

Abstract
Background:
The T cell-inflamed phenotype correlates with efficacy of immune-checkpoint blockade while non-T cell-inflamed tumors infrequently benefit. Tumor-intrinsic WNT/β-catenin signaling mediates immune exclusion in melanoma, but association with the non-T cell-inflamed tumor microenvironment in other tumor types is not well understood.
Methods:
Using The Cancer Genome Atlas (TCGA), a T cell-inflamed gene expression signature segregated samples within tumor types. Activation of WNT/β-catenin signaling was inferred using three approaches: somatic mutations or somatic copy number alterations (SCNAs) in β-catenin signaling elements including CTNNB1, APC, APC2, AXIN1, AXIN2; pathway prediction from RNAseq gene expression; and inverse correlation of β-catenin protein levels with the T cell-inflamed gene expression signature.
Results:
Across TCGA, 3137/9244 (33.9%) tumors were non-T cell-inflamed while 3161/9244 (34.2%) were T cell-inflamed. Non-T cell-inflamed tumors demonstrated significantly lower expression of T cell inflammation genes relative to matched normal tissue, arguing for loss of a natural immune phenotype. Mutations of β-catenin signaling molecules in non-T cell-inflamed tumors were enriched three-fold relative to T cell-inflamed tumors. Across 31 tumors, 28 (90%) demonstrated activated β-catenin signaling in the non-T cell-inflamed subset by at least one method. This included target molecule expression from somatic mutations and/or SCNAs of β-catenin signaling elements (19 tumors, 61%), pathway analysis (14 tumors, 45%), and increased β-catenin protein levels (20 tumors, 65%).
Conclusions:
Activation of tumor-intrinsic WNT/β-catenin signaling is enriched in non-T cell-inflamed tumors. These data provide a strong rationale for development of pharmacologic inhibitors of this pathway with the aim of restoring immune cell infiltration and augmenting immunotherapy.
Introduction
Immunotherapies targeting immune checkpoints have contributed to a marked improvement in treatment outcomes in patients with advanced cancer. In melanoma, anti-cytotoxic T lymphocyte antigen 4 (CTLA-4) and anti-programmed death 1 (PD-1) antibodies have demonstrated robust response rates with years of durability in some patients(1,2) and improvement in overall survival(3,4). Significant clinical activity of PD-1-targeting agents has led to FDA approval in multiple additional cancer entities. Despite this broad activity, only a subset of patients benefits from treatment within each cancer subtype, and molecular mechanisms to explain primary resistance in these patients remain incompletely understood.
High expression of specific immune cell genes in the tumor microenvironment, described as the T cell-inflamed phenotype, has been observed to correlate with response to multiple immunotherapies including therapeutic vaccines and checkpoint blocking antibodies(1,5–10). Conversely, the non-T cell inflamed tumor microenvironment appears to closely associate with lack of clinical benefit to immunotherapy, particularly with anti-PD-1 antibodies(9,10). Categorization of tumors using transcriptional profiles marking the T cell-inflamed gene expression signature is advantageous as it can define biologically relevant patient sub-populations and set a framework in which to investigate hypothetical mechanisms for primary immunotherapy resistance.
Although multiple molecular mechanisms could theoretically disfavor a T cell-inflamed microenvironment, several lines of investigation have indicated that specific oncogenic molecular aberrations can be sufficient to drive this immune exclusion phenotype in some cases. Tumor cell-intrinsic WNT/β-catenin signaling in melanoma was the first somatic alteration associated with the non-T cell-inflamed tumor microenvironment in patients, and was demonstrated to be causal using a genetically-engineered mouse model(11). The mechanism of this effect appears to be through transcriptional repression of key chemokine genes that leads to lack of Batf3-lineage dendritic cell recruitment and subsequent failure to prime and recruit CD8+ T cells(11,12). This effect is dominant in the tumor microenvironment and leads to loss of therapeutic efficacy of checkpoint blockade, tumor antigen vaccination, and adoptive T cell transfer immunotherapy approaches preclinically(11,12). While the above studies of tumor-intrinsic WNT/β-catenin signaling have been evaluated in the context of melanoma, the impact of this pathway in driving the non-T cell-inflamed tumor microenvironment in other tumor types are increasingly being recognized. In syngeneic murine models of B16F10 melanoma, 4T1 mammary carcinoma, Neuro2A neuroblastoma, and Renca renal adenocarcinoma, blocking β-catenin pathway signaling via RNA interference resulted in influx of CD8+ T cells and increase in interferon-γ-associated gene targets(13). Subsequent combination with immunotherapy yielded complete regressions in the majority of treated animals. More broadly, roles for WNT/β-catenin signaling impacting on the immune system via development and function, active immune exclusion by tumor cells and cancer immunosurveillance are being recognized and accepted across cancer types(14).
To investigate the influence of WNT/β-catenin signaling across cancers, we performed an integrative analysis of The Cancer Genome Atlas (TCGA) separating individual tumors by T cell-inflamed status and identifying β-catenin pathway activation on three levels. We find that most tumor types within TCGA are enriched for activation of WNT/β-catenin signaling in non-T cell-inflamed tumors. These observations suggest pharmacologic targeting of this pathway could have broad implications for improving immunotherapy efficacy.
Materials and Methods
TCGA cancer datasets
Level 3 gene expression data (release date February 4, 2015), level 4 somatic mutation data (release date January 28, 2016), and level 3 somatic copy number data (release date January 28, 2016) were downloaded for 31 solid tumor types from The Cancer Genome Atlas (TCGA) preprocessed by Broad’s TCGA team (https://gdac.broadinstitute.org/). To make gene expression values comparable among cancer types, raw read counts mapped to gene features were processed by upper quartile normalization followed by log2 transformation across all samples. For mutation data, silent mutations were excluded from all analysis. For copy number (CN) alterations, we focused on high-level changes including putative biallelic (or higher) CN gain (+2) or putative biallelic loss (−2) as defined in GISTIC2 data files entitled “all_thresholded_by_gene.txt” of each tumor type (15). The high-level SCNAs represent events where focal CN gain (or loss) are higher (or lower) than the maximum (or minimum) median arm-level CN gain (or loss), hence indicate more conservative thresholds and are less likely to be artifacts or false positives compared to low-level SCNAs given the purity and ploidy of tumor samples. Acute myeloid leukemia, diffuse large B-cell lymphoma, and thymoma were excluded because of high tumor cell-intrinsic immune cell transcripts. Skin cutaneous melanoma had both primary and metastatic samples available hence counted as two histologies, whereas the other 29 cancers had only primary tumors available. A total of 9,555 tumor samples and 742 normal samples were initially downloaded. After excluding the three tumor types, 9244 tumors and 683 matched normal were included in the analysis.
