

Abstract
Nitric oxide (NO) is an endogenous, diffusible, transcellular messenger shown to affect regulatory and signaling pathways with impact on cell survival. Exposure to NO can impart direct post-translational modifications on target proteins such as nitration and/or nitrosylation. As an alternative, after interaction with oxygen, superoxide, glutathione, or certain metals, NO can lead to S-glutathionylation, a post-translational modification potentially critical to signaling pathways. A novel glutathione S-transferase π (GSTπ)-activated pro-drug, O2-{2,4-dinitro-5-[4-(N-methylamino)benzoyloxy]phenyl}1-(N,N-dimethylamin-o)diazen-1-ium-1,2-diolate (PABA/NO), liberates NO and elicits toxicity in vitro and in vivo. We now show that PABA/NO induces nitrosative stress, resulting in undetectable nitrosylation, limited nitration, and high levels of S-glutathionylation. After a single pharmacologically relevant dose of PABA/NO, S-glutathionylation occurs rapidly (<5 min) and is sustained for ~7 h, implying a half-life for the deglutathionylation process of approximately 3 h. Two-dimensional SDS-polyacrylamide gel electrophoresis and immunoblotting with a monoclonal antibody to S-glutathionylated residues indicated that numerous proteins were S-glutathionylated. Subsequent matrix-assisted laser desorption ionization/time of flight analysis identified 10 proteins, including β-lactate dehydrogenase, Rho GDP dissociation inhibitor β, ATP synthase, elongation factor 2, protein disulfide isomerase, nucleophosmin-1, chaperonin, actin, protein tyrosine phosphatase 1B (PTP1B), and glucosidase II. In addition, we showed that sustained S-glutathionylation was temporally concurrent with drug-induced activation of the stress kinases, known to be linked with cell death pathways. This is consistent with the fact that PABA/NO induces S-glutathionylation and inactivation of PTP1B, one phosphatase that can participate in deactivation of kinases. These effects were consistent with the presence of intracellular PABA/NO or metabolites, because cells overexpressing MRP1 were less sensitive to the drug and had reduced levels of S-glutathionylated proteins.
The human genome is composed of <30,000 genes, yet complexity of protein structure/function seems distinctly more layered. In the proteomics era, it has become clear that the dogma of genetic determinism is distinctly influenced by a number of processes, including polymorphic variation, gene splicing events, exon shuffling, protein domain rearrangements, and a number of post-translational modifications that contribute to alterations in tertiary and quaternary protein structure. Among these, phosphorylation, glycosylation, methylation, and acetylation can account for a large proportion of modifications. Cellular homeostasis is an intricate balance of survival and death signals, the control of which can occur through post-translational modification of target proteins. Protein phosphorylation is regulated by kinase/phosphatase interplay, frequently allowing an adaptive response to a changing environment. In addition to the more studied post-translational modifications, cysteine residues localized in a basic environment can have low pKa values, making them targets for the addition of glutathione; i.e., they can be S-glutathionylated. This modification can regulate cellular response to stresses such as reactive oxygen (ROS) and nitrogen (RNS) species (Shelton et al., 2005). Figure 1 shows how ROS and RNS can interact with cysteine residues, resulting in the formation of S-glutathionylated proteins. This reaction can serve to protect the protein from further oxidative or nitrosative damage or can effect a change in conformation and/or charge that may alter protein function. Critical in ascribing any regulatory function (i.e., equivalence to phosphorylation/dephosphorylation) to this process is the reversibility of S-glutathionylation by small-molecule, cysteine-rich proteins such as glutaredoxin and/or sulfiredoxin (Beer et al., 2004; Findlay et al., 2005; Shelton et al., 2005).
Fig. 1.

Nitrosative stress leads to the formation of S-nitrosation and S-glutathionylation modifications. Both posttranslational modifications alter protein function.
It seems plausible that both endogenous and exogenous ROS/RNS can cause S-glutathionylation. For example, endogenous ROS can be by-products of lipid peroxidation and the electron transport chain, whereas exogenous sources include X-ray and UV irradiation. Elevated levels of nitric oxide (NO) provide the primary source of RNS. NO is an endogenous diffusible transcellular messenger shown to participate in survival and death pathways (Moncada et al., 1997) and can alter protein function directly through post-translational modifications (nitration or nitrosylation) or indirectly through interactions with oxygen, superoxide, thiols, and heavy metals, the products of which can ultimately lead to S-glutathionylation (Fig. 1) (for review, see Chung et al., 2001).
