

Abstract
Alkylating chemotherapy is a central component of the management of glioblastoma (GBM). Among the factors that regulate the response to alkylation damage, NF-кB acts to both promote and block cytotoxicity. In this study, we used genome-wide expression analysis in U87 GBM to identify NF-кB-dependent factors altered in response to temozolomide (TMZ) and found the long non-coding RNA (lncRNA) MALAT1 as one of the most significantly upregulated. Additionally, we demonstrated that MALAT1 expression was co-regulated by p50 (p105) and p53 via novel кB- and p53-binding sites in the proximal MALAT1 coding region. TMZ treatment inhibited p50 recruitment to its cognate element as a function of Ser329 phosphorylation while concomitantly increasing p53 recruitment. Moreover, luciferase reporter studies demonstrated that both кB and p53 cis-elements were required for efficient transactivation in response to TMZ. Depletion of MALAT1 sensitized patient-derived GBM cells to TMZ cytotoxicity, and in vivo delivery of nanoparticle encapsulated anti-MALAT1 siRNA increased the efficacy of TMZ in mice bearing intracranial GBM xenografts. Despite these observations, in situ hybridization of GBM specimens and analysis of publically available datasets revealed that MALAT1 expression within GBM tissue was not prognostic of overall survival. Together, these findings support MALAT1 as a target for chemosensitization of GBM and identify p50 and p52 as primary regulators of this ncRNA.
INTRODUCTION
Resistance to DNA damage-induced cytotoxicity plays a major role in the poor response of many glioblastoma (GBM) patients to chemotherapy. The oral alkylator temozolomide (TMZ) is the most commonly used chemotherapeutic for the management of GBM yet, despite its routine use, overall response to TMZ remains modest (1). While upstream repair factors such as methylguanine DNA-methyltransferase (MGMT) that attenuate the efficacy of TMZ are well established, the downstream pathways that modulate cytotoxicity and chemoresistance remain poorly elucidated. Nuclear factor-кB (NF-кB) is an important regulator of the cytotoxic response to DNA damage and acts in a context and subunit-specific manner to attenuate and augment cell death (2,3). There are five primary NF-кB subunits, p50 (NF-кB1, p105), p52 (NF-кB2, p100), p65 (relA), relB, and crel, that act as dimers to regulate the downstream NF-кB response.
In GBM, p50 makes up a significant portion of the nuclear NF-кB dimer (4). While p50 is required for TMZ-induced cytotoxicity (3,5), certain p50-dependent genes have the opposite effect and attenuate killing (6). TMZ induces p50 S329 (S328) phosphorylation and this modification regulates p50 binding to its cognate elements (7). p50 lacks a transactivation domain (TAD) and regulates downstream signaling in association with various co-regulating factors. The tumor suppressor protein, p53, is intimately involved in modulating the NF-кB response (8,9), and deregulation of the p53 pathway has been reported in up to 85% of GBM (10). p53 signaling is induced by DNA damage and is involved in both promoting and attenuating cytotoxicity (11).
Long non-coding RNAs (lncRNAs) are molecules larger than 200 nucleotides that are not translated into proteins. They are involved in a wide range of cellular processes including chromatin remodeling, transcriptional regulation, and post-transcriptional processing (12,13). In the field of cancer biology, lncRNAs mediate both oncogenic and tumor-suppressive pathways, and aberrant expression of several lncRNAs has been observed in cancer including glioma (14–16). LncRNAs play an important role in promoting chemoresistance (17,18) and several lncRNAs have been reported to modulate GBM progression through p53-dependent oncogenic activity (19,20).
Metastasis associated lung adenocarcinoma transcript 1 (MALAT1), also known as nuclear-enriched transcript 2 (NEAT2), is a conserved 8.7 kb lncRNA located on chromosome 11q13. MALAT1 was originally identified as a predictor of metastasis in non-small cell lung cancer and subsequently shown to be up-regulated in many malignancies including GBM (21–23). MALAT1 localizes to nuclear speckles where it participates in alternative splicing of pre-mRNA to enable diversification of gene function (24–26). Although the cellular localization, interacting partners, and functional role of MALAT1 have been extensively examined (24,25,27,28), less is known about the mechanism by which MALAT1 expression is regulated. In GBM, MALAT1 was shown to promote chemoresistance by regulating microRNAs and by inducing transformation to a mesenchymal phenotype (29–31). Additionally, a recent study indicated that systemic knockdown of MALAT1 augments cytotoxicity by TMZ in GBM (32). These findings suggest that MALAT1 may be a fruitful target for chemosensitization and underline the need to better define its regulation in GBM.
Given the importance of p50 in the response to alkylating chemotherapy, we examined p50-dependent gene expression in isogenic GBM cells proficient and deficient in p50 following treatment with TMZ. MALAT1 was identified as a p50-dependent gene upregulated following TMZ treatment. We show that MALAT1 is co-regulated by p50 and p53 and demonstrate that targeting MALAT1 increased the efficacy of TMZ in experimental GBM.
MATERIALS AND METHODS
Cell lines, reagents, recombinant proteins and plasmids
Human glioma cell lines U87, A172, and U251 cells were purchased from American Type Culture Collection (ATCC). The patient-derived GSCs, GBM34 and GBM44, were obtained from Dr. Mariano Viapiano (Brigham and Women’s Hospital, Boston, MA) and were previously described (33). Immortal Nfkb1+/+ and Nfkb1−/− mouse embryonic fibroblasts (MEFs) were cultured and stable re-expression of p50 isoforms performed as previously described (3). U87 glioma cells expressing sh-control or sh-p105, targeting the C-terminal of p105 to enable re-expression of p50 constructs, were also previously described (3). Cell lines were authenticated by routine morphological and growth analysis and also by western blotting. All cell lines were screened for the presence of mycoplasma using the ATCC Universal Mycoplasma Detection Kit (Catalogue # 30-1012K) every four months. Cells were used within four passages of being thawed. Full-length recombinant human p53 protein was obtained from Abcam (ab82201, Cambridge, MA, USA). His-p50 was bacterially-expressed and purified as previously described (3). TMZ was obtained from Sigma-Aldrich (St. Louis, MO, USA).
RNA interference
The following siRNA constructs were obtained: si-MALAT1-(1) (SASI_Hs02_00377093, Sigma-Aldrich), si-MALAT1-(2) (n272231, Thermo Fisher) si-p53 #1 (sc-29435, Santa Cruz Biotechnology, Dallas, TX, USA), si-p53 #2 (M-003329-03, GE Dharmacon, Lafayette, CO, USA), si-p50 (sense: GUCACUCUAACGUAUGCAUU, GE Dharmacon), si-p65 (sense: GAUUGAGGAGAAACGUAAAUU, GE Dharmacon) and si-Control (D-001210-03, GE Dharmacon). All siRNA constructs were transfected at a concentration of 200 nM for 48 hours or as noted in the legend using Oligofectamine (Invitrogen, Life Technologies, Grand Island, NY, USA).
For construction of stable shRNA clones, oligos targeting nucleotides 8224 to 8244 of MALAT1 (NCBI Reference Sequence: NR_002819.2) (target seq: TAGCGGAAGCTGATCTCCAAT) were obtained (Integrated DNA Technologies, Coralville, IO, USA) and cloned into pLKO.1 (Clontech, Mountain View, CA, USA). The control sh-RNA construct was obtained from Dr. David Sabatini (Addgene plasmid #1864, Addgene, Cambridge, MA, USA) and contains a scrambled sequence in the pLKO.1 vector (34). Lentiviral particles were generated with the packaging system from Addgene (pMD2.G and psPAX) and U87 cells infected with lentivirus and selected with puromycin for 5 days before use. Infections were performed at a Multiplicity of Infection (MOI) of 5 as previously optimized (6). Following transduction, cells were selected with puromycin and qPCR performed to determine acceptable transduction efficiency and knockdown of MALAT1.
RNA extraction, microarray analysis and data processing
For gene expression analysis, sh-p105 and sh-control U87 cells were plated, in triplicate, and treated with vehicle or 100 μM TMZ for 16 hours. This time point and concentration of TMZ were previously demonstrated to allow maximal inhibition of NF-кB DNA binding (5). In addition, 100 μM TMZ achieves a therapeutically relevant dose of TMZ in the range of the plasma level achieved during chemotherapy (35,36). Cells were then harvested and total RNA extraction performed using TRIzol (Invitrogen), added directly to cells in culture and lysates collected using a cell lifter. RNA extraction was carried out per manufacturer’s instructions and RNA purity and concentration assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Samples were then stored at −80 °C until microarray analysis.
Affymetrix® GeneChip Human Genome U133 Plus 2.0 Arrays (HG-U 133 Plus 2.0, Santa Clara, CA, USA) containing 47,000 gene-level transcripts were used for gene expression analysis on 5 μg RNA from biologic triplicates. Array hybridization and scanning were performed in the UCLA Bioinformatics Institute Core Laboratory Facility (Los Angeles, CA, USA) according to Affymetrix protocols. The raw data were processed using the Affymetrix Power Tools. The probe intensities were summarized using the robust multi-array average (RMA) algorithm (37), and quantile normalized across all samples. Differentially expressed genes (DEGs) were identified using the linear models approach in the limma package in R/Bioconductor. False discovery rate (FDR) < 5% (0.05) was used as a cut off in a typical analysis and controlled using the Benjamini-Horchberg procedure (38). Functional annotations were based on Gene Ontology terms (e.g. biological processes) and the NIH/DAVID (Database for Annotation, Visualization and Integrated Discovery) tool (39). Microarray data have been submitted to the NCBI Gene Expression Omnibus (GEO) [accession number: GSE65363].