Identification of non-T cell-inflamed and T cell-inflamed tumor groups
Using a defined T-cell inflamed gene expression signature consisting of 160 genes (“concordant gene list”, described in (16)), the tumor samples were categorized into three groups: non–T-cell inflamed, T-cell inflamed, and intermediate following previous protocols (16). In brief, a quantitative scoring system was developed to categorize tumors into three groups: non-T cell-inflamed, T cell-inflamed, and intermediate, based on the expression profile of the T cell-inflamed gene expression signature. First, gene expression values were converted to a score Si = μi ± βiσi (i = 1,2, … n), where μ and σ represent the mean and standard deviation (sd) of the i th gene’s expression across all samples, respectively. n is the total number of genes. β represents the distance between the i th gene’s expression in a sample and its mean in the unit of sd (equivalent to a z-score). In this system, the threshold for non-T cell-inflamed and T cell-inflamed tumors was β0 = 0.1. For each gene, if its coefficient βi > β0, then this gene is concluded as ‘upregulated’ in this sample and assigned score as +1; likewise, if its coefficient βi < −β0, then this gene is concluded as ‘downregulated’ in this sample and assigned score as −1; otherwise, this gene is concluded as ‘unchanged’ and assigned score as 0. Next, the summation of the score across all 160 genes from the signature was computed to determine the group of a patient. As each gene score has values of −1, 0, or +1, the summation of all gene scores ranges from −160 (if all genes are downregulated) to +160 (if all genes are upregulated), where tumors of < −80 score summation were categorized as non-T cell-inflamed, > +80 score summation as T-cell inflamed, and the rest as intermediate.
Differential gene expression detection and pathway activation prediction
Within each individual cancer, genes significantly differentially expressed between non-T cell-inflamed and T cell-inflamed tumor groups were identified by Linear Models for Microarray and RNA-Seq Data (limma) voom algorithm with precision weights (17), filtered by FDR-adjusted p<0.05, and fold change ≥ 1.5 or ≤ −1.5. Pathways significantly altered by the differentially expressed genes (DEGs) were detected by IPA® (QIAGEN Inc., Germany) using the curated Ingenuity Knowledge Base (accessed November 2015). Upstream transcriptional regulators and their change of direction were predicted by the cumulative effect of target molecules up or downregulated under condition (18). Those of overlap p<0.05 (measuring the enrichment of target molecules in the dataset) were selected for further analysis. For each individual tumor sample, a β-catenin pathway activation score was calculated requiring at least 50% of the cancer-specific target molecules (Supplementary Table 2) to be upregulated (relative to its median expression across all samples from this individual cancer) in non-T cell-inflamed tumor group relative to inflamed. For cancers where the total number of target molecules is less than 10, at least 5 molecules are required to be upregulated in order to determine a patient from this cancer to be activated; for cancers where the total number of target molecules is less than 5, all molecules must be upregulated in order to conclude that a patient is activated.
Estimation of stroma and immune cell enrichment from RNAseq data
The relative enrichment of 64 stroma and immune cell types in tumor microenvironment was estimated by xCell (19) (v1.1.0) from the bulk-tissue RNAseq data. For each cell type, xCell assigns enrichment scores across all samples by integrating single-sample gene set enrichment analysis (ssGSEA) and deconvolution methods. All TCGA tumors were processed by xCell at once to compute per-sample stroma score and immune score, and then compared between β-catenin activated (activation score > 0.5, high) and non-activated samples (activation score ≤ 0.5, low) by non-parametric Mann-Whitney U test with FDR-correction for multiple comparisons.
RPPA data analysis
Level 3 reverse phase protein array (RPPA) antibody-level protein abundance data (release date January 28, 2016; patch July 14, 2016) produced by MD Anderson Cancer Center were downloaded from The Cancer Genome Atlas (TCGA) preprocessed by Broad’s TCGA team (https://gdac.broadinstitute.org/) (accessed April 26, 2018). β-catenin protein level was estimated using median-centered normalized values corresponding to antibody ‘beta-Catenin’ from the data files without gene annotation. The gene-annotated RPPA data files were not used in this analysis due to potential gene symbol mismatch for beta-catenin. Within each tumor type, a one-sided Pearson’s correlation (R function cor.test, alternative = “less”) test was performed within each tumor type between β-catenin protein level and T-cell inflamed gene expression from normalized RNA-seq data, followed by FDR-correction for multiple testing.
Statistical analysis
For analysis of contingency tables including comparison of sample frequency between groups activated by β-catenin pathway, Fisher’s exact test was used. Gene expression comparison between groups were performed using linear regression models implemented in limma voom. For multiple comparisons, p-value was adjusted using Benjamini-Hochberg FDR correction for multiple testing (20). Pearson’s correlation r was used for measuring statistical dependence between normalized and log2-transformed expression level of different genes. p<0.05 was considered statistically significant and denoted as follows: * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Statistical analysis was performed using R and Bioconductor.
