

Abstract
Piperidines and fluorine-substituents are both independently indispensable components in pharmaceuticals, agrochemicals and materials. Logically, the incorporation of fluorine atoms into piperidine scaffolds is therefore an area of tremendous potential. However, synthetic approaches towards the formation of these architectures are often impractical. The diastereoselective synthesis of substituted monofluorinated piperidines often requires substrates with pre-defined stereochemistry. That of multifluorinated piperidines is even more challenging, and often needs to be carried out in multistep syntheses. In this report, we describe a straightforward process for the one-pot rhodium-catalyzed dearomatization–hydrogenation (DAH) of fluoropyridine precursors. This strategy enables the formation of a plethora of substituted all-cis-(multi)fluorinated piperidines in a highly diastereoselective fashion through pyridine dearomatization followed by complete saturation of the resulting intermediates by hydrogenation. Fluorinated piperidines with defined axial/equatorial orientation of fluorine-substituents were successfully applied in the preparation of commercial drugs analogues. Additionally, fluorinated PipPhos as well as fluorinated ionic liquids were obtained by this DAH process.
The predictable and modular assembly of complex molecular structures is one of the main challenges in synthetic organic chemistry, which therefore necessitates the development of new synthetic strategies with improved selectivity and efficiency1–4. Among the many elegant chemical transformations that have been developed to meet this end, the metal-catalyzed hydrogenation of unsaturated compounds is arguably one of the most powerful methods. However, many unsolved challenges related to the reactivity and selectivity for the hydrogenation of simple planar aromatic ring systems still remain5–8.
Saturated nitrogen heterocycles are among the most significant structural components of pharmaceuticals and natural products9. To this end, a recent analysis of FDA-approved drugs revealed that 59% of small molecule drugs contain at least one N-heterocycle, of which saturated piperidine is the most prevalent (Fig. 1a)10. In a similar fashion to N-heterocycles, fluorine substituents are frequently found in drugs, agrochemicals and materials science (Fig. 1a). Furthermore, the incorporation of fluorine into lead drug candidates has been recognized as a powerful strategy to improve their pharmacokinetic and physicochemical properties and hence increase the likelihood of success in clinical trials11–14. Therefore, it is not surprising that around 20% of all marketed drugs and 40% of the current top ten best-selling drugs today contain at least one fluorine substituent15,16. With these considerations in mind, it seems only logical to assume that the incorporation of fluorine into aliphatic N-heterocycles is therefore of significant interest to the scientific community (Fig. 1a). For instance, it was recently shown that the pKa of a KSP inhibitor could be altered by the introduction of an axial or equatorial fluorine substituent into the piperidine core16. From these studies, the axial diastereomer (MK-0731) was selected for clinical trials over the equatorial homologue (Fig. 1a).
Figure 1. The preparation of all-cis-(multi)fluorinated piperidines by the DAH process.

a, Merging common elements of drugs. The goal is the incorporation of fluorine into piperidine derivatives. The basicity of kinesin spindle protein (KSP) inhibitor was affected by the orientation of the fluorine atom. The more basic axial isomer (MK-0731) was selected for clinical evaluation. b, Known retrosynthetic routes for the preparation of monofluorinated piperidines. LG: leaving group. c, The multistep synthesis of cis-3,5-difluoropiperidine hydrochloride. A 1,3-diaxial behavior of the fluorine atoms was observed. d, Our proposed DAH process is a combined dearomatization and hydrogenation that gives access to all-cis-(multi)fluorinated piperidine building blocks.