Datasets
Raw gene expression and clinical data from the Repository for Molecular Brain Neoplasia Data (REMBRANDT) and TCGA datasets were accessed, analyzed, and downloaded from GlioVis data portal (gliovis.bioinfo.cnio.es) in October 2017 (40). Kaplan Meier survival curves were plotted based on the MALAT1 expression level.
кB-site and p53 binding site analysis
For p53 2,500 bps up- and down-stream of the human MALAT1 coding sequence were interrogated using p53MH (41), JASPAR (42) and MATCH (43) software and a single binding site (BS) that overlapped in all three searches was identified in the coding sequence (NCBI Ref. Seq: NR_002819.2). 2,500 bps up- and down-stream of the murine MALAT1 coding sequence were also interrogated using JASPAR. One site was identified in the range of (−2003 to −1989) with a sequence of ACAAGTTAAGGCTTT that contained 80% homology with the human p53 binding site.
For NF-кB, JASPAR, MATCH, and TFSEARCH (44) were used and a single decameric sequence, that overlaps in all three searches and has 85% homology with the canonical B-site was identified upstream from the p53 BS of the human MALAT1 coding sequence. 2,500 bps up- and down-stream of the murine MALAT1 coding sequence were also interrogated using JASPAR. One site was identified in the range of (−378 to −368) with a sequence of GGGAATGTCC that contained 82% homology with the human кB-site.
Quantitative real-time polymerase chain reaction (qPCR) and quantitative chromatin immunoprecipitation (qChIP)
Total RNA was isolated and qPCR performed as described (3). The primers used were: MALAT1 (human and mouse, sense GGATCCTAGACCAGCATGCC, antisense AAAGGTTACCATAAGTAAGT), MDM2 (sense AACCACCTCACAGATTCCAG, antisense TCAAGGTGACACCTGTTCTC), PLK2 (sense CCACCATTCGCACTCG, antisense CGGCGTAGACTTTGTTATTT), TP53I3 (sense TCTCTATGGTCTGATGGG, antisense AAGTAAGTTCCAGAAAA), LMP2 (sense ATGCTGACTCGACAGCCTTT, antisense GCAATAGCGTCTGTGGTGAA), and GAPDH (sense CTTCACCACCATGGAGAAGGC, antisense GGCATGGACTGTGGTCATGAG). Relative expression data is shown as the average of each experiment run in triplicate.
qChIP was performed following IP with the indicated antibodies as described (3). qPCR was carried out using primers for human MALAT1 that span the region encompassing the putative кB-site (sense CGCAACTGGCCTCTCCTGCC, anti-sense CTCGTCGCTGCGTCCCAAGG) or p53 BS (sense GCTAGGAAACAAAAAGCTAAGGGCA, anti-sense CCTTCTAACTTCTGCACCACCAGA). qPCR was carried out using primers for murine MALAT1 that span the region encompassing the putative кB-site (sense GGAGAGAGAAACAGGCAGC, anti-sense GGAAGACAGTGGGCATTTGG) or p53 BS (sense AAGCACCTCAGCTCAAGTCC, anti-sense AGCCACATACAAGATTGAAGCT). The change in DNA enrichment for each IP condition was determined relative to input DNA. To control for non-specific binding, the anti-p50, anti-p52, anti-p65 and anti-p53 data was subtracted from the anti-H1 results (anti-IgG showed no binding) as previously described (7).
Immunoblot and electrophoretic mobility shift assay (EMSA)
Immunoblotting was performed using whole cell lysate as previously described (7). Primary antibodies used include: anti-p50 (sc7178, Santa Cruz Biotechnology), anti-p65 (#8242, Cell Signaling, Beverly, MA, USA), anti-gapdh (sc-137179,), anti-p53 (sc71818,). Alexa-Fluor 680 and Alexa-Fluor 800 fluorescent dye-conjugated secondary antibodies (Invitrogen) were used for visualization with Odyssey Infrared system (LI-COR, Lincoln, NB, USA). For EMSA, nuclear fraction was isolated, or pure protein obtained, and assay performed as described (3). Supershift assays were performed using antibody cocktails specific to the indicated NF-кB subunit or p53. Competition was performed by pre-incubating the mixture with cold specific and non-specific DNA probe.
Luciferase assay
A 2400bp region of the MALAT1 proximal promoter and region bearing the putative p53 and кB BSs was generated by SwitchGear Genomics (Active Motif, Carlsbad, CA, USA) and transferred into the pGl4.20 luciferase reporter vector (Promega, Madison, WI, USA). Cells were co-transfected with the reporter and Renilla reniformis and relative luciferase activity measured after treatment using the Dual-Luciferase Reporter® Assay kit (Promega) as previously described (5). All experiments were performed in triplicate. The QuikChange Lightning Site Directed Mutagenesis Kit (Agilent, Santa Clara, CA, USA) was used to mutate the B-site (GCAGGTCCCC to tCAGGTCata) and the p53 BS (AAACAAGCTAAGACAAGTAT to AtcCAAGCTAAGACAAtcga).
Clonogenic and trypan blue assays
Cells were plated and allowed to attach overnight and assays performed as previously described after treatment with TMZ or vehicle (3,45).
Nanoparticle production and characterization
NPs were provided by LNK Chemsolutions LLC, Lincoln, NB, USA. These were manufactured and characterized as previously described (46). siRNAs were incorporated into NPs in a similar fashion to other agents and the product maintained under sterile conditions.
Animal studies
Six to seven-week old male nude mice (Harlan Laboratories, Madison, USA) were used in accordance with guidelines of the Institutional Animal Care and Use Committee (IACUC) of the University of Chicago. For intracranial studies, 5×105 U87 cells were injected into the right striatum as previously described (45), and animals randomized into 6 groups (n = 6 per group). 10 μL of vehicle or NPs carrying si-Control or si-MALAT1 at a concentration of 5 nM were injected intracranially (day 4 and 7) following tumor inoculation as previously described (6). Mice were also treated with intraperitoneal (i.p.) vehicle or TMZ on day 4 (5 mg/kg), 7 (5 mg/kg) and 10 (2.5 mg/kg) (total TMZ dose: 12.5 mg/kg). Animals were followed until terminal, sacrificed and brains harvested for verification of tumor.
For hindlimb studies, 7.5×106 U87 cells were injected into the right hindlimb and animals randomized (n = 5 per group) when tumors reached an average of 100 mm3 (day 0). Mice were treated with a single daily intratumoral (i.t.) injection of 10 μL of si-Control or si-MALAT1 NPs over 5 consecutive days, and with i.p. TMZ (total TMZ dose: 12.5 mg/kg) or vehicle over 3 alternating days. Control animals were injected i.t. or i.p. with vehicle. Tumor volume was measured every 2 - 3 days and fractional tumor volume calculated (V/ V0 where V0 = volume of tumor on day 0, V = volume of tumor on day measured). Animals were followed until tumors reached 2 cm3. Tumors were then harvested for verification of MALAT1 knockdown. Briefly, tumors underwent manual homogenization and total RNA extraction was performed using TRIzol (Invitrogen) added directly to homogenized suspension. RNA extraction was carried out per manufacturer’s instructions and RNA purity and concentration assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). qPCR was performed following mRNA isolation using the primers described above.
Tissue Microarray Construction
Tissue Microarray (TMA) was generated as previously described (47). Briefly, patients who presented to the University of Chicago with a diagnosis of glioma between 2007-2013 were retrospectively enrolled under an Institutional Review Board (IRB) protocol. Consent was waived under IRB approval given patients were deceased. 34 GBM patients were found to have adequate clinical data and tumor tissue for inclusion into TMA and their clinical characteristics are shown (Supplementary Table 1). Survival time was determined as the time from initial diagnosis until death, or last follow up for survivors. For construction of TMAs, FFPE donor blocks were examined by a neuropathologist and representative tumor regions identified. 1mm cores were punched and TMAs were made using a Beech Instrument Tissue Arrayer. Cores were placed in duplicate with control samples that included normal tissue and low grade glioma specimens. Interspersed between. Sections were cut at 4 μm thickness and initially examined under light microscopy following hematoxylin and eosin staining. TMAs were also examined for IDH1 mutation by staining with a clinical antibody to IDH1-R132H (antibody clone H09, Dianova).
