.336(5)°, V = 1873.8(8) Å3, Z = 4, ρcalc = 1.490 Mg/m3, μ = 0.122 mm−1, and F(000) = 872. Data reduction was performed using SAINT (v6.36A)9 and XPREP (v6.12).10 Corrections were applied for Lorentz, polarization, and absorption effects using SADABS (v2.03).11 The structure was solved and refined with the aid of the programs in the SHELXTL-plus (v6.10) system of programs.12 The full-matrix least-squares refinement on F2 included atomic coordinates and anisotropic thermal parameters for all non-H atoms. The H atoms were included using a riding model. The final R value (R1) was 0.0629 for 1930 observed (I > 2σI) reflections and 0.1240 for all 3419 reflections using 271 parameters, and goodness-of-fit was 1.020.
For 6c, a yellow needle of dimensions 0.04 × 0.06 × 0.36 mm3 was mounted on glass fiber using a small amount of epoxy. Data were collected on a Bruker three-circle diffractometer equipped with a SMART 6000 CCD detector. The crystals were irradiated using a rotating anode Cu Kα source (λ = 1.54178) with incident beam Göbel mirrors. Data collection was performed and the unit cell was initially refined using SMART (v5.625).8 The crystals were triclinic, P1h, with a = 11.2367(11) Å, b = 12.8743(10) Å, c = 25.143(2) Å, α = 89.451(5)°, β = 88.172(6)°, γ = 87.516(5)°, V = 3631.9(6) Å3, Z = 8, ρcalc = 1.483 Mg/m3, μ = 1.063 mm−1, and F(000) = 1672. Data reduction was performed using SAINT (v6.36A)9 and XPREP (v6.12).10 Corrections were applied for Lorentz, polarization, and absorption effects using SADABS (v2.03).11 The structure was solved and refined with the aid of the programs in the SHELXTL-plus (v6.10) system of programs.12 The full-matrix least-squares refinement on F2 included atomic coordinates and anisotropic thermal parameters for most non-H atoms. The H atoms were included using a riding model. The final R value (R1) was 0.0916 for 5783 observed (I > 2σI) reflections and 0.2693 for all 10 961 reflections using 1130 parameters, and goodness-of-fit was 1.033.
Supplementary Material
Acknowledgment.
This project was funded in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, by the National Cancer Institute under Contract No. NO1-CO-12400, and by the National Institute on Drug Abuse under Contract No. Y1-DA-1002, as well as NIH Grant R01 CA84496 and a Translational Research Award to P.J.S. from the Leukemia and Lymphoma Society.
Footnotes
Supporting Information Available: Complete crystal structure reports for 6a and 6c, as well as a figure showing the superposition of distinct conformational orientations in crystalline 6c; IC50 data for compounds 6a-d in the HL-60 and NIH3T3 cell line screens; kinetic data used to derive the rate constants of Table 1; chemiluminescence traces confirming that NO was generated on reaction of 6a–d with GSH. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Findlay VJ; Townsend DM; Saavedra JE; Buzard GS; Citro ML; Keefer LK; Ji X; Tew KD Tumor Cell Responses to a Novel Glutathione S-Transferase-Activated Nitric Oxide-Releasing Prodrug. Mol. Pharmacol 2004, 65, 1070–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Saavedra JE; Srinivasan A; Bonifant CL; Chu J; Shanklin AP; Flippen-Anderson JL; Rice WG; Turpin JA; Davies KM; Keefer LK The Secondary Amine/Nitric Oxide Complex Ion R2N[N(O)NO]− as Nucleophile and Leaving Group in SNAr Reactions. J. Org. Chem 2001, 66, 3090–3098. [DOI] [PubMed] [Google Scholar]
- (3).Shami PJ; Saavedra JE; Wang LY; Bonifant CL; Diwan BA; Singh SV; Gu Y; Fox SD; Buzard GS; Citro ML; Waterhouse DJ; Davies KM; Ji X; Keefer LK JS-K, a Glutathione/Glutathione S-Transferase-Activated Nitric Oxide Donor of the Diazeniumdiolate Class with Potent Antineoplastic Activity. Mol. Cancer Ther 2003, 2, 409–417. [PubMed] [Google Scholar]
- (4).Keefer LK; Nims RW; Davies KM; Wink DA “NONOates” (1-Substituted Diazen-1-ium-1,2-diolates) as Nitric Oxide Donors: Convenient Nitric Oxide Dosage Forms. Methods Enzymol. 1996, 268, 281–293. [DOI] [PubMed] [Google Scholar]
- (5).Houben J; Brassert W Über die Einwirkung von alkoholischem Chlorwasserstoff auf m-Methylnitrosamin-benzoesäure. Chem. Ber 1910, 43, 206–213. [Google Scholar]
- (6).Monks A; Scudiero D; Skehan P; Shoemaker R; Paull K; Vistica D; Hose C; Langley J; Cronise P; Vaigro-Wolff A; Gray-Goodrich M; Campbell H; Mayo J; Boyd M Feasibility of a High-Flux Anticancer Drug Screen Using a Diverse Panel of Cultured Human Tumor Cell Lines. J. Natl. Cancer Inst 1991, 83, 757–766. [DOI] [PubMed] [Google Scholar]
- (7).Jia L; Tomaszewski J; Jothianandan D; Furchgott RF; Keefer L; Shami P EDRF–Like Activity and Anticancer Spectrum of JS-K, a Novel Diazeniumdiolate Nitric Oxide Donor. Proc. Am. Assoc. Cancer Res 2003, 44, 1059. [Google Scholar]
- (8).SMART, v5.625; Bruker AXS Inc., Madison, WI, 2001. [Google Scholar]
- (9).SAINT, v6.36A; Bruker AXS Inc., Madison, WI, 2002. [Google Scholar]
- (10).XPREP, v6.12; Bruker AXS Inc., Madison, WI, 2001. [Google Scholar]
- (11).SADABS, v2.03; Bruker AXS Inc., Madison, WI, 2000. [Google Scholar]
- (12).SHELXTL, v6.10; Bruker AXS Inc., Madison, WI, 2000. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.