Dysregulation of endogenous NO can lead to the release of RNS and ROS, each of which has been implicated in a number of human pathologies, including neurodegenerative disorders, cystic fibrosis, aging, and cancer (van der Vliet et al., 1997; Ilic et al., 1999; Tieu et al., 2003; Townsend et al., 2003). This observation has led to investigations of the capacity of NO to induce cytotoxicity, with particular reference to antitumor activities (Cui et al., 1994). The coalescence of NO and GST biology led to the design and synthesis of a novel anticancer pro-drug (PABA/NO) (Saavedra et al., 2006). The GSTπ isozyme is expressed at elevated levels in a variety of human tumors and is linked with the development of resistance to a number of anticancer agents (O’Brien et al., 2000; Townsend and Tew, 2003). Such characteristics have been instrumental in the development of a GSTπ-activated phosphorodiamidate pro-drug, Telcyta, that has undergone preclinical evaluation and has just entered phase III clinical trial (Rosario et al., 2000). Catalytic activation of PABA/NO by GSTπ releases NO that elicits antitumor activity both in vitro and in vivo. The release of NO also causes an activation of stress response pathways that involve p38 and c-jun-NH2-terminal kinase (JNK), eventually leading to apoptosis. In the present study, the capacity of PABA/NO to induce post-translational modifications such as nitration, nitrosylation, and S-glutathionylation was viewed in the context of the cytotoxic properties and mechanism of action of the drug.
Materials and Methods
Reagents and Cell Culture.
PABA/NO was prepared as described previously (see also structure revision in Saavedra et al., 2006). The following reagents were commercially available: peroxynitrite and purified PTP1B (Upstate Biotechnologies, Lake Placid, NY), hydrogen peroxide, oxidized glutathione, and the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) re-agent (Sigma, St. Louis, MO), S-glutathionylated HeLa cell lysate (ViroGen Corp, Watertown, MA), and the Bradford reagent (Bio-Rad Laboratories, Hercules, CA). Cell lines were maintained in Dulbecco’s modified Eagle’s medium (NIH3T3 and NIH3T3-MRP) or RPMI medium (HL60 and HL60-ADR) supplemented with 10% fetal calf serum, 100 μg/ml streptomycin, 100 U/ml penicillin, and 2 mM l-glutamine at 5% CO2 and 37°C.
Cytotoxicity Assays.
Cells (104) were seeded in 96-well plates in 50 μl of medium. Increasing drug concentrations were added to a final volume of 100 μl and maintained in drug for 72 h. After drug exposure, cell viability was assayed with the use of the MTT conversion assay. Each drug concentration was represented in quadruplicate and the experiments were repeated in three independent experiments. Mean values and S.E. were computed for each group.
Protein Preparation.
Cells (5 × 105 per treatment group) were collected and washed with 1× phosphate-buffered saline. Cell pellets were suspended in lysis buffer containing 20 mM Tris-HCl, pH 7.5, 15 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, and 1 mM β-glycerophosphate with freshly added phosphatase inhibitors (5 mM NaF and 1 mM Na3VO4) and a protease inhibitor cocktail (Sigma). Cells were incubated for 30 min on ice, sonicated three times for 10 s each and centrifuged for 30 min at 10,000g at 4°C. Protein concentrations in the supernatant were determined with the Bradford (1976) method using IgG as a standard.
Two-Dimensional SDS-PAGE.
For the first dimension, 100 μg of whole cell protein was resuspended in 8 M urea with 0.5% carrier ampholytes and run on immobilized pH gradients covering exponentially the pH range from 3 to 10. For the second dimension, the immobilized pH gradient strips were separated on 10% polyacryl-amide gels. Nonreducing conditions were used for electrophoresis (Righetti et al., 1991). Protein spots of interest were excised and subject to trypsin digestion. Peptide mass was analyzed by MALDI-TOF mass spectrometry at the Proteomics Core Facility of the Medical University of South Carolina. Protein identification was performed using software from the National Center for Biotechnology Information protein database.
Immunoblot Analysis.
For analysis of S-glutathionylation and nitration, equivalent amounts of protein were separated under non-denaturing conditions on 10% SDS-polyacrylamide gels; unmodified proteins were separated under denaturing conditions. Gels were transferred overnight onto nitrocellulose membranes (Bio-Rad, Hercules, CA). Nonspecific binding was reduced by incubating the membrane in 10% blocking buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10% bovine serum albumin, 0.1% Tween 20, 1× protease inhibitors, 5 mM NaF, and 1 mM Na3VO4 for 1 h. Membranes were incubated with specific antibodies (5% blocking buffer) overnight at 4°C, washed three times with PBS for 15 min, and incubated with the appropriate secondary antibody conjugated to horseradish peroxidase for 1 h. The membranes were washed three times and developed with enhanced chemiluminescence detection reagents (Bio-Rad). The relative abundance of proteins were evaluated using Quantity One software (ver. 4.5.2; Bio-Rad) and plotted as arbitrary units in relation to actin.
