

Abstract
Keloids are benign fibrous overgrowths that occur as a result of abnormal wound healing following cutaneous injury. MicroRNAs (miRNAs/miRs) are short non-coding RNAs that serve critical roles in numerous important biological processes, such as cell proliferation, differentiation and apoptosis. However, their role in keloid development remains largely unknown. In the present study, the role of miR-30a-5p, a miRNA regulated by Trichostatin A (TSA), in apoptosis within cultured keloid fibroblasts was investigated. An MTT assay was used to detect the proliferation of cultured keloid fibroblasts treated with TSA. Cell apoptosis and cell cycle phases were analyzed using flow cytometry. In addition, an miRNA microarray was performed to compare expression profiles between cultured keloid fibroblasts treated with or without 1,000 nM TSA. Reverse transcription-quantitative polymerase chain reaction analysis was conducted to estimate miRNA expression levels. The direct target of miR-30a-5p was identified using a dual-luciferase reporter assay. Western blotting was employed to assess protein expression levels in keloid fibroblasts. The results demonstrated that TSA inhibited the proliferation of keloid fibroblasts in a time- and dose-dependent manner. The miRNA microarray revealed alterations in the expression of numerous miRNA sequences in response to TSA when compared with controls. Notably, the expression of miR-30a-5p was downregulated in keloid tissues. In addition, overexpression of miR-30a-5p induced apoptosis by targeting B-cell lymphoma 2, which was similar to that observed in response to TSA. These results provide important information regarding a novel miR-30a-5p-mediated signaling pathway induced by TSA treatment, and suggest a potential use for TSA and miR-30a-5p as effective therapeutic strategies for keloids.
Keywords: Trichostatin A, microRNA-30a-5p, keloid fibroblast, B-cell lymphoma 2
Introduction
Keloids are benign dermal proliferative tumours that develop as a result of abnormal wound-healing processes following skin injury. Keloids are characterized by fibroblast proliferation, excessive deposition of extracellular matrix (ECM), particularly collagen and fibronectin, and increased infiltration of inflammatory cells (1–3). Although it has been established that excess deposition of ECM components, including collagen (4) by fibroblasts, is responsible for keloids (5), the aetiology and mechanisms underlying these effects remain poorly understood. Keloids do not regress over time, and there is a high recurrence rate following surgical excision.
MicroRNAs (miRNAs/miRs) are short non-coding RNAs that serve critical roles in a number of important biological processes, including cell proliferation, differentiation and apoptosis (6–8). To date, hundreds of miRNAs have been identified to be dysregulated in various diseased tissues; however, only a fraction has been functionally characterized. miRNAs are known to regulate skin development, and alterations in gene expression and have been associated with skin pathologies, including inflammatory disorders (8) and malignant lesions (9,10). The primary function of miRNAs in skin fibrosis is to regulate the expression of genes involved in its pathogenesis and maintenance (11). However, the functional role of miRNAs in the pathogenesis of keloids remains largely unknown.
Trichostatin A (TSA) is a classical and widely used histone deacetylase (HDAC) inhibitor (12). Inhibition of HDAC activity using HDAC inhibitors results in cell growth inhibition. Thus, inhibition of HDAC activity through the use of agents such as TSA has been developed as a form of targeted therapy, which has demonstrated promising antitumor effects in multiple malignancies, including skin and colon cancer (13,14). It has also been reported that TSA activity is associated with the development and progression of certain chronic diseases characterized by skin fibrosis (14). Therefore, it was hypothesized that selective alterations in the miRNA expression profile caused by TSA treatment may result in keloid fibroblast proliferation and the accumulation of ECM.
In the current study, miR-30a-5p was observed to induce apoptosis in keloid fibroblasts. In addition, the results demonstrated that miR-30a-5p directly targeted and negatively regulated B-cell lymphoma 2 (BCL2) by binding to its 3′-untranslated region (UTR), which resulted in the induction of apoptosis. Notably, these effects were similar to those observed in response to TSA exposure. These data provide novel and important information that may help to elucidate the factors and mechanisms underlying TSA exposure and miR-30a-5p in keloid fibroblasts.
Materials and methods
Samples
Keloid tissue samples were obtained from 15 patients, and healthy skin samples were obtained from 5 patients, all admitted to the Department of Dermatology at No. 1 Hospital of China Medical University (Shenyang, China) between June 2016 and June 2017. The diagnosis of keloid samples was confirmed by pathological examination. All protocols were approved by the Ethics Committee of the No. 1 Hospital of China Medical University.
