

Abstract
Anticancer drug development using the platform of glutathione (GSH), glutathione S-transferases (GST) and pathways that maintain thiol homeostasis has recently produced a number of lead compounds. GSTπ is a prevalent protein in many solid tumors and is overexpressed in cancers resistant to drugs. It has proved to be a viable target for pro-drug activation with at least one candidate in late-stage clinical development. In addition, GSTπ possesses noncatalytic ligand-binding properties important in the direct regulation of kinase pathways. This has led to the development and testing of agents that bind to GSTπ and interfere with protein–protein interactions, with the phase II clinical testing of one such drug. Attachment of glutathione to acceptor cysteine residues (glutathionylation) is a post-translational modification that can alter the structure and function of proteins. Two agents in preclinical development (PABA/NO, releasing nitric oxide on GST activation, and NOV-002, a pharmacologically stabilized pharmaceutical form of GSSG) can lead to glutathionylation of a number of cellular proteins. The biological significance of these modifications is linked with the mechanism of action of these drugs. In the short term, glutathione-based systems should continue to provide viable targets and a platform for the development of novel cancer drugs.
Introduction
Drug discovery in cancer has evolved significantly in the past few years. High-throughput screening and cancer-specific target discrimination have essentially supplanted the classical synthetic chemistry structure–activity approaches to identify new lead compounds. Pathways that involve proteins aberrantly expressed in cancer cells are optimal as targets for drug intervention. Increased expression of the GSTπ isozyme (the most ubiquitous and prevalent GST in nonhepatic tissues) has been linked to both drug resistance and the malignant phenotype of many solid tumors (Tew, 1994). In addition, GSTπ has been found to be an endogenous regulator of c-Jun NH2-terminal kinase (JNK) (Adler et al., 1999a). On the basis of such differential expression and unusual signaling function in tumors, it was concluded that GSTπ might be an opportunistic drug target that could provide for an enhanced therapeutic index in the treatment of cancer.
The Kcat values for GSTπ-mediated GSH conjugation reactions with a number of anticancer drug substrates have been measured and are not impressive. Although rate and extent of conjugation for some alkylating drugs can be enhanced by GSTπ catalysis (Ciaccio et al., 1991), it has never seemed unreasonable to presume that some GSTπ functionality other than catalysis may be of consequence to the biological importance of the protein. The recent description of protein–protein interactions between GSTπ and JNK serve to extend the basic principles of the ligand-binding properties of GST isozymes. Indeed, early characterization of the GSTs centered on their capacity to act as a ligand in association with other proteins, particularly nonsubstrate ligands such as heme and bilirubin (Litwack et al., 1971). Although comparatively large macromolecules, they do not encompass the dimensions of a kinase such as JNK, and, not surprisingly, interactions between these two proteins are complex, with apparent association constants in the nanomolar range (Wang et al., 2001). The following sections describe how the evolving understanding of GST functions in cells can be adapted to the principles of drug design and development.
GSH and GST in Cell Signaling
Adding a further layer of complexity to the understanding of GST function is the fact that proteins rarely act in isolation in a cellular milieu. Rather, essential protein–protein interactions govern how cellular events unfold (Golemis et al., 2002). This process has proved to be significant to the regulation of JNK signaling by GSTπ (Adler et al., 1999a; Wang et al., 2001). This same paradigm seems to hold for additional redox proteins and associated kinases such as thioredoxin and apoptosis signal-regulating kinase, ASK1 (Saitoh et al., 1998), implying the possible existence of a general regulatory mechanism for kinases that may involve GSH and associated pathways (Adler et al., 1999b; Davis et al., 2001). In addition, emergent literature suggests that direct glutathionylation of critical signaling molecules may also serve as a trigger for cellular events that are influenced by oxidative stress (Adachi et al., 2004; Cross and Templeton, 2004). It would seem that GSH and GSTs have roles that extend much further than simple detoxification reactions.
Other small redox-active proteins such as peroxiredoxin have the potential to heterodimerize with GSTπ. Oxidative stress-mediated signaling events can be activated by GST-mediated catalytic addition of GSH to a critical cysteine residue in a sterically protected region of peroxiredoxin (Manevich et al., 2004). This observation broadens the functional importance of GSTπ into yet another arena, emphasizing how redox-active proteins have roles that are more than just removal of reactive oxygen species but are central to the signaling processes required in the cell’s response to stress. Changes in redox conditions can trigger cellular responses through a number of different pathways. The nature and extent of the ROS insult may determine the threshold of the cellular response manifest as proliferation, stress response and damage repair or apoptosis. With further understanding, the link between thiol-active proteins, GSTs, and stress-activated protein kinases exemplified by JNK and ASK may become an expansive series of interconnected pathways. In an unstressed cellular environment, JNK is kept in an inactive mode by the presence of one or more repressors. Under conditions of oxidative stress, GSTπ dissociates from JNK and forms dimers and/or multimeric complexes (Adler et al., 1999a). Meanwhile, the liberated JNK regains its functional capacity to be phosphorylated and to phosphorylate c-Jun. This process can activate the stress cascade involving the numerous sequential downstream kinases. However, in GSTπ-overexpressing cells, constitutively active MEKK1 effectively phosphorylated both MKK4 (immediately upstream of JNK) and JNK but did not result in jun phosphorylation, confirming the specificity of the GSTπ/JNK association (Yin et al., 2000). Mouse embryo fibroblast cell lines (MEF) from mice (Henderson et al., 1998) engineered to be null for GSTπ expression have high basal levels of JNK activity that can be reduced if these cells are transfected with GSTπ cDNA. In addition, treatment of GSTπ wild-type cells (but not null cells) with a specific GSTπ inhibitor, TLK199, causes activation of JNK activity. Also, human HL60 cells chronically exposed to this inhibitor develop tolerance to the drug and also overexpress JNK, presumably as a means of compensating for the constancy of GSTπ inhibition and the perceived chronic stress (Ruscoe et al., 2001).
These combined data provide evidence that GSTπ has a nonenzymatic, regulatory role in controlling cellular response to external stimuli. MEFs from GST−/− mice have a 24-h doubling time compared with 36 h for wild type (Ruscoe et al., 2001). TLK199, although designed to be a modulator of drug resistance through preferential inhibition of GSTπ (Morgan et al., 1996), was found to possess additional pharmacological activity as a myeloproliferative agent (Gate et al., 2004). Each study provides support for the concept that GSTπ has a role in regulation of proliferative pathways. Although GSTπ regulates JNK activity through protein–protein interactions, the influence of GST on GSH–GSSG homeostasis could also be a contributory factor. For example, the GSH binding site of GSTs (G-site) may be an important sequestration site for cellular GSH with concomitant impact on cellular redox status.