Current synthetic routes for the synthesis of relatively simple monofluorinated piperidine derivatives are usually based on electrophilic fluorination and thus require the careful preparation of pre-functionalized precursors. The diastereoselective synthesis of substituted monofluorinated piperidines can be achieved by nucleophilic substitution (Fig. 1b). However, the difficult pre-decoration of substrates with defined stereochemistry is required17,18. Other synthetic routes based on radical intermediates have been recently reported, with a minor focus on the generation of fluorinated piperidines19,20. Furthermore, the preparation of multifluorinated piperidines is rarely disclosed in the literature and often requires tedious multistep syntheses21. It should also be noted that, to the best of our knowledge, there are no known methods for the direct diastereoselective synthesis of multifluorinated piperidines (Fig. 1c). In light of the above, we considered that a more straightforward synthetic route to access these motifs via the hydrogenation of fluoropyridine precursors would represent a significant synthetic advancement. However, this strategy would require several challenges to be overcome, such as avoiding catalyst deactivation by the Lewis-basic heterocycles and the hydrodefluorination side reactions7. Strategies to circumvent catalyst poisoning by employing pyridinium salts instead of pyridines are known22,23. However, the extension of this method to fluorinated compounds has yet to be disclosed, presumably due to competing hydrodefluorination side reactions24. Thus, a direct synthetic protocol for the hydrogenation of fluorinated nitrogen containing heteroarenes, in particular fluoropyridine derivatives, remains elusive.
Herein, we describe a protocol for the highly selective one-pot dearomatization-hydrogenation (DAH) of fluorinated pyridines, providing convenient and diastereoselective access to cis-fluorinated piperidine building blocks (Fig. 1d). Furthermore, this synthetic protocol was successfully extended to multifluorinated pyridines to access the all-cis-multifluorinated piperidines with excellent diastereoselectivity.
Results and discussion
Our studies towards the hydrogenation of fluoropyridines began by testing a variety of catalysts known to be capable of arene hydrogenation for the reduction of 3-fluoro and 3,5-difluoropyridine6,7,25. However, these initial attempts were unsuccessful due to either catalyst poisoning or uncontrolled hydrodefluorination side reactions (see Supplementary Section 3). We therefore tried to develop an efficient process that could both prevent catalyst poisoning and enable fluoropyridine hydrogenation without the loss of the fluorine atoms. Considering this, we envisioned that a borane reagent could be used to first dearomatize the pyridine ring system by forming a mixture of dienes that could be more easily hydrogenated whilst also preventing catalyst poisoning by protecting the Lewis-basic nitrogen.
Initial studies towards our envisioned approach were conducted using 4,4,5,5-tetramethyl-1,3,2-dioxaborolane (HBpin), a known additive in the rhodium-catalyzed dearomatization of pyridine26,27, and [Rh(COD)Cl]2 [Rh-1] as catalyst for the hydrogenation of 3-fluoropyridine in THF at 25 °C. Indeed, the desired product was obtained in both good yield and chemoselectivity (Supplementary Table 2). Other rhodium complexes were then examined, from which Rh–CAAC28 complex [Rh-2] was found to be optimal. The reaction conditions were further optimized utilizing complex [Rh-2] by screening different solvents and concentrations. The influence of different hydride sources as well as the reaction pressure to ensure high efficiency, were also investigated (Supplementary Section 3). With the optimized conditions in hands, we began scoping studies for the hydrogenation of fluoropyridines (Table 1). Notably, the substrates were obtained either from commercial sources or via a single step synthesis