Antibodies.
Antibodies were purchased from the following sources: anti-active-JNK and p38 (Promega, Madison, WI), anti-p38, anti-extracellular signal-regulated kinase, and phosphospecific extracellular signal-regulated kinase (Santa Cruz Biotechnology, Santa Cruz, CA), anti-JNK2 (BD Biosciences PharMingen, San Diego, CA), anti-nitrotyrosine (Upstate Biotechnology), anti-actin and anti-His (Invitrogen, Carlsbad, CA), anti-SSG (ViroGen Corp., Watertown, MA).
In Vitro S-Glutathionylation of PTP1B.
His-tag-purified PTP1B (1 μg; Upstate Biotechnology) was incubated at 37°C for 0 to 30 min with 100 μM PABA/NO, 10 mM glutathione, 50 mM KPO4 buffer. The samples were separated by SDS-PAGE under nonreducing conditions. The gels were transferred overnight to nitrocellulose membranes and immunoblotted with anti-S-glutathionylation and anti-His antibodies.
Results
Detection of S-Glutathionylation.
Because drug treatment can lead to multiple post-translational changes, the specificity of the monoclonal antibodies used to detect S-glutathionylation was first tested. HL60 cells were treated for 30 min with peroxynitrite (a known inducer of nitration) and PABA/NO. We used S-glutathionylated HeLa cell extracts provided by Virogen Corp as a positive control for the anti-S-glutathionylation antibody and a negative control for the anti-nitrotyrosine antibody. Figure 2 shows that neither S-glutathionylation (Fig. 2A) nor nitration (Fig. 2B) were detected in untreated cell lysates. Nitration of proteins was observed as a consequence of peroxynitrite treatment. A positive reaction for S-glutathionylation was observed in the control HeLa cell extract and in the PABA/NO-treated cells. These data were consistent with specific antibody reactivity and validate the sensitivity and specificity comparisons for detection of S-glutathionylated proteins.
Fig. 2.

Anti-S-glutathionylation antibodies do not cross-react with nitration modifications. Cells in logarithmic growth were treated for 30 min with inducers of oxidative and nitrosative stress. Protein lysate (30 μg) was separated on a nonreducing SDS-PAGE and analyzed by immunoblot using: anti-S-glutathionylation antibody (A) and anti-nitrotyrosine antibody (B). Lane 1, RPN800 molecular markers; lane 2, untreated cells; lane 3, 5 mM glutathione; lane 4, 195 μM peroxynitrite; lane 5, 195 μM peroxynitrite + 5 mM glutathione; lane 6 30 μM PABA/NO; lane 7, 30 μM PABA/NO + 5 mM glutathione; and lane 8, S-glutathionylated HeLa cell lysate.
PABA/NO Induces S-glutathionylation of Proteins in a Dose- and Time-Dependent Manner.
Toxicity of PABA/NO and three potential hydrolysis products of PABA/NO (JS-42–25, JS-43–19, and 4-methylaminobenzoic acid) was measured in HL60 cells along with an analog of PABA/NO incapable of releasing NO (JS-39–93) (Table 1). The IC50 value of PABA/NO was 33 ± 2.9 μM, whereas the metabolites and NO-free control showed a 3-fold increase in the IC50 value. HL60 cells treated with 0 to 250 μM concentrations of the metabolites and NO-free control failed to S-glutathionylate proteins (data not shown). These data suggest that intact PABA/NO is essential to elicit the cytotoxic effects and post-translational modification. The approximate IC50 concentration (30 μM) was used to examine the temporal modification of proteins. SDS-PAGE and immunoblot analysis showed that PABA/NO treatment induced S-glutathionylation of numerous proteins within 5 min (Fig. 3) and that in the presence of drug, this was sustained >6 h (Fig. 5), implying that deglutathionylation occurs in the absence of drug with a half-life of approximately 3 h.
TABLE 1. Cytotoxicity and S-glutathionylation properties of PABA/NO and related compounds.
HL60 cells were treated with varying concentrations of PABA/NO and its metabolites. Cell viability was measured after 72 h with the use of the MTT assay. The IC50 mean value of three independent experiments ± S.D. is shown. Immunoblot analyses of lysates treated with 0 to 250 μM PABA/NO or its metabolites for 2 h show that S-glutathionylation is specific to intact PABA/NO.
| Drug | Structure | MM | IC50 | Glutathionylation |
|---|---|---|---|---|
| kDa | μM | |||
| PABA/NO | 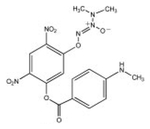 |
420 | 33.16 ± 2.9 | Yes |
| JS-39-93 | 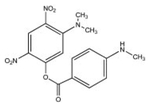 |
360 | 84.2 ± 0.2 | No |
| JS-42-25 | 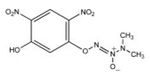 |
287 | 93.6 ± 0.3 | No |
| JS-43-19 | 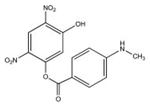 |
333 | 100.1 ± 2.6 | No |
| 4-Methylamino-benzoic acid | 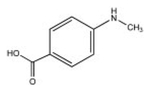 |
151 | 88.6 ± 3.5 | No |
MM, molecular mass
Fig. 3.