In Situ Hybridization
TMAs were de-paraffinized by immersion in xylene (10 min twice) and then rehydrated through serial ethanol (100%, 75%, 50%, and 25%) for 5 min each followed by immersion in DEPC-treated PBS for 5 min. Slides were then digested with proteinase K (20 mg/ml) for 20 min at 37˚C and acetylated in 0.25% (v/v) acetic anhydride in 0.1 M triethanolamine (pH 8.0). Sections were prehybridized in 50% (v/v) deionized formamide in 2X SSC at 58˚C for 60 min and then hybridized overnight at 58˚C in a humidified chamber with 0.5 mg/ ml DIG-labeled RNA probe in hybridization buffer containing 50% deionized formamide, 10% (w/v) dextran sulfate, 1X Denhardt’s solution, 1 mg/ml yeast tRNA, 0.6M NaCl, 10mMTris-HCl (pH 7.4), and 1 mM EDTA (pH 8.0). After hybridization, slides were treated with RNase A (20 mg/ml) for 30 min at 37˚C and washed with 0.1X SSC at 58˚C for 30 min. Hybridized probes were detected with alkaline phosphatase-conjugated anti-DIG antibody (Roche, Indianapolis, IN) and color reaction was developed with NBT/BCIP (Roche) according to the manufacturer’s instructions.
Probe construction: A 436 bp DNA fragment (nucleotides 7484-7919 of reference sequence NR_002819, see below) was amplified by PCR and inserted into the pGEM-T vector (Promega). Sense and antisense probes were then generated with MEGAscript T7 Transcription Kit (sense probe) and MEGAscript SP6 Transcription Kit (Thermo scientific) for the anti-sense probe as described in manufacturer’s protocol. The sequence of the MALAT1 probe region is: GGCAGGAGAGACAACAAAGCGCTATTATCCTAAGGTCAAGAGAAGTGTCAGCCTCACCTGATTTTTATTAGTAATGAGGACTTGCCTCAACTCCCTCTTTCTGGAGTGAAGCATCCGAAGGAATGCTTGAAGTACCCCTGGGCTTCTCTTAACATTTAAGCAAGCTGTTTTTATAGCAGCTCTTAATAATAAAGCCCAAATCTCAAGCGGTGCTTGAAGGGGAGGGAAAGGGGGAAAGCGGGCAACCACTTTTCCCTAGCTTTTCCAGAAGCCTGTTAAAAGCAAGGTCTCCCCACAAGCAACTTCTCTGCCACATCGCCACCCCGTGCCTTTTGATCTAGCACAGACCCTTCACCCCTCACCTCGATGCAGCCAGTAGCTTGGATCCTTGTGGGCATGATCCATAATCGGTTTCAAGGTAACGATGGTGTCGAGG
ISH Scoring: Staining was scored in a semi-quantitative fashion based on a four-tier system: 0 (no hybridization), 1 (<25% positive cells), 2 (25- 50 % positive) and 3 (>50% positive). The score was then converted into a binary grade where a score of 0 or 1 was deemed low and a score of 2 or 3 deemed high. Only nuclear hybridization was considered positive and scoring performed in a blinded manner.
Statistical Analysis
In vitro data are expressed as a mean ±SD and significance determined as P < 0.05 using a 2-tailed student’s t-test. For survival studies, Kaplan-Meier curves were plotted, and the log-rank test was performed for comparison of cohorts. For hindlimb studies tumor growth is expressed as fractional tumor growth V/ V0 +SD and significance determined as P < 0.05. Datasets were analyzed as indicated in the legends.
Data availability
All data generated and analyzed during the current study are included in this published article and its Supplementary Information files or are available from the corresponding author on reasonable request.
RESULTS
MALAT1 is induced by DNA damage in a p50/p53 dependent manner
To identify p50/p105-dependent factors modified in response to TMZ, we used isogenic U87 cells stably expressing a short-hairpin (sh) targeting the c-terminal of p105 or a non-coding sequence (sh-control). Following treatment with TMZ, differential gene expression was determined. Cells were treated with 100 μM TMZ, the concentration reported in the plasma of patients treated with TMZ (35), for 16 hours, the time point of maximal change in NF-кB DNA binding in prior work (5). 133 transcripts were significantly (FDR < 0.05) altered following TMZ treatment (Figure 1A and Supplementary Tables 2 and 3). The primary pathways modulated included the p53 response, DNA damage signaling and regulation of cell death (Supplementary Figure 1A-C). Using a very stringent cutoff (FDR < 0.01), we identified eight transcripts significantly altered in a p50/p105-dependent fashion following TMZ treatment (Figure 1B and Supplementary Table 4). Five of these factors either had no identified gene product or coded for a histone protein. The expression of the remaining three genes was examined using qPCR. While PLK2 and MALAT1 were induced by TMZ, TP53I3 was not even though it was p50/p105-dependent (Figure 1C). Of these two genes, MALAT1 was conspicuous because it was the sole lncRNA in the entire p50/p105-regulated gene set and because it had previously been associated with resistance to TMZ in GBM (29,31,48).
Figure 1. Gene expression analysis.
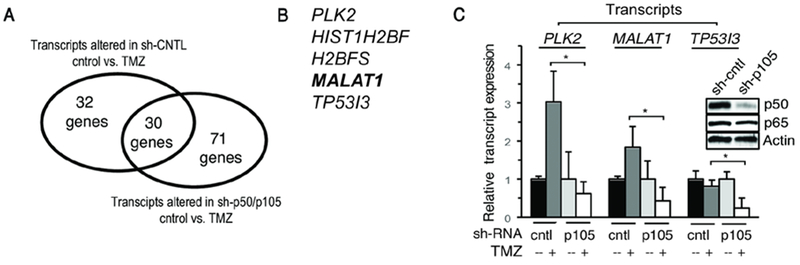
(A) Number of transcripts significantly altered in response to TMZ (100 μM, 16 hrs) in U87 cells expressing indicated shRNA (FDR<0.05). (B) List of genes most significantly altered (FDR < 0.01) following TMZ treatment in cells expressing sh-control but not sh-p50/p105. (C) RNA expression in U87 cells expressing sh-control or sh-p105 following treatment with TMZ (100 μM, 16 hrs). Data show mean expression relative to GAPDH, ±SD of triplicate samples from two biological experiments normalized to vehicle. Inset: immunoblot in U87 cells expressing control or p105 shRNA. *, p< 0.05.
To further examine the requirement of p50/p105, a distinct siRNA targeting this subunit was used confirming the requirement of p50/p105 for expression of MALAT1 by TMZ (Figure 2A). In addition, the kinetics of induction of MALAT1 by TMZ were determined (Figure 2B and C). Induction of MALAT1 expression by TMZ was subsequently demonstrated in a second GBM cell line (Supplementary Figure 2A). As RNA silencing results in depletion of both p50 and p105, to specifically look at mature p50, we overexpressed p50 in MEFs derived from Nfkb1 knockout (Nfkb1−/−) mice. Importantly, the nucleotide sequence of MALAT1 is highly conserved between man and mouse (26). While TMZ induced MALAT1 expression in wildtype (Nfkb1+/+) MEFs, deletion of Nfkb1 blocked this induction (Figure 2D). Reconstitution of p50 in Nfkb1−/− MEFs restored induction of MALAT1 by TMZ, confirming that p50 alone, and not the entire p105, was sufficient for induction of MALAT1 by TMZ (Figure 2D). In addition, as TMZ induces phosphorylation of p50 Ser329 (3,7), we examined whether an unphosphorylateable p50 mutant (S329A) altered induction of MALAT1. In contrast to the wildtype p50 (p50wt), p50S329A did not restore expression of MALAT1 by TMZ (Figure 2D) indicating that S329 phosphorylation is required for induction of MALAT1. Finally, given that a prior report demonstrated that p65 can bind the MALAT1 promoter (49), we examined whether p65 is required for induction of MALAT1. Knockdown of p65 did not alter induction of MALAT1 by TMZ (Supplementary Figure 2B).
Figure 2. MALAT1 is induced in a p50/p53-depedent manner.
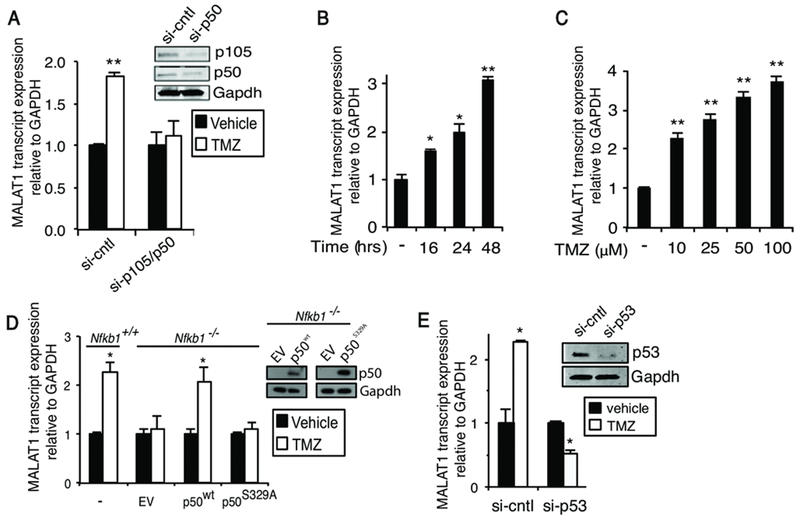
qPCR analysis of MALAT1 expression. (A) U87 cells transfected with the indicated si-RNA (si-cntl: scrambled sequence) and treated as shown (100 μM TMZ, 24 hours). Inset: immunoblot in U87 cells expressing control or p105 si-RNA. (B) U87 cells treated with 100 μM TMZ for the indicated time. (C) U87 cells treated for 48 hours with TMZ. (D) Nfkb1+/+ and Nfkb1−/− MEFs untransfected or transfected with empty vector (EV), p50wt or p50S329A and treated as indicated (TMZ, 100 μM, 24 hours). Inset: immunoblot in Nfkb1−/− MEFs expressing EV, S329A or wt-p50. (E) U87 cells transfected with si-RNA against p53 and treated as shown (100 μM TMZ, 24 hours). Inset: immunoblot in U87 cells transected with p53 si-RNA. Data show mean expression relative to GAPDH, ±SD of triplicate samples from three biological experiments normalized to vehicle. *, p< 0.05; **, p< 0.01.