Cell culture
Human keloid tissues (n=8) were collected from patients who underwent surgery at the Center for Plastic Surgery of No. 1 Hospital of China Medical University between June 2016 and June 2017. The skin specimens were washed in PBS containing 100 U/ml penicillin-streptomycin (Beyotime Institute of Biotechnology, Haimen, China) and incubated with 2.5 mg/ml dispase II (Roche Applied Science, Penzberg, Germany) overnight at 4°C. The following day, the specimens were washed with PBS, and the dermis was manually separated from the epidermis, cut into small 1-mm sections and seeded in a culture flask. Fibroblasts were cultured in Dulbecco's modified Eagle's medium (DMEM; Corning Inc., Corning, NY, USA) supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The cultures were maintained at 37°C in an atmosphere with 5% CO2. The medium was refreshed three times/week. Cells at passages 3–5 were used for the purposes of this study.
Cell proliferation assay
Cell proliferation was determined using a standard MTT assay. In brief, keloid fibroblasts were first seeded at a density of 5×103 cells/well into 96-well culture plates, and cultured in DMEM containing 10% FBS for 24 h. The medium was removed and replaced with DMEM containing 10% FBS and different concentrations (0, 250, 500, 1,000 or 1,500 nM) of TSA (Enzo Life Sciences, Inc., Farmingdale, NY, USA). Following incubation for a further 24, 48 or 72 h, 20 µl MTT reagent (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was added, and the cells were incubated at 37°C for 4 h. A total of 150 µl dimethyl sulfoxide was added to stop the reaction. The effect of TSA on cell viability was determined by measuring the absorbance at 490 nm.
Cell cycle analysis
Cells were washed three times with cold PBS and then fixed in ice-cold 70% ethanol at 4°C overnight. The cells were washed with cold PBS and each sample was incubated with 500 µl of propidium iodide (PI; 50 µg/ml; Sigma-Aldrich; Merck KGaA) and 50 µl RNase A (50 µg/ml) at 37°C for 20 min in the dark. Analyses were performed using a BD LSRFortessa Cell Analyzer (BD Biosciences, San Jose, CA, USA) and the percentage of cells in the G0/G1, S or G2/M cell cycle phases was analyzed using ModFit software version 3.0 (BD Biosciences). Experiments were repeated three times.
Apoptosis analysis
The number of apoptotic cells was quantified using an Annexin-V-allophycocyanin (APC) Apoptosis Detection kit (cat. no. 88-8007-74; Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. Early apoptotic cells were defined as Annexin-V-positive and PI-negative cells. Analyses were performed using a BD LSRFortessa Cell Analyzer (BD Biosciences) and FlowJo version 10.0.7 software (FlowJo LLC, Ashland, OR, USA). All experiments were repeated three times.
Total RNA extraction and reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Dermal tissue or dermal fibroblasts were first crushed on ice and total RNA extracted using the miRNeasy mini kit (Qiagen GmbH, Hilden, Germany), according to the manufacturer's protocol. Fibroblasts at passages 3–5 were treated with TSA or transfected with mimics and total RNA extracted using the miRNeasy mini kit (Qiagen GmbH, Hilden, Germany), according to the manufacturer's protocol. cDNA was synthesized from RNA using the High Capacity cDNA Reverse Transcription kit or TaqMan MicroRNA Reverse transcription kit (Thermo Fisher Scientific, Inc.). Each RT reaction using the High Capacity cDNA Reverse Transcription kit was performed in a 96 well Thermal Cycler (Applied Biosystems, USA) for 10 min at 25°C, 37°C for 120 min, 85°C for 5 min. Each RT reaction using the TaqMan MicroRNA Reverse transcription kit was performed in a 96 well Thermal Cycler (Applied Biosystems, USA) for 30 min at 16°C, 42°C for 30 min, 85°C for 5 min. Primer sequences are listed in Table I. Realtime PCR of mRNA was performed using GoTaq qPCR Master Mix reagent (Promega, WI, USA) and a 7900HT Fast Real-Time PCR system (Thermo Fisher Scientific, Inc.) with following program: Stage 1, 95°C for 2 min; Stage 2, 40 cycles of 95°C for 15 sec, 60°C for 1 min. Analysis of miRNA expression by qPCR was performed using TaqMan miRNA assays (Thermo Fisher Scientific, Inc.) and a 7900HT Fast Real-Time PCR system (Thermo Fisher Scientific, Inc.) with following program: Hold 1, 50°C for 2 min; Hold 2, 95°C for 10 min; Hold 3, 40 cycles of 95°C for 15 sec, then 60°C for 1 min. Relative expression was calculated using the 2−ΔΔCq method or ΔCq method (15). Confirmation of TSA-induced miRNA clusters in keloid fibroblasts were normalized to the control gene U6 small nuclear RNA and analysis of relative gene Expression data performed with the 2−ΔΔCq method. Expression of miR-30a-5p in keloid tissue as determined by use of RT-qPCR. miR-30a-5p PCR data were compared with small nucleolar RNA, C/D box 48 and analysis of relative gene expression data performed using the 2−ΔΔCq method.