Results
Solid tumor histologies can be segregated based on the degree of the T cell-inflamed tumor microenvironment gene expression signature
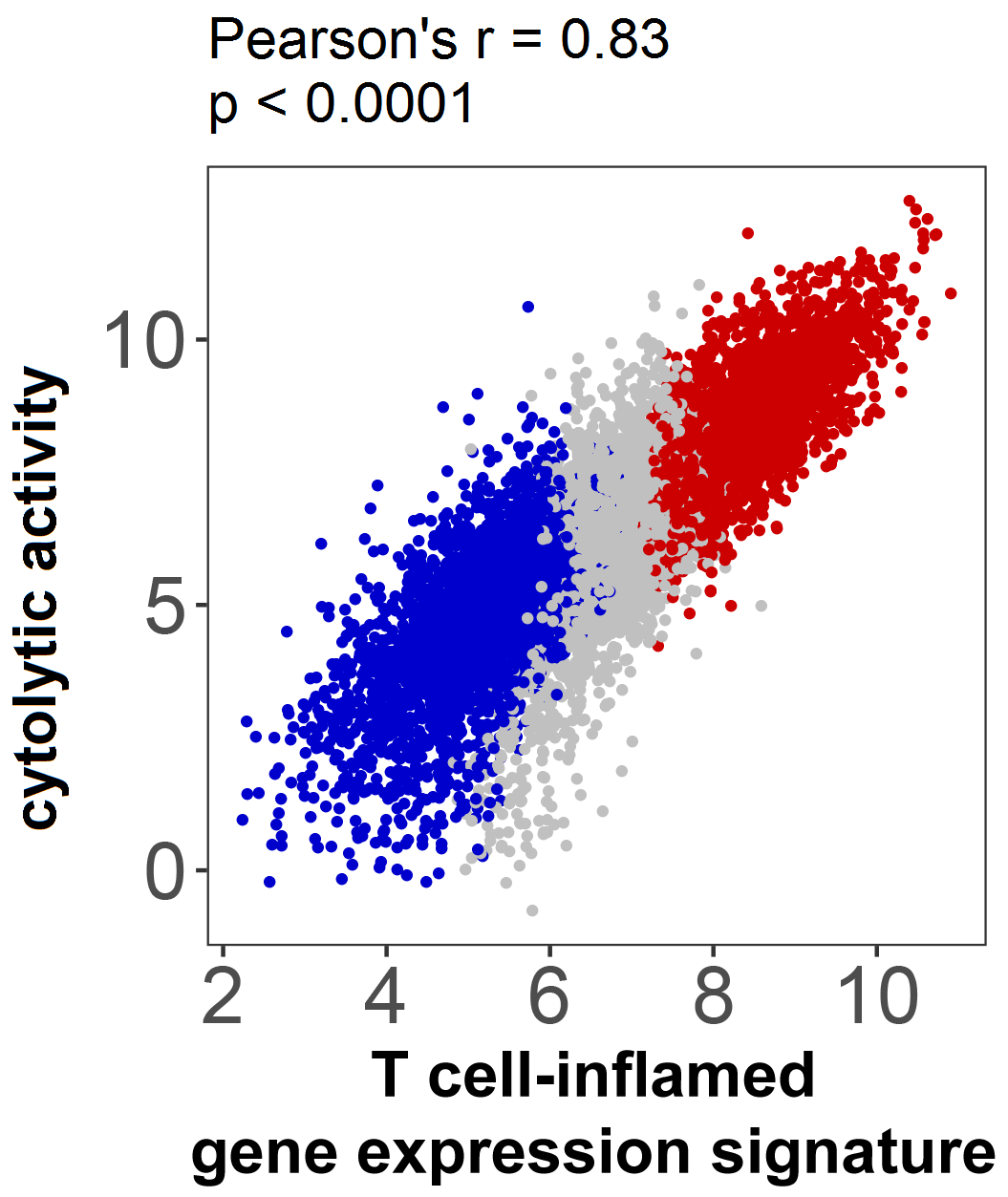
Strong associations have been observed between the presence of T cell-infiltration and/or a T cell-inflamed gene expression signature and response to immune-checkpoint blockade(10,21). These data imply that a non-T cell-inflamed tumor microenvironment is associated with therapeutic resistance, and understanding the molecular basis of this phenotype should point towards candidate targets for expanding efficacy(10). To assess the landscape of the non-T cell-inflamed tumor microenvironment across human cancers, we analyzed the RNA-seq data from 31 solid tumors contained within The Cancer Genome Atlas (TCGA) (Supplementary Table 1). We segregated samples into T cell-inflamed, intermediate, or non-T cell-inflamed using a T cell-inflamed filtration score based on categorical definition (–1, 0, 1) of each gene from our previously defined 160-gene T cell-inflamed expression signature (see Methods for further details)(16). This gene expression signature shows a high level of correlation with a previously published immune cytolytic activity score used in other studies(22) (Pearson’s r=0.83, p=3.57e-09; Supplementary Figure 1).
Combining all cancer types within TCGA, 3137/9244 (33.9%) samples showed a non-T cell-inflamed gene expression signature, with 2946/9244 (31.9%) scoring as intermediate and 3161/9244 (34.2%) displaying the T cell-inflamed phenotype (Supplementary Figure 2). Cancer types were ranked by the fraction of samples showing the non-T cell-inflamed tumor microenvironment per tumor type (Figure 1A). Paraganglioma, uveal melanoma, and adrenocortical tumors showed the highest frequency of non-T cell-inflamed tumors, whereas lung adenocarcinoma, mesothelioma, and clear cell kidney cancer showed the lowest frequency of non-T cell-inflamed samples. Relevant to clinical practice, this pattern approximates the activity of anti-PD-1/PD-L1 monotherapy in which little if any activity has been observed in uveal melanoma(23), whereas substantial clinical activity has been demonstrated in lung cancer, kidney cancer, and mesothelioma(24–26).
Figure 1. Landscape of T cell inflammation across 31 human solid tumors.

(A) The fraction of non-T cell-inflamed (blue), intermediate (grey) and T cell-inflamed (red) tumors in each cancer, sorted by the fraction of non-T cell-inflamed tumor high to low, with the most non-inflamed cancer on the left (Paraganglioma) and the most inflamed cancer on the right (Kidney, clear cell). The number of samples in each cancer is shown on the x-axis. (B) Distribution of T cell-inflamed gene expression in non-T cell-inflamed (blue), intermediate (grey) and T cell-inflamed (red) tumors, as well as in normal samples (green). Cancers are shown on the x-axis in the same order as in (A), and only 14 cancers with ≥ 10 normal samples are shown. The expression level of the T cell-inflamed gene expression signature (defined as, the average expression of genes from the signature in a sample) is shown on the y-axis. Each data point represents one tumor or normal sample.
It was of interest to assess whether the T cell-inflamed gene expression signature was a phenotype that was gained when comparing cancer tissue to normal tissue, or whether the non-T cell-inflamed phenotype represented a loss of immune cell infiltration that is a component of normal tissue homeostasis. In most cases, loss of immune phenotype was observed in the malignant tissue. In tumor types of TCGA where adjacent normal tissue was available, non-T cell-inflamed tumors generally showed a significantly lower T cell-inflamed gene expression relative to normal tissue controls (p=1.10e-111, two-sided Welch Two Sample t-test) and the T cell-inflamed tumors showed a significantly higher T cell-inflamed gene expression relative to normal tissue (p=2.23e-106). Within each individual tumor type, a similar pattern was observed for most cancers (Figure 1B) with the exception of the three types of kidney cancer (chromophobe, clear cell and renal papillary cell carcinoma), where those normal tissues showed a low T cell-inflamed gene expression that was indistinguishable from that of non-T cell-inflamed tumors (p >0.05, ranges from 0.07 to 0.62). Though generally responsive to anti-PD-1 immunotherapy, the immunobiology of clear cell kidney cancer may vary from other tumor types based on significant clinical activity despite low mutational density or neoantigen load(27), and the unexpected negative prognostic value of a higher CD8+ T cell density at baseline(28).
Having previously identified activated WNT/β-catenin signaling in 48% of non-T cell-inflamed metastatic melanoma samples(11), we investigated whether activation of this pathway was associated with the non-T cell-inflamed phenotype in other cancer histologies. β-catenin pathway activation was addressed on three levels: somatic mutations or copy number alterations in CTNNB1 and other regulators predicted to yield pathway activation, expression of downstream target genes indicative of β-catenin signaling, and β-catenin protein levels as assessed by reverse phase protein array (RPPA).