It was notable that p53 signaling was one of the pathways most significantly activated by TMZ (Supplementary Figure 1B). As p53 has been reported to modulate MALAT1 expression, we examined the role of p53 in this response. Remarkably, and in contrast to p53 overexpression (32,50), knockdown of p53 using two distinct si-RNA constructs blocked induction of MALAT1 by TMZ (Figure 2E and Supplementary Figure 2B). Consistent with this, we found that in U251 GBM cells that are p53 mutant (51), TMZ did not induce MALAT1 expression (Supplementary Figure 2C). These results indicate that MALAT1 is induced by TMZ in a manner dependent on p50 and p53.
TMZ modulates recruitment of p50 and p53 to the MALAT1 coding sequence
The above findings, and prior reports (32,49,50), suggested that MALAT1 is transcriptionally regulated by p53 and NF-кB. We therefore searched the promoter and coding region of human MALAT1 (NCBI Gene ID: 378938) for potential p53 and кB BSs. For p53, 2,500 base-pairs up- and down-stream of the MALAT1 coding sequence were interrogated using several programs: p53MH (41), JASPAR (42), and MATCH (52). Only one p53 BS, within the MALAT1 coding sequence (NCBI Ref. Seq: NR_002819.2), was present in all three searches. This p53 BS was distinct from that described in a previous report (Supplementary Figure 3A) (50). For NF-кB, the latter two programs and TFSEARCH (44) were used and a single decameric sequence present in all three searches with 85% homology to the canonical кB-site was identified. This кB-site was in the proximal MALAT1 coding sequence (Figure 3A). As both BSs were distinct from previously reported p53 and NF-кB BSs (Supplementary Figure 3A) (49,50), we examined whether purified p50 and p53 proteins actually bound to these putative sites. Each protein was incubated with an oligonucleotide sequence bearing its respective BS and a concentration-dependent increase in binding was noted at each BS (Figure 3B). Next, to examine endogenous p50 and p53 DNA binding, nuclear extracts were harvested from U87 cells following treatment with TMZ. Gel shift with either the p53 or p50 BS led to the appearance of a specific band (Figure 3C). Importantly, supershift analysis demonstrated that both p53 and p50 were present in these bands and bound their respective putative BSs at baseline (Figure 3C). While TMZ increased binding of p53 to the p53 probe (Figure 3C, compare lanes 2 and 3), it decreased binding of NF-кB/p50 to the B probe (Figure 3C, compare lanes 7 and 8), an observation supported by the decrease in p50 supershift following treatment with TMZ (Figure 3C, compare lanes 9 and 10). To examine changes in transcription factor binding in vivo, we used qChIP with primers spanning each specific BS. Both p53 and p50 were recruited to chromatin in the region of their respective BSs and, while TMZ inhibited recruitment of p50 to the кB-site, it induced recruitment of p53 to the p53 BS (Figure 3D).
Figure 3. TMZ modulates binding of p53 and NF-кB to кB and p53 binding sites.
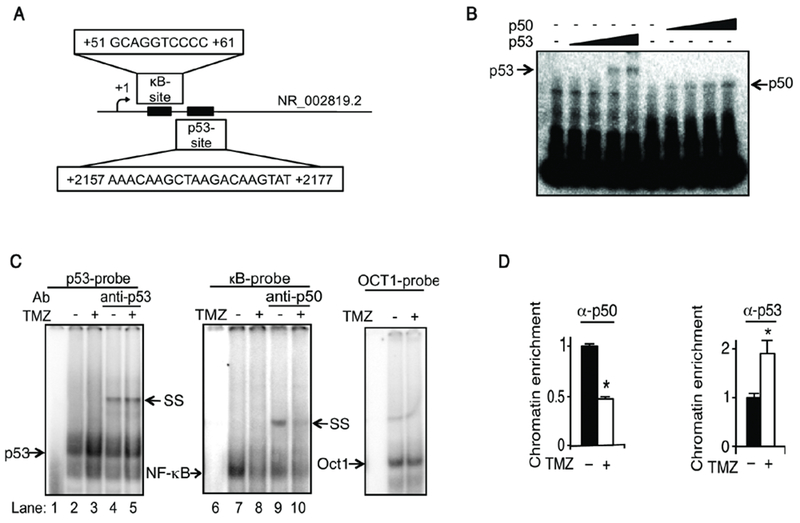
(A) Schematic representation of the кB and p53 binding sites in human MALAT1. (B) EMSA using the кB and p53 probes with increasing concentrations of purified His-p50 or p53 (0 - 300 ng). (C) EMSA using nuclear extracts from U87 cells treated with vehicle or TMZ (100 μM TMZ, 16 hours). Supershift (SS) with anti-p50 and anti-p53. Oct1 EMSA demonstrates equal lysate loading. (D) qChIP using primers spanning the кB and p53 BSs in U87 cells treated as shown (100 μM TMZ, 16 hours). Data represent chromatin enrichment of the indicated protein, relative to input DNA after controlling for non-specific binding using anti-histone H1 (positive control) and anti-IgG, normalized to vehicle, mean ±SD of triplicate samples shown. *, p< 0.05.
Given the requirement of p50 for expression of MALAT1 by TMZ, we examined binding in cells depleted of this subunit. The primary NF-кB dimer bound to the MALAT1 кB-site was p50/p65 and TMZ attenuated this binding (Figure 4A). Knockdown of p105/p50 led to cross-compensation by the p52 subunit resulting in formation of p52/p65 dimers (Figure 4A, lanes 9 and 10). Also, loss of p50 blocked the ability of TMZ to inhibit NF-кB DNA binding (Figure 4A, compare lanes 2 and 7). ChIP studies with stable sh-RNA expressing cells demonstrated increased chromatin enrichment of the p52 subunit in cells depleted of p105/p50 (Figure 4B), a finding consistent with the reported propensity of p52 to compensate for loss of p50 (53,54). Moreover, consistent with the gel shift data, in sh-p105/p50-expressing cells TMZ did not alter the chromatin recruitment of p52 (Figure 4B, center panel). Notably, with loss of p105/p50, there was a significant reduction in TMZ-induced recruitment of p53 to the MALAT1 regulatory region (Figure 4B, right panel). The minor increase in p53 binding in sh-p105 cells following treatment was likely due to the residual p50 in these cells (Figure 2A, inset). There was no change in p65 enrichment following treatment (Supplementary Figure 3B). In addition, as p50 has been shown to induce p53 expression (55), we examined p53 expression in cells depleted of p50. In GBM cells, depletion of p50 did not alter p53 expression (Supplementary Figure 3C).
Figure 4. Recruitment to the кB and p53 binding sites is dependent on p105/p50.
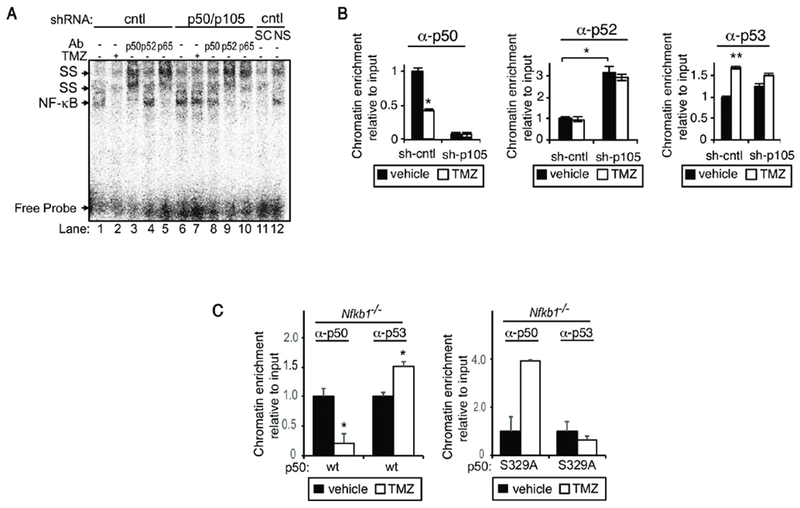
(A) EMSA using nuclear extract from U87 cells stably expressing sh-p105/p50 or sh-control treated with vehicle or TMZ (100 μM, 16 hours). SS with the indicated antibody. Competition performed with specific (SC) and non-specific (NS) cold probe identifies the NF-кB band. (B) qChIP using primers spanning the human кB and p53 BSs in sh-cntl and sh-p105/p50 U87 cells. Cells were treated as shown (100 μM TMZ, 16 hours) and IP performed with the indicated antibodies. (C) qChIP in Nfkb1−/− MEFs expressing either p50wt or p50S329A using primers spanning murine BSs in MALAT1. qChIP data represent chromatin enrichment of the indicated protein, relative to input DNA after controlling for non-specific binding using anti-histone H3 (positive control) and anti-IgG, normalized to vehicle, mean ± SD of triplicate samples, repeated with similar results. *, p< 0.05; **, p< 0.01.