following literature procedures. Upon the completion of the reactions, trifluoroacetic anhydride was added as a trapping agent to prevent loss of the volatile fluorinated piperidine derivatives. For less reactive substrates, an increase of catalyst loading and/or reaction temperature improved the yield of the final product. We also observed that in some cases, an excess of pinacol borane could reduce the amount of the undesired hydrodefluorinated side-product. Generally, high yields and excellent diastereomeric ratios were obtained. Furthermore, the isolation of the major diastereomer by standard column chromatography was often possible. Deprotection of the TFA-analogues afforded the piperidine hydrochlorides almost quantitatively. Accordingly, the cis-3,5-difluoropiperidine hydrochloride (4), previously requiring a 6-step synthesis21, is now obtained in a two-step process as a single diastereomer (d.r. >99:1) after deprotection of TFA-analogue 3. This reaction was also readily carried out on a gram-scale affording 1.57 g of 3 in good yield and with excellent diastereoselectivity (72%, d.r. >99:1, Sigma-Aldrich product no. 903817). The cis-selectivity as well as the 1,3-diaxial behavior of the fluorine atoms in 4 was confirmed by NMR studies and was consistent with previously published reports (Supplementary Section 4). The same behavior was also observed for the hydrochloride analogue of 1. The observation of the large axial preference for 3- and 3,5-difluoropiperidine hydrochloride ring systems was first discussed by Lankin, Snyder et al. and was rationalized by the occurrence of charge dipole interactions (C–F⋯HN+)21,29. As our protocol provides access to a variety of new substituted fluorinated piperidines, we wondered whether the axial preference will also be preserved in the presence of bulky substitutes on the piperidine ring system (Table 1). Comprehensive NMR studies (including NOEs, HetNOEs, high and low temperature experiments) for the TFA-analogues as well as the hydrochloride-analogues revealed that in most cases the axial preference is preserved (see Supplementary Section 4 for more details). Interestingly, even in highly crowded ring systems (15–18), fluorine atoms still prefer occupying axial positions (Table 1). However, when the TFA-analogue of the piperidine ring system contains a substituent adjacent to the nitrogen, the conformers favor equatorial fluorine (9, 11 and 13) in sharp contrast to the conformational behavior of their HCl-analogues (10, 12 and 14). Notably, in both the TFA- and HCl-analogues of cis-3-fluoro-5-methylpiperidine (5 and 6), the equatorial orientation is dominant. It should be noted that the vicinal 3J(F,H) coupling constants provide useful insight into the conformational structure, as large values of 3J(F,Ha) indicate axial preference and small values of 3J(F,Ha) indicate equatorial preference (for more details, see Supplementary Section 4)30.
Table 1. Scope and conformational behavior of all-cis-(multi)fluorinated piperidines.
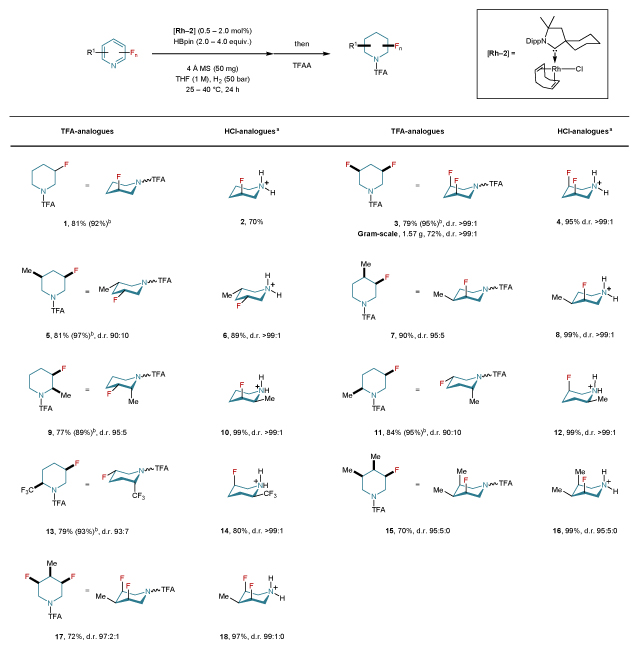 |
Reactions were carried out on a 0.25–10.0 mmol scale. Yields were determined after column chromatography (TFA-analogues) or precipitation (HCl-analogues). Diastereomeric ratio (d.r.) values were determined by 19F NMR or GC analysis prior to purification. The conformational behavior was determined by NMR studies. For details concerning catalyst loading, amount of HBpin and temperature, see Supplementary Section 4. aSee general procedure B in Supplementary Section 4 for the deprotection of TFA-fluoropiperidines to generate the fluoropiperidine hydrochloride analogues. bNMR yields are provided in parentheses and were determined by 19F NMR spectroscopy with hexafluorobenzene as internal standard prior to the addition of TFAA. HBpin: 4,4,5,5-tetramethyl-1,3,2-dioxaborolane; Dipp: 2,6-diisopropylphenyl; THF: tetrahydrofuran; TFAA: trifluoroacetic anhydride; TFA: trifluoroacetyl; MS: molecular sieves; Me: methyl.
