

Abstract
Purpose:
Thyroid cancer cell lines are valuable models but have been neglected in pan-cancer genomic studies. Moreover, their misidentification has been a significant problem. We aim to provide a validated dataset for thyroid cancer researchers.
Experimental Design:
We performed next-generation sequencing and analyzed the transcriptome of 60 authenticated thyroid cell lines and compared our findings with the known genomic defects in human thyroid cancers.
Results:
Unsupervised transcriptomic analysis showed that 94% of thyroid cell lines clustered distinctly from other lineages. Thyroid cancer cell line mutations recapitulate those found in primary tumors (e.g., BRAF, RAS or gene fusions). Mutations in the TERT promoter (83%) and TP53 (71%) were highly prevalent. There were frequent alterations in PTEN, PIK3CA and of members of the SWI/SNF chromatin remodeling complex, mismatch repair, cell cycle checkpoint, histone methyl- and acetyltransferase functional groups. Copy number alterations (CNA) were more prevalent in cell lines derived from advanced vs. differentiated cancers, as reported in primary tumors, although the precise CNAs were only partially recapitulated. Transcriptomic analysis showed that all cell lines were profoundly dedifferentiated, regardless of their derivation, making them good models for advanced disease. However, they maintained the BRAFV600E vs. RAS-dependent consequences on MAPK transcriptional output, which correlated with differential sensitivity to MEK inhibitors. Paired primary tumor-cell line samples showed high concordance of mutations. Complete loss of p53 function in TP53 heterozygous tumors was the most prominent event selected during in vitro immortalization.
Conclusions:
This cell line resource will help inform future pre-clinical studies exploring tumor-specific dependencies.
INTRODUCTION
Cell lines are useful pre-clinical models to study cancer mechanisms and to test novel therapies. The collection of thyroid cancer-derived cell lines is significantly smaller compared to other common tumor types, and has been poorly characterized. None of the cell lines in the NCI-60 panel are of thyroid origin, and there are only 18 thyroid cancer cell lines - some of which are redundant or of dubious origin - out of the 1,100 specimens assessed by the Cancer Cell Line Encyclopedia (CCLE) (1,2). Moreover, misidentification and cross-contamination of thyroid cancer cell lines has bedeviled the field. We previously profiled 40 cell lines, only 23 of which were found to be unique and likely of thyroid origin based on genetic fingerprinting, Sanger sequencing of the main drivers and detectable expression of the thyroid lineage markers PAX8 and NKX2.1 (3). Therefore, there is a critical need for a properly curated thyroid cancer cell line resource for the research community.
Thyroid cancer cell line genotyping has thus far been restricted to a few of the canonical drivers of the disease. Next-generation sequencing (NGS) has revolutionized the characterization of cancer specimens, both in terms of authentication and genetic makeup. It has also paved the way to assess whether cell lines faithfully recapitulate the features of the tumors from which they originate, and whether specific traits arise or are enriched during selection in culture (4,5).
Here, we performed targeted cancer gene NGS and expression array profiling of 60 cell lines, representing virtually every thyroid cancer-derived line established to date. We identified a wide spectrum of somatic mutations, gene fusions, copy number alterations (CNAs) and expression changes that in part recapitulate those reported in papillary (PTC), follicular (FTC), poorly-differentiated (PDTC), anaplastic (ATC) and medullary thyroid cancers (MTC) (6–11). Thyroid cancer cell lines mostly share the mutational features of ATCs, from which more than half were derived, and constitute good models for studies of driver-dependency. Transition to in vitro culture profoundly affects CNAs, global expression patterns and the differentiation state of the cells, suggesting that other models may be more suitable to test for therapeutic strategies exploring events controlling thyroid specification and differentiated function. In addition, sequencing of paired primary tumors and patient-derived xenografts (PDX) provided valuable insights into thyroid cancer microevolution, showing that the drivers are uniformly enriched towards a heterozygous or homozygous state in the cell lines, whereas genes such as TP53 are selected during in vitro culture.
METHODS
Cell line origin and culture conditions
Thyroid cancer cell lines included in this study were developed in our laboratories ((12,13) and unpublished), acquired directly from the originator when possible, or from repositories. We studied 60 cell lines, from which we excluded ML-1 and THJ-11T. THJ-11T yielded low-quality sequencing data. Our analysis of two independent vials of the ML-1 cell line stored in our laboratories showed evidence of contamination from BHT-101 cells, therefore the ML-1 gene expression profile from the CCLE was used in these studies. For mutational analyses, we present data on 58 cell lines. All cell lines were maintained at 37°C and 5% CO2 in humidified atmosphere and grown in the recommended media.
Single nucleotide variant calling
MSK-IMPACT targeted sequencing was performed in 83 specimens, including 60 cell lines, 12 primary tumors, 3 PDX and 8 paired normal tissues. 42 samples were assessed for exonic mutations of 341 cancer genes. For 41 samples, a newer MSK-IMPACT version covering 69 additional genes (total n=410) was used (14). Information about the platform version used for each sample (IMPACT-341/410) is included in Suppl. Table S2. Single nucleotide variants (SNVs) and short indels (<30 bp in length) were automatically annotated by the MSK-IMPACT pipeline, as previously described (7,14). Full details on variant filtering are described in the Supplementary Methods. Mutation plots were generated using the OncoPrinter (v1.0.1) tools available at the cBioPortal (http://cbioportal.org) (15,16).
Chromosomal rearrangements were called for genes whose introns were covered by MSK-IMPACT, which included all previously reported fusions in thyroid tumors, with the exception of NTRK1 and NTRK3.
Copy number alterations
Copy number alterations were called from MSK-IMPACT, by comparing sequence reads of targeted regions in tumors relative to a standard diploid normal sample, as described (14). Focal, single-arm level and whole chromosome CNAs were identified using the GISTIC 2.0 tool (17), as detailed in the Supplementary Methods. CNAs were visualized in the Integrative Genomics Viewer (IGV), version 2.3.57 (18).
Gene expression
Transcriptome-wide gene expression data for thyroid cancer cell lines was generated using Affymetrix Human Genome U133 Plus 2.0 microarrays. The quality control was performed using the arrayQualityMetrics package (19) from Bioconductor 3.5 in R. The outliers were detected using between array comparisons, MA plots and by analyzing array intensity distributions. 56 microarray profiles passed quality control and were used for downstream analysis. Gene expression values for 10 cell lines analyzed in duplicate (8505C, B-CPAP, C-643, Hth74, KTC-1, SW1736, T238, T243, TPC-1, TTA-1) were averaged. Background subtraction and quantile normalization were performed with Affymetrix Power Tools (http://media.affymetrix.com/support/developer/powertools/changelog/index.html). Probe sets were collapsed to genes using GSEA v2.1.0 software (20). Gene expression data for thyroid cancer cell lines is summarized in Suppl. Table S7.