S329 phosphorylation was required for expression of MALAT1 (Figure 2D), therefore, we examined the role of this residue for the changes in p50 and p53 chromatin recruitment. We scanned murine MALAT1 (NCBI Gene ID: 378938) for potential кB and p53 BSs using JASPAR and identified a кB-site with 82% homology to the human sequence and a p53 BS with 80% homology to human. Nfkb1−/− MEFs were infected with lentiviral vectors expressing either p50wt or p50S329A and chromatin recruitment of p50 or p53 examined. As with U87 cells, TMZ blocked p50 and induced p53 recruitment in cells expressing p50wt (Figure 4C, left panel). However, in the presence of p50S329A, TMZ neither inhibited p50 binding nor induced p53 recruitment. These findings are consistent with prior work demonstrating that TMZ-induced S329 phosphorylation blocks p50 recruitment to certain кB sequences (7), and indicate that phosphorylation of S329 is necessary for the increase in p53 chromatin recruitment to the MALAT1 regulatory region.
MALAT1 кB and p53 cis-elements are functional
To examine the functional relevance of these BSs, a luciferase reporter consisting of 2,400 basepairs of the human MALAT1 proximal promoter and p53 and p50 BSs was constructed and each BS mutated individually and together (Figure 5A). TMZ induced expression from the wildtype (wt) reporter that was maximal at 16 hours (Figure 5B). Mutation of either the p53 BS or the кB-site alone blocked the increase in activity induced by TMZ (Figure 5C). Mutation of the кB-site alone led to increased reporter activity in the absence of TMZ (Figure 5C) suggesting that in unstimulated cells this кB-site inhibits transcriptional activity. Knockdown of p105/p50 blocked luciferase expression by TMZ from the wt reporter (Figure 5D). Moreover, when p50wt was re-expressed in sh-p105-expressing cells, TMZ induced luciferase expression from a reporter containing the wt but not the mutant B site (Figure 5E). However, consistent with the requirement of S329 for induction of MALAT1, expression of p50S329A did not enable luciferase expression from the reporter with the wt B site (Figure 5E). Together, these findings demonstrate that the putative p53 and кB BSs are functional and are required for the increase in luciferase expression by TMZ.
Figure 5. MALAT1 кB and p53 binding sites are functional.
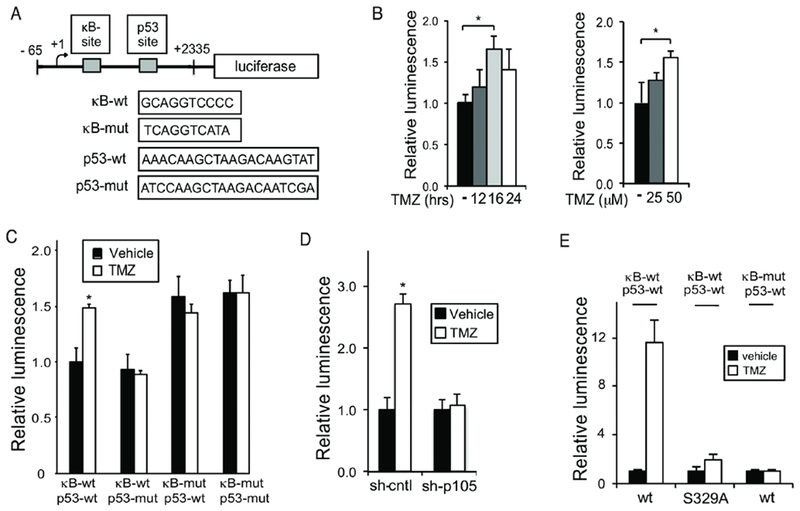
(A) Schematic of the 2.4 kbp luciferase reporter containing putative human p53 and p50 BSs. (B-E) Luciferase assays. Data show mean luciferase expression relative to renilla, ±SD of triplicate samples. (B) U87 cells treated with 100 μM TMZ for different times as noted (left) or U87 cells treated with TMZ for 16 hours (right). (C) U87 cells transfected with the indicated reporter construct and treated as shown (100 μM TMZ, 16 hours). (D) sh-cntl and sh-p105/p50 U87 cells treated as in C. (E) U87 cells stably expressing sh-p105/p50 were transfected with p50wt or p50S329A and the indicated кB reporter (p53-wt). Cells were treated as in C. Data represent mean ±SD of triplicate samples, repeated with similar results. *, p < 0.05, **p < 0.01.
Knockdown of MALAT1 sensitizes GBM cells to TMZ
High serum MALAT1 has been associated with resistance to TMZ (48); however, MALAT1 has been reported to both increase and decrease cell proliferation and tumor growth (29,32,56,57). Given these conflicting observations, we examined whether knockdown of MALAT1 modulates the efficacy of TMZ. Although MALAT1 is a nuclear lncRNA, si-RNA reduced its expression by 50% (Supplementary Figure 4A). Knockdown of MALAT1 enhanced the efficacy of TMZ in U87 cells as determined by clonogenic survival assay (Figure 6A). Similarly, in GBM34 cells, a patient derived glioma stem-like cell (GSC) (33), depletion of MALAT1 increased TMZ-induced killing (Figure 6B). In addition, although TMZ did not induce MALAT1 in U251 cells, knockdown of MALAT1 still augmented the effect of TMZ in these cells (Supplementary Figure 4B), likely due to the depletion of basal MALAT1. These findings were replicated in A172 cells and GBM44 patient-derived GSCs (Supplementary Figures 4C and D). Importantly, a second si-RNA targeting MALAT1 recapitulated these results (Supplementary Figures 4E-G). We also constructed a lentiviral vector expressing sh-MALAT1 that targeted a distinct region and depleted MALAT1 expression by 70% (Figure 6C). Expression of this vector in GBM cells sensitized them to TMZ (Figure 6D). These findings indicate that MALAT1 promotes resistance to TMZ in GBM.
Figure 6. Knockdown of MALAT1 enhances the anti-glioma effect of TMZ.
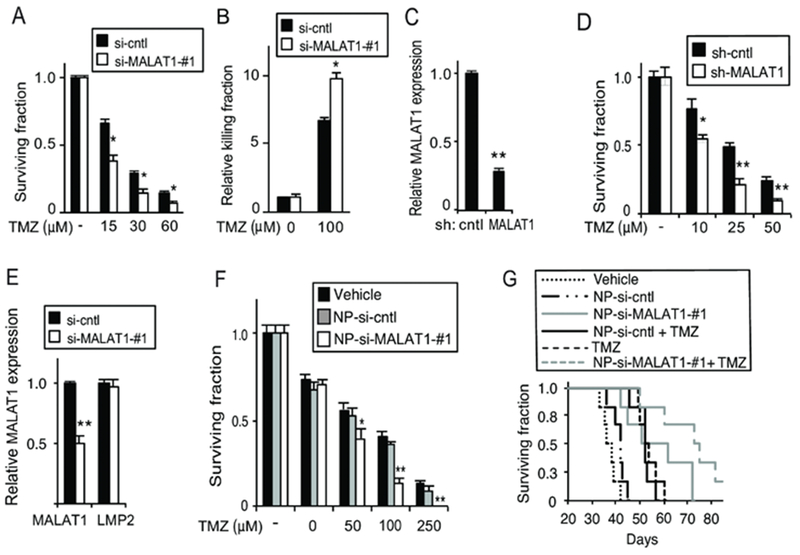
(A) Clonogenic assay in U87 cells transfected with si-cntrl or si-MALAT1-#1 treated as shown. (B) Trypan blue assay in GBM34 GSCs transfected with si-cntrl or si-MALAT1-#1 following treatment with TMZ for 72 hours. (C) MALAT1 expression in U87 cells following infection with the indicated sh-RNA construct. (D) Clonogenic assay in U87 cells expressing sh-control or sh-MALAT1 treated with TMZ. (E) qPCR analysis of MALAT1 and LMP2 mRNA expression, relative to GAPDH, in U87 cells treated with nanoparticles carrying si-control (NP-si-cntrl) or si-MALAT1 (NP-si-Malat1). (F) Clonogenic assay in U87 cells following treatment with the indicated nanoparticles. (G) Kaplan-Meier survival curves of mice bearing intracranial U87 GBM xenografts (n= 6 mice per group) following treatment with TMZ (days 4, 7 and 10) and/or the indicated NP. P < 0.02, Log-rank: TMZ + NP-si-MALAT1-#1 vs. TMZ + NP-si-cntl or NP-si-MALAT1-#1 alone. Trypan blue, clonogenic, qPCR data represent mean ±SD of triplicate samples, repeated with similar findings. *, p< 0.05; **, p< 0.01 relative to control.