Table I.
Primers for reverse transcription-quantitative polymerase chain reaction analysis with the SYBR® Green system.
| mRNA | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| GAPDH | AAGAGCACAAGAGGAAGAGAGAGAC | GTCTACATGGCAACTGTGAGGAG |
| BCL2 | GGATTGTGGCCTTCTTTGAG | CCAAACTGAGCAGAGTCTTC |
| COL1A1 | AGCCAGCAGATCGAGAACAT | TCCTTGGGGTTCTTGCTGAT |
BCL2, B-cell lymphoma 2; COL1A1, collagen type I α 1.
Western blotting
Cells were first washed with ice-cold PBS, and lysed with phenylmethylsulfonyl fluoride (Beyotime Institute of Biotechnology). Following centrifugation at 5,000 × g for 15 min at 4°C, the protein concentration was determined using a bicinchoninic acid protein assay kit (cat. no. 23225; Thermo Fisher Scientific, Inc.). Protein lysates (50 µg) were loaded onto 10% SDS-PAGE gels before being transferred to polyvinylidene difluoride membranes. The membranes were blocked with TBS with Tween 20 and 5% non-fat milk for 1 h at room temperature, and subsequently incubated with collagen I antibody (cat. no. BA0325; dilution, 1:200; Wuhan Boster Biological Technology, Ltd., Wuhan, China), BCL2 antibody (cat. no. ab196495; dilution, 1:1,000; Abcam, Cambridge, MA, USA), GAPDH (cat. no. 10494-1-AP; dilution, 1:2,000; ProteinTech Group, Inc., Chicago, IL, USA) or β-actin (cat. no. ab90641; dilution, 1:5,000; Abcam) overnight at 4°C. The membranes were subsequently incubated with a horseradish peroxidase-conjugated secondary antibody (cat. no. A0181; dilution, 1:1,000; Beyotime Institute of Biotechnology) at room temperature for 1 h, and detected using enhanced chemiluminescence (Amersham™ ECL™ Prime Western Blotting Detection Reagent; GE Healthcare Life Sciences, Little Chalfont, UK) and a MF-Chemi BIS 2.0 Imaging Systems(NDR Bio Imaging Systems, Israel) the following day. Grayscale value analysis of protein was performed by Image-Pro Plus 6.0 software (Media Cybernetics, Inc., Rockville, MD, USA). The ratio of the target protein to the internal reference protein was calculated for each group.
Transient transfection
All oligonucleotides were synthesized by Ambion (Thermo Fisher Scientific, Inc.). Transfections were performed using Lipofectamine® RNAiMAX (cat. no. 13778-075; Thermo Fisher Scientific, Inc.). Cells in the growth phase were seeded at 2×105 cells/well in six-well plates. To assess the transfection efficiency of miR-30a-5p mimics, cells were transfected with 40, 60 or 80 nM mimics for 24, 48 or 72 h, respectively. Transfections of 60 nM miR-30a-5p mimics or negative control mimics for 72 h was selected for all experiments.
TaqMan low-density miRNA RT-qPCR array and miRNA analyses
The miRNA array was performed using three different paired samples. RNA was first reverse transcribed using the TaqMan miRNA Reverse Transcription kit and TaqMan miRNA Multiplex RT assays, respectively; Thermo Fisher Scientific, Inc.) with following program: Stage 1, 40 cycles of 16°C for 2 min, 42°C for 1 min, 50°C for 1 sec; Stage 2, 85°C for 5 min. Expression was profiled using a TaqMan Human MicroRNA array card A, B (V2.1 and V3.0, respectively) and a 7900HT Fast Real-Time PCR System (Thermo Fisher Scientific, Inc.), according to the manufacturer's recommended protocol. Briefly, the array was loaded and run using the 384-well Taqman Low Density Array default thermal-cycling conditions with following program: Stage 1, 94.5°C for 10 min; Stage 2, 40 cycles of 97°C for 30 sec, 59.7°C for 1 min on a 7900HT Fast Real-Time PCR System. The quantification cycle (Cq) values were obtained with 900HT System Fast Real-Time PCR SDS software version 2.3 (Applied Biosystems; Thermo Fisher Scientific, Inc.), and the data were analyzed using RQ manager 1.2 software (Applied Biosystems; Thermo Fisher Scientific, Inc.). U6 small nuclear RNA was selected as a control. The -ΔCq[-(Cq-CqU6)] values were calculated, and heatmap analyses were performed with hierarchical clustering (16).