There are indications that GSH and associated enzymes play a role in cellular immunity. For example, GSH levels in antigen-presenting cells determine whether a Th1 or Th2 pattern of response predominates (Peterson et al., 1998). The Th1 response is characterized by production of interleukin-12 (IL-12) and interferon α and the enhancement of delayed hypersensitivity response; Th2 by IL-4 and IL-10 production and up-regulation of a number of antibody responses. The molecular basis for this difference is not known, but it is also significant that patients with HIV receiving n-acetylcysteine (a bioavailable precursor of GSH biosynthesis) have longer survival times than untreated controls. In diseases such as HIV, Th2 predominance is a critical component of immune response and, thus, GSH levels in antigen-presenting cells may play an integral role in determining disease progression (Herzenberg et al., 1997). The question now extends to a possible link between JNK and immune response/myeloproliferation. To this end, T cells from a JNK1 null mouse hyperproliferated, exhibited decreased activation, and induced cell death and preferentially differentiated into Th2 cells (Dong et al., 1998). Despite the redundancy accorded to this system by the continued production of JNK2, it seems reasonable to conclude that the JNK1 signaling pathway may be playing a role in T-cell receptor–initiated Th cell proliferation, differentiation, and apoptosis. By association, GSTπ must also be involved.
There are other examples where ROS or electrophilic insults stimulate stress response pathways. Thioredoxins are a family of redox proteins of approximately 12 kDa responsible for mediating numerous cytoplasmic functions largely influenced by the Cys 73 residue of the monomeric protein. Dimerization at this site mitigates many of the redox-dependent functions of the protein. Recent data implicate a secreted form of thioredoxin in control of cell growth, where the redox function is essential for growth stimulation (Powis et al., 1998). Tumor cells transfected with thioredoxin demonstrate increased growth and decreased sensitivity to drug-induced apoptosis. As mentioned earlier, thioredoxin has also been shown to bind to ASK1 to inhibit its activity as a kinase (Saitoh et al., 1998). This inhibition is attenuated by ROS, which causes dimerization of thioredoxin (Gotoh and Cooper, 1998). Thioredoxin regulation of the glucocorticoid receptor through association at the DNA-binding domain has also been reported (Makino et al., 1999), and this too is influenced by ROS. These provide examples analogous to the GSTπ/JNK association and perhaps suggest a more broad ranging role for thiol–disulfide regulation of proliferation.
In summary, alterations in redox balance by exposure to ROS cause dose-dependent changes in GSH/GSSG ratios that can potentially influence a number of target proteins by causing oxidation and disulfide exchange reactions at specific cysteine residues. Specifically for GSTπ, the conversion from monomer to dimer (or multimers) causes dissociation from JNK with resultant activation of the kinase. With subsequent phosphorylation of c-Jun and enhanced transcription of AP-1–responsive genes, the stage is set for signal transduction for stress response, proliferation, or apoptosis. Cysteine residues of GSTπ have been shown to be sensitive to oxidation by H2O2 (Shen et al., 1993). The model shown in Fig. 1 illustrates how oxidative stress can transmit through a GST “switch” into the kinase cascade of pathways. Of the numerous literature reports of drug-induced perturbations in GSH/GST levels, few consider whether the biological implications extend further than an enhanced rate of catalytic thioether product formation with a prescribed electrophilic center of an administered drug. The early nomenclature of “ligandin” (Litwack et al., 1971) may prove to have been prescient, particularly regarding the interaction with kinases. This property forms the basis for much of the rationale for drug design in directly inhibiting GSTπ and is described in the following section.
Fig. 1.
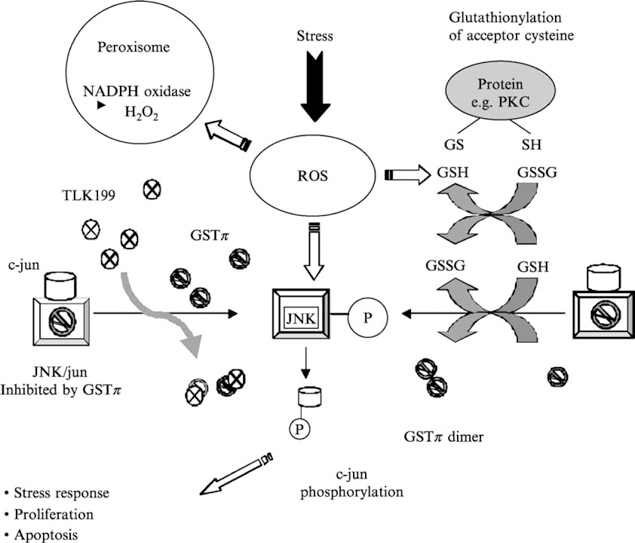
Model scheme showing a means by which oxidative stress can be transmitted through a GST “switch” connecting to kinase cascades influencing cell signaling. In the example shown, TLK199 can cause a disassociation of the GST–JNK complex, activating JNK. PKC is an example of a protein susceptible to glutathionylation.
GSTπ as a Drug Target
In the past, modulation by inhibition of GST has been attempted as a means to improve response to cancer drugs. Use of, for example, ethacrynic acid, although effective in its experimental effects on various GST isozymes, was not successful enough in the clinic to merit continued development. The dose-limiting toxicity of fluid imbalance was triggered by the diuretic properties of the drug (O’Dwyer et al., 1991). However, one consequence of this approach was the conceptual design of a peptidomimetic inhibitor of GSTπ, TLK199 (γ-glutamyl-S-(benzyl)cysteinyl-R(-)phenyl glycine diethyl ester; Fig. 2). In concept, the drug was envisioned as a plausible means to sensitize drug-resistant tumors that overexpress GSTπ (Morgan et al., 1996). However, preclinical and mechanism of action studies with this agent revealed an unexpected effect in animals, namely that the drug caused myeloproliferative activity (Gate et al., 2004; Ruscoe et al., 2001). Given the link between GSTπ and the kinase pathways, a model for how TLK199 can produce proliferative effects in the marrow compartment has been proposed (Gate et al., 2004). Initial clues were provided from the observation of increased myeloproliferation in GSTπ-deficient mice compared with wild-type animals (Ruscoe et al., 2001). A general increase in white blood cell counts in GSTπ−/− mice but no change in leukocyte composition was observed. Spleen cell counts were higher in knock out animals, and this was associated with a twofold increase in B lymphocytes, whereas T lymphocytes and NK cell counts were similar in both animal strains. In contrast, no difference in thymocyte counts and thymus subset composition was observed. Red blood cell and platelet counts were also higher in GSTπ−/− mice. Taken together, these data inferred that the absence of GSTπ expression potentiates hematopoiesis by influencing the proliferation and/or the differentiation of hematopoietic cells.
Fig. 2.