Somatic mutations of CTNNB1 and upstream repressors are enriched in non-T cell-inflamed tumors
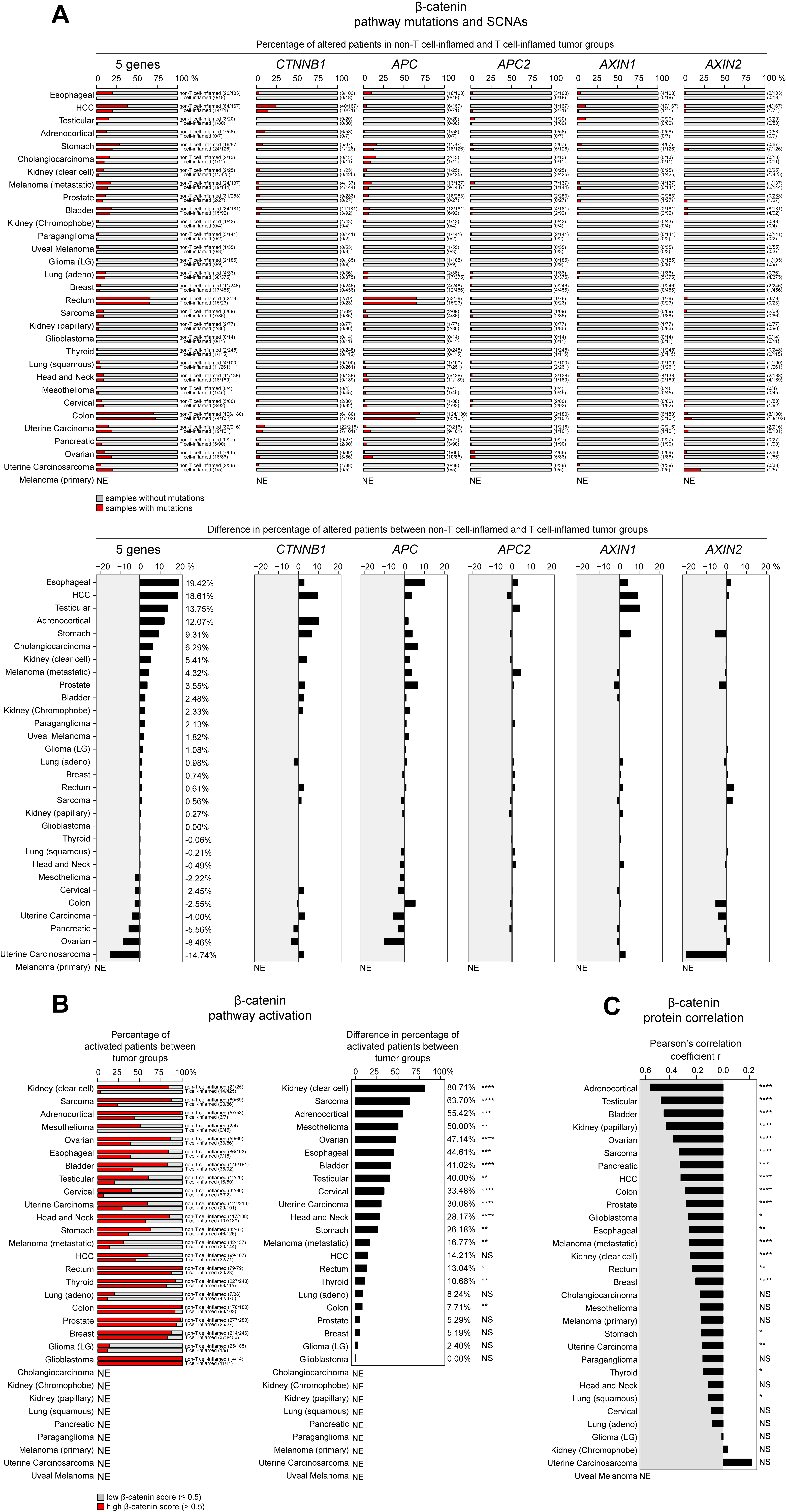
To interrogate tumors for β-catenin pathway activation, non-synonymous mutations (NSSMs) across TCGA samples in the CTNNB1, APC, APC2, AXIN1, and AXIN2 genes were identified and investigated for abundance within the non-T cell-inflamed versus the T cell-inflamed cohorts. This analysis was restricted to defined mutations predicted to mediate pathway activation. For CTNNB1, we defined relevant mutations as those affecting exon 3 (amino acid position 29 to 49) corresponding to the phosphorylation site by GSK3β which normally leads to proteolytic degradation of β-catenin (Figure 2A). Across the aggregate TCGA cohort, we observed a statistically significant three-fold enrichment (p<0.0001, two-sided Fisher’s exact test) of these mutations in non-T cell-inflamed versus T cell-inflamed tumors (Figure 2B, black bars). Mutations were most commonly missense mutations effecting serine or threonine residues as previously published(29). For other β-catenin pathway genes we included potential damaging mutations such as nonsynonymous or stopgain single nucleotide variants (SNVs), variants that affect splicing site, as well as frameshift/non-frameshift small insertions and deletions (indels). We additionally analyzed high-level somatic copy number (CN) gain for CTNNB1 and CN loss for APC, APC2, AXIN1, and AXIN2 (defined as +/−2 in GISTIC2 data files; see Methods). Similarly to as activation mutations, CTNNB1 CN gain occurred at a higher frequency in non-T cell-inflamed tumors than T cell-inflamed tumors (Figure 2B, grey bars).
Figure 2. Somatic mutation and high-level SCNA profile in β-catenin signaling in non-T cell-inflamed and T cell-inflamed tumor groups.

(A) Overview of potential coding somatic mutations in CTNNB1 predicted to cause protein sequence changes. Each circle represents each amino acid change, with the height of the connection line represents the total number of samples carrying this mutation. Color of the circle represents mutation type, with missense mutations in green (nonsynonymous SNVs), truncating mutations in red (nonsense SNVs, frameshift deletions/insertions, splicing site variants), and in-frame mutations in blue (in-frame insertions/deletions/substitutions). Sites affected by multiple mutation types are colored in purple. Silent mutations (synonymous SNVs) are not shown. Protein domains are shown as dark grey boxes on the protein schema, HEAT_2 = HEAT repeats, Arm = Armadillo/beta-catenin-like repeat as defined in Pfam database. (B) Fraction of non-T cell-inflamed and T cell-inflamed tumors carrying β-catenin pathway activation mutations in exon 3 or high-level copy number gain. The number above each bar represents the number of altered samples in each group. (C) Landscape of activation mutations (exon 3) or high-level copy number gain in CTNNB1, and damaging mutations or high-level copy number loss in APC, APC2, AXIN1 and AXIN2 across non-T cell-inflamed tumors. The percentage of samples carrying mutations or SCNAs in each gene is presented to the left side, and the sample number is shown to the right side. Color represents predicted functional consequence of somatic mutations (red: activation, green: damaging) or SCNAs (gold: high-level CN gain, blue: high-level CN loss). CN = copy number. (D) Difference in the percentage of β-catenin mutated or copy number altered patients between non-T cell-inflamed and T cell-inflamed tumor groups per cancer. Cancers are shown to the left side and sorted by percentage difference in patients carrying mutations between tumor groups (top to bottom: larger to smaller percentage differences). 19 cancers with higher percentage of β-catenin mutated patients in non-T cell-inflamed tumor group relative to T cell-inflamed group are shown.