PABA/NO induces S-glutathionylation in a dose- and time-dependent manner. A, HL60 cells were treated with varying concentrations of PABA/NO. Cell viability was measured at 72 h with the MTT assay. B,1.5 × 106 HL60 cells were treated with varying concentrations of PABA/NO for 1 h. C, 1.5 × 106 HL60 cells were treated with 30 μM PABA/NO for 0 to 120 min. After treatment, 20 μg of lysate was separated by SDS-PAGE and analyzed for S-glutathionylation by immuno-blot.
Fig. 5.

Temporal S-glutathionylation and activation of kinases after PABA/NO treatment. HL60 cells were treated for 1 h with 30 μM PABA/NO and resuspended in complete media, and aliquots were removed hourly for 6 h. Each time point was analyzed by immunoblot (top), and the corresponding relative abundance of proteins was plotted as the ratio to actin (bottom) for S-glutathionylation; the relative abundance of two bands, 37 kDa (■) and 18 kDa (●) are plotted in arbitrary units relative to actin (A); nitration (B), and phosphorylated and unmodified kinases and actin as a loading control(C). Phosphorylated p38 (●), JNK (■), and jun (▲) are plotted in arbitrary units relative to actin.
In previous studies, we showed the toxicity of PABA/NO to be significantly diminished in NIH/3T3 cells overexpressing MRP1, with an IC50 value of 50 versus 11 μM in wild-type (Findlay, 2004). We used these cells to address whether PABA/NO-induced S-glutathionylation is independent of toxicity (Fig. 4). The data show that a 30 μM PABA/NO treatment in NIH3T3 wild-type cells results in rapid S-glutathionylation of a variety of proteins within 15 min. However, S-glutathionylation in 3T3-MRP cells was not observed until ~1 h. A dose- and time-dependent delay of S-glutathionylation in resistant cells was concurrent with a decrease in cytotoxicity. These data are consistent with our hypothesis that a glutathione-containing metabolite of PABA/NO elicits the cytotoxic effects and acts as both a substrate for MRP1 efflux and a donor for S-glutathionylation.
Fig. 4.

S-Glutathionylation is delayed in NIH3T3 cells over-expressing MRP1. NIH3T3 (A) and NIH3T3/MRP1 (B) cells were treated with 30 μM PABA/NO for 0 to 120 min.
Activation of the MAP Kinase Pathway.
PABA/NO-induced cytotoxicity could be mediated through downstream effects on kinase cascades (Findlay et al., 2004). To assess whether S-glutathionylation was linked to this effect, HL60 cells were treated with 30 μM PABA/NO for 1 h, drug was removed, and cells were analyzed for temporal S-glutathionylation and MAP kinase pathway activation during the recovery phase (Fig. 5). Maximal S-glutathionylation was observed at the time point when drug was removed (1 h). A diminishment over 6 h followed and led to undetectable levels at 6 h (Fig. 5A). Activation of kinases linked to stress response and cell death pathways was evaluated by measuring phosphorylated p38, JNK, and c-jun. After immunoblot with phosphoantibodies, the membranes were stripped and probed for the unmodified protein. Although no changes in total protein were observed, phosphorylation of p38 and JNK was maximal 1 h after PABA/NO treatment. The relative abundances of active p38 and JNK were comparable with untreated lysates at 2 and 3 h, respectively. Phosphorylation of c-jun increased after drug exposure, reached a maximum at 2 h, and declined to basal levels at 6 h.
Protein Nitration.
Nitration of proteins was evaluated with the anti-nitrotyrosine antibody (Fig. 5B). Three proteins were detected. The immunoreactive spots were excised from a two-dimensional SDS-PAGE gel, digested with trypsin, and their identity was analyzed by MALDI-TOF mass spectrometry. The proteins were identified as actin (41 kDa), ATP synthase (51 kDa), and protein disulfide isomerase (57 kDa). Nitration did not decrease throughout the experiment, consistent with irreversible nature of this post-translational modification. The data shown in Fig. 5 are representative of at least five experiments, all of which showed a temporal relationship between S-glutathionylation and activation of these kinases.
Identification of S-Glutathionylated Proteins.