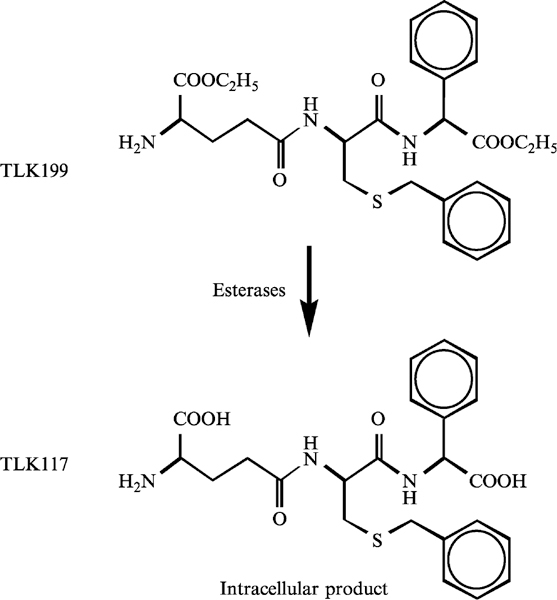
Structure of a peptidomimetic inhibitor of GSTπ, TLK199 (γ-glutamyl-S-(benzyl) cysteinyl-R(-)phenyl glycine diethyl ester) and the de-esterified active inhibitor of GSTπ, TLK117.
In in vitro hematopoiesis experiments, IL-3, GM-CSF, or G-CSF induced more colonies in GSTπ−/− cells than in wild type. TLK199 stimulated colony formation in wild-type, but not in knock out, animals. The JNK inhibitor SP600125 decreased marrow colonies produced by cytokine treatment of knock out animals, and JNK phosphorylation was endogenously elevated in bone marrow cells from GSTπ−/− animals. These data are consistent with GSTπ acting as a physiological inhibitor of JNK, where TLK199 disassociates GSTπ from JNK, allowing kinase phosphorylation and subsequent activation of the kinase cascade (see Fig. 1; Adler et al., 1999a; Ruscoe et al., 2001). Inhibition of JNK, abrogates the increased phosphorylation of this kinase observed in the presence of TLK199 and consequently reduces the myeloproliferative effect of the drug. Because the cell compositions of the colonies were similar in both mouse strains after exposure to cytokines, the increase is a function of proliferation, rather than differentiation, of hematopoietic cells. Taken together, these data suggest that JNK plays an integral role in the elevated myeloproliferation observed in GSTπ-deficient mice and the myelostimulant properties of TLK199. This is consistent with reports indicating a possible role for JNK in control of proliferation. Yang and colleagues have shown that the inhibition of JNK1 or JNK2 expression in human prostate carcinoma was associated with a decrease in cell proliferation (Yang et al., 2003). Similarly, it has been observed that JNK activity was required for rat liver regeneration by increasing cyclin D1 expression and allowing G0–G1 transition (Schwabe et al., 2003). In addition, JNK phosphorylates and induces the transactivation of the transcription factors c-Jun, JunB, and JunD (Kallunki et al., 1996). Overexpression or activation of Jun and JunD has been linked with cell proliferation and transformation (Shaulian and Karin, 2001), and JNK activation has been linked to induction of apoptosis (Tournier et al., 2000). More recent data have suggested that, after UV, JNK1 was more likely to be proapoptotic, whereas JNK2 was associated with survival (Hochedlinger et al., 2002). The discrimination between the survival and apoptotic functions of JNK also seems to correlate with the level and duration of the enzyme activation. A strong and sustained activation is associated with apoptosis, whereas a weaker and transient phosphorylation is correlated with proliferation (Shaulian and Karin, 2001). For example, in mouse hematopoietic BaF3 cells, JNK activity was three times lower when cells were exposed to mitogenic concentrations of IL-3 than to cytotoxic concentrations of anisomycin (Terada et al., 1997). From such reports, it seems likely that regulation of JNK activity by GSTπ should be a viable target for drug intervention. As a consequence, company-sponsored (Telik Inc., Palo Alto, CA) clinical trials of TLK199 (now named Telintra) in patients with myelodysplastic (MDS) syndrome have now been instigated. An ongoing multicenter phase II trial has produced interim results as of the last quarter of 2004. For 34 patients with MDS, clinically significant improvement in one or more blood cell lineages was observed in 61.5% of patients from all major FAB subtypes (Callander, 2004). Responses were associated with decreased requirements for red blood cell, platelet, and growth factor support, in some cases leading to transfusion independence. Although these results are preliminary in nature, they do provide encouragement for the translational relevance of targeting GSTπ and for the mechanism of action data for TLK199.
GST-Activated Prodrugs
TLK286 (Telcyta)
The latent prodrug, TLK286 [γ,-glutamyl-α-amino-β(2-ethyl-N,N,N′,N′-tetrakis (2-chloroethyl)phosphorodiamidate)-sulfonyl-propionyl-(R)-(-) phenylglycine] was synthesized as the lead candidate from a group of rationally designed glutathione analogs designed, once again, to exploit high GSTπ levels associated with malignancy, poor prognosis, and the development of drug resistance (Hayes and Pulford, 1995; Tew, 1994). Thus, selective targeting of susceptible tumor phenotypes is a strategy that should result in the “release” of more active drug in malignant cells compared with normal tissue, thereby achieving an improved therapeutic index.
In TLK286, the sulfhydryl of a glutathione conjugate has been oxidized to a sulfone. The tyrosine-7 in GSTπ promotes a β-elimination reaction that cleaves the compound (see Fig. 3). The cleavage products are a glutathione analog and a phosphorodiamidate, which in turn spontaneously forms aziridinium species, the actual alkylating moieties. The cytotoxic moiety has tetrafunctional alkylating properties, similar in concept to bifunctional nitrogen mustards. Each can react with cellular nucleophiles with a short half-life (Lyttle et al., 1994; Satyam et al., 1996). The other part of the molecule contains the glutathione backbone and an electrophilic vinyl sulfone moiety. The contribution of this component to drug efficacy or toxicity is not entirely clear. Glutathione conjugates of the sulfone are possible, and these could be substrates for transporters such as MRP1. In addition, the vinyl sulfone could be a factor in chain reactions leading to lipid peroxidation and even production of hydrogen peroxide (Comporti, 1989), considerations that could explain the enhanced expression of catalase in the HL60 cell line selected for resistance to TLK286 (Rosario et al., 2000).
Fig. 3.