Despite the generality of the reaction, we also discovered some limitations. For example, while 2-and 4-fluoropyridine derivatives readily underwent hydrogenation, the hydrodefluorinated products were identified as the major species, presumably due to unavoidable defluorination of the unstable conjugated intermediates. Further optimization of 4-fluoropyridine precursors allowed the access to a variety of all-cis-4-fluoropiperidine derivatives (19–24) with high diastereoselective ratios but in reduced yields. Their conformation was also determined by comprehensive NMR studies for both TFA- and HCl-analogues (Table 2). Interestingly, in most cases the axial orientation is dominant.
Table 2. Scope and conformational behavior of all-cis-4-fluoropiperidines.
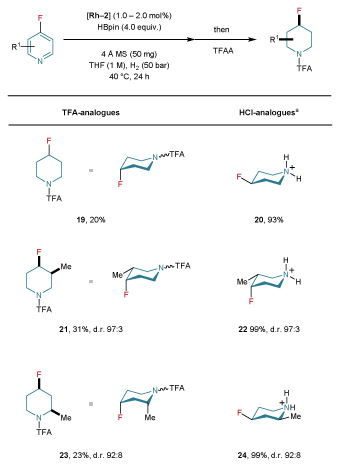 |
Reactions were carried out on a 0.5–1.0 mmol scale. Yields were determined after column chromatography (TFA-analogues) or precipitation (HCl-analogues). Diastereomeric ratio (d.r.) values were determined by 19F NMR or GC analysis prior to purification. The conformational behavior was determined by NMR studies. For details concerning catalyst loading, see Supplementary Section 4. aSee general procedure B in Supplementary Section 4 for the deprotection of TFA-fluoropiperidines to generate the fluoropiperidine hydrochloride analogues.
We next sought to demonstrate the utility of this methodology for the preparation of highly valuable and versatile fluorinated building blocks, and at the same time to demonstrate the mild nature of the conditions employed (Table 3). A variety of functional groups, that allow further elaboration of the molecular structure, were well-tolerated. Among these are tert-butyl(dimethyl)silyl (TBS)-protected alcohols (25–27), tert-butyloxycarbonyl (Boc)-protected amines (28–31), methoxy (36), pinacol boronic ester (37), and trimethylsilyl groups (38). Different trapping agents such as TFAA, (Boc)2O, or simple addition of MeOH were chosen to ensure straightforward isolation of the final products. The fluorinated analogues of the (Boc)-protected 4-aminopiperidine (34) and its HCl-analogue (35), a common core in pharmaceuticals, were also obtained in high yields and diastereoselectivities. The conformational behavior in solution of the new fluorinated building blocks was determined by NMR and was found to be consistent with the simplified substitution analogues in table 1 (see Supplementary Section 4). The cis-selectivity and equatorial preference of the fluorine in 29 were also confirmed by X-ray crystallographic analysis. Orthogonally protected building blocks can be obtained through our strategy, albeit in low yields due to the deprotection of the Boc group upon addition of TFAA (29, 31). The multi-substituted monofluorinated piperidine (39) as well as multifluorinated moieties bearing additional functional groups (40–44) were all obtained in a highly diastereoselective manner. In all of these cases, the axial orientation of the fluorine atoms is dominant. The scope of this reaction was also extended to other heterocycles (45–48). Interestingly, we could even selectively reduce the pyridine ring system, in the presence of other phenyl rings (46–48).
Table 3. Scope of all-cis-(multi)fluorinated piperidine building blocks.