PABA/NO treatment leads to rapid S-glutathionylation of numerous proteins in HL60 cells. To gain insight into the biological importance, 100 μg of protein equivalents of whole-cell lysates from HL60 cells were separated by two-dimensional SDS-PAGE. Coomassie-stained gels of treated and control cells confirmed equivalent loading (Fig. 6, A and C). In untreated cells, S-glutathionylated proteins were not detected, whereas PABA/NO treatment produced >20 modified proteins (Fig. 6D). Ten immunoreactive spots were excised from the gel, digested with trypsin, and identified by matrix-assisted laser desorption ionization/time of flight mass spectrometry (Table 2). S-Glutathionylated proteins are listed according to their subcellular localization: β lactate dehydrogenase, Rho GDP dissociation inhibitor β, elongation factor 2, PTP1B, and actin (cytoplasm); nucleophosmin (nucleus); protein disulfide isomerase and glucosidase II (endoplasmic reticulum); and ATP synthase β subunit and chaperonin (mitochondria).
Fig. 6.

Two-dimension analysis of HL60 cells untreated (C) and treated (D) with 30 μM PABA/NO. One hundred micrograms of protein lysate were separated under nondenaturing conditions and immunoblotted with the anti-S-glutathionylation antibody. Coomassie-stained gels of untreated (A) and treated (B) represent equivalent loading.
TABLE 2.
MALDI-TOF identification of S-glutathionylated proteins in HL60 cells treated with PABA/NO
| Protein | NCBI Accession Number | MM | pI | Protein Confidence Interval |
|---|---|---|---|---|
| Da | % | |||
| β-Lactate dehydrogenase | 49259212 | 36,516 | 6.2 | 100 |
| Rho GDP dissociation inhibitor β | 56676393 | 22,973 | 5.1 | 99.6 |
| Nucleophosmin-1 | 40353734 | 29,446 | 4.64 | 100 |
| Protein disulfide isomerase | 2098329 | 57,080 | 5.9 | 100 |
| ATP synthase β subunit | 1374715 | 51,170 | 4.9 | 100 |
| Chaperonin | 41399285 | 61,016 | 5.5 | 100 |
| Glucosidase II | 2274968 | 106,832 | 5.74 | 100 |
| Elongation factor 2 | 4503483 | 95,277 | 6.42 | 100 |
| Actin | 15277503 | 41,736 | 5.29 | 99.9 |
| PTP1B | 18031 | 49,966 | 5.8 | 99.9 |
MM, molecular mass.
In Vitro S-glutathionylation of PTP1B.
PABA/NO treatment caused S-glutathionylation of PTP1B in vivo (Table 2) and in vitro (Fig. 7). We have shown that PABA/NO treatment reduces PTP1B activity from 13 ± 7 to 3.7 ± 1.2 nmol/min/mg (Findlay et al., 2005). These data are in agreement with those from other studies showing that reversible S-glutathionylation of the phosphatase PTP1B inhibits activity and thereby affects kinase signaling (Barrett et al., 1999).
Fig. 7.

PTP1B (1 μg) was treated for varying times with 100 μM PABA/NO in the presence of 10 mM glutathione. Immunoblot analysis with anti-S-glutathionylation and anti-His antibodies confirm in vitro modification of PTP1B.
Discussion
PABA/NO is a diazeniumdiolate in which the N-methyl-para-aminobenzoic acid substituent is bound to the dinitrobenzene ring through the carboxyl oxygen (Saavedra et al., 2006). GST-mediated activation via the Meisenheimer complex intermediate results in a number of metabolites and the release of NO, the species considered to be critical to pharmacological activity. Our data suggest that PABA/NO produces detectable nitration of three proteins. Although these modifications may be pertinent to the cytotoxic efficacy of the drug, they are quantitatively quite limited in extent compared with cells treated with peroxynitrite. Nitration is an irreversible modification that could decrease the half-life and therefore lower the detection of target proteins, which might be further compromised by the lack of sensitivity of the commercially available antibody. However, our data show categorically that PABA/NO is a potent inducer of S-glutathionylation, whereas peroxynitrite is not. We propose that the lack of nitrated proteins is a reflection of the fact that hydrolysis products of PABA/NO cause higher levels of cysteine modifications. PABA/NO-induced S-glutathionylation of numerous proteins occurred within minutes of drug treatment and was sustained in the absence of the drug for ~6 h. The reduced and delayed S-glutathionylation in NIH3T3-MRP1 provides support for the conclusion that active metabolite of PABA/NO is responsible for causing S-glutathionylation of target proteins. In this and prior studies, we demonstrated that despite equivalent GSTπ expression, S-glutathionylation and cytotoxicity are diminished in MRP1-overexpressing cells compared with the parental line. Taken together, these data imply that a glutathione conjugate of PABA/NO can act both as a substrate for MRP1 efflux and as a donor for S-glutathionylation. Whether the metabolite is the proximal donor for the S-glutathionylation reaction has yet to be determined.