MALAT1 is a chemosensitizing target, not a prognostic factor, in GBM
To determine whether depletion of MALAT1 can be used in established GBM, we encapsulated an siRNA targeting MALAT1 in a nanoparticle vector previously used for the treatment of experimental GBM (46). Compared to control siRNA (NP-si-cntl), nanoparticles encapsulating si-MALAT1 (NP-si-MALAT1) resulted in approximately 50% knockdown of MALAT1 specifically (Figure 6E). Combination treatment of GBM cells with NP-si-MALAT1 and TMZ led to a significant decrease in clonal survival relative to NP-si-cntl and TMZ (Figure 6F). Subsequently, intracranial GBM xenografts were established and NPs delivered by direct intracranial injection. The combination of TMZ and NP-si-MALAT1 resulted in increased animal survival compared to control (Figure 6G, p < 0.02, Log-rank NP-si-MALAT1+TMZ vs. all other groups). A similar finding was seen when hindlimb xenografts were treated with TMZ and NPs, demonstrating that tumor growth is significantly inhibited by combination TMZ and NP-si-MALAT1 (Supplementary Figure 5A). Importantly, nanoparticles encapsulating si-RNA inhibited MALAT1 expression in vivo (Supplementary Figure 5B). These findings indicate that depletion of MALAT1 combines effectively with TMZ to treat experimental GBM.
To evaluate whether MALAT1 is prognostic in GBM, as was previously suggested (58), we performed in situ hybridization (ISH) of GBM specimens. Two tissue microarrays (TMA) containing 34 separate GBM specimens that have been previously described (47) were used and a specific anti-sense probe targeting a 436-nucleotide fragment of reference sequence, NR_002819, constructed. The signal intensity of the anti-sense probe was evaluated and the lack of signal with the sense probe demonstrated (Supplementary Figure 6). TMAs were then hybridized with the anti-sense probe and signal intensity graded in a blinded manner on a four-tier scale (0,1, 2 or 3). This signal intensity was converted into a binary score of high or low (Figure 7A). The clinical characteristics of the patients are noted (Supplementary Table 1). No significant difference in overall survival was seen between patients with high and low MALAT1 hybridization score (Figure 7B). To further examine the potential prognostic role of MALAT1, we looked at RNA sequencing data from The Cancer Genome Atlas (TCGA). Consistent with the ISH data, MALAT1 expression level in TCGA GBM patients was unable to separate patients into survival groups (Figure 7C) (GlioVis data portal for visualization and analysis of brain tumor expression datasets) (40). This result was recapitulated in data from the REMBRANDT dataset (P = 0.55, log-rank, HR = 1.1, GlioVis data portal). In addition, analysis of combined GBM and lower grade glioma (LGG) patients from TCGA demonstrated that not only was MALAT1 expression level uninformative of survival (Supplementary Figure 7), but also that there was no significant difference in expression between different grades of glioma (Supplementary Figure 8A). Interestingly, a significant difference in MALAT1 expression was observed between primary and recurrent gliomas (Supplementary Figure 8B). These results indicate that, while MALAT1 was not prognostic in GBM, its expression was elevated in recurrent tumors.
Figure 7. MALAT1 expression is not prognostic of overall survival in GBM.
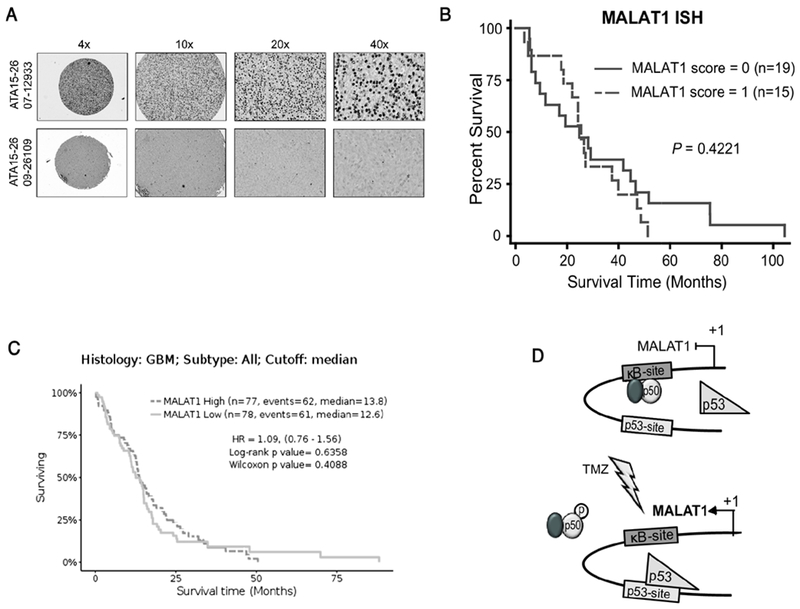
(A) Representative MALAT1 in-situ hybridization staining in GBM. Low staining (bottom) and high staining (top). (B) Kaplan-Meier survival curves in thirty-four GBM patients separated by their MALAT1 ISH staining score. Significance was analyzed by Log-rank method. (C) Kaplan-Meier survival curves in all GBM patients from TCGA based on RNA-SEQ expression separated at median value. (D) Model illustrating the mechanism by which TMZ induces MALAT1 expression. p50-containing dimers occupy the кB-site at rest (upper). Following treatment (lower), phosphorylation of p50 Ser329 leads to decreased p50 dimer binding and a concomitant increased p53 chromatin recruitment resulting in increased MALAT1 expression.
DISCUSSION
Here, using genome-wide analysis we identified MALAT1 as a p50-dependent gene upregulated following treatment of GBM cells with TMZ. MALAT1 was the sole p50-dependent lncRNA induced by alkylation damage. While MALAT1 expression was previously reported to be elevated in glioma cells resistant to TMZ (29), the mechanism underlying its induction was unclear. Interestingly, it was recently noted that MALAT1 was not induced in GBM cells by other chemotherapeutics (59), suggesting that MALAT1 is not generally upregulated by DNA damaging therapy.
Mechanistically, we found that MALAT1 was induced by DNA alkylation damage in a manner dependent on p50 and p53. We identified novel кB and p53 BSs downstream of the MALAT1 transcription start site that bound their respective transcription factors. While TMZ decreased the chromatin recruitment of p50, it concomitantly increased the recruitment of p53. Although it was possible that these reciprocal changes were coincidental, their co-dependence in promoting expression of MALAT1 by TMZ was supported by the finding that in the absence of p50, p53 recruitment did not increase and TMZ failed to induce MALAT1. Moreover, with loss of p50, p52 bound the кB-site and remained bound following treatment. We previously reported that TMZ induces p50 phosphorylation at Ser329 resulting in inhibition of NF-кB binding and activity (3). Consistent with this, we found that expression of an S329A mutant p50 blocked both the decrease in p50 and the increase in p53 binding in response to TMZ. The requirement for loss of p50 binding for MALAT1 expression was supported by luciferase studies where mutation of the кB-site alone, which would result in the loss of p50 binding, led to increased reporter activity in untreated cells. These findings support a model whereby phosphorylation of p50 in response to TMZ leads to its release from chromatin and the concomitant increase in p53 binding that promotes MALAT1 expression (Figure 7D).
Although regulation of MALAT1 by p53 and NF-кB was previously studied, the data are somewhat conflicting. Whereas we find that in GBM cells and MEFs MALAT1 was induced by a mechanism requiring p53, it was recently reported that overexpression of p53 in erythroid myeloid lymphoid (EML) cells decreased MALAT1 via a putative p53 BS different from the p53 site identified here (50). This inhibitory effect of p53 overexpression on MALAT1 was also noted in GBM cells (32). These differences in the role of p53 in regulating MALAT1 expression are likely because p53 overexpression induces changes that are different to the p53-dependent alterations seen with TMZ treatment. In the setting of TMZ, MALAT1 expression is induced by the interaction of p53 with NF-кB, a response not seen when p53 is overexpressed by itself. Interestingly, in support of the requirement of p53, a previous comprehensive microarray study demonstrated that MALAT1 was one of multiple lncRNAs induced in a p53-dependent manner by oncogenic K-ras (60). With respect to NF-кB, a prior report identified a different кB-site within the MALAT1 promoter (49). Although direct binding of NF-кB to that consensus site was not examined, LPS induced p65 recruitment to that region of the promoter and mutation of the putative site blocked LPS-induced expression from a luciferase reporter. Together, these findings suggest that like protein coding genes, expression of MALAT1 is transcriptionally modulated in both a cell type and stimulus-dependent manner. Moreover, the co-dependence on NF-кB and p53 further emphasizes the multi-dimensional manner by which these transcription factors regulate downstream gene expression (8,9). Finally, it is notable that histones were among the genes initially identified on differential gene expression analysis (Supplementary Table 4). Histone modifications have previously been reported to modulate NF-кB activity and contribute to TMZ resistance in GBM (61). Interestingly, the interaction of MALAT1 with chromatin-modulating factors was previously reported to play a critical role in regulating cellular growth programs (25). Whether, MALAT1 regulates histones to mediate the response to TMZ is a potential mechanism that will require further analysis.