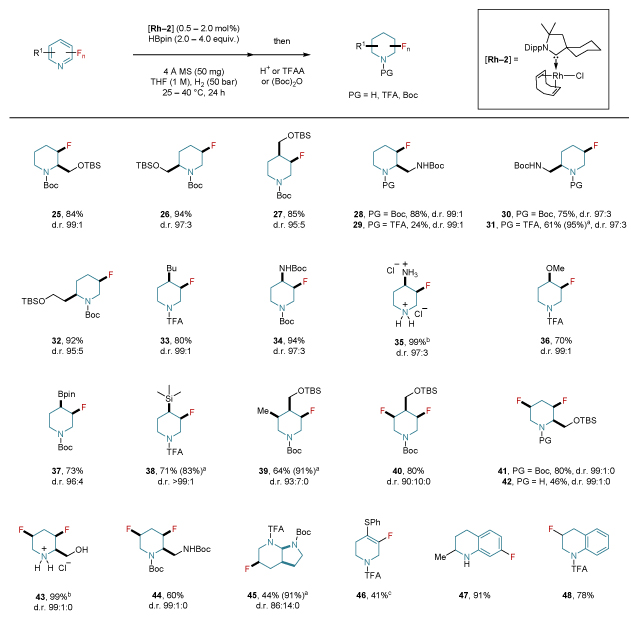 |
Reactions were carried out on a 0.25–0.5 mmol scale. Yields were determined after column chromatography (TFA- or Boc-analogues) or precipitation (HCl-analogues). Diastereomeric ratio (d.r.) values were determined by 19F NMR or GC analysis prior to purification. The conformational behavior was determined by NMR studies. For details concerning catalyst loading, amount of HBpin and temperature, see Supplementary Section 4. aNMR yields are provided in parentheses and were determined by 19F NMR spectroscopy with hexafluorobenzene as internal standard prior the addition of the trapping agent. bSee general procedure C in Supplementary Section 4 for the deprotection of Boc-fluoropiperidines to generate the fluoropiperidine hydrochloride analogues. c60% conversion. PG: protecting group; (Boc)2O: di-tert-butyl dicarbonate; Boc: tert-butyloxycarbonyl; TBS: tert-butyl(dimethyl)silyl; Bpin: 4,4,5,5-tetramethyl-1,3,2-dioxaboroyl; Bu: n-butyl; Ph: phenyl.
Preliminary mechanistic experiments were performed in order to gain a better understanding of the DAH process (Fig. 2). In the absence of pinacol borane, no product was detected (Supplementary Section 3). Traces of the proposed borylated-dearomatized intermediates could be observed by 1H and 19F NMR studies of the crude reaction mixture of 3,5-difluoropyridine after 5 h. However, our attempts to isolate the intermediates were unsuccessful, presumably due to their rapid hydrogenation. On the other hand, NMR analysis of the crude reaction showed that HBpin was consumed during the reaction (Fig. 2b), suggesting that it has a crucial role in the DAH process. Hydrogenation of 3,5-difluoropyridine using molecular deuterium was also performed generating the isotopologues of the 3,5-difluoropiperidine 4-D (Fig. 2c). Deuterium scrambling can be rationalized due to the formation of imine-enamine intermediates as well as the D-incorporation into HBpin that was also observed under our reaction conditions (Fig. 2d).
Figure 2. Selected mechanistic experiments for the DAH process.

a, Complete hydrodefluorination of 2-fluoropydrine was observed. b, The amount of HBpin was determined after the reaction by NMR analysis using benzene as internal standard. c, Deuterium scrambling was observed by employing molecular deuterium. The structure of 4-D was confirmed by NMR and ESI-MS studies. d, The D-incorporation for HBpin was observed under the reaction conditions. For more details concerning additional mechanistic studies, see Supplementary Section 6.
To illustrate the utility of our method and further demonstrate the value of the prepared building blocks, we synthesized several fluorinated analogues of commercially available drugs, PipPhos ligand and ionic liquids (Fig. 3a–c). The monofluorinated analogue of Melperone (49) as well as mono- and difluorinated analogues of Diphenidol (50, 51) were prepared using the corresponding piperidinium salts by nucleophilic substitution. The monofluorinated analogue of Dyclonine (52), Eperisone (53), and Cycrimine (55) were all prepared starting from fluorinated piperidine hydrochloride derivatives utilizing a modified Mannich reaction with 1,3-dioxolane. Moreover, the synthesis of mono- and difluorinated analogues of Cloperastine (57, 59) were also achieved (Fig. 3a–b, see Supplementary Section 7 for more details). Additionally, the difluorinated phosphoramidite analogue of PipPhos (60) was accessed starting from 3,5-difluoropiperidine hydrochloride (4) in a one-pot process (Fig. 3c). Interestingly, NMR analysis of 60 revealed that the fluorine atoms adopt equatorial orientation in polar and non-polar solvents. However, X-ray crystallographic analysis of F2-PipPhos (60) showed that the 1,3-diaxial behavior is dominant in the solid state. In materials science, fluorinated ionic liquids (FILs) are gaining attention due to their unique physical properties31. Therefore, we incorporated our new building blocks into analogues of previously known ionic liquids (61, 62) (Fig. 3c).