Across non-T cell-inflamed tumors we observed 15.21% of samples as harboring mutations predicted to activate β-catenin signaling, somatic CN alterations (SCNAs) in pathway genes or both (Figure 2C; Supplementary Figure 3A–B) as opposed to 10.06% in T cell-inflamed tumors (Supplementary Figure 3C–E). These mutations and high-level SCNAs were predominately non-overlapping and were dominated by activating mutations in CTNNB1 and damaging mutations in APC. Beyond exon 3, we identified 18 activation mutations located in exon 6, 7 or 8 based on known experimental evidence reported from the literature that occur on amino acid position 292 (30), 335 (31), 383 (30), and 387 (31) (Supplementary Figure 4). Consistent with those disrupting GSK3β-phosphorylation sites in exon 3, those additional mutations showed a strong anti-correlation with the T cell-inflamed expression signature (10 in non-T cell-inflamed, 7 in intermediate, and 1 in T cell-inflamed group), potentially by reducing APC/AXIN1/AXIN2 binding or affecting ubiquitin-mediated protein degradation pathways. We acknowledge our analysis is likely an underestimated assessment of somatic mutations that drive β-catenin signaling activation, considering that mutations in genes other than CTNNB1, APC, APC2, AXIN1, and AXIN2 may also be contributors (32,33). Also, it is possible that SCNAs could be influenced by the presence of non-tumor cells in the tissue. Across individual tumors, 19 cancer types showed a higher fraction of β-catenin pathway mutations, high-level SCNAs or both associated with non-T cell-inflamed as compared with T cell-inflamed tumors (Figure 2D; Supplementary Figure 5). Additionally, we compared the expression of the T cell-inflamed gene expression signature with the fraction of β-catenin pathway altered samples (mutations or high-level SCNA changes in CTNNB1, APC, APC2, AXIN1, and AXIN2) per tumor type (Supplementary Figure 6). We found a weak and statistically in-significant correlation across TCGA and within specific tumor types, suggesting that direct genomic activation of β-catenin by mutation or high-level SCNAs may not be the main driver of non-T cell-inflamed phenotype. However, the results should be interpreted with caution considering the limited sample size for β-catenin pathway mutations or high-level SCNAs within each tumor type.
Activation of a β-catenin transcriptional program correlates with the non-T cell-inflamed tumor microenvironment across tumor types
To describe the frequency of β-catenin pathway activation at the level of target gene expression, we identified genes significantly differentially expressed in non-T cell-inflamed tumors relative to T cell-inflamed, and that overlapped with known downstream target molecules of CTNNB1 as upstream regulator from the curated Ingenuity Knowledge Base™ (QIAGEN Inc., Germany), yielding a β-catenin activation score for each patient. Downstream target molecules in this analysis are collated from the literature as being regulated by CTNNB1 (18). We chose to accept a β-catenin activation score of > 0.5 and scored individual samples as β-catenin activated or not activated. The percentage of activated samples in non-T cell-inflamed versus T cell-inflamed was calculated for each tumor type. Of the 31 solid tumors from TCGA, 14 tumor types met the definition of WNT/β-catenin activation by pathway analysis and demonstrated a greater proportion of samples with β-catenin activation in the non-T cell-inflamed versus T cell-inflamed (FDR-adjusted p<0.05, two-sided Fisher’s exact test; Figure 3A and B).
Figure 3. β-catenin activation in non-T cell-inflamed and T cell-inflamed tumor groups.

14 cancers with significantly higher percentage of β-catenin activated patients in non-T cell-inflamed tumor group relative to T cell-inflamed group and at least 5 target molecules are shown (also listed in Supplementary Table 2). (A) The percentage of β-catenin activated patients in each tumor group per cancer (β-catenin activation score > 0.5), with the subset of tumors with β-catenin signaling activated and total number of tumor samples shown to the right side of each bar inside parentheses. (B) Difference in the percentage of β-catenin activated patients between non-T cell-inflamed and T cell-inflamed tumor groups per cancer. In both panels, cancers are shown to the left side and sorted by percentage difference in the activated patients between tumor groups (top to bottom: larger to smaller percentage differences). The significance of such difference was computed using Fisher’s exact test, two-sided. p < 0.05 was considered statistically significant, and significance is indicated by: * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, after FDR-correction for multiple comparisons.
The individual target molecules identified by pathway analysis varied by tumor type though some were shared between tumors (Supplementary Table 2). A total of 201 β-catenin target molecules were identified in at least one tumor type with the top shared molecules in Supplemental Figure 7 showing those shared by at least 3 tumor types for CTNNB1 as upstream regulator. Representative shared molecules across clear cell kidney, ovarian and stomach cancer include BMP7, WNT11, and TDGF1 which each demonstrates statistically significant inverse correlations with the T cell-inflamed gene expression signature and upregulation of expression in non-T cell-inflamed tumors (Supplemental Figure 8). In particular, shared WNT molecules, such as WNT11, were found to be significantly upregulated in kidney renal papillary cell carcinoma, ovarian serous cystadenocarcinoma, stomach adenocarcinoma, and testicular germ cell tumors, suggesting that WNT11 may be a candidate therapeutic target in those cancer types (Supplemental Figure 9).
To evaluate the potential contribution of stroma cells on the β-catenin pathway activation score, we analyzed all TCGA tumors at once to compute per-sample stroma score and immune score (19) from the RNAseq data, and then compared the enrichment scores between β-catenin activated (activation score > 0.5, high) and non-activated samples (activation score ≤ 0.5, low) (Supplemental Figure 10). From the 14 tumor types where WNT/β-catenin activation by pathway analysis was observed, significant stroma score differences were detected in 3 tumor types (esophageal carcinoma, ovarian serous cystadenocarcinoma, testicular germ cell tumors) at FDR-adjusted p<0.05 (Supplemental Figure 10, upper panel). Esophageal carcinoma showed reduced stroma score in the β-catenin activated samples, however, the other two tumor types showed the opposite trend. In contrast, all 14 tumor types consistently showed a reduction in the immune score comparing the β-catenin activated to non-activated samples, 13 of which reached the significance level of 0.05, with esophageal carcinoma at p=0.080 (Supplemental Figure 10, bottom panel). Taken together, those results do not appear to support a dominant role for the contribution of stromal cells to β-catenin pathway activation across tumor types.
To assess whether somatic mutations of β-catenin pathway genes were associated with upregulated expression of downstream target genes, we overlapped samples carrying β-catenin pathway gene mutations or high-level SCNAs with samples with a high β-catenin activation score. From the 14 tumor types where WNT/β-catenin activation by pathway analysis was observed, we identified 291/348 (83.6%) as mutated or copy number altered and activated in non-T cell-inflamed as compared with 127/225 (56.4%) in T cell-inflamed tumors (Supplemental Figure 11). This demonstrates a significantly higher concordance between mutated samples and activated samples with a low level of T cell inflammation (p<0.0001, odds ratio=3.93, two-sided Fisher’s exact test).
β-catenin protein level inversely correlates with the T cell-inflamed tumor phenotype across tumor types
β-catenin staining by immunohistochemistry has previously been shown to inversely correlate with staining for CD8+ T cells in melanoma and bladder cancer(11,34). It was therefore of interest to analyze available RPPA data for β-catenin protein levels from TCGA and examine a potential inverse correlation with the T cell-inflamed gene expression signature. Indeed, taking the aggregate data from all 31 TCGA tumors, a statistically significant inverse correlation was observed between β-catenin protein abundance and the T cell-inflamed gene expression (p<0.0001). Among individual cancer types, this inverse correlation was observed for 20 cancers subtypes at a significance level of FDR-adjusted p<0.05 (Figure 4A). Representative tumor types including bladder (Pearson’s r= −0.44, p<0.0001), kidney renal papillary cell carcinoma (Pearson’s r= −0.42, p<0.0001), ovarian serous cystadenocarcinoma (Pearson’s r= −0.37, p<0.0001) and hepatocellular carcinoma (Pearson’s r= −0.32, p<0.0001) are shown in Figure 4B. All individual tumor types are included in Supplementary Figure 12. Note that adrenocortical carcinoma showed the strongest anti-correlation (Pearson’s r= −0.55) but was not included in Figure 4B due to relatively smaller sample size (46 samples with both RNA-seq and RPPA data available). Results for all analyses across all tumor types investigated above are shown as Supplementary Figure 13.