We included in the analysis publicly available microarray profiles (all done on HG-U133 Plus 2.0 platform) for 1037 cell lines of various cancer types from CCLE (2) (http://www.broadinstitute.org/ccle/home). The expression data for four thyroid cancer cell lines analyzed in triplicate by GlaxoSmithKline (GSK) was downloaded from the NCI’s Cancer Bioinformatics Grid (https://cabig-stage.nci.nih.gov/community/caArray_GSKdata/). To distinguish cell lines from different studies we added prefixes “CU_” (our data), “CCLE_” and “GSK_” to gene expression profiles. We also used published microarray gene expression profiles for ATCs, PDTCs ((7), GSE76039), PTCs and normal thyroid tissue ((21), GSE3467). Specific gene expression analyses are detailed in Supplementary Methods.
RESULTS
Samples and overall approach
We studied 60 thyroid cancer cell lines, including 12 that were recently established (13). Two cell lines (THJ-11T and ML-1) were excluded from the mutational analysis.
The remaining 58 cell lines (Table 1), were unique by STR fingerprinting (Suppl. Table S1), and derived from the following thyroid tumor types: 12 PTCs, 8 FTCs, 3 PDTCs, 31 ATCs and 2 MTCs. We also characterized one cell line derived from normal thyroid tissue immortalized with SV40 large T antigen (Nthy-ori-3–1) (22), and one cell line generated from a benign adenomatoid nodule (CUTC6). The 58 cell lines represented 55 individuals, since FTC-133/FTC-236/FTC-238 and SDAR1/SDAR2 were established from the same patients, respectively. We also sequenced 12 primary tumors from which the cell lines were derived, as well as 3 PDX.
Table 1.
Characteristics of the 58 thyroid cell lines highlighted in the study
| Cell Line | Original Thyroid Tumor Type | Key genetic drivers |
|---|---|---|
| 8305C | Anaplastic | BRAF p.V600E |
| 8505C | Anaplastic | BRAF p.V600E |
| ACT-1 | Anaplastic | NRAS p.Q61K |
| ASH-3 | Anaplastic | NRAS p.Q61R |
| B-CPAP | Papillary | BRAF p.V600E |
| BHT-101 | Anaplastic | BRAF p.V600E |
| C-643 | Anaplastic | HRAS p.G13R |
| CAL-62 | Anaplastic | KRAS p.G12R |
| CUTC48 | Papillary | CCDC6-RET fusion |
| CUTC5 | Papillary | BRAF p.V600E |
| CUTC6 | Adenomatoid nodule | NRAS p.Q61K |
| CUTC60 | Anaplastic | BRAF p.V600E |
| CUTC61 | Follicular | HRAS p.Q61R |
| EAM306 | Follicular | NRAS p.Q61R |
| FTC-133 | Follicular | NF1 p.C167*, PTEN p.R130*, TP53 p.R273H |
| FTC-236 | Follicular | PTEN p.R130*, TP53 p.R273H |
| FTC-238 | Follicular | PTEN p.R130*, TP53 p.R273H |
| HTC-C3 | Anaplastic | BRAF p.V600E |
| HTh104 | Anaplastic | BRAF p.V600E |
| HTh7 | Anaplastic | NRAS p.Q61R |
| HTh74 | Anaplastic | NF1 p.L732fs |
| HTh83 | Anaplastic | HRAS p.Q61R |
| IHH-4 | Anaplastic | BRAF p.V600E |
| JEM493 | Anaplastic | HRAS p.Q61R |
| K1 (GLAG-66) | Papillary | BRAF p.V600E |
| KAT-18 | Anaplastic | Unknown |
| KHM-5M | Anaplastic | BRAF p.V600E |
| KMH-2 | Anaplastic | NRAS p.Q61R |
| KTC-1 | Papillary | BRAF p.V600E |
| KTC-2 | Anaplastic | BRAF p.V600E |
| LAM1 | Papillary | BRAF p.V600E |
| LAM136 | Papillary | BRAF p.V600E |
| MDA-T120 | Papillary | BRAF p.V600E |
| MDA-T32 | Papillary | BRAF p.V600E |
| MDA-T41 | Papillary | BRAF p.V600E |
| MDA-T85 | Papillary | BRAF p.V600E |
| MZ-CRC-1 | Medullary | RET M918T |
| Nthy-ori-3-1 | Normal thyroid | N/A |
| OCUT-1 | Anaplastic | BRAF p.V600E |
| OCUT-2 | Anaplastic | BRAF p.V600E |
| SDAR1 | Follicular | PTEN V54fs, TP53 p.R282P |
| SDAR2 | Follicular | PTEN V54fs, TP53 p.V217fs |
| SW1736 | Anaplastic | BRAF p.V600E |
| T235 | Anaplastic | BRAF p.V600E |
| T238 | Anaplastic | BRAF p.V600E |
| T241 | Anaplastic | PTEN D252fs |
| T243 | Poorly-Differentiated | MSH2 p.Q130fs, microsatellite instability |
| T351 | Poorly-Differentiated | NF1 p.Q28* |
| TCO-1 | Anaplastic | BRAF p.V600E |
| THJ-16T | Anaplastic | MKRN1-BRAF fusion |
| THJ-21T | Anaplastic | BRAF p.V600E |
| THJ-29T | Anaplastic | FGFR2-OGDH fusion |
| THJ529 | Poorly-Differentiated | BRAF p.V600E |
| THJ560 | Anaplastic | BRAF p.V600E |
| TPC-1 | Papillary | CCDC6-RET fusion |
| TT | Medullary | RET C634W |
| TT2609-CO2 | Follicular | NRAS p.Q61R |
| TTA-1 | Anaplastic | Unknown |
All cell lines and paired tissues were analyzed using MSK-IMPACT, a NGS platform targeting 341/410 cancer genes (14), allowing us to call point mutations, short indels and CNAs. Gene expression profiling was performed in a subset of 44 cell lines to assess transcriptomic changes.
Point mutations and short indels
Cell lines harbored a median number of 10 mutations (interquartile range [IQR]=7–14) in the 341 cancer genes studied in all specimens, after filtering out variants reported in the ExAC database. The annotated full list of variants identified is shown in Supplementary Table S2. Figure 1 highlights the main genetic alterations found in the 58 cell lines, curated based on genes harboring somatic mutations in human thyroid cancers (6,7). Overall, mutations in the thyroid cancer cell lines faithfully recapitulated those previously reported in primary tumors.
Figure 1. Cancer genome alterations in 58 thyroid cancer cell lines.

Cell line names, normal tissue availability (paired normal or pooled control), patient’s age, gender, original tumor type and derivation are shown in the top panel. Genes are listed on the left of the oncoprint, and the percentage of samples harboring genetic alterations in those genes is shown on the right. Genes are clustered in functional groups, where indicated. The number of variants identified in each cell line is shown in the bottom-most panel. Color codes for mutational and clinicopathological features are listed in the boxes on the right.
Twenty-eight out of 58 of the thyroid cancer cell lines harbored V600E hotspot activating mutations in BRAF. All but one of the BRAF-mutant cell lines were derived from PTCs or ATCs. Mutant allelic frequencies (MAF) of BRAF p.V600E were close to 50% in most cell lines, supporting the clonal nature of this mutation in heterozygosis. 8505C, B-CPAP and THJ-21T cell lines displayed only mutant BRAF p.V600E reads; a closer look at the CNA profile showed that all three samples showed signs of a heterozygous loss of the BRAF locus, suggesting that the wild-type allele might be absent. The THJ-16T cell line displayed a MKRN1-BRAF fusion, as previously reported in thyroid tumors (23).