Figure 3. Application of the all-cis-(multi)fluorinated piperidines building blocks.

a, The preparation of fluorinated analogues of commercial drugs starting from fluoropiperidine hydrochloride derivatives. b, Fluorinated analogues of additional commercial drugs. For reaction conditions, see Supplementary Section 7. c, The preparation of fluorinated analogues of PipPhos and ionic liquids. For detailed reaction conditions, see Supplementary Section 7. Et3N: triethylamine; MeCN: acetonitrile; DMF: dimethylformamide; (R)-BINOL-PCl: (R)-1,1'-binaphthyl-2,2'-dioxychlorophosphine; LiTFSI: bis(trifluoromethane) sulfonimide lithium salt; AgOTf: silver trifluoromethanesulfonate.
Conclusions
In conclusion, we have developed a straightforward strategy to access all-cis-(multi)fluorinated piperidines from the corresponding fluoropyridine precursors in a highly diastereoselective manner. This process proceeds via a rhodium-catalyzed pyridine dearomatization event followed by complete saturation of the resulting intermediates by hydrogenation. We envision that the newly developed methodology will be of immediate interest in medicinal, agrochemical and materials science.
Methods
General procedure for the dearomatization-hydrogenation of fluoropyridine derivatives
An oven-dried reaction vessel (4 or 9 mL screw-cap vial) equipped with a stirring bar was allowed to cool to room temperature under vacuum. Then activated 4 Å MS (crushed, 50 mg), [Rh-2] (and solid substrates, 1.0 equiv.) were added under air. The vial was then depressurized and pressurized with argon gas three times prior the addition of dry tetrahydrofuran (1 M) (and liquid substrates – distilled over CaH2, 1.0 equiv.). Upon the addition of 4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2.0 – 4.0 equiv. as indicated), the glass vial was placed in a 150 mL stainless steel autoclave under argon atmosphere. The autoclave was pressurized and depressurized with hydrogen gas three times before the indicated pressure was set. The reaction mixture was stirred at 25–40 °C for 24 h. After the autoclave was carefully depressurized, trifluoroacetic anhydride (3.0 equiv.) and CH2Cl2 (0.5 mL) were added to the crude mixture and stirring was continued for 10 min at room temperature. Alternatively, di-tert-butyl dicarbonate (3.0 equiv.), triethyl amine (3.0 equiv.) and CH2Cl2 (0.5 mL) were added to the reaction mixture and stirring was continued for 2 h at room temperature. The crude was then filtered over fritted funnel and the remaining solid was washed with ethyl acetate (2×5 mL). The combined solution was concentrated under reduced pressure and submitted to column chromatography (pentane/ethyl acetate or pentane/dichloromethane) to obtain the final product. The indicated diastereoselectivities were determined by GC analysis or from the 19F NMR spectrum immediately after the reaction. NMR yield was calculated using hexafluorobenzene (20 μL, 0.173 mmol) as internal standard.
Supplementary Material
Experimental procedures, extensive optimization data, comprehensive NMR analysis and MS data are available in the Supplementary Information (SI).