In order to target MALAT1 therapeutically, siRNA was encapsulated in a polymeric vector and delivered via stereotactic injection into GBM xenografts. Notably, a recent report also demonstrated that targeted MALAT1 knockdown improved the efficacy of TMZ against intracranial GBM (32). In that study, si-RNA targeting MALAT1 was delivered via intravenous injection. We used direct intratumoral injection to minimize the systemic effects of MALAT1 depletion. These studies suggest that MALAT1 is a potential target for chemosensitization and raised the question of whether MALAT1 expression is also informative of patient outcome in GBM as it is in other malignancies (21,62). We examined MALAT1 expression both in GBM tissue by ISH and in publically available databases. In contrast to protein coding genes whose mRNA expression may not accurately reflect protein level, ncRNA expression more directly indicates the level of the functional end product of the gene. We found that tumoral MALAT1 expression level did not have prognostic value. In addition, no significant difference in MALAT1 expression was seen between gliomas of different grade. These findings are in contrast to results from a previous study that examined MALAT1 by qPCR in a series of grade I-IV gliomas where it was reported that MALAT1 expression correlated with grade and was an independent predictor of survival (58).
It is important to highlight potential limitations of this study. First, we found that loss of p65 did not block induction of MALAT1 by TMZ. While this finding may be because TMZ acts via p50 and not p65 (3), it is also possible that cross-compensation by other NF-кB subunits, for example crel (54), may facilitate the continued expression of MALAT1 in the absence of p65. Second, MALAT1 was identified as a mechanism of resistance based on screening studies focused on the NF-кB pathway. Such an approach likely excludes identification of other important pathways of TMZ resistance downstream of MGMT. Clearly, resistance to TMZ is multifactorial involving not only cell intrinsic processes but also factors in the GBM microenvironment (63–65). Finally, although we did not observe a prognostic role for MALAT1 in GBM, it is important to note that we identified MALAT1 following TMZ treatment. However, in most glioma databases, tumor tissue is procured prior to treatment initiation. Therefore, despite the ability of MALAT1 to attenuate cytotoxicity, its expression level in ‘treatment-naive’ specimens would not necessarily be expected to correlate with survival. Indeed, analysis of recurrent glioma tissue demonstrated increased MALAT1 expression compared to primary, treatment-naive tumors. Interestingly, a recent report noted that serum MALAT1 was higher in GBM patients that responded poorly to TMZ (48). Although this study did not report a correlation between tumor MALAT1 and survival, given the ease of obtaining serum samples, analyzing serum MALAT1 level following TMZ treatment may be a potential strategy for predicting response to TMZ.
Supplementary Material
SIGNIFICANCE.
These findings identify NF-кB and p53 as regulators of the lncRNA MALAT1 and suggest MALAT1 as a potential target for the chemosensitization of GBM.
ACKNOWLEDGEMENTS
We thank Lingjiao Zhang for assistance. This work was supported by NIH grant R01CA136937 (B.Y.), CA014599 from the University of Chicago Comprehensive Cancer Center (B.Y.) and R44CA135906 (R.S., L.N. and B.Y.) and by the Ludwig Center for Metastasis Research. D.V. was a Howard Hughes Medical Research Fellow.
Footnotes
Conflict of Interest: LNK Chemsolutions, LLC (R.S., L.N. and G.F.L.) have commercial interests in the nanoparticles described.
REFERENCES
- 1.Hegi ME, Diserens A-C, Gorlia T, Hamou M-F, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med [Internet]. 2005;352:997–1003. [DOI] [PubMed] [Google Scholar]
- 2.Perkins ND. The diverse and complex roles of NF-кB subunits in cancer. Nat Rev Cancer. 2012;12:121–32. [DOI] [PubMed] [Google Scholar]
- 3.Schmitt AM, Crawley CD, Kang S, Raleigh DR, Yu X, Wahlstrom JS, et al. p50 (NF-kappaB1) is an effector protein in the cytotoxic response to DNA methylation damage. Mol Cell [Internet]. 2011;44:785–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raychaudhuri B, Han Y, Lu T, Vogelbaum MA. Aberrant constitutive activation of nuclear factor kappaB in glioblastoma multiforme drives invasive phenotype. J Neurooncol. 2007;85:39–47. [DOI] [PubMed] [Google Scholar]
- 5.Yamini B, Yu X, Dolan ME, Wu MH, Darga TE, Kufe DW, et al. Inhibition of nuclear factor-kappaB activity by temozolomide involves O6-methylguanine induced inhibition of p65 DNA binding. Cancer Res [Internet]. 2007;67:6889–98. [DOI] [PubMed] [Google Scholar]
- 6.Mansour NM, Bernal GM, Wu L, Crawley CD, Cahill KE, Voce DJ, et al. Decoy receptor DcR1 Is Induced in a p50/Bcl3-Dependent manner and attenuates the efficacy of temozolomide. Cancer Res. 2015;75:2039–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crawley CD, Raleigh DR, Kang S, Voce DJ, Schmitt AM, Weichselbaum RR, et al. DNA damage-induced cytotoxicity is mediated by the cooperative interaction of phospho-NF-кB p50 and a single nucleotide in the кb-site. Nucleic Acids Res. 2013;41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perkins ND. NF-kappaB: tumor promoter or suppressor? Trends Cell Biol [Internet]. 2004;14:64–9. [DOI] [PubMed] [Google Scholar]
- 9.Schneider G, Kramer OH. NFkappaB/p53 crosstalk-a promising new therapeutic target. Biochim Biophys Acta [Internet]. 2011;1815:90–103. [DOI] [PubMed] [Google Scholar]
- 10.Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma & supp. Cell. 2013;155:462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wei W, Ji S. Cellular senescence: Molecular mechanisms and pathogenicity. J. Cell. Physiol. 2018. page 9121–35. [DOI] [PubMed] [Google Scholar]
- 12.Fang Y, Fullwood MJ. Roles, Functions, and Mechanisms of Long Non-coding RNAs in Cancer. Genomics, Proteomics Bioinforma. 2016. page 42–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–9. [DOI] [PubMed] [Google Scholar]
- 14.Barsyte-Lovejoy D, Lau SK, Boutros PC, Khosravi F, Jurisica I, Andrulis IL, et al. The c-Myc oncogene directly induces the H19 noncoding RNA by allele-specific binding to potentiate tumorigenesis. Cancer Res. 2006;66:5330–7. [DOI] [PubMed] [Google Scholar]
- 15.Park JY, Lee JE, Park JB, Yoo H, Lee S-H, Kim JH. Roles of Long Non-Coding RNAs on Tumorigenesis and Glioma Development. Brain tumor Res Treat [Internet]. 2014;2:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han L, Zhang K, Shi Z, Zhang J, Zhu J, Zhu S, et al. LncRNA profile of glioblastoma reveals the potential role of lncRNAs in contributing to glioblastoma pathogenesis. Int J Oncol. 2012;40:2004–12. [DOI] [PubMed] [Google Scholar]
- 17.Xue X, Yang YA, Zhang A, Fong KW, Kim J, Song B, et al. LncRNA HOTAIR enhances ER signaling and confers tamoxifen resistance in breast cancer. Oncogene. 2016;35:2746–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu Y, Yu DD, Hu Y, Yan D, Chen X, Cao HX, et al. Genome-wide profiling of long non-coding RNA expression patterns in the EGFR-TKI resistance of lung adenocarcinoma by microarray. Oncol Rep. 2016;35:3371–86. [DOI] [PubMed] [Google Scholar]
- 19.He Z, Wang Y, Huang G, Wang Q, Zhao D, Chen L. The lncRNA UCA1 interacts with miR-182 to modulate glioma proliferation and migration by targeting iASPP. Arch Biochem Biophys. 2017;623–624: 1–8. [DOI] [PubMed] [Google Scholar]
- 20.Zhao H, Peng R, Liu Q, Liu D, Du P, Yuan J, et al. The lncRNA H19 interacts with miR-140 to modulate glioma growth by targeting iASPP. Arch Biochem Biophys. 2016;610:1–7. [DOI] [PubMed] [Google Scholar]
- 21.Ji P, Diederichs S, Wang W, Böing S, Metzger R, Schneider PM, et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22:8031–41. [DOI] [PubMed] [Google Scholar]
- 22.Yoshimoto R, Mayeda A, Yoshida M, Nakagawa S. MALAT1 long non-coding RNA in cancer. Biochim Biophys Acta - Gene Regul Mech. 2016;1859:192–9. [DOI] [PubMed] [Google Scholar]
- 23.Vassallo I, Zinn P, Lai M, Rajakannu P, Hamou M-F, Hegi ME. WIF1 re-expression in glioblastoma inhibits migration through attenuation of non-canonical WNT signaling by downregulating the lncRNA MALAT1. Oncogene [Internet]. 2015;35:1–10. [DOI] [PubMed] [Google Scholar]