Mutations in NRAS, HRAS and KRAS genes occurred in 14%, 9% and 2% of cell lines, respectively, and were mutually exclusive with BRAF (Fisher’s p-value=0.005, Figure 1). Two cell lines harbored both BRAF and RAS mutations. 8305C had a likely passenger NRAS frameshift mutation (NRAS p.F90fs, MAF=5% vs. BRAF p.V600E, MAF=47%). MDA-T85 cells harbored oncogenic BRAF p.V600E (48%) and HRAS p.Q61K (47%) mutations (24), although the latter was not detected in the original tumor (not shown).
Loss-of-function mutations in the neurofibromin 1 gene (NF1) were found in three cell lines without alterations in BRAF, RAS or gene fusion events (Fisher’s p-value=0.012). Missense variants in the thyroid stimulating hormone receptor gene (TSHR) occurred in 4 cell lines, but only p.I486F, found in BRAF-mutant SW1736 cells, has been reported as a gain-of-function somatic mutation found in autonomously functioning follicular carcinomas and toxic thyroid adenomas, and proven to activate both the cAMP and inositol phosphate pathways (25–27).
Mutations in the proximal promoter of TERT (telomerase reverse transcriptase) were the most common genetic alterations, occurring in 83% of cell lines. Canonical mutations at c.−124C>T (71%) and c.−146C>T (23%) accounted for the majority of TERT alterations, but three cell lines (B-CPAP, 8505C and T238) had additional promoter changes. FTC-133, Hth7, Hth74, TCO-1, OCUT-2, SDAR1 and SDAR2 cell lines were homozygous for TERT mutations. TERT CNA profile showed evidence of TERT amplification in Hth74, TCO-1, OCUT-2 and SDAR2, whereas Hth7 had a deletion, presumably of the wild-type copy, and FTC-133 and SDAR1 profiles were consistent with uniparental diploidy (Suppl. Fig. S1).
The TP53 gene was altered in 71% of cell lines, showing frequent truncating events and pathogenic missense mutations. Complete loss-of-function of p53 was strongly favored in culture: 36/47 TP53 mutations were present in homozygosis or hemizygosis (as suggested by the frequent losses of TP53 locus in the CNA profiling), and six cell lines harbored two TP53 mutations/each.
Eight cell lines (14%) had mutations in PTEN, seven of which were truncating. Seven out of eight PTEN mutations occurred in TP53-mutant cell lines, a combination reported to induce ATC in genetically engineered mouse (GEM) models (28). PIK3CA mutations were seen in 12% of cell lines. Known gain-of-function mutations in the helical and kinase protein domains co-occurred with BRAF mutation (Fisher’s p-value=0.02), as reported in ATCs (7), also sufficient to induce ATC in GEM models (29). These two key effectors of the PI3K/AKT/mTOR pathway highlight its importance in a subset of thyroid cancers. Missense variants in other members of this pathway (AKT2, MTOR, PIK3C2G, PIK3C3, PIK3CB, PIK3CG, PIK3R1, PIK3R2, PIK3R3, RICTOR, RPS6KA4, RPS6KB2, RPTOR and TSC2) were found at low frequencies, but, besides AKT1 p.E17K gain-of-function mutation in IHH-4 cells, their oncogenic properties are unclear.
Mutations in the translation initiation factor EIF1AX were exclusively found in RAS-mutant cell lines ACT-1, C-643 and Hth83 (Fisher’s p-value=0.02), and occurred either at the N-terminal region or in a hotspot splicing acceptor site on exon 6, as reported in thyroid cancers (6,7).
Truncating mutations in NF2 were found in five cell lines (9%), with MAFs showing total loss-of-function for all five. Copy number profile of chromosome arm 22q suggested NF2 hemizygosis for TCO-1 and MDA-T120 cells. Truncating mutations in other tumor suppressor genes, such as RB1 and MEN1, were also found at low frequencies.
The DNA repair gene ATM, and mismatch repair (MMR) members MLH1, MSH2 and MSH6 were altered in approximately 15% of cell lines, typically via truncating mutations, which were mutually exclusive with BRAF, RAS mutations and gene fusions. T243 and FTC-133/FTC-236/FTC-238, which had complete loss-of-function alterations in MMR genes, showed a higher number of mutations compared with cells retaining MMR wild-type activity (median, IQR=38.5, 35.75–63.75 vs. 9.5, 6.75–13; Mann-Whitney p-value<0.0001) pointing to hypermutation as an underlying oncogenic mechanism, as we demonstrated in aggressive differentiated thyroid cancer (DTC) and ATC (8). Of interest, MMR mutations only affected the stability of the T243 cell line STR profile.
We found loss-of-function mutations in the cell cycle checkpoint gene CHEK2 and in cyclin-dependent kinase inhibitor genes CDKN1A (p21), CDKN1B (p27) and CDKN2A (p16). CDKN2A mutations (12%) and deletions of the CDKN2A locus at 9q21.3 were particularly frequent in cell lines (27/58), as reported in advanced thyroid cancers (8).
Genetic alterations in members of the SWI/SNF chromatin remodeling complex, such as ARID1A, ARID1B, ARID2, SMARCA4, SMARCD1, PBRM1 and ATRX were found in 18/58 (31%) cell lines. Some of these were loss-of-function mutations, whereas others were missense variants of unknown significance. Other genes involved in epigenetic regulation were frequently mutated: histone methyltransferases (HMTs), such as KMT2A, KMT2C, KMT2D and SETD2, which were found in 18/58 (31%) cell lines, and histone acetyltransferases CREBBP and EP300, found in 10% and 14% of cell lines, with frequent loss-of-function events.
Missense and truncating mutations in PTCH1, a gene encoding the patched 1 receptor, which represses hedgehog signaling in its unliganded form, were present in 7% of cell lines, although they were all subclonal events (MAFs<15%). Other genes occasionally mutated in cell lines included NOTCH3, FANCA1, AR, MDC1, RAC1, NOTCH4, ROS1, TET2, ERBB2, GRIN2A, STAG2, FAT1 and MED12.
The two MTC-derived cell lines, TT and MZ-CRC-1, had known activating mutations in RET at C634W and M918T, respectively. TT cells also harbored a subclonal mutation at TP53 p.S127F (MAF=2%) and a truncating alteration in the transcriptional repressor TBX3, whereas MZ-CRC-1 cell line displayed a homozygous truncating event in the SWI/SNF gene PBRM1, a homozygous splicing mutation in MAX (MYC associated factor X), and a missense mutation in PIK3CA of unknown oncogenic consequences.
Nthy-ori-3–1 cells, derived from normal human thyroid follicular cells (22), harbored missense variants in KMT2A, POLE and CHEK2, all of which have unspecified functional effects.