S-Glutathionylation preceded activation of the MAP kinases and followed a decay rate that was similar to that of kinase dephosphorylation. PTP1B is an important cellular tyrosine phosphatase that is a negative regulator of the insulin-stimulated kinase pathway. However, several studies have shown that PTP1B participates in various signal transduction pathways (for review, see Zhang et al., 2002). Other laboratories have shown that when PTP1B is S-glutathionylated on Cys215, it is rendered catalytically inactive (Barrett et al., 1999; Li and Whorton, 2003). Other studies in our laboratory have shown that deglutathionylation of PTP1B by sulfiredoxin can reinstate enzyme activity (Findlay et al., 2005). In at least one model of drug action that is consistent with our present data, PTP1B S-glutathionylation after PABA/NO allows the activation of the kinase pathway that leads to apoptosis. The deglutathionylation of PTP1B (and other targeted proteins) after 6 h leads to phosphatase activation and subsequent dephosphorylation of the kinase cascade, consistent with Fig. 6.
The decay of S-glutathionylation after removal of PABA/NO is not accompanied by any diminution of protein stain intensity in individual spots on gels. This suggests that S-glutathionylated proteins are not targeted for degradation. Our data imply a half-life for S-glutathionylation of approximately 3 h. It seems plausible that this modification may serve to protect important cysteine residues and to prevent degradation. When thiol groups are exposed to ROS, the flexible valency of the sulfur can allow the formation of sulfenic, sulfinic, or sulfonic acids. Only the latter of these seems irreversible with resultant proteolysis (Paget and Buttner, 2003). With PABA/NO, the proximal donor of the S-glutathionylation could be one of a number of drug metabolites. As shown in Fig. 1, nitrosoglutathione, oxidized glutathione, glutathione sulfenate, and glutathione disulfide monoxide and dioxide have all been characterized as plausible donors of the GSH peptide (-SG) (Tao and English, 2004). Nitrosoglutathione is a likely substrate for MRP1, and its efflux would be consistent with the reduced levels of S-glutathionylation in cells overexpressing MRP1. Glutathione sulfenate and glutathione disulfide monoxide and dioxide would be plausible downstream products of PABA/NO metabolism. The rapid onset of S-glutathionylation predicts that PABA/NO has a short decomposition half-life, at least with respect to liberation of NO. Indeed, this is commensurate with the measured half-life of the drug with respect to NO release.
Alterations in redox balance by exposure to RNS or ROS cause dose-dependent changes in glutathione/oxidized glutathione ratios that can potentially influence a number of target proteins by causing oxidation and disulfide exchange reactions at specific cysteine residues. The importance of modifying cysteine residues is not necessarily restricted to redox regulation but now seems to be a plausible event that can lead to changes in signaling processes. By adding the glutathione tripeptide to a target protein, an additional negative charge is introduced (as a consequence of the glu residue), and a change in protein conformation is made likely. A recent report (Manevich et al., 2004) showed that GSTπ is capable of acting in a catalytic manner to S-glutathionylate peroxiredoxin, a non–selenium-dependent lipid peroxidase that converts lipid hydroperoxides to corresponding alcohols. The catalytically important cysteine residue on peroxiredoxin is sterically inaccessible within the globular dimeric complex, and GSTπ facilitates glutathione transfer to this site. The resultant enzyme activation serves a cellular regulatory role, particularly with respect to response to oxidative stress. Actin is a prevalent structural protein, the very abundance of which makes it a primary target for S-glutathionylation. Indeed, hydrogen peroxide-induced ROS seemed to result almost exclusively in S-glutathionylation of actin. SGlutathionylation of actin has been shown to be physiologically relevant. Under normal conditions, a proportion of G-actin is S-glutathionylated at Cys374. Because deglutathionylation increases the rate of polymerization, by extrapolation, this modification limits polymerization into F-actin (Wang et al., 2001). Inhibition of deglutathionylation suppresses growth factor-mediated actin polymerization (Wang et al., 2003). As elegantly discussed by Shelton et al. (2005), redox modification of actin fulfils many of the criteria important in defining a regulatory role for reversible S-glutathionylation.