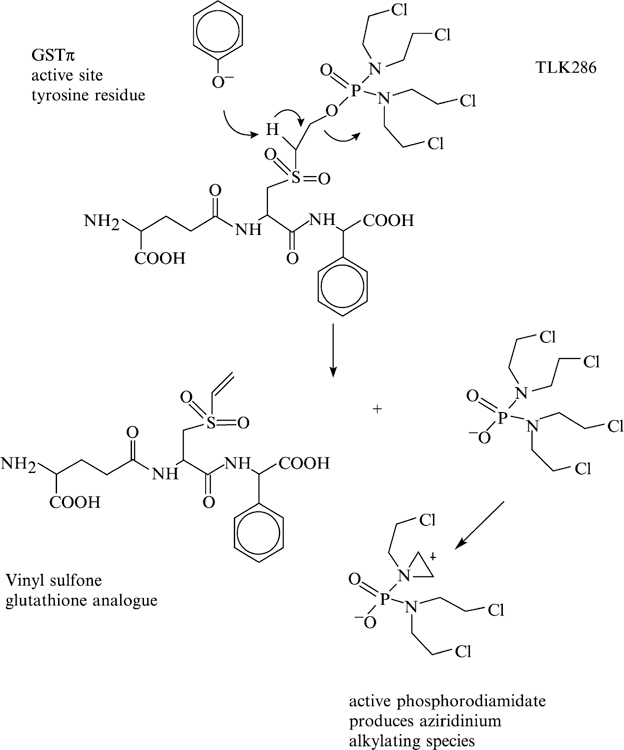
Structure of TLK286 and its activation by GSTπ.
TLK286 exhibits activity against a variety of tumors and tumor cell lines. In vitro studies have shown that elevated GSTπ in stably transfected cell lines correlates with increased sensitivity to TLK286 cells. Similarly, drug-resistant cell lines that overexpress GSTπ are more sensitive to TLK286 (Morgan et al., 1996). In an ex vivo clonogenic assay against human solid tumors, TLK286 showed activity against 15 of 21 lung tumors and 11 of 20 breast tumors tested. In addition, effective antitumor activity was found in vivo using xenografted human tumors in nude mice, with only mild bone marrow toxicity (Morgan et al., 1998).
Three distinct model systems have been used to study the pharmacology of TLK286 with the ultimate goal of gaining proof-of-principle with respect to mechanism of action. The first required establishment of a TLK286 acquired–resistant cell line, a task that proved more difficult to achieve than would normally have been expected. Although cells frequently survived the initial low concentration–selecting treatment and partially repopulated the culture, recovery to full viability was difficult to attain. Considering the relative ease with which HL60 cells can usually be made resistant to anticancer drugs, this result was somewhat surprising. This may indicate that resistance to TLK286 is governed by multiple factors or that survival response pathways are not readily invoked after chronic drug exposure. A characteristic of GSTπ as a GSH-conjugating enzyme is the generally low catalytic efficiency with broad substrate “specificity.” The observation that GSTπ is directly involved in the regulation of JNK-mediated stress response emphasizes the ligand binding, noncatalytic function for the protein. In turn, this may provide a partial explanation for the high GSTπ levels seen in many tumors, where kinase cascade pathways involving JNK may be imbalanced. Although for TLK286 the β-elimination reaction catalyzed by GSTπ does not inactivate the protein, it may serve to compartmentalize it away from the JNK ligand–binding function. This may influence the stoichiometry that controls kinase-mediated proliferative/apoptotic pathways and may be a contributory factor in the difficulty experienced in establishing a TLK286 resistant cell line. With perseverance, eventually a fivefold resistant cell line was established. Phenotypically, decreased expression of GSTπ in resistant cells supported a mechanism of action based on the rational design of the drug (Rosario et al., 2000). Two other experimental systems served to corroborate these results. For example, increased resistance to TLK286 in the MEF cell lines derived from GSTP1-1−/− mice (Henderson et al., 1998) was consistent with reduced activation of the drug. In addition, increased sensitivity in a NIH3T3 cell line transfected to overexpress GSTπ also confirmed a direct involvement of the isozyme in determining cytotoxicity.
The resistant cell line also expressed increased glutathione levels, a mechanism commonly associated with resistance to a range of alkylating agents (Tew, 1994). Enhanced GSH levels were not a consequence of induced overexpression of the primary enzymes responsible for de novo (γ-glutamyl cysteine synthetase; γ) or salvage (γ-transpeptidase; γ-GT) synthesis of the tripeptide. Similarly, MRP expression was unaltered in resistant cells. A co-coordinated, increased expression of γ-GCS, GSTP1-1, and MRP1 has been shown in cells selected for resistance to ethacrynic acid, a drug with Michael addition properties (Ciaccio et al., 1996). TLK286 produces aziridinium moieties characteristic of other nitrogen mustards but distinct in electrophilic properties from Michael acceptors. This difference may account for the absence of evidence for inducible expression of the cadre of GSH-related detoxification gene products in HL60/TLK286-resistant cells. In considering the other major metabolites of TLK286, it is less clear how the release of the vinyl sulfone impacts on the pharmacology of TLK286. Its electrophilic characteristics would predict reactivity with cellular nucleophiles, and because GS conjugates are primary substrates for MRP (Keppler et al., 1997), a GS–vinyl sulfone could prove to be an effective substrate for MRP. Thus, although the vinyl sulfone component of the drug is unlikely to be important to the therapeutic alkylating activity, it may prove to have some pharmacological significance.
Approximately 8 years encompassed the design, synthesis, and pretesting of TLK286. As such, many of the preclinical studies discussed previously were instrumental in leading to clinical trial design. Perhaps the most informative progress to report is the recent clinical results presented at the 2004 American Society of Clinical Oncology meeting. In a series of abstracts, a 46% objective response rate was reported for a combination of Telcyta and liposomal doxorubicin in patients with platinum-refractory ovarian cancer; a 56% objective response rate for Telcyta and Carboplatin in platinum-resistant ovarian cancer; and a 27% objective response rate for combinations of Telcyta and Docetaxel in platinum-resistant non-small cell lung cancer (NSCLC). Telcyta is also under active testing in a clinical phase III setting for a number of disease states, including non-small cell, ovarian, and colon cancers.
PABA-NO
High GSTπ expression levels in human cancers and drug-resistant disease, together with the knowledge that nitric oxide (NO) has therapeutic potential, also provided the rationale for the design of the NO-releasing GST-activated pro-drug, O2-[2,4-dinitro-5-(N-methyl-N-4-carboxyphenylamino)phenyl] 1-N,N-dimethylamino)diazen-1-ium-1,2-diolate; THF, tetrahydrofuran (PABA/NO; (Saavedra et al., 2004)). GSTπ-catalyzed conjugation of GSH to PABA/NO releases a diazeniumdiolate ion, with subsequent release of NO (Fig. 4). Other NO donors of the diazeniumdiolate class are known to release NO in an enzyme-catalyzed manner, with activation by cytochrome P450’s (Saavedra et al., 1997) or esterases (Saavedra et al., 2000).
Fig. 4.