Gene fusions
RET/PTC1 rearrangements were detected in the TPC-1 (as described (30)) and CUTC48 cell lines (Suppl. Table S3). Both fusion genes were generated by intrachromosomal inversions that fused RET tyrosine kinase domain to the CCDC6 gene. MKRN1-BRAF and FGFR2-OGDH fusions were detected in THJ-16T and THJ-29T cells, respectively, as reported (31). FGFR2-OGDH was also detected in the primary tumor from which THJ-29T cell line was derived, and MKRN1-BRAF was identified by manually inspecting the mapping of the sequences to the reference genome around the breakpoints. All four rearrangements were present in cell lines without BRAF or RAS mutations. No other high-confidence calls for in-frame fusion events were identified.
Somatic copy number alterations
Somatic CNAs in thyroid cancer cell lines were frequent and widespread. It is unclear whether these may have arisen in vitro or if they were present in the tumors of origin. PTCs are known to be largely diploid, whereas CNAs are much more common in PDTCs and ATCs (6,7). Remarkably, cell lines derived from ATCs showed greater CNAs than those derived from PTCs (Suppl. Fig. S2).
We identified 16 recurrent focal CNAs across cell lines by GISTIC analysis (Table 2, Suppl. Fig. S3, Suppl. Table S4). Generally, the magnitude of copy-number losses was greater than those of copy-number gain.
Table 2.
Recurrent copy number alterations identified in 58 thyroid cancer cell lines.
| Region | Chromosomal Coordinates (hg19) | Region Size (Mb) | q value |
|---|---|---|---|
| Copy number gains | |||
| 5p15.33 | chr5:1-4089200 | 4.1 | 3.3E-02 |
| 7p22.1 | chr7:2640379-6439787 | 3.8 | 3.7E-02 |
| 8q24.21 | chr8:120071899-131128147 | 11.1 | 3.3E-02 |
| 11q13.2 | chr11:64577353-77034190 | 12.5 | 3.3E-02 |
| 11q22.1 | chr11:100850203-108108386 | 7.3 | 3.3E-02 |
| 16q24.3 | chr16:89250883-90354753 | 1.1 | 7.4E-02 |
| 20p12.2 | chr20:7601014-15799746 | 8.2 | 3.3E-02 |
| Copy number losses | |||
| 3p24.1 | chr3:29250697-30667217 | 1.4 | 7.6E-03 |
| 3p13 | chr3:71007446-75156467 | 4.1 | 2.5E-02 |
| 4p16.3 | chr4:1-12341653 | 12.3 | 3.0E-03 |
| 4q35.2 | chr4:187367540-187529941 | 0.2 | 5.8E-03 |
| 6q25.1 | chr6:139292684-150013096 | 10.7 | 5.6E-02 |
| 7q31.1 | chr7:103122646-116359002 | 13.2 | 9.3E-03 |
| 9p21.3 | chr9:18995151-36840594 | 17.8 | 4.9E-30 |
| 13q12.11 | chr13: 21004738-43633863 | 22.6 | 9.7E-06 |
| 18q12.3 | chr18:39535292-43025490 | 3.5 | 4.6E-02 |
To study the effect of CNAs on gene expression we compared microarray data for cell lines with and without CNAs for each gene located in the affected regions (Suppl. Table S5). One-hundred thirty-four genes were differentially expressed (Suppl. Table S6, adjusted p-value<0.05) and the direction of change corresponded to the CNA type (133/134, overexpression-gene amplification and underexpression-gene deletion) supporting high quality of our CNA calling approach. Remarkably, amplifications at 7p22.1, 8q24.21 and 11q22, which are common events in several cancers (32–34), correlated with significant overexpression of their target oncogenes RAC1, MYC and YAP1, respectively (Suppl. Table S6). Deletion at 9p21.3, which includes CDKN2A/CDKN2B (32), was highly recurrent in thyroid cancer cell lines, and correlated with lower expression of CDKN2B (Suppl. Table S6). The following cell lines showed copy number values at 9p21.3 locus ≤ −1.0-fold change: C643, Cal-62, HTC-C3, HTh74, HTh83, IHH-4, GLAG-66 (K1), KTC-1, KTC-2, OCUT-1, OCUT-2, TCO-1, THJ21T, TTA-1, MZ-CRC-1, THJ560, THJ529, LAM1 and HTh7.
Gene expression profiling
Hierarchical clustering of gene expression data
We performed hierarchical clustering of transcriptome-wide gene expression in thyroid cancer cell lines (Suppl. Table S7), together with the expression data publicly available for cancer cell lines of different origins from the CCLE and GSK databases (n=1041). The replicate expression profiles for thyroid cancer cell lines between our study and other databases typically clustered together, indicating high quality and reproducibility of the data.
Most thyroid cancer cells from our study, CCLE and GSK formed a tissue-specific cluster (Figure 2, red background). This was not caused by a batch effect, because expression profiles from different studies (e.g., BHT-101 and CAL-62) clustered together within the thyroid cancer group. Several FTC-derived cells, including ML-1 and replicates of FTC-133 and TT2609-CO2 cell lines formed a separate cluster. In addition, we confirmed that cell lines previously misidentified as of thyroid origin (e.g., ARO81, DRO90) (3) clustered according to their true identities and separately from thyroid cell lines.
Figure 2. Cluster analysis of combined gene expression data from this study (“CU”, University of Colorado), CCLE and GSK.

Color background indicates clusters consisting predominantly of cell lines originating from the same primary site (such as thyroid, in red). Thyroid cell lines clustering outside the main thyroid group are indicated by red arrows, whereas cell lines from other origins clustering within the thyroid group are designated by blue dots.
Although 45/48 thyroid cancer cell lines clustered together, supporting their lineage identity, a few did not (Fig 2, red arrows). Two replicates of HTh74 cells clustered with central nervous system tumor cell lines. HTh74 cells do not express the thyroid-lineage transcription factor PAX8, and harbor loss-of-function NF1 and TERT promoter mutations, which are common events in glioblastomas and thyroid cancers. As ATCs are profoundly dedifferentiated, the aberrant clustering may represent transcriptional outputs of the cancer drivers that are common to both lineages. The CCLE expression profiles for FTC-238, SW579 and CGTH-W-1 also clustered outside of the main thyroid cancer group. We conclude that the CCLE isolates of CGTH-W-1 and SW579, which are identical based on STR profiling (not shown), are misidentified. Finally, the MTC cell line TT clustered with small cell lung cancer cell lines, which is biologically plausible as both tumors originate from neuroendocrine cells. Conversely, the main thyroid cluster contained some CCLE non-thyroid cell lines (Fig 2, blue dots).
We also performed hierarchical clustering of gene expression profiles for thyroid cancer cell lines only from our study. There was no clear separation of profiles based on the histologic subtype of the primary tumor or primary oncogene(s) (Suppl. Fig. S4, see color labels).
Interestingly, when comparing global gene expression, only 2 genes were found to be differentially expressed between PTC- and ATC-derived cell lines (Suppl. Table S8, limma, adjusted p-value<0.05): VAPB (VAMP associated protein B and C) and STK4 (serine/threonine kinase 4). In contrast, gene expression of normal thyroid, PTC and ATC tumors was very different (top 1000 differentially expressed genes are listed in Suppl. Table S9).