Using a variety of methods, other investigators have identified a number of distinct S-thiolated proteins that may be involved in cell regulation (Baty et al., 2002; Eaton et al., 2002; Fratelli et al., 2002). Using the monoclonal antibody technique, our data showed that ~20 distinct proteins were modified after PABA/NO treatment. This is the first evidence that the cytoxicity of an antitumor agent may elicit concomitant S-glutathionylation of proteins. This is a reasonably small proportion of the total proteome and suggests that there would be value in identifying those members of this group that might be involved in regulation/signaling, particularly as related to NO cytotoxicity and the mechanism of action of PABA/NO. To this end, we identified 10 S-glutathionylated proteins (Table 2). Of particular significance to PABA/NO-induced apoptosis is ATP synthase β subunit. This enzyme is located in the inner mitochondrial membrane and catalyzes ATP formation through the energy of proton flux during oxidative phosphorylation. Nitration of numerous proteins, including ATP synthase, was proposed to contribute to the pathogenesis of familial amyotrophic lateral sclerosis (Casoni et al., 2005). In a separate study, inhibition of β-oxidative respiration with trinitazidine resulted in a dose-dependent decrease in ATP levels and an induction of apoptosis (Andela et al., 2005). These authors postulated that inhibition of β-oxidative respiration is a therapeutic window associated with the anticancer activity of peroxisome proliferator-activated receptor-γ agonists. The present data suggest that ATP synthase is an indirect target of the RNS generated by GST-mediated activation of PABA/NO and may contribute to the antitumor activity of the drug.
Acknowledgments
We thank the Medical University of South Carolina Mass Spectrometry Facility.
This research was funded through the National Cancer Institute grant CA53783, the Intramural Research Program of the National Institutes of Health/National Cancer Institute Center for Cancer Research, and National Cancer Institute contract 1-CO-12400.
ABBREVIATIONS:
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- PABA/NO
O2-{2,4-dinitro-5-[4-(N-methylamino)benzoyloxy]-phenyl}1-(N,N-dimethylamino)diazen-1-ium-1,2-diolate
- JNK
c-jun-NH2-terminal kinase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- PAGE
polyacrylamide gel electrophoresis
- MALDI-TOF
matrix-assisted laser desorption ionization/time of flight
- MAP
mitogen-activated protein
- MRP1
multidrug resistance protein 1
- PTP1B
protein tyrosine phosphatase 1B
- GST
glutathione S-transferase
- JS-39-93
1,3-dinitro-4-(N,N-dimethylamino)-6-(p-methylaminobenzoyloxy)benzene
- JS-42-25
O2-(2,4-dinitro-5-hydroxyphenyl) 1-(N,N-dimethylamino)diazen-1-ium-1,2-diolate
- JS-43-19
1,3-dinitro-4-hydroxy-6-(p-methylaminobenzoyloxy)benzene
References
- Andela VB, Altuwaijri S, Wood J, and Rosier RN (2005) Inhibition of beta-oxidative respiration is a therapeutic window associated with the cancer chemo-preventive activity of PPARgamma agonists. FEBS Lett 579:1765–1769. [DOI] [PubMed] [Google Scholar]
- Barrett WC, DeGnore JP, Konig S, Fales HM, Keng YF, Zhang ZY, Yim MB, and Chock PB (1999) Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry 38:6699–6705. [DOI] [PubMed] [Google Scholar]
- Baty JW, Hampton MB, and Winterbourn CC (2002) Detection of oxidant sensitive thiol proteins by fluorescence labeling and two-dimensional electrophoresis. Proteomics 2:1261–1266. [DOI] [PubMed] [Google Scholar]
- Beer SM, Taylor ER, Brown SE, Dahm CC, Costa NJ, Runswick MJ, and Murphy MP (2004) Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: implications for mitochondrial redox regulation and antioxidant DEFENSE. J Biol Chem 279:47939–47951. [DOI] [PubMed] [Google Scholar]
- Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. [DOI] [PubMed] [Google Scholar]
- Casoni F, Basso M, Massignan T, Gianazza E, Cheroni C, Salmona M, Bendotti C, and Bonetto V (2005) Protein nitration in a mouse model of familial amyotrophic lateral sclerosis: Possible multifunctional role in the pathogenesis. J Biol Chem 280:16295–16304. [DOI] [PubMed] [Google Scholar]
- Chung HT, Pae HO, Choi BM, Billiar TR, and Kim YM (2001) Nitric oxide as a bioregulator of apoptosis. Biochem Biophys Res Commun 282:1075–1079. [DOI] [PubMed] [Google Scholar]
- Cui S, Reichner JS, Mateo RB, and Albina JE (1994) Activated murine macrophages induce apoptosis in tumor cells through nitric oxide-dependent or -independent mechanisms. Cancer Res 54:2462–2467. [PubMed] [Google Scholar]