Acknowledgements
We gratefully acknowledge the Hans-Jensen-Minerva Foundation (Z.N.) and the Deutsche Forschungsgemeinschaft IRTG 2027 (M.W.) and the European Research Council (ERC Advanced Grant Agreement no. 788558) for generous financial support. We thank Dr. M. P. Wiesenfeldt, M. Teders, Dr. M. J. James and Dr. M. van Gemmeren for helpful discussions. Dr. C. G. Daniliuc is acknowledged for X-ray crystallographic analysis. 1-(cis-3,5-difluoropiperidin-1-yl)-2,2,2-trifluoroethan-1-one (3) can now be obtained from Sigma-Aldrich (product #903817). Correspondence and requests for materials should be addressed to F.G.
Footnotes
Data availability
Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 1845054 (29) and 1845055 (60). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All other data supporting the findings of this study are available within the Article and its Supplementary Information, or from the corresponding author upon reasonable request.
Author contributions
Z.N., M.W., C.S. and F.G. designed, performed and analyzed experiments. K.B. performed and analyzed NMR data. Z.N. and F.G. prepared the manuscript with contributions from all authors.
Competing interests
Z.N., C.S. and F.G. are inventors on German patent application 10 2018 104 201.9 held by WWU Muenster that covers the DAH process for the synthesis of all-cis-(multi)fluorinated aliphatic heterocycles.
References
- 1.Trost BM. Selectivity: a key to synthetic efficiency. Science. 1983;219:245–250. doi: 10.1126/science.219.4582.245. [DOI] [PubMed] [Google Scholar]
- 2.Baran PS, Maimone TJ, Richter JM. Total synthesis of marine natural products without using protecting groups. Nature. 2007;446:404–408. doi: 10.1038/nature05569. [DOI] [PubMed] [Google Scholar]
- 3.Young IS, Baran PS. Protecting-group-free synthesis as an opportunity for invention. Nat Chem. 2009;1:193–205. doi: 10.1038/nchem.216. [DOI] [PubMed] [Google Scholar]
- 4.Wender PA, Miller BL. Synthesis at the molecular frontier. Nature. 2009;460:197–201. doi: 10.1038/460197a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao D, Candish L, Paul D, Glorius F. N-Heterocyclic carbenes in asymmetric hydrogenation. ACS Catal. 2016;6:5978–5988. [Google Scholar]
- 6.Wei Y, Rao B, Cong X, Zeng X. Highly selective hydrogenation of aromatic ketones and phenols enabled by cyclic (amino)(alkyl)carbene rhodium complexes. J Am Chem Soc. 2015;137:9250–9253. doi: 10.1021/jacs.5b05868. [DOI] [PubMed] [Google Scholar]
- 7.Wiesenfeldt MP, Nairoukh Z, Li W, Glorius F. Hydrogenation of fluoroarenes: Direct access to all-cis-(multi)fluorinated cycloalkanes. Science. 2017;357:908–912. doi: 10.1126/science.aao0270. [DOI] [PubMed] [Google Scholar]
- 8.Wiesenfeldt MP, Knecht T, Schlepphorst C, Glorius F. Silylarene hydrogenation: A strategic approach enabling direct access to versatile silylated saturated carbo- and heterocycles. Angew Chem Int Ed. 2018;57:8297–8300. doi: 10.1002/anie.201804124. [DOI] [PubMed] [Google Scholar]
- 9.O’Hagan D. Pyrrole, pyrrolidine, pyridine, piperidine and tropane alkaloids. Nat Prod Rep. 2000;17:435–446. doi: 10.1039/a707613d. [DOI] [PubMed] [Google Scholar]
- 10.Vitaku E, Smith DT, Njardarson JT. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J Med Chem. 2014;57:10257–10274. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- 11.Müller K, Faeh C, Diederich F. Fluorine in pharmaceuticals: Looking beyond intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 12.Shah P, Westwell AD. The role of fluorine in medicinal chemistry. J Enzyme Inhib Med Chem. 2007;22:527–540. doi: 10.1080/14756360701425014. [DOI] [PubMed] [Google Scholar]