Thyroid differentiation score (TDS)
We calculated the 16-gene thyroid differentiation score (TDS) panel, defined by the TCGA in PTCs (6), in our cell line dataset, and compared it to normal thyroid and thyroid tumors. The TDS for ATC and thyroid cancer cell lines was significantly lower than for normal thyroid, PTC and PDTC (Kruskal-Wallis, post-hoc Tukey-Kramer, p<0.005; Figure 3A, Suppl. Table S10). There was no difference in TDS between ATC and cell lines originated from thyroid cancers of various histologic types, supporting that all thyroid cell lines are dedifferentiated regardless of the tumor of origin. The difference in TDS of PTC and PDTC was not statistically significant (p = 0.99). Some PTC-derived cell lines (e.g., CUTC48, KTC1) retained marginal expression of discrete TDS genes, such as PAX8 and FOXE1, compared to their ATC-derived counterparts.
Figure 3. Thyroid differentiation score (TDS) and BRAFV600E-RAS score (BRS) in thyroid tumors and cell lines.
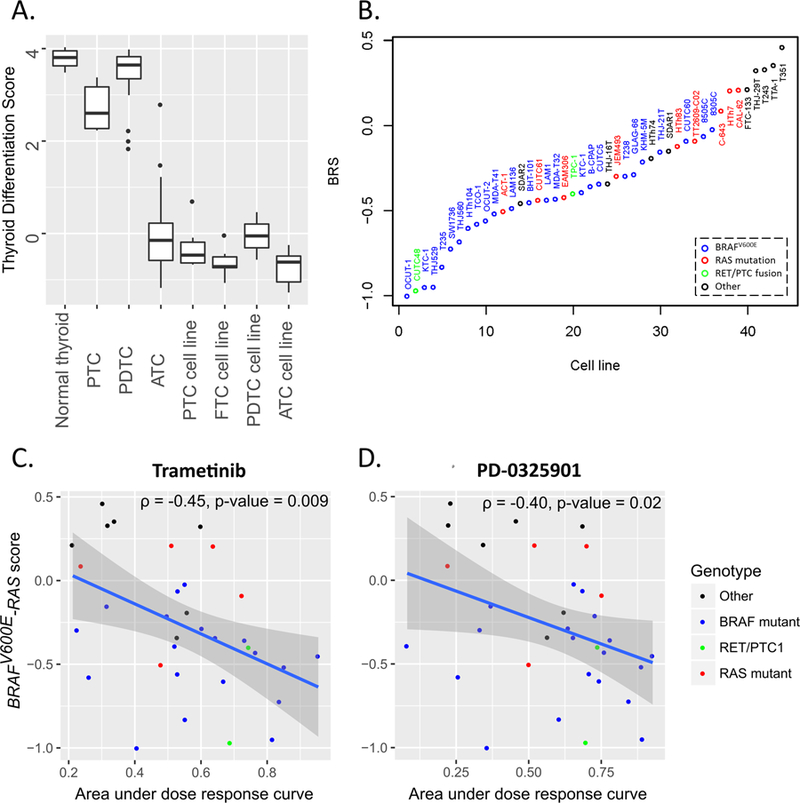
A. TDS of normal thyroid, thyroid cancers and thyroid cancer cell lines; B. BRS for thyroid cancer cell lines. Cell line oncogenes are labeled with different colors: blue – BRAFV600E, red – RAS, green – RET/PTC1, black – wild-type for BRAF, RAS and RET genes; C–D. Correlation of BRAFV600E-RAS score with the sensitivity to the MEK inhibitors trametinib (C) and PF-0325901 (D) in vitro. The sensitivity to MEK inhibitors is measured as area under the dose response curve (greater values indicate greater drug sensitivity). Cell line oncogenes are labeled with different colors: blue – BRAFV600E, red – RAS, green – RET/PTC1, black – wild-type for BRAF, RAS and RET genes.
The contribution of individual genes to the TDS signature varies (Suppl. Fig. S6). Most genes in the signature follow an expected pattern: high expression in normal thyroid and PTC, and low expression in ATC and cell lines. However, SLC5A8 and THRB mRNA levels are higher in undifferentiated cells, and GLIS3 does not change across groups. We thus propose TDS13 (a refined signature without those three genes) as a measure of thyroid cell differentiation. To validate TDS13, we applied it to the TCGA-PTC dataset, and found that lower TDS13 score is associated with higher histologic grade (Kruskal-Wallis, p-value=3.6E-08), greater American Joint Committee on Cancer stage (p-value=7.4E-06), presence of extrathyroidal extension (p-value=4.4E-12), tall cell histologic subtype (Tukey-Kramer, p-value=1.6E-04), and higher risk for persistent or recurrent disease (35) (p-value=1.5E-13). TDS13 and TDS16 performed comparably, but conclusions based on TDS13 were made with greater calculated probability (lower p-value, Suppl. Table S11).
BRAFV600E-RAS score (BRS): MAPK signaling and response to MAPK inhibitors
We next evaluated whether key driver-dependent gene expression characteristics, as defined by the TCGA-derived BRAFV600E-RAS score (BRS), persist in thyroid cancer cell lines. We adapted the 71-gene BRS for our microarray data (see Suppl Methods and Suppl. Fig. S7). Consistent with the purpose and design of BRS, BRAFV600E-mutant cell lines had lower BRS, when compared to RAS-mutant cells (Figure 3B, one-way ANOVA, p=0.0001, post-hoc t-test p=0.008). Cells with wild-type BRAF, RAS and RET/PTC1 had the highest BRS (post-hoc t-test p=4E-05 and 0.03 when compared to BRAF- and RAS-mutant cell lines, respectively). RET-rearranged TPC-1 and CUTC48 cell lines were BRAF-like, consistent with the TCGA-PTC findings (6).
We hypothesized that BRS reflects MAPK-pathway dependency of thyroid cancer cells: BRAF-mutants are most dependent, RAS-mutant being intermediate, and the cells without activating mutations in MAPK pathway (highest BRS) are least dependent on MAPK-pathway. To investigate this, 33 cell lines were tested for their sensitivity to the MEK inhibitors trametinib and PD0325901 (Figure 3C–D, Suppl. Table S12). BRS negatively correlated with the sensitivity to trametinib (ρ=−0.45, p=0.009) and PD0325901 (ρ=−0.40, p=0.02). As expected BRS was lower in BRAF-mutant cell lines (median=−0.44) than in BRAF-wild-type cell lines (median=−0.13, Kruskal-Wallis, p=0.002). However, the sensitivity to trametinib (median area under the dose response curve of 0.55 and 0.52 for BRAF-mutant and BRAF-wild type cell lines, respectively) and PD0325901 (median area under the dose response curve of 0.69 and 0.54 for BRAF-mutant and BRAF-wild type cell lines, respectively) was not statistically different (Kruskal-Wallis, p>0.05) indicating that BRS is a better biomarker of MEK inhibitor sensitivity than BRAF mutation status.