Figure 4. Inverse correlation between β-catenin protein level and T cell-inflamed gene expression.

(A) Pearson’s correlation per tumor type. 20 cancers with significant inverse correlation are shown. (B) Dot plots of β-catenin protein on Y axis and T cell-inflamed gene expression on X axis in bladder urothelial carcinoma, kidney papillary, ovarian serous cystadenocarcinoma, liver hepatocellular carcinoma (HCC) shown with linear regression correlations using Pearson’s test. The significance of such difference was computed using Pearson’s correlation, one-sided test. p < 0.05 was considered statistically significant, and significance is indicated by: * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, after FDR-correction for multiple comparisons.
Conclusions/Discussion
In our current analysis of the T cell-inflamed versus non-T cell-inflamed tumor microenvironment across the solid tumors as represented in TCGA, we observed that 33.9% of tumors were non-T cell-inflamed, with most of these tumors showing lower immune gene expression compared to matched normal tissue. This observation suggests that the lack of T cell-based tissue inflammation is a frequent occurrence in primary cancers and that loss of this biologic process could be a component of carcinogenesis. The development of the non-T cell-inflamed tumor microenvironment therefore might be regulated by oncogenic events. Analysis of molecular evidence for WNT/β-catenin pathway activation as one potentially causal pathway revealed that as many as 28/31 (90%) of tumor types across TCGA showed an inverse correlation with a T cell-inflamed gene expression signature. We inferred β-catenin pathway activation using three approaches: 1) somatic pathway mutations or high-level SCNAs, where 19 cancers showed a higher proportion of mutated samples in non-T cell-inflamed group relative to inflamed; 2) pathway target gene expression, where 14 cancers showed predicted β-catenin activation based on upregulation of known target molecules in non-T cell-inflamed group relative to inflamed 3) β-catenin protein level by RPPA, where 20 cancers showed significant anti-correlation between β-catenin protein level and T-cell inflamed gene expression. As a result, we categorized cancer subtypes into tier 1 to 3, with increasing support combining the three approaches (Table 1). In summary, we conclude that WNT/β-catenin pathway activation is frequently associated with poor spontaneous T cell infiltration across most human cancers.
Table 1.
Summary of evidence for β-catenin signaling activation non-T cell-inflamed tumors from the 31 cancer subtypes using three approaches.
| Cancer | Name | Joint evidence of activation in non-T cell-inflamed tumors | Somatic mutation or high-level SCNA enrichment | Pathway gene expression activation | Higher CTNNB1 protein level |
|---|---|---|---|---|---|
| ACC | Adrenocortical | Tier 3 | Y | Y | Y |
| BLCA | Bladder | Tier 3 | Y | Y | Y |
| ESCA | Esophageal | Tier 3 | Y | Y | Y |
| KIRC | Kidney (clear cell) | Tier 3 | Y | Y | Y |
| SARC | Sarcoma | Tier 3 | Y | Y | Y |
| SKCMmets | Melanoma (metastatic) | Tier 3 | Y | Y | Y |
| READ | Rectum | Tier 3 | Y | Y | Y |
| STAD | Stomach | Tier 3 | Y | Y | Y |
| TGCT | Testicular | Tier 3 | Y | Y | Y |
| BRCA | Breast | Tier 2 | Y | N | Y |
| COAD | Colon | Tier 2 | N | Y | Y |
| KIRP | Kidney (papillary) | Tier 2 | Y | N | Y |
| LIHC | HCC | Tier 2 | Y | N | Y |
| OV | Ovarian | Tier 2 | N | Y | Y |
| PRAD | Prostate | Tier 2 | Y | N | Y |
| UCEC | Uterine Carcinoma | Tier 2 | N | Y | Y |
| CESC | Cervical | Tier 1 | N | Y | N |
| CHOL | Cholangiocarcinoma | Tier 1 | Y | N | N |
| GBM | Glioblastoma | Tier 1 | N | N | Y |
| HNSC | Head and Neck | Tier 1 | N | Y | N |
| KICH | Kidney (Chromophobe) | Tier 1 | Y | N | N |
| LGG | Glioma (LG) | Tier 1 | Y | N | N |
| LUAD | Lung (adeno) | Tier 1 | Y | N | N |
| LUSC | Lung (squamous) | Tier 1 | N | N | Y |
| PAAD | Pancreatic | Tier 1 | N | N | Y |
| PCPG | Paraganglioma | Tier 1 | Y | N | N |
| THCA | Thyroid | Tier 1 | N | N | Y |
| UVM | Uveal Melanoma | Tier 1 | Y | N | NE[3] |
| MESO | Mesothelioma | No tier | N[1] | N | N |
| SKCMprimary | Melanoma (primary) | No tier | NE[2] | N | N |
| UCS | Uterine Carcinosarcoma | No tier | N | N | N |
Mesothelioma have GDAC-Firehose preprocessed level 3 copy number data available; somatic mutation data were not available.
Melanoma primary tumors do not have GDAC-Firehose preprocessed level 3 somatic mutation or copy number data available, hence shown as non-evaluable (NE).
Uveal melanoma has limited number of RPPA samples (n=12), hence the correlation between T cell-inflamed gene expression signature and its β-catenin protein level was non-evaluable (NE).
Y: presence of evidence. N: lack of evidence.
Of note, the three methods for analyzing WNT/β-catenin pathway activation did not give exactly concordant results. It is formally possible that some of the signal of both gene expression and β-catenin protein level may be derived from stromal cells in addition to cancer cells. Activating mutations in the β-catenin gene itself, as well as inactivating mutations in negative regulators of the β-catenin pathway, are likely restricted to the tumor cells. But all cell types theoretically have the potential to activate β-catenin signaling, including T cells and other immune cells. The non-T cell-inflamed tumors that lack CD8+ T cells also have few dendritic cells, so it is unlikely that increased β-catenin activation in those tumors is related to adaptive immunity. However, we observed limited contribution of stromal cells when comparing a stroma score between β-catenin pathway activated and non-activated samples. Given that the most abundant cell type in these tumors is very likely the tumor cells themselves, these data support tumor-intrinsic β-catenin as a major correlate of the non-T cell-inflamed tumor microenvironment. Future biomarker development to document the degree of β-catenin pathway activation should incorporate somatic mutations and SCNAs of the pathway coupled with immunohistochemical analysis of CD8+ T cells along with stabilized β-catenin protein levels within tumor cells.