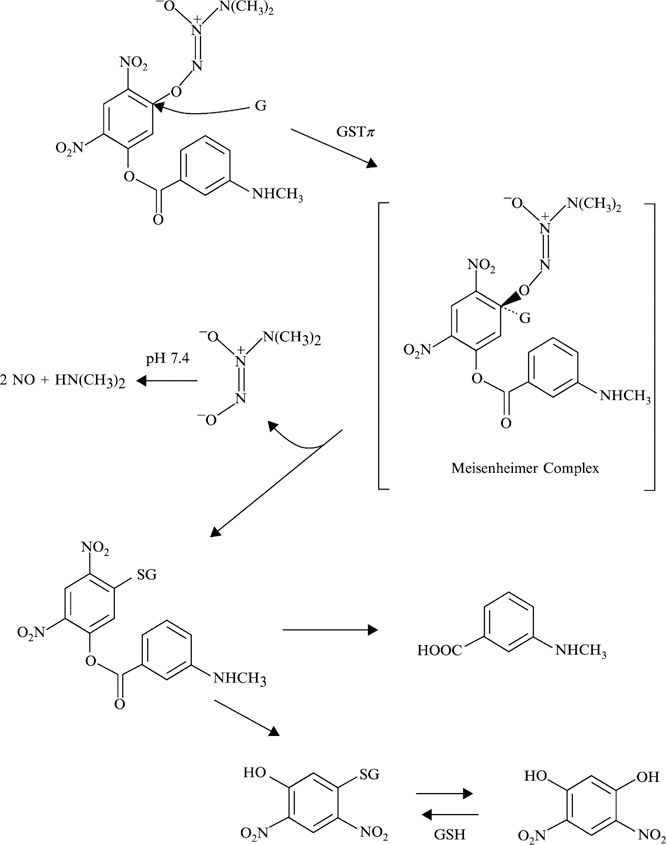
Structure of an NO-releasing GST-activated pro-drug, O2-[2,4-dinitro-5-(N-methyl-N-4-carboxyphenylamino)phenyl] 1-N,N-dimethylamino)diazen-1-ium-1,2-diolate; THF, tetrahydrofuran (PABA/NO). Metabolism and release of nitric oxide are based on the schema outlined elsewhere (Keefer, 2005).
To confirm the GSTπ activation requirements of PABA/NO, a number of model systems have been used. For example, mouse embryo fibroblasts from GSTπ−/− mice showed a decreased sensitivity to PABA/NO. These data are consistent with those for activation of TLK286 (Rosario et al., 2000). The quantitative effect on cytotoxicity of eliminating GSTπ−/− is influenced by the expression of other GST isoforms that can also activate PABA/NO at different rates. In addition, slow spontaneous activation of PABA/NO can occur through noncatalytic GSH conjugation. Other components involved in metabolism and detoxification pathways were analyzed in a model system with stably transfected GSTπ, γGCS, and MRP1. A role for GST and GSH in the activation of PABA/NO was confirmed (Findlay et al., 2004). Forced expression of γGCS and MRP1 also provided insight into cellular resistance mechanisms towards PABA/NO. Unlike TLK286, increased resistance to PABA/NO was conferred by the overexpression of MRP1, supporting the concept that this transporter (an efflux pump with affinity for GS conjugates) is involved in the removal of PABA/NO and/or active metabolite(s). Inhibition of LTC4 transport (a known substrate for MRP1) was observed in the presence of both PABA/NO and GSH but not with either alone. Furthermore, GSTπ stimulated PABA/NO-induced inhibition of LTC4 transport, suggesting that metabolites of PABA/NO may be substrates for, and effluxed by, the transporter (Findlay et al., 2004).
The in vivo efficacy of PABA/NO has been shown in tumor-bearing animals (Findlay et al., 2004; Shami et al., 2003). For example, nontoxic doses of PABA/NO led to a significant growth delay in a human ovarian cancer model in SCID mice, with results comparable to those seen with cisplatin (the standard of care for management of ovarian cancer). Of interest, selective GST activation of PABA/NO may produce a therapeutic advantage, because GSTπ overexpression has been associated (although not necessarily causally linked) with resistance to platinum drugs (Townsend and Tew, 2003). PABA/NO exerts an apoptotic effect by induction of p38 and JNK. As noted earlier, this observation carries greater significance because GSTπ functions as an endogenous negative regulatory switch for these same regulatory kinase pathways (Adler et al., 1999a). NO has diverse roles in a variety of physiological processes, and augmentation of endogenous with exogenous NO provides the foundation for a broad range of therapeutic applications (Keefer, 2003; Napoli and Ignarro, 2003). NO has been reported to modulate the apoptotic process in a number of cell types. Bifurcating pathways of inhibition or induction of cell death seem to be tissue specific and to depend on the amount, duration, and site of NO production (Umansky and Schirrmacher, 2001). In support of both cellular antioxidant and prooxidant actions of NO in vivo, it has been reported that low doses of NO protect cells against peroxide-induced death, whereas higher doses result in increased killing (Joshi et al., 1999). PABA-NO has a dose- and time-dependent effect for drug-induced activation of the kinases, and MRP1 abrogates and/or delays the effect, primarily as a consequence of reducing the effective intracellular concentration of the drug (Findlay et al., 2004). Whether kinase activation occurs as a result of direct NO interaction (e.g., nitrosylation/nitration of residues in JNK) or by the impact of PABA/NO (or metabolites including GSNO) effects on the GSTπ–JNK complex remains to be shown. PABA/NO remains in early preclinical testing but has the potential disadvantage of poor solubility and stability in aqueous solution (Srinivasan, 2005). However, reasonable in vivo antitumor data suggest that it is a good lead compound for further structure activity and drug discovery efforts (Keefer, 2003).
Modulation of Glutathione and Glutathionylation
One of the more interesting conundrums to emerge from the completion of the genome project is the realization that humans are a composite of <30,000 genes, and yet the complexity of protein structure/function seems distinctly more layered. In the burgeoning era of proteomics, it becomes clear that the central dogma of genetic determinism can be influenced by a number of processes that include polymorphic variants, gene-splicing events, exon shuffling, protein domain rearrangements, and the large number of posttranslational modifications that contribute to alterations in tertiary and quaternary protein structure. Among these, phosphorylation, glycosylation, methylation, and acetylation can account for a large proportion of modifications, and indeed, there is evidence to suggest that GSTs may be subject to either glycosylation (Kuzmich et al., 1991) or phosphorylation (Lo et al., 2004). More recently, the addition of glutathione to available cysteine residues (glutathionylation; Fig. 5) has been shown to be of consequence to protein function. The importance of modifying cysteine residues is not necessarily restricted to redox regulation but now seems to be a plausible event that can lead to changes in signaling processes, particularly as a response to a divergent number of stress responses. By adding the GSH tripeptide to a target protein, an additional negative charge is introduced (as a consequence of the glu residue), and a change in protein conformation is made likely. The implication from this analysis is that cells actively participate in the stochastic production of multiple protein building blocks with the intent of realizing functional nonredundancy. A recent report (Manevich et al., 2004) showed that GSTπ is capable of acting in a catalytic manner to glutathionylate peroxiredoxin, a non-selenium–dependent lipid peroxidase that converts lipid hydroperoxides to corresponding alcohols. The catalytically important cysteine residue on peroxiredoxin is sterically inaccessible within the globular dimeric complex, and GSTπ facilitates GSH transfer to this site. The resultant enzyme activation serves a cellular regulatory role, particularly with respect to response to oxidative stress. It will be interesting for future research to determine how widespread the phenomenon of GSTπ catalyzed glutathionylation may be.