Microevolution of thyroid cancer
To study possible genetic alterations selected/acquired as part of the in vitro adaptation of thyroid cancer cells, we sequenced 12 primary tumors from which cell lines were derived, as well as 3 PDX, and compared the presence and respective allelic frequencies of specific mutations.
Figure 4 shows the MAFs for key mutations in primary tumor/cell line and/or PDX-paired samples from 11 patients. BRAF or RAS mutations were invariably enriched in vitro, regardless of their frequency in primary tumors, suggesting that they were truncal events within typically impure specimens, such as ATCs. For example, in CUTC5 cells, derived from a pleural effusion of a PTC patient (likely with a low thyroid cellular content), BRAF p.V600E was clonal (MAF=0.44), but unnoticed in the primary sample. However, manual review of the sequencing reads detected BRAF p.V600E (MAF=0.02) in the pleural effusion (Fig 4A, Suppl. Fig. S7), indicative of the strong selective advantage of this driver for in vitro growth. Other mutations in CUTC5 cells were similarly enriched: TP53 p.C135W (1% in primary vs. 99% in cell line) and ARID1A p.E1108* (3% vs. 49%). MAFs for BRAF and RAS alterations were ~50% in all cell lines, except for THJ-21T (BRAF p.V600E MAF=0.99, Fig 4B), in which the CNA profile suggests heterozygous loss of the BRAF wild-type copy.
Figure 4. Evolution of allelic frequencies from thyroid primary tumors to cell lines and patient-derived xenografts (PDX).

Graphic representation of the alternative allele frequencies (“Alt Allele Freq”) for selected mutations in 11 primary tumors and their derived cell lines and/or PDXs. Cell line names are displayed on top of each graph, and mutations are color-coded, as shown. Gray arrows indicate the relation between specimens (e.g., cell line established from primary tumor). A. CUTC5; B. THJ-21T; C. THJ-29T; D. THJ-16T; E. CUTC60; F. CUTC61; G. LAM136; H. EAM306; I. THJ529; J. THJ560; K. SDAR1/2. Abbreviations: PE= pleural effusion; PTC= papillary thyroid cancer; FTC= follicular thyroid cancer; PDTC= poorly-differentiated thyroid cancer; ATC= anaplastic thyroid cancer; PDX= patient-derived xenograft; Met= metastatic tissue.
TERT promoter mutations were predominantly enriched in vitro and in PDXs (Suppl. Fig. S8A), with frequencies of typically ~50% or higher (THJ-21T, THJ-29T, THJ560 and SDAR1/2). In the latter, CNA profiles showed signs of TERT amplification. Mutations in TP53 were strongly selected in vitro. Of the 14 TP53 mutations found in the 11 cell lines with primary tumor and/or PDX available, 7 were present in the original tumor (including CUTC5, MAF=1%) and 7 had no detectable mutations in the primary specimen (Fig 4 and Suppl. Fig. S8B). The cell lines derived from apparent TP53 wild-type samples may have developed de novo TP53 mutations in vitro, or may have resulted from a primary tumor subclone that was below the sequencing detection limits. Copy number profiles for CUTC5, THJ-21T, EAM306, THJ560, CUTC61 and THJ529 cells were consistent with LOH as the mechanism of TP53 complete loss-of-function, whereas other cell lines (e.g., CUTC60, THJ-16T) might have alternatively developed uniparental disomy. The plasticity of thyroid cells to abolish p53 function is exemplified by the SDAR1/2 samples (Fig 4K), in which two independent cell lines were derived from a primary FTC and a neck metastasis from the same patient, respectively. TP53 p.V217fs, was present in the original FTC (MAF=13%) and enriched in the SDAR2 cell line (MAF=96%), whereas SDAR1 cells acquired another mutation (TP53 p.R282P, MAF=89%). Other loss-of-function mutations in known tumor suppressor genes, such as CDKN2A in THJ-29T, NF2 in CUTC60 and PTEN in SDAR1/2 (Fig 4A, C, E, K), were consistently enriched, with evidence of LOH events for the first two mutations. Finally, CUTC60, THJ529 and THJ560 (Fig 4E, I, J) provided good examples of thyroid cancer microevolution in vitro vs. direct engraftment in animals: CUTC60 and THJ560 showed clonal selection of BRAF, TERT and TP53 mutations in all specimens, whereas THJ529 displayed a divergent evolution illustrated by a TP53 mutation present in the cell line but absent in the primary and PDX.
Regarding CNAs, despite insufficient tumor purity in most primary tumors, we could identify conserved CNAs between primary ATCs with higher tumor cell content and their respective cell lines. For example, CUTC61 specimens showed a conserved gain at 1q32-q44, whereas THJ-29T displayed shared losses of chromosomes 9 and 13 (Suppl. Fig. S9). There was also evidence of occasional convergent focal changes occurring in cell lines and PDX but absent in the primary samples, such as deep deletions of CDKN2A in THJ560 (Suppl. Fig. S10).
DISCUSSION
A decade ago we reported that 17/40 commonly used thyroid cancer cell lines were either redundant or misidentified with other tumor types (3). In the ensuing time, new cell lines have been established, providing new thyroid cancer in vitro models (12,24,36). Although cell line misidentification remains a concern, the generalization of NGS technologies have greatly facilitated authentication. In the present study, we applied a comprehensive genomic and transcriptomic approach to study virtually every unique thyroid cancer-derived cell line currently being used in research laboratories. We characterized the cancer genome and defined key expression features of these cells, which will allow researchers to better plan and interpret their results. We also provided insights into thyroid cancer microevolution by sequencing paired primary tumors and PDXs for a subset of cell lines.
With respect to main driver alterations, thyroid cancer cell lines harbored mutually-exclusive activating mutations in BRAF (50%), RAS genes (22%), gene fusions affecting RET and FGFR2 (5%) and truncating events in NF1 (5%). Mutations in the TERT promoter (83%) and TP53 (71%) were the most frequent events, as in ATC (7,10,37). Mutations in PI3K/AKT pathway effectors PTEN and PIK3CA were found in 14 and 12% of cell lines, respectively. Variants in genes belonging to the SWI/SNF chromatin remodeling complex, mismatch repair, histone methyl- and acetyltransferase functional groups were also common in these specimens. Overall, driver alterations in cell lines recapitulated those observed in primary tumors. For instance, all RET/PTC fusions were identified in PTC-derived cell lines, whereas every FTC-derived cell line harbored oncogenic mutations in either RAS or PTEN genes (6,9).
The sequencing of paired primary tumor-cell line-PDX was particularly instructive. The oncogenes believed to arise as early events in tumor development were enriched in a manner consistent with the purity of the original sample. By contrast, TP53 mutations, which are markedly enriched in ATC (7), were clearly selected for in vitro. In several cases, the TP53 mutation was not detected in the primary sample, indicating a de novo mutation arising in vitro, or expansion of a tumor subclone present at a frequency below the resolution of the NGS approach (average depth=500X). Other events detected in cell lines but not in primary specimens, such as copy number changes, should be interpreted in the same manner. These discrepancies between primary tumors and cell lines should alert researchers to interpret their in vitro results cautiously.