- 13.O’Hagan D. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem Soc Rev. 2008;37:308–319. doi: 10.1039/b711844a. [DOI] [PubMed] [Google Scholar]
- 14.Thiehoff C, Rey YP, Gilmour R. The fluorine gauche effect: A brief history. Isr J Chem. 2017;57:92–100. [Google Scholar]
- 15.Wang J, et al. Fluorine in pharmaceutical industry: Fluorine-containing drugs introduced to the market in the last decade (2001–2011) Chem Rev. 2014;114:2432–2506. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- 16.Gillis EP, Eastman KJ, Hill MD, Donnelly DJ, Meanwell NA. Applications of fluorine in medicinal chemistry. J Med Chem. 2015;58:8315–8359. doi: 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- 17.Li X, et al. Process development for scale-up of a novel 3,5-substituted thiazolidine-2,4-dione compound as a potent inhibitor for estrogen-related receptor 1. Org Process Res Dev. 2014;18:321–330. [Google Scholar]
- 18.Goldberg NW, Shen X, Li J, Ritter T. AlkylFluor: Deoxyfluorination of alcohols. Org Lett. 2016;18:6102–6104. doi: 10.1021/acs.orglett.6b03086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu W, et al. Oxidative aliphatic C–H fluorination with fluoride ion catalyzed by a manganese porphyrin. Science. 2012;337:1322–1325. doi: 10.1126/science.1222327. [DOI] [PubMed] [Google Scholar]
- 20.Ventre S, Petronijevic FR, MacMillan DWC. Decarboxylative fluorination of aliphatic carboxylic acids via photoredox catalysis. J Am Chem Soc. 2015;137:5654–5657. doi: 10.1021/jacs.5b02244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Snyder JP, Chandrakumar NS, Sato H, Lankin DC. The unexpected diaxial orientation of cis-3,5-difluoropiperidine in water: A potent CF- - -NH charge-dipole effect. J Am Chem Soc. 2000;122:544–545. [Google Scholar]
- 22.Glorius F, Spielkamp N, Holle S, Goddard R, Lehmann CW. Efficient asymmetric hydrogenation of pyridines. Angew Chem Int Ed. 2004;43:2850–2852. doi: 10.1002/anie.200453942. [DOI] [PubMed] [Google Scholar]
- 23.Zhou Y-G. Asymmetric hydrogenation of heteroaromatic compounds. Acc Chem Res. 2007;40:1357–1366. doi: 10.1021/ar700094b. [DOI] [PubMed] [Google Scholar]
- 24.Whittlesey MK, Peris E. Catalytic hydrodefluorination with late transition metal complexes. ACS Catal. 2014;4:3152–3159. [Google Scholar]
- 25.Dyson PJ. Arene hydrogenation by homogeneous catalysts: Fact or fiction? Dalton Trans. 2003;0:2964–2974. [Google Scholar]
- 26.Park S, Chang S. Catalytic dearomatization of N-heteroarenes with silicon and boron compounds. Angew Chem Int Ed. 2017;56:7720–7738. doi: 10.1002/anie.201612140. [DOI] [PubMed] [Google Scholar]
- 27.Oshima K, Ohmura T, Suginome M. Regioselective synthesis of 1,2-dihydropyridines by rhodium-catalyzed hydroboration of pyridines. J Am Chem Soc. 2012;134:3699–3702. doi: 10.1021/ja3002953. [DOI] [PubMed] [Google Scholar]
- 28.Jazzar R, et al. Intramolecular “hydroiminiumation” of alkenes: Application to the synthesis of conjugate acids of cyclic alkyl amino carbenes (CAACs) Angew Chem Int Ed. 2007;46:2899–2902. doi: 10.1002/anie.200605083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lankin DC, Chandrakumar NS, Rao SN, Spangler DP, Snyder JP. Protonated 3-fluoropiperidines: An unusual fluoro directing effect and a test for quantitative theories of solvation. J Am Chem Soc. 1993;115:3356–3357. [Google Scholar]
- 30.Silla JM, et al. Gauche preference of β-fluoroalkyl ammonium salts. J Phys Chem A. 2014;118:503–507. doi: 10.1021/jp410458w. [DOI] [PubMed] [Google Scholar]
- 31.Pereiro AB, et al. Fluorinated ionic liquids: Properties and applications. ACS Sustainable Chem Eng. 2013;1:427–439. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.