Fig. 5.
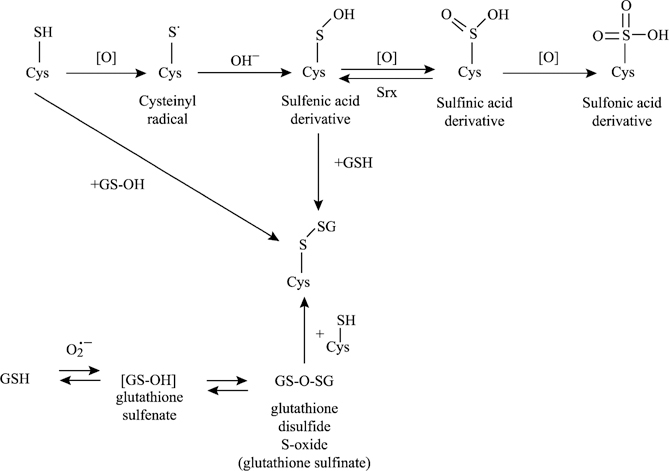
Possible mechanisms of ROS-induced protein glutathionylation. ROS may induce glutathionylation of protein thiols by many different routes. Those highlighted here include the direct oxidation of protein cysteines to generate reactive protein thiol intermediates such as the reactive cysteinyl radical or sulfenic acid, which can further react with GSH to form a mixed disulfide. Alternately, a mixed disulfide is formed through reaction with oxidized forms of GSH (i.e., GS-OH or GS[O]SG).
In the past, modulation of GSH and GST has been attempted as a means to improve response to cancer drugs. Use of, for example, BSO and ethacrynic acid, although effective in their experimental effects on each system, was not successful enough in the clinic to merit continued development (Bailey et al., 1997; O’Dwyer et al., 1991). One consequence of these approaches was the conceptual design of a peptidomimetic inhibitor of GSTπ discussed earlier. More recently, Novelos Inc. has developed a platinum-stabilized coordination complex of oxidized glutathione called NOV-002 (Fig. 6). It is oxidized glutathione complexed with cis-platinum at an approximate 1000:1 ratio. Standard animal and patient dosing with NOV-002 results in a cumulative total of cis-platinum that is equivalent to <2% of a typical standard of care in oncology single-dose cis-platinum. As such, it seems unlikely that the platinum component contributes substantially to the pharmacology of the compound. In some manner, the platinum (and perhaps the cis-amine groups) serves to stabilize the GSSG. In rodents, the serum and tissue levels of GSSG are profoundly affected by this stabilizing influence where standard doses will routinely produce a measurable elevation in circulating GSSG. Clinical data have been already been generated in Russia. Evidence of efficacy has been reported in 340 patients with diseases such as NSCLC, colorectal, pancreatic, and breast cancer. In particular, a 55% 1-year survival rate in patients with NSCLC (17% in control arm) improved the Karnofsky score for quality of life, and improved hematopoietic parameters (with results not dissimilar to those generated for TLK199) have been demonstrated. Lymphocyte counts were elevated in the NOV-002–treated animals, where the numbers of both CD25+ and CD16+/CD56+ cells increased approximately fourfold after long-term treatment (60 days) with the drug.
Fig. 6.
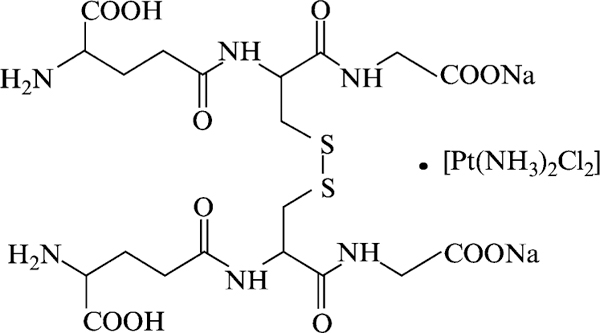
Structure of a platinum-stabilized coordination complex of oxidized glutathione, NOV-002.
Concentrations of NOV-002 between 10–100 mM induce glutathionylation of proteins in target cells. Although this concentration range may seem high, it is achievable in the plasma and tissues of rodents and has no obvious toxicities in the animals. NOV-002 affects the phosphorylation of both ERK and p38, two kinases with critical regulatory roles in governing cell proliferation and apoptotic pathways. In vitro data for NOV-002 seem to mimic those where GSSG is used, emphasizing that this moiety is, indeed, the active component of the drug. Once again, the similarity between NOV-002 and the data for TLK199 seem to indicate the general importance of GSH and GST pathways in governing proliferation in normal tissues and apoptosis in cancer cells.
NOV-002 has undergone significant clinical testing in Russia, and these trials are now being repeated in the United States. Treatment was associated with increases in circulating lymphocyte, monocyte, T-cell, and NK cell counts, and patient response rates were affected through reduced morbidity and the capacity to tolerate longer periods of standard chemotherapy. Quite independent of the clinical data, there are important implications for mechanism of action for NOV-002 that stem from the possibility that GSSG can act as a donor of glutathione (Fig. 5) in glutathionylation of receptive cysteine residues in target proteins.