The frequency of truncating mutations and magnitude of losses in cyclin-dependent kinase inhibitor genes CDKN2A/CDKN2B in cell lines was comparable to those reported in aggressive DTC and ATC (8). In a subset of cases, these events might have been selected in vitro (e.g., THJ560). Loss of CDKN2C (p18) was observed in MTC-derived TT cells, in line with reports showing that p18 loss cooperates with RET oncogenic mutations in MTC tumorigenesis and progression (38–40).
CNAs were generally more prevalent in cell lines compared to primary tumors, although our analysis showed that those derived from DTCs tended to be more diploid than those coming from PDTC/ATCs. We identified 16 recurrent CNA regions, including some well-known pathogenic CNAs in thyroid cancer cell lines. Of interest are amplifications of 5p15.33, 7p22.1, 8q24.21, 11q13.2 and deletion of 9p21.3, which cause CNAs and corresponding expression changes of the target genes. The amplification of 5p15.33 locus may represent a mechanism of TERT gene activation alternative or complementary to the widespread TERT promoter mutations. RAC1 amplification is noteworthy in view of recently reported activating RAC1 point mutations in thyroid cancer (8), and correlation with resistance to MAPK-directed therapies in melanoma (41). The amplifications of TERT, RAC1, MYC, YAP1 and deletion of CDKN2B were found in our genomic analysis of advanced thyroid tumors (8). Therefore, cell lines recapitulate key CNAs in thyroid cancers and are useful models to study the functional significance of these genetic events.
Our expression profiling showed thyroid cancer cell lines are profoundly dedifferentiated, as demonstrated by low TDS comparable to that of ATC specimens. The dedifferentiation likely occurs when tumor cells adapt to the in vitro growth conditions. By comparing gene expression pattern of cell lines and primary tumors, we were able to refine TDS and proposed a reduced TDS13 signature, which might serve as a clinically relevant biomarker in view of the highly significant association of low TDS13 with all histologic and clinical markers of tumor aggressiveness in the TCGA-PTC cohort. We also confirmed that BRS discriminates BRAF and RAS-mutant cells in a dataset different than the TCGA. BRS negatively correlated with the sensitivity to MEK inhibitors in vitro, supporting that the score reflects cell line reliance on MAPK pathway activation downstream of BRAF (42).
In conclusion, we performed comprehensive genetic characterization of the largest assembled panel of thyroid cancer cell lines, and found that they have many of the same point mutations, gene fusions and CNAs observed in PTC, aggressive DTC and ATC (6–8). The analysis of gene expression showed that, without exception, thyroid cancer cell lines had a profound loss of markers of thyroid differentiation, regardless of their derivation. Although the precise mechanistic connection between p53 loss-of-function and the differentiation state of thyroid cells has not been worked out, these events are strongly associated, and may explain in part these results. Interestingly, despite the loss of thyroid differentiated gene expression, the vast majority of thyroid cancer cell lines clustered as a group distinct from other cancer lineages, attesting to their origin. Cell lines exhibit properties more akin to ATCs. However, these cells still show dependence on their driver for their biology and viability, as exemplified by the relationship between BRAF/RAS mutation and the BRS score, and their response to selective inhibitors of key effectors in the MAPK pathway.
Supplementary Material
TRANSLATIONAL RELEVANCE.
Human cancer cell lines are valuable models to study cancer biology and therapeutic dependencies. Experiments with thyroid cancer cell lines have been problematic due to cell line misidentification. Here we provide a comprehensive characterization of cancer gene mutations, copy number alterations and transcriptomic changes of nearly all unique thyroid cancer cell lines that are currently in use, highlighting their key features, which largely recapitulate the genomic lesions of the primary tumors. We show that they remain dependent on their drivers (i.e., BRAF vs. RAS) and select for other genetic events (e.g., TP53 and CDKN2A losses, TERT promoter mutations). However, they are uniformly dedifferentiated in vitro regardless of the differentiation state of the tumor from which they were derived, and do not retain transcriptomic markers of differentiated thyroid cancer. We expect this resource to help design more rational mechanism-based studies in the thyroid cancer field.
Acknowledgments
Financial support: This work was supported by NCI grant RC1CA147371 (JAF, BRH, JAK and RES) and the Mary Rossick Kern and Jerome H Kern Endowment (BRH).
Footnotes
Conflict of interest: The authors declare no potential conflicts of interest
REFERENCES
- 1.Abaan OD, Polley EC, Davis SR, Zhu YJ, Bilke S, Walker RL, et al. The exomes of the NCI-60 panel: a genomic resource for cancer biology and systems pharmacology. Cancer Res 2013;73(14):4372–82 doi 10.1158/0008-5472.CAN-12-3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483(7391):603–7 doi 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schweppe RE, Klopper JP, Korch C, Pugazhenthi U, Benezra M, Knauf JA, et al. Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. J Clin Endocrinol Metab 2008;93(11):4331–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao N, Liu Y, Wei Y, Yan Z, Zhang Q, Wu C, et al. Optimization of cell lines as tumour models by integrating multi-omics data. Brief Bioinform 2017;18(3):515–29 doi 10.1093/bib/bbw082. [DOI] [PubMed] [Google Scholar]
- 5.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nature communications 2013;4:2126 doi 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research N. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014;159(3):676–90 doi 10.1016/j.cell.2014.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Landa I, Ibrahimpasic T, Boucai L, Sinha R, Knauf JA, Shah RH, et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest 2016;126(3):1052–66 doi 10.1172/JCI85271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pozdeyev N, Gay L, Sokol ES, Hartmaier RJ, Deaver KE, Davis SN, et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin Cancer Res 2018. doi 10.1158/1078-0432.CCR-18-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoo SK, Lee S, Kim SJ, Jee HG, Kim BA, Cho H, et al. Comprehensive Analysis of the Transcriptional and Mutational Landscape of Follicular and Papillary Thyroid Cancers. PLoS Genet 2016;12(8):e1006239 doi 10.1371/journal.pgen.1006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ibrahimpasic T, Xu B, Landa I, Dogan S, Middha S, Seshan V, et al. Genomic Alterations in Fatal Forms of Non-Anaplastic Thyroid Cancer: Identification of MED12 and RBM10 as Novel Thyroid Cancer Genes Associated with Tumor Virulence. Clin Cancer Res 2017;23(19):5970–80 doi 10.1158/1078-0432.CCR-17-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agrawal N, Jiao Y, Sausen M, Leary R, Bettegowda C, Roberts NJ, et al. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J Clin Endocrinol Metab 2013;98(2):E364–9 doi 10.1210/jc.2012-2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marlow LA, D’Innocenzi J, Zhang Y, Rohl SD, Cooper SJ, Sebo T, et al. Detailed molecular fingerprinting of four new anaplastic thyroid carcinoma cell lines and their use for verification of RhoB as a molecular therapeutic target. J Clin Endocrinol Metab 2010;95(12):5338–47 doi 10.1210/jc.2010-1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marlow LA, Rohl SD, Miller JL, Knauf JA, Fagin JA, Ryder M, et al. Methodology, criteria and characterization of patient-matched thyroid cell lines and patient-derived tumor xenografts. J Clin Endocrinol Metab 2018. doi 10.1210/jc.2017-01845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. The Journal of molecular diagnostics : JMD 2015;17(3):251–64 doi 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2(5):401–4 doi 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling 2013;6(269):pl1 doi 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 2011;12(4):R41 doi 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nature biotechnology 2011;29(1):24–6 doi 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kauffmann A, Gentleman R, Huber W. arrayQualityMetrics--a bioconductor package for quality assessment of microarray data. Bioinformatics (Oxford, England) 2009;25:415–6 doi 10.1093/bioinformatics/btn647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 2005;102:15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He H, Jazdzewski K, Li W, Liyanarachchi S, Nagy R, Volinia S, et al. The role of microRNA genes in papillary thyroid carcinoma. Proc Natl Acad Sci U S A 2005;102(52):19075–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lemoine NR, Mayall ES, Jones T, Sheer D, McDermid S, Kendall-Taylor P, et al. Characterisation of human thyroid epithelial cells immortalised in vitro by simian virus 40 DNA transfection. Br J Cancer 1989;60(6):897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smallridge RC, Chindris AM, Asmann YW, Casler JD, Serie DJ, Reddi HV, et al. RNA sequencing identifies multiple fusion transcripts, differentially expressed genes, and reduced expression of immune function genes in BRAF (V600E) mutant vs BRAF wild-type papillary thyroid carcinoma. J Clin Endocrinol Metab 2014;99(2):E338–47 doi 10.1210/jc.2013-2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henderson YC, Ahn SH, Ryu J, Chen Y, Williams MD, El-Naggar AK, et al. Development and characterization of six new human papillary thyroid carcinoma cell lines. J Clin Endocrinol Metab 2015;100(2):E243–52 doi 10.1210/jc.2014-2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Camacho P, Gordon D, Chiefari E, Yong S, DeJong S, Pitale S, et al. A Phe 486 thyrotropin receptor mutation in an autonomously functioning follicular carcinoma that was causing hyperthyroidism. Thyroid 2000;10(11):1009–12 doi 10.1089/thy.2000.10.1009. [DOI] [PubMed] [Google Scholar]
- 26.Trulzsch B, Krohn K, Wonerow P, Chey S, Holzapfel HP, Ackermann F, et al. Detection of thyroid-stimulating hormone receptor and Gsalpha mutations: in 75 toxic thyroid nodules by denaturing gradient gel electrophoresis. J Mol Med (Berl) 2001;78(12):684–91. [DOI] [PubMed] [Google Scholar]
- 27.Parma J, Van Sande J, Swillens S, Tonacchera M, Dumont J, Vassart G. Somatic mutations causing constitutive activity of the thyrotropin receptor are the major cause of hyperfunctioning thyroid adenomas: identification of additional mutations activating both the cyclic adenosine 3’,5’-monophosphate and inositol phosphate-Ca2+ cascades. Mol Endocrinol 1995;9(6):725–33 doi 10.1210/mend.9.6.8592518. [DOI] [PubMed] [Google Scholar]
- 28.Antico Arciuch VG, Russo MA, Dima M, Kang KS, Dasrath F, Liao XH, et al. Thyrocyte-specific inactivation of p53 and Pten results in anaplastic thyroid carcinomas faithfully recapitulating human tumors. Oncotarget 2011;2(12):1109–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charles RP, Silva J, Iezza G, Phillips WA, McMahon M. Activating BRAF and PIK3CA mutations cooperate to promote anaplastic thyroid carcinogenesis. Molecular cancer research : MCR 2014;12(7):979–86 doi 10.1158/1541-7786.MCR-14-0158-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishizaka Y, Ushijima T, Sugimura T, Nagao M. cDNA cloning and characterization of ret activated in a human papillary thyroid carcinoma cell line. Biochem Biophys Res Commun 1990;168(2):402–8. [DOI] [PubMed] [Google Scholar]
- 31.Kasaian K, Wiseman SM, Walker BA, Schein JE, Zhao Y, Hirst M, et al. The genomic and transcriptomic landscape of anaplastic thyroid cancer: implications for therapy. BMC Cancer 2015;15:984 doi 10.1186/s12885-015-1955-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010;463(7283):899–905 doi 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garnis C, Lockwood WW, Vucic E, Ge Y, Girard L, Minna JD, et al. High resolution analysis of non-small cell lung cancer cell lines by whole genome tiling path array CGH. Int J Cancer 2006;118(6):1556–64 doi 10.1002/ijc.21491. [DOI] [PubMed] [Google Scholar]
- 34.Lorenzetto E, Brenca M, Boeri M, Verri C, Piccinin E, Gasparini P, et al. YAP1 acts as oncogenic target of 11q22 amplification in multiple cancer subtypes. Oncotarget 2014;5(9):2608–21 doi 10.18632/oncotarget.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haugen BR, Alexander EK, Bible KC, Doherty G, Mandel SJ, Nikiforov YE, et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2015;26:thy.2015.0020 doi 10.1089/thy.2015.0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Onoda N, Nakamura M, Aomatsu N, Noda S, Kashiwagi S, Hirakawa K. Establishment, characterization and comparison of seven authentic anaplastic thyroid cancer cell lines retaining clinical features of the original tumors. World journal of surgery 2014;38(3):688–95 doi 10.1007/s00268-013-2409-7. [DOI] [PubMed] [Google Scholar]
- 37.Kunstman JW, Juhlin CC, Goh G, Brown TC, Stenman A, Healy JM, et al. Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Hum Mol Genet 2015;24(8):2318–29 doi 10.1093/hmg/ddu749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Veelen W, Klompmaker R, Gloerich M, van Gasteren CJ, Kalkhoven E, Berger R, et al. P18 is a tumor suppressor gene involved in human medullary thyroid carcinoma and pheochromocytoma development. Int J Cancer 2009;124(2):339–45 doi 10.1002/ijc.23977. [DOI] [PubMed] [Google Scholar]
- 39.van Veelen W, van Gasteren CJ, Acton DS, Franklin DS, Berger R, Lips CJ, et al. Synergistic effect of oncogenic RET and loss of p18 on medullary thyroid carcinoma development. Cancer Res 2008;68(5):1329–37 doi 10.1158/0008-5472.CAN-07-5754. [DOI] [PubMed] [Google Scholar]
- 40.Grubbs EG, Williams MD, Scheet P, Vattathil S, Perrier ND, Lee JE, et al. Role of CDKN2C Copy Number in Sporadic Medullary Thyroid Carcinoma. Thyroid 2016;26(11):1553–62 doi 10.1089/thy.2016.0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watson IR, Li L, Cabeceiras PK, Mahdavi M, Gutschner T, Genovese G, et al. The RAC1 P29S hotspot mutation in melanoma confers resistance to pharmacological inhibition of RAF. Cancer Res 2014;74(17):4845–52 doi 10.1158/0008-5472.CAN-14-1232-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pratilas CA, Hanrahan AJ, Halilovic E, Persaud Y, Soh J, Chitale D, et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res 2008;68(22):9375–83 doi 10.1158/0008-5472.CAN-08-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.