Identification of molecular factors associated with the non-T cell-inflamed tumor microenvironment has important relevance for future development of combination immunotherapy regimens. Mechanistic studies using genetically-engineered mice have demonstrated that tumor cell-intrinsic β-catenin activation was sufficient to prevent spontaneous T cell priming and infiltration into the tumor microenvironment, rendering those tumor-bearing mice resistant to combination checkpoint blockade therapy(11,12). The molecular mechanism of this effect was mapped to defective recruitment and activation of Batf3-lineage dendritic cells, although additional mechanisms also may contribute. The lack of intra-tumoral Batf3 dendritic cells in β-catenin-expressing melanomas also rendered those tumors resistant to vaccination and adoptive T cell transfer, arguing that this type of molecular aberration also could be relevant to other therapeutic modalities being explored clinically(11,12).
The WNT/β-catenin pathway, therefore, should be a high priority molecular target for new drug development, in an effort to restore T cell infiltration and potentially expand immunotherapy efficacy in the clinic. Several specific inhibitors have been developed and brought forward into early phase clinical trials; however, none have yet had sufficient monotherapy activity to advance further into registration trials(35). β-catenin signaling is broadly utilized by many cell types, and on-target, off-tumor effects likely limit the potential therapeutic window. However, for cancer applications, most agents developed to date have likely been screened for the ability to slow cancer cell proliferation and/or induce cancer cell apoptosis. The immune evasive mechanisms of β-catenin activation, including suppression of chemokines and cytokine gene expression by tumor cells(11), may offer an opportunity to develop agents with more restricted activity that predominantly augment immunity while sparing other essential cellular functions.
In addition to WNT/β-catenin signaling as a major driver of immune exclusion, data are accumulating that other oncogenic events also can contribute to immune exclusion (36,37). Loss of PTEN in melanoma has been associated with the non-T cell-inflamed tumor microenvironment, and in that context inhibition of phosphoinositide 3-kinase (PI3K) signaling synergized with immune-checkpoint blockade(38). PTEN/PI3K signaling has also been correlated with resistance to immunotherapy from a patient with uterine leiomyosarcoma who had previously had a complete response for two years prior to losing benefit from anti-PD-1 therapy(39). In bladder cancer, activating mutations in FGFR3, as well as molecular evidence for PPAR-γ activation, have been associated with the non-T cell-inflamed tumor microenvironment(34). Inasmuch as FGFR inhibitors are currently in clinical development this may be the first pathway targeted to entertain the hypothesis that blockade of an immune-evasive oncogene pathway may improve immunotherapy efficacy in human cancer patients, as these agents will be combined with PD-1/PD-L1 blocking Abs as well (ClinicalTrials.gov Identifier: NCT02393248). Additionally, some tumors may possess a complicated interplay of multiple oncogenic events that synergize to mediate more potent immune exclusion, as has been reported for colorectal cancers showing activation of WNT/β-catenin, MYC and RAS(33,40,41).
We acknowledge that our analysis may have some limitations. This current work was based on in silico analysis of TCGA. While the T cell-inflamed tumor microenvironment has been shown to predict response to multiple immunotherapies (as well as lack of response in non-T cell inflamed tumors)(5,10), the clinical outcomes of individual patients from TCGA were deliberately not encompassed in our analysis. Such a study is best performed prospectively in cohorts of patients receiving systemic therapy with the same immunotherapeutic agent, e.g. anti-PD-1. Our study is also limited in that the range of potential somatic genomic changes in the β-catenin pathway was restricted to point mutations and small insertions and deletions, as well as copy number variants, while other types of genomic aberrations such as large-scale structural variants or gene fusions remain to be explored. Nonetheless, this set of data provides sufficient motivation to study baseline WNT/β-catenin pathway activation as a potential primary resistance pathway to checkpoint blockade in prospective clinical trials in defined patient cohorts.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Statement of Translational Relevance:
Tumors can be categorized by gene expression based on the presence or absence of a T cell-inflamed tumor microenvironment and this phenotype correlates with response to immune-checkpoint blockade. WNT/β-catenin signaling in melanoma was the first tumor cell-intrinsic oncogenic signaling pathway linked to an immune exclusion phenotype. Here we have profiled the correlation of WNT/β-catenin signaling and the T cell-inflamed tumor microenvironment across The Cancer Genome Atlas. We have analyzed activated WNT/β-catenin signaling by somatic mutations, copy number alterations, gene expression, and reverse phase protein array showing a pan-cancer association of this signaling pathway with immune-exclusion. These data highlight a high priority unmet need for the development of selective Wnt/β-catenin pathway inhibitors as rational combination partners for immune-checkpoint blockade and identify tumor types for priority investigation.
Acknowledgments
Funding Support: JL: Department of Defense Career Development Award (W81XWH-17-1-0265), the Arthur J Schreiner Family Melanoma Research Fund, the J. Edward Mahoney Foundation Research Fund, Brush Family Immunotherapy Research Fund and Buffet Fund for Cancer Immunotherapy with support from The Center for Research Informatics by the Biological Sciences Division at the University of Chicago with additional support provided by The Institute for Translational Medicine/Clinical and Translational Award (NIH 5UL1TR002389–02), and The University of Chicago Comprehensive Cancer Center Support Grant (NIH P30CA014599). The bioinformatics analysis was performed on High-Performance Computing clusters at Center for Research Informatics, Biological Sciences Division. We thank M. Jarsulic for technical assistance on the HPC clusters. SS was a postdoctoral fellow of the Cancer Research Institute. TG: American Cancer Society-Jules L. Plangere Jr. Family Foundation Professorship in Cancer Immunotherapy, and R35 CA210098 from the NIH.
Footnotes
Disclosures: JL: DSMB: TTC Oncology; SAB: 7 Hills, Actym, Alphamab Oncology, Array, BeneVir, Mavu, Tempest; Consultancy: Aduro, Astellas, AstraZeneca, Bayer, Bristol-Myers Squibb, Castle, CheckMate, Compugen, EMD Serono, IDEAYA, Immunocore, Janssen, Jounce, Merck, NewLink, Novartis, RefleXion, Spring Bank, Syndax, Tempest, Vividion, WntRx; Research Support: (clinical trials unless noted) AbbVie, Array (Scientific Research Agreement; SRA), Boston Biomedical, Bristol-Myers Squibb, Celldex, CheckMate (SRA), Compugen, Corvus, EMD Serono, Evelo (SRA), Delcath, Five Prime, FLX Bio, Genentech, Immunocore, Incyte, Leap, MedImmune, Macrogenics, Novartis, Pharmacyclics, Palleon (SRA), Merck, Tesaro, Xencor
Travel: Array, AstraZeneca, Bayer, BeneVir, Bristol-Myers Squibb, Castle, CheckMate, EMD Serono, IDEAYA, Immunocore, Janssen, Jounce, Merck, NewLink, Novartis, RefleXion
Patents: (both provisional) Serial #15/612,657 (Cancer Immunotherapy), PCT/US18/36052 (Microbiome Biomarkers for Anti-PD-1/PD-L1 Responsiveness: Diagnostic, Prognostic and Therapeutic Uses Thereof)
RB declares no disclosures or conflicts of interest
RS has received consulting fees or honoraria from Bristol-Myers Squibb, Eisai, Exelixis, AstraZeneca, Puma and has research support from Bayer, Bristol-Myers Squibb, Eisai, MIrati, CytomX.