References
- Adachi T, Pimentel DR, Heibeck T, Hou X, Lee YJ, Jiang B, Ido Y, and Cohen RA (2004). S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J. Biol. Chem 279, 29857–29862. [DOI] [PubMed] [Google Scholar]
- Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, Tew KD, Pincus MR, Sardana M, Henderson CJ, Wolf CR, Davis RJ, and Ronai Z (1999a). Regulation of JNK signaling by GSTp. EMBO J 18, 1321–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler V, Yin Z, Tew KD, and Ronai Z (1999b). Role of redox potential and reactive oxygen species in stress signaling. Oncogene 18, 6104–6111. [DOI] [PubMed] [Google Scholar]
- Bailey HH, Ripple G, Tutsch KD, Arzoomanian RZ, Alberti D, Feierabend C, Mahvi D, Schink J, Pomplun M, Mulcahy RT, and Wilding G (1997). Phase I study of continuous-infusion L-S,R-buthionine sulfoximine with intravenous melphalan. J. Natl. Cancer Inst 89, 1789–1796. [DOI] [PubMed] [Google Scholar]
- Callander N, Ochoa-Bayona J, Piro L, Guba S, Shapiro G, Williams S, Oliff I, Burris H, Jameson A, Patel K, Brown G, FAderl S, Estrov Z, and Emanuel P (2004). Hematologic improvement following treatment with TLK199 (Telintra™), a novel glutathione analog inhibitor of GST P1–1, in myelodysplastic syndrome (MDS): Interim results of a dose-ranging phase 2a study. Session Type: Poster Session 582-I. American Society Hematology
- Ciaccio PJ, Shen H, Kruh GD, and Tew KD (1996). Effects of chronic ethacrynic acid exposure on glutathione conjugation and MRP expression in human colon tumor cells. Biochem. Biophys. Res. Commun 222, 111–115. [DOI] [PubMed] [Google Scholar]
- Ciaccio PJ, Tew KD, and LaCreta FP (1991). Enzymatic conjugation of chlorambucil with glutathione by human glutathione S-transferases and inhibition by ethacrynic acid. Biochem. Pharmacol 42, 1504–1507. [DOI] [PubMed] [Google Scholar]
- Comporti M (1989). Three models of free radical-induced cell injury. Chem. Biol. Interact 72, 1–56. [DOI] [PubMed] [Google Scholar]
- Cross JV, and Templeton DJ (2004). Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem. J 381, 675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis W Jr., Ronai Z, and Tew KD (2001). Cellular thiols and reactive oxygen species in drug-induced apoptosis. J. Pharmacol. Exp. Ther 296, 1–6. [PubMed] [Google Scholar]
- Dong C, Yang DD, Wysk M, Whitmarsh AJ, Davis RJ, and Flavell RA (1998). Defective T cell differentiation in the absence of Jnk1. Science 282, 2092–2095. [DOI] [PubMed] [Google Scholar]
- Findlay VJ, Townsend DM, Saavedra JE, Buzard GS, Citro ML, Keefer LK, Ji X, and Tew KD (2004). Tumor cell responses to a novel glutathione S-transferase-activated nitric oxide-releasing prodrug. Mol. Pharmacol 65, 1070–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gate L, Majumdar RS, Lunk A, and Tew KD (2004). Increased myeloproliferation in glutathione S-transferase pi-deficient mice is associated with a deregulation of JNK and Janus kinase/STAT pathways. J. Biol. Chem 279, 8608–8616. [DOI] [PubMed] [Google Scholar]
- Golemis EA, Tew KD, and Dadke D (2002). Protein interaction-targeted drug discovery: Evaluating critical issues. Biotechniques 32, 636–638, [DOI] [PubMed] [Google Scholar]
- Gotoh Y, and Cooper JA (1998). Reactive oxygen species- and dimerization-induced activation of apoptosis signal-regulating kinase 1 in tumor necrosis factor-alpha signal transduction. J. Biol. Chem 273, 17477–17482. [DOI] [PubMed] [Google Scholar]
- Hayes JD, and Pulford DJ (1995). The glutathione S-transferase supergene family: Regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit. Rev. Biochem. Mol. Biol 30, 445–600. [DOI] [PubMed] [Google Scholar]
- Henderson CJ, Smith AG, Ure J, Brown K, Bacon EJ, and Wolf CR (1998). Increased skin tumorigenesis in mice lacking pi class glutathione S-transferases. Proc. Natl. Acad. Sci. USA 95, 5275–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzenberg LA, De Rosa SC, Dubs JG, Roederer M, Anderson MT, Ela SW, and Deresinski SC (1997). Glutathione deficiency is associated with impaired survival in HIV disease. Proc. Natl. Acad. Sci. USA 94, 1967–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochedlinger K, Wagner EF, and Sabapathy K (2002). Differential effects of JNK1 and JNK2 on signal specific induction of apoptosis. Oncogene 21, 2441–2445. [DOI] [PubMed] [Google Scholar]
- Joshi MS, Ponthier JL, and Lancaster JR Jr. (1999). Cellular antioxidant and pro-oxidant actions of nitric oxide. Free Radic. Biol. Med 27, 1357–1366. [DOI] [PubMed] [Google Scholar]
- Kallunki T, Deng T, Hibi M, and Karin M (1996). c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell 87, 929–939. [DOI] [PubMed] [Google Scholar]
- Keefer LK (2003). Progress toward clinical application of the nitric oxide-releasing diazeniumdiolates. Annu. Rev. Pharmacol. Toxicol 43, 585–607. [DOI] [PubMed] [Google Scholar]
- Keefer LK (2005). Nitric Oxide (NO)- and Nitroxyl (HNO)-Generating Diazeniumdiolates (NONOates): Emerging Commercial Opportunities. Curr. Top. Med. Chem 5, 625–636. [DOI] [PubMed] [Google Scholar]
- Keppler D, Leier I, and Jedlitschky G (1997). Transport of glutathione conjugates and glucuronides by the multidrug resistance proteins MRP1 and MRP2. Biol. Chem 378, 787–791. [PubMed] [Google Scholar]
- Kuzmich S, Vanderveer LA, and Tew KD (1991). Evidence for a glycoconjugate form of glutathione S-transferase pI. Int. J. Pept. Protein Res 37, 565–571. [DOI] [PubMed] [Google Scholar]
- Litwack G, Ketterer B, and Arias IM (1971). Ligandin: A hepatic protein which binds steroids, bilirubin, carcinogens and a number of exogenous organic anions. Nature 234, 466–467. [DOI] [PubMed] [Google Scholar]
- Lo HW, Antoun GR, and Ali-Osman F (2004). The human glutathione S-transferase P1 protein is phosphorylated and its metabolic function enhanced by the Ser/Thr protein kinases, cAMP-dependent protein kinase and protein kinase C, in glioblastoma cells. Cancer Res 64, 9131–9138. [DOI] [PubMed] [Google Scholar]
- Lyttle MH, Satyam A, Hocker MD, Bauer KE, Caldwell CG, Hui HC, Morgan AS, Mergia A, and Kauvar LM (1994). Glutathione-S-transferase activates novel alkylating agents. J. Med. Chem 37, 1501–1507. [DOI] [PubMed] [Google Scholar]