- 24.Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39:925–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang L, Lin C, Liu W, Zhang J, Ohgi KA, Grinstein JD, et al. NcRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell. 2011;147:773–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hutchinson JN, Ensminger AW, Clemson CM, Lynch CR, Lawrence JB, Chess A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics [Internet]. 2007;8:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernard D, Prasanth K V, Tripathi V, Colasse S, Nakamura T, Xuan Z, et al. A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J. 2010;29:3082–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eißmann M, Gutschner T, Hämmerle M, Günther S, Caudron-Herger M, Groß M, et al. Loss of the abundant nuclear non-coding RNA MALAT1 is compatible with life and development. RNA Biol. 2012;9:1076–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Yuan X, Yan D, Li D, Guan F, Dong Y, et al. Long Non-Coding RNA MALAT1 Decreases the Sensitivity of Resistant Glioblastoma Cell Lines to Temozolomide. Cell Physiol Biochem [Internet]. 2017. [cited 2017 Aug 22];42:1192–201. [DOI] [PubMed] [Google Scholar]
- 30.Zhou X, Liu S, Cai G, Kong L, Zhang T, Ren Y, et al. Long non coding RNA MALAT1 promotes tumor growth and metastasis by inducing epithelial-mesenchymal transition in oral squamous cell carcinoma. Sci Rep. 2015;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cai T, Liu Y, Xiao J. Long noncoding RNA MALAT1 knockdown reverses chemoresistance to temozolomide via promoting microRNA-101 in glioblastoma. Cancer Med. 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim S- S, Harford JB, Moghe M, Rait A, Pirollo KF, Chang EH. Targeted nanocomplex carrying siRNA against MALAT1 sensitizes glioblastoma to temozolomide. Nucleic Acids Res [Internet]. 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsen AR, Long PM, Driscoll HE, Davies MT, Teasdale BA, Penar PL, et al. Triacetin-based acetate supplementation as a chemotherapeutic adjuvant therapy in glioma. Int J Cancer. 2014;134:1300–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sarbassov DD, Guertin D a, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. [DOI] [PubMed] [Google Scholar]
- 35.Hammond LA, Eckardt JR, Baker SD, Gail Eckhardt S, Dugan M, Forral K, et al. Phase I and pharmacokinetic study of temozolomide on a daily-for-5-days schedule in patients with advanced solid malignancies. J Clin Oncol. 1999;17:2604–13. [DOI] [PubMed] [Google Scholar]
- 36.Baker SD, Wirth M, Statkevich P, Reidenberg P, Alton K, Sartorius SE, et al. Absorption, metabolism, and excretion of 14C-temozolomide following oral administration to patients with advanced cancer. Clin Cancer Res. 1999;5:309–17. [PubMed] [Google Scholar]
- 37.Irizarry RA, Bravo HC, Irizarry RA, Irizarry R, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics [Internet]. 2003;4:249–64. [DOI] [PubMed] [Google Scholar]
- 38.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing [Internet]. J. R. Stat. Soc. B. 1995. page 289–300. [Google Scholar]
- 39.Huang DW, Lempicki R a, Sherman BT. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 40.Bowman RL, Wang Q, Carro A, Verhaak RGW, Squatrito M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro. Oncol. 2017. page 139–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoh J, Jin S, Parrado T, Edington J, Levine a J, Ott J. The p53MH algorithm and its application in detecting p53-responsive genes. Proc Natl Acad Sci U S A. 2002;99:8467–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, et al. JASPAR 2014: An extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014;42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wingender E, Chen X, Fricke E, Geffers R, Hehl R, Liebich I, et al. The TRANSFAC system on gene expression regulation. Nucleic Acids Res [Internet]. 2001;29:281–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel O V., et al. Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 1998;26:362–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamini B, Yu X, Gillespie GY, Kufe DW, Weichselbaum RR. Transcriptional targeting of adenovirally delivered tumor necrosis factor α by temozolomide in experimental glioblastoma. Cancer Res. 2004;64:6381–4. [DOI] [PubMed] [Google Scholar]
- 46.Bernal GM, LaRiviere MJ, Mansour N, Pytel P, Cahill KE, Voce DJ, et al. Convection-enhanced delivery and in vivo imaging of polymeric nanoparticles for the treatment of malignant glioma. Nanomedicine Nanotechnology, Biol Med. 2014;10:149–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu L, Bernal GM, Cahill KE, Pytel P, Fitzpatrick CA, Mashek H, et al. BCL3 expression promotes resistance to alkylating chemotherapy in gliomas. Sci Transl Med. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen W, Xu X- K, Li J- L, Kong K- K, Li H, Chen C, et al. MALAT1 is a prognostic factor in glioblastoma multiforme and induces chemoresistance to temozolomide through suppressing miR-203 and promoting thymidylate synthase expression. Oncotarget [Internet]. 2017;8:22783–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao G, Su Z, Song D, Mao Y, Mao X. The long noncoding RNA MALAT1 regulates the lipopolysaccharide-induced inflammatory response through its interaction with NF-кB. FEBS Lett. 2016. page 2884–95. [DOI] [PubMed] [Google Scholar]
- 50.Ma X-Y, Wang J-H, Wang J-L, Ma CX, Wang X-C, Liu F-S. Malat1 as an evolutionarily conserved lncRNA, plays a positive role in regulating proliferation and maintaining undifferentiated status of early-stage hematopoietic cells. BMC Genomics [Internet]. 2015;16:676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brázdová M, Quante T, Tögel L, Walter K, Loscher C, Tichý V, et al. Modulation of gene expression in U251 glioblastoma cells by binding of mutant p53 R273H to intronic and intergenic sequences. Nucleic Acids Res. 2009;37:1486–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wingender E, Chen X, Fricke E, Geffers R, Hehl R, Liebich I, et al. The TRANSFAC system on gene expression regulation. Nucleic Acids Res. 2001;29:281–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmitt AM, Crawley CD, Kang S, Raleigh DR, Yu X, Wahlstrom JS, et al. P50 (NF-кB1) is an effector protein in the cytotoxic response to DNA methylation damage. Mol Cell. 2011;44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoffmann A, Leung TH, Baltimore D. Genetic analysis of NF-кB/Rel transcription factors defines functional specificities. EMBO J. 2003;22:5530–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu Y, Zhang D, Huang H, Li J, Zhang M, Wan Y, et al. NF-кB1 p50 promotes p53 protein translation through miR-190 downregulation of PHLPP1. Oncogene. 2014;33:996–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Han Y, Wu Z, Wu T, Huang Y, Cheng Z, Li X, et al. Tumor-suppressive function of long noncoding RNA MALAT1 in glioma cells by downregulation of MMP2 and inactivation of ERK/MAPK signaling. Cell Death Dis. 2016;7:e2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Z, Xu C, Ding B, Gao M, Wei X, Ji N. Long non-coding RNA MALAT1 promotes proliferation and suppresses apoptosis of glioma cells through derepressing Rap1B by sponging miR-101. J Neurooncol. 2017;134:19–28. [DOI] [PubMed] [Google Scholar]
- 58.Ma KX, Wang HJ, Li XR, Li T, Su G, Yang P, et al. Long noncoding RNA MALAT1 associates with the malignant status and poor prognosis in glioma. Tumor Biol. 2015;36:3355–9. [DOI] [PubMed] [Google Scholar]
- 59.Liu Q, Sun S, Yu W, Jiang J, Zhuo F, Qiu G, et al. Altered expression of long non-coding RNAs during genotoxic stress-induced cell death in human glioma cells. J Neurooncol. 2015;122:283–92. [DOI] [PubMed] [Google Scholar]
- 60.Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142:409–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.yang Li Z, zhong Li Q, Chen L, dong Chen B, Wang B, jun Zhang X, et al. Histone Deacetylase Inhibitor RGFP109 Overcomes Temozolomide Resistance by Blocking NF-кB-Dependent Transcription in Glioblastoma Cell Lines. Neurochem Res. 2016;41:3192–205. [DOI] [PubMed] [Google Scholar]
- 62.Shen L, Chen L, Wang Y, Jiang X, Xia H, Zhuang Z. Long noncoding RNA MALAT1 promotes brain metastasis by inducing epithelial-mesenchymal transition in lung cancer. J Neurooncol. 2015;121:101–8. [DOI] [PubMed] [Google Scholar]
- 63.Grek CL, Sheng Z, Naus CC, Sin WC, Gourdie RG, Ghatnekar GG. Novel approach to temozolomide resistance in malignant glioma: connexin43-directed therapeutics. Curr. Opin. Pharmacol. 2018. page 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lathia J, Mack S, Mulkearns-Hubert E, Valentim C, Rich J. Cancer stem cells in glioblastoma. Genes Dev. 2015;15:29:1203–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zeng AL, Yan W, Liu YW, Wang Z, Hu Q, Nie E, et al. Tumour exosomes from cells harbouring PTPRZ1-MET fusion contribute to a malignant phenotype and temozolomide chemoresistance in glioblastoma. Oncogene. 2017;36:5369–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated and analyzed during the current study are included in this published article and its Supplementary Information files or are available from the corresponding author on reasonable request.