SS is holding a patent on WNT/b-catenin targeting to enhance anti-tumor immune responses (PCT15/155,099), serves on the SAB on Venn Therapeutics, Tango Therapeutics and consults for TAKEDA, Replimune, Ribon, Torque and Arcus.
T.F.G. has received consultancy fees from Merck, Roche-Genentech, Abbvie, Bayer, Jounce, Aduro, Fog Pharma, Adaptimmune, FivePrime, and Sanofi. T.F.G. has received research support from Roche-Genentech, BMS, Merck, Incyte, Seattle Genetics, Celldex, Ono, Evelo, Bayer, Aduro. T.F.G. has intellectual property/licensing agreements with Aduro, Evelo, and BMS. T.F.G is a co-founder/shareholder with Jounce.
References:
- 1.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. The New England journal of medicine 2012;366(26):2443–54 doi 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. The New England journal of medicine 2010: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. The New England journal of medicine 2015. doi 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 4.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. The New England journal of medicine 2015;373(1):23–34 doi 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer research 2009;69(7):3077–85 doi 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gajewski TF, Louahed J, Brichard VG. Gene signature in melanoma associated with clinical activity: a potential clue to unlock cancer immunotherapy. Cancer J 2010;16(4):399–403 doi 10.1097/PPO.0b013e3181eacbd8. [DOI] [PubMed] [Google Scholar]
- 7.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 2013;14(10):1014–22 doi 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol 2013;25(2):268–76 doi 10.1016/j.coi.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 9.Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother 2011. doi 10.1007/s00262-011-1172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. The Journal of clinical investigation 2017;127(8):2930–40 doi 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015;523(7559):231–5 doi 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 12.Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017;31(5):711–23 e4 doi 10.1016/j.ccell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ganesh S, Shui X, Craig KP, Park J, Wang W, Brown BD, et al. RNAi-Mediated beta-Catenin Inhibition Promotes T Cell Infiltration and Antitumor Activity in Combination with Immune Checkpoint Blockade. Molecular therapy : the journal of the American Society of Gene Therapy 2018;26(11):2567–79 doi 10.1016/j.ymthe.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galluzzi L, Spranger S, Fuchs E, Lopez-Soto A. WNT Signaling in Cancer Immunosurveillance. Trends in cell biology 2018. doi 10.1016/j.tcb.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol 2011;12(4):R41 doi 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y, et al. Density of immunogenic antigens does not explain the presence or absence of the T-cell-inflamed tumor microenvironment in melanoma. Proceedings of the National Academy of Sciences of the United States of America 2016;113(48):E7759–E68 doi 10.1073/pnas.1609376113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014;15(2):R29 doi 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kramer A, Green J, Pollard J Jr., Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014;30(4):523–30 doi 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18(1):220 doi 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res 2001;125(1–2):279–84. [DOI] [PubMed] [Google Scholar]
- 21.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515(7528):568–71 doi 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160(1–2):48–61 doi 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Algazi AP, Tsai KK, Shoushtari AN, Munhoz RR, Eroglu Z, Piulats JM, et al. Clinical outcomes in metastatic uveal melanoma treated with PD-1 and PD-L1 antibodies. Cancer 2016;122(21):3344–53 doi 10.1002/cncr.30258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. The New England journal of medicine 2015;373(2):123–35 doi 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. The New England journal of medicine 2015;373(19):1803–13 doi 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alley EW, Lopez J, Santoro A, Morosky A, Saraf S, Piperdi B, et al. Clinical safety and activity of pembrolizumab in patients with malignant pleural mesothelioma (KEYNOTE-028): preliminary results from a non-randomised, open-label, phase 1b trial. Lancet Oncol 2017;18(5):623–30 doi 10.1016/S1470-2045(17)30169-9. [DOI] [PubMed] [Google Scholar]
- 27.Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol 2017;18(8):1009–21 doi 10.1016/S1470-2045(17)30516-8. [DOI] [PubMed] [Google Scholar]
- 28.Bromwich EJ, McArdle PA, Canna K, McMillan DC, McNicol AM, Brown M, et al. The relationship between T-lymphocyte infiltration, stage, tumour grade and survival in patients undergoing curative surgery for renal cell cancer. Br J Cancer 2003;89(10):1906–8 doi 10.1038/sj.bjc.6601400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Provost E, Yamamoto Y, Lizardi I, Stern J, D’Aquila TG, Gaynor RB, et al. Functional correlates of mutations in beta-catenin exon 3 phosphorylation sites. J Biol Chem 2003;278(34):31781–9 doi 10.1074/jbc.M304953200. [DOI] [PubMed] [Google Scholar]
- 30.von Kries JP, Winbeck G, Asbrand C, Schwarz-Romond T, Sochnikova N, Dell’Oro A, et al. Hot spots in beta-catenin for interactions with LEF-1, conductin and APC. Nat Struct Biol 2000;7(9):800–7 doi 10.1038/79039. [DOI] [PubMed] [Google Scholar]
- 31.Pilati C, Letouze E, Nault JC, Imbeaud S, Boulai A, Calderaro J, et al. Genomic profiling of hepatocellular adenomas reveals recurrent FRK-activating mutations and the mechanisms of malignant transformation. Cancer Cell 2014;25(4):428–41 doi 10.1016/j.ccr.2014.03.005. [DOI] [PubMed] [Google Scholar]
- 32.Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell reports 2016;15(4):857–65 doi 10.1016/j.celrep.2016.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer discovery 2018;8(6):730–49 doi 10.1158/2159-8290.CD-17-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sweis RF, Spranger S, Bao R, Paner GP, Stadler WM, Steinberg G, et al. Molecular Drivers of the Non-T-cell-Inflamed Tumor Microenvironment in Urothelial Bladder Cancer. Cancer immunology research 2016;4(7):563–8 doi 10.1158/2326-6066.CIR-15-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Voronkov A, Krauss S. Wnt/beta-catenin signaling and small molecule inhibitors. Curr Pharm Des 2013;19(4):634–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gajewski TF, Corrales L, Williams J, Horton B, Sivan A, Spranger S. Cancer Immunotherapy Targets Based on Understanding the T Cell-Inflamed Versus Non-T Cell-Inflamed Tumor Microenvironment. Adv Exp Med Biol 2017;1036:19–31 doi 10.1007/978-3-319-67577-0_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nature Reviews Cancer 2018. doi 10.1038/nrc.2017.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer discovery 2015. doi 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.George S, Miao D, Demetri GD, Adeegbe D, Rodig SJ, Shukla S, et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity 2017;46(2):197–204 doi 10.1016/j.immuni.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nature medicine 2015;21(11):1350–6 doi 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lal N, White BS, Goussous G, Pickles O, Mason MJ, Beggs AD, et al. KRAS Mutation and Consensus Molecular Subtypes 2 and 3 Are Independently Associated with Reduced Immune Infiltration and Reactivity in Colorectal Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2018;24(1):224–33 doi 10.1158/1078-0432.CCR-17-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.