- Makino Y, Yoshikawa N, Okamoto K, Hirota K, Yodoi J, Makino I, and Tanaka H (1999). Direct association with thioredoxin allows redox regulation of glucocorticoid receptor function. J. Biol. Chem 274, 3182–3188. [DOI] [PubMed] [Google Scholar]
- Manevich Y, Feinstein SI, and Fisher AB (2004). Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc. Natl. Acad. Sci. USA 101, 3780–3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AS, Ciaccio PJ, Tew KD, and Kauvar LM (1996). Isozyme-specific glutathione S-transferase inhibitors potentiate drug sensitivity in cultured human tumor cell lines. Cancer Chemother. Pharmacol 37, 363–370. [DOI] [PubMed] [Google Scholar]
- Morgan AS, Sanderson PE, Borch RF, Tew KD, Niitsu Y, Takayama T, Von Hoff DD, Izbicka E, Mangold G, Paul C, Broberg U, Mannervik B, Henner WD, and Kauvar LM (1998). Tumor efficacy and bone marrow-sparing properties of TER286, a cytotoxin activated by glutathione S-transferase. Cancer Res 58, 2568–2575. [PubMed] [Google Scholar]
- Napoli C, and Ignarro LJ (2003). Nitric oxide-releasing drugs. Annu. Rev. Pharmacol. Toxicol 43, 97–123. [DOI] [PubMed] [Google Scholar]
- O’Dwyer PJ, LaCreta F, Nash S, Tinsley PW, Schilder R, Clapper ML, Tew KD, Panting L, Litwin S, Comis RL, and Ozols RF (1991). Phase I study of thiotepa in combination with the glutathione transferase inhibitor ethacrynic acid. Cancer Res 51, 6059–6065. [PubMed] [Google Scholar]
- Peterson JD, Herzenberg LA, Vasquez K, and Waltenbaugh C (1998). Glutathione levels in antigen-presenting cells modulate Th1 versus Th2 response patterns. Proc. Natl. Acad. Sci. USA 95, 3071–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powis G, Kirkpatrick DL, Angulo M, and Baker A (1998). Thioredoxin redox control of cell growth and death and the effects of inhibitors. Chem. Biol. Interact 111–112, 23–34. [DOI] [PubMed] [Google Scholar]
- Rosario LA, O’Brien ML, Henderson CJ, Wolf CR, and Tew KD (2000). Cellular response to a glutathione S-transferase P1–1 activated prodrug. Mol. Pharmacol 58, 167–174. [DOI] [PubMed] [Google Scholar]
- Ruscoe JE, Rosario LA, Wang T, Gate L, Arifoglu P, Wolf CR, Henderson CJ, Ronai Z, and Tew KD (2001). Pharmacologic or genetic manipulation of glutathione S-transferase P1–1 (GSTpi) influences cell proliferation pathways. J. Pharmacol. Exp. Ther 298, 339–345. [PubMed] [Google Scholar]
- Saavedra JE, Billiar TR, Williams DL, Kim YM, Watkins SC, and Keefer LK (1997). Targeting nitric oxide (NO) delivery in vivo. Design of a liver-selective NO donor prodrug that blocks tumor necrosis factor-alpha-induced apoptosis and toxicity in the liver. J. Med. Chem 40, 1947–1954. [DOI] [PubMed] [Google Scholar]
- Saavedra JE, Bohle DS, Smith KN, George C, Deschamps JR, Parrish D, Ivanic J, Wang YN, Citro ML, and Keefer LK (2004). Chemistry of the diazeniumdiolates. O- versus N-alkylation of the RNH[N(O)NO](-) ion. J. Am. Chem. Soc 126, 12880–12887. [DOI] [PubMed] [Google Scholar]
- Saavedra JE, Shami PJ, Wang LY, Davies KM, Booth MN, Citro ML, and Keefer LK (2000). Esterase-sensitive nitric oxide donors of the diazeniumdiolate family: In vitro antileukemic activity. J. Med. Chem 43, 261–269. [DOI] [PubMed] [Google Scholar]
- Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, and Ichijo H (1998). Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 17, 2596–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyam A, Hocker MD, Kane-Maguire KA, Morgan AS, Villar HO, and Lyttle MH (1996). Design, synthesis, and evaluation of latent alkylating agents activated by glutathione S-transferase. J. Med. Chem 39, 1736–1747. [DOI] [PubMed] [Google Scholar]
- Schwabe RF, Bradham CA, Uehara T, Hatano E, Bennett BL, Schoonhoven R, and Brenner DA (2003). c-Jun-N-terminal kinase drives cyclin D1 expression and proliferation during liver regeneration. Hepatology 37, 824–832. [DOI] [PubMed] [Google Scholar]
- Shami PJ, Saavedra JE, Wang LY, Bonifant CL, Diwan BA, Singh SV, Gu Y, Fox SD, Buzard GS, Citro ML, Waterhouse DJ, Davies KM, Ji X, and Keefer LK (2003). JS-K, a glutathione/glutathione S-transferase-activated nitric oxide donor of the diazeniumdiolate class with potent antineoplastic activity. Mol. Cancer Ther 2, 409–417. [PubMed] [Google Scholar]
- Shaulian E, and Karin M (2001). AP-1 in cell proliferation and survival. Oncogene 20, 2390–2400. [DOI] [PubMed] [Google Scholar]
- Shen H, Tsuchida S, Tamai K, and Sato K (1993). Identification of cysteine residues involved in disulfide formation in the inactivation of glutathione transferase P-form by hydrogen peroxide. Arch. Biochem. Biophys 300, 137–141. [DOI] [PubMed] [Google Scholar]
- Terada K, Kaziro Y, and Satoh T (1997). Ras-dependent activation of c-Jun N-terminal kinase/stress-activated protein kinase in response to interleukin-3 stimulation in hematopoietic BaF3 cells. J. Biol. Chem 272, 4544–4548. [DOI] [PubMed] [Google Scholar]
- Tew KD (1994). Glutathione-associated enzymes in anticancer drug resistance. Cancer Res 54, 4313–4520. [PubMed] [Google Scholar]
- Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, and Davis RJ (2000). Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288, 870–874. [DOI] [PubMed] [Google Scholar]
- Townsend D, and Tew K (2003). Cancer drugs, genetic variation and the glutathione-S-transferase gene family. Am. J. Pharmacogenomics 3, 157–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umansky V, and Schirrmacher V (2001). Nitric oxide-induced apoptosis in tumor cells. Adv. Cancer Res 82, 107–131. [DOI] [PubMed] [Google Scholar]
- Wang T, Arifoglu P, Ronai Z, and Tew KD (2001). Glutathione S-transferase P1-1 (GSTP1-1) inhibits c-Jun N-terminal kinase (JNK1) signaling through interaction with the C terminus. J. Biol. Chem 276, 20999–21003. [DOI] [PubMed] [Google Scholar]
- Yang YM, Bost F, Charbono W, Dean N, McKay R, Rhim JS, Depatie C, and Mercola D (2003). C-Jun NH(2)-terminal kinase mediates proliferation and tumor growth of human prostate carcinoma. Clin. Cancer Res 9, 391–401. [PubMed] [Google Scholar]
- Yin Z, Ivanov VN, Habelhah H, Tew K, and Ronai Z (2000). Glutathione S-transferase p elicits protection against H2O2-induced cell death via coordinated regulation of stress kinases. Cancer Res 60, 4053–4057. [PubMed] [Google Scholar]