- Eaton P, Wright N, Hearse DJ, and Shattock MJ (2002) Glyceraldehyde phosphate dehydrogenase oxidation during cardiac ischemia and reperfusion. J Mol Cell Cardiol 34:1549–1560. [DOI] [PubMed] [Google Scholar]
- Findlay VJ, Morris TE, Townsend DM, and Tew KD (2005) A role for human sulfiredoxin in stress response and redox regulation of protein glutathionylation, in Proceedings of the American Association for Cancer Research 96th Annual Meeting; 2005 Apr 16–20; Anaheim, CA. [Google Scholar]
- Findlay VJ, Townsend DM, Saavedra JE, Buzard GS, Citro ML, Keefer LK, Ji X, and Tew KD (2004) Tumor cell responses to a novel glutathione S-transferase-activated nitric oxide-releasing prodrug. Mol Pharmacol 65:1070–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratelli M, Demol H, Puype M, Casagrande S, Eberini I, Salmona M, Bonetto V, Mengozzi M, Duffieux F, Miclet E, et al. (2002) Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc Natl Acad Sci USA 99:3505–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic TV, Jovanovic M, Jovicic A, and Tomovic M (1999) Oxidative stress indicators are elevated in de novo Parkinson’s disease patients. Funct Neurol 14:141–147. [PubMed] [Google Scholar]
- Li S and Whorton AR (2003) Regulation of protein tyrosine phosphatase 1B in intact cells by S-nitrosothiols. Arch Biochem Biophys 410:269–279. [DOI] [PubMed] [Google Scholar]
- Manevich Y, Feinstein SI, and Fisher AB (2004) Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc Natl Acad Sci USA 101:3780–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada S, Higgs A, and Furchgott R (1997) International Union of Pharmacology nomenclature in nitric oxide research. Pharmacol Rev 49:137–142. [PubMed] [Google Scholar]
- O’Brien M, Kruh GD, and Tew KD (2000) The influence of coordinate overexpression of glutathione phase II detoxification gene products on drug resistance. J Pharmacol Exp Ther 294:480–487. [PubMed] [Google Scholar]
- Paget MS and Buttner MJ (2003) Thiol-based regulatory switches. Annu Rev Genet 37:91–121. [DOI] [PubMed] [Google Scholar]
- Righetti PG, Gianazza E, Bianchi-Bosisio A, Sinha P, and Kottgen E (1991) Isoelectric focusing in immobilized pH gradients: applications in clinical chemistry and forensic analysis. J Chromatogr 569:197–228. [DOI] [PubMed] [Google Scholar]
- Rosario LA, O’Brien ML, Henderson CJ, Wolf CR, and Tew KD (2000) Cellular response to a glutathione S-transferase P1–1 activated prodrug. Mol Pharmacol 58:167–174. [DOI] [PubMed] [Google Scholar]
- Saavedra JE, Srinivasan A, Buzard GS, Davies KM, Waterhouse DJ, Inami K, Wilde TC, Citro ML, Cuellar M, Deschamps JR, et al. (2006) PABA/NO as an anticancer lead: analogue synthesis, structure revision, solution chemistry and reactivity toward glutathione. J Med Chem, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton MD, Chock PB, and Mieyal JJ (2005) Glutaredoxin: role in reversible protein S-glutathionylation and regulation of redox signal transduction and protein trans-location. Antioxid Redox Signal 7:348–366. [DOI] [PubMed] [Google Scholar]
- Tao L and English AM (2004) Protein S-glutathiolation triggered by decomposedS-nitrosoglutathione. Biochemistry 43:4028–4038. [DOI] [PubMed] [Google Scholar]
- Tieu K, Ischiropoulos H, and Przedborski S (2003) Nitric oxide and reactive oxygen species in Parkinson’s disease. IUBMB Life 55:329–335. [DOI] [PubMed] [Google Scholar]
- Townsend DM, Tew KD, and Tapiero H (2003) The importance of glutathione in human disease. Biomed Pharmacother 57:145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend D and Tew K (2003) Cancer drugs, genetic variation and the glutathione-S-transferase gene family. Am J Pharmacogenomics 3:157–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vliet A, Eiserich JP, Marelich GP, Halliwell B, and Cross CE (1997) Oxidative stress in cystic fibrosis: does it occur and does it matter? Adv Pharmacol 38:491–513. [DOI] [PubMed] [Google Scholar]
- Wang J, Boja ES, Tan W, Tekle E, Fales HM, English S, Mieyal JJ, and Chock PB (2001) Reversible glutathionylation regulates actin polymerization in A431 cells. J Biol Chem 276:47763–47766. [DOI] [PubMed] [Google Scholar]
- Wang J, Tekle E, Oubrahim H, Mieyal JJ, Stadtman ER, and Chock PB (2003) Stable and controllable RNA interference: Investigating the physiological function of glutathionylated actin. Proc Natl Acad Sci USA 100:5103–5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZY, Zhou B, and Xie L (2002) Modulation of protein kinase signaling by protein phosphatases and inhibitors. Pharmacol Ther 93:307–317. [DOI] [PubMed] [Google Scholar]