Verification of BCL2 as a direct target gene of miR-30a-5p
The predicted (TargetScan Human 7.1 and miRanda) or confirmed gene targets of these miRNAs were provided. A dual-luciferase reporter assay was performed using 293T cells. Briefly, the putative miR-30a-5-p binding sites within the 3′-UTR of the BCL2 gene were amplified and cloned into the GV272 vector (GeneChem, Inc., Shanghai, China), and the miR-30a-5p gene was amplified and cloned into the GV251 vector (Shanghai GeneChem, Inc.). Cells (1×105) were seeded in 24-well plates and co-transfected with 100 ng wild-type or mutated BCL2 3′-UTR constructs and 400 ng negative control or miR-30a-5p plasmids using X-tremegene HP (Roche Diagnostics, Basel, Switzerland for ≤48 h. Luciferase activity was determined using the Dual-luciferase Reporter Assay System (cat. no. E1910; Thermo Fisher Scientific, Inc.) following transfection for 48 h. Briefly, Dual-Glo® Luciferase Assay Reagent was added to the plate, the firefly luminescence measured, Dual-Glo® Stop & Glo® Reagent added to the plate and Renilla luminescence measured. The ratio of firefly:Renilla luminescence for each well was calculated. The sample well ratio to the ratio from control wells was normalized. Since the miRNA functions primarily by targeting the 3′-UTR of the target gene, this region may be cloned into a luciferase vector and positioned before the luciferase reporter gene. Luciferase activity in mimic or negative control-transfected cells was subsequently measured. Modifications in gene expression are reflected in the change in luciferase activity, and may quantitatively reflect the inhibitory effect of miRNA on the target gene. With TRAF6-3′UTR as a positive control, the expression of luciferase in the group was significantly decreased (P<0.05), indicating that there was no problem in the whole transfection detection system.
Statistical analysis
The results are expressed as the mean ± standard deviation of at least three separate experiments, each performed in triplicate. Differences between groups were analyzed using a two-tailed Student's t-test, Mann-Whitney U test or one-way analysis of variance with Tukeys post hoc test using the software GraphPad Prism 7.0 (GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
TSA inhibits the growth of keloid fibroblasts in a time- and dose-dependent manner
An initial dose titration was performed to determine the appropriate concentration of TSA to be used in subsequent experiments. Keloid fibroblasts were treated with media containing increasing doses (0, 250, 500, 1,000 and 1,500 nM) of TSA. The effects of TSA on cell viability were monitored using an MTT proliferation assay. As presented in Fig. 1, keloid fibroblasts treated with TSA demonstrated a statistically significant reduction in cell growth following incubation with TSA for 24, 48 or 72 h. Accordingly, TSA inhibited keloid fibroblast growth in a time- and dose-dependent manner. The proliferation of keloid fibroblasts treated with either 1,000 or 1,500 nM TSA was significantly inhibited and morphological alterations were observed when compared with the controls. Overall, cells treated with 1,000 nM TSA tolerated the treatment well, and preserved their viability compared with the control. Therefore, 1,000 nM TSA was used as the working dose for all subsequent experiments.
Figure 1.

TSA inhibits the growth of keloid fibroblasts in a time- and dose-dependent manner. (A) Treatment with 1,000 nM TSA altered the morphology of keloid fibroblasts at 24, 48 or 72 h in culture (×100 magnification). Keloid fibroblast phenotypes were examined by phase-contrast microscopy for changes in morphology. (B) The MTT assay indicated that TSA inhibited the cell viability of keloid fibroblasts at concentrations of 250, 500, 1,000, 1,500 nM as observed after 24, 48 or 72 h in culture compared with the control. Results are presented as the mean ± standard deviation of three independent experiments (n=8). One-way analysis of variance with Tukey's post-hoc test was used to compare the groups. *P<0.05, **P<0.01 and ***P<0.001 vs. respective control. TSA, trichostatin A.
Apoptosis of keloid fibroblasts is upregulated following TSA treatment
To investigate the effects of TSA on apoptosis, cultured keloid fibroblasts were incubated with 1,000 nM TSA for 24, 48 or 72 h, and Annexin V-APC/PI staining for apoptosis detection was performed, followed by flow cytometry. The results demonstrated that keloid fibroblast apoptosis was upregulated following TSA treatment at all three time points tested (P<0.01; Fig. 2). In addition, the apoptosis rate in cells pretreated with TSA for 72 h was increased by 12.5% relative to the controls.
Figure 2.

TSA promotes the apoptosis of keloid fibroblasts in a time-dependent manner. (A) Annexin V and PI staining was performed to test for cell apoptosis ratios in response to 1,000 nM TSA treatment at 24, 48 or 72 h in culture. (B) Data are displayed as bar graphs. Results are presented as the mean ± standard deviation of three independent experiments (n=8). A two-tailed Student's t-test was used to compare the groups. **P<0.01 and ***P<0.001 vs. respective control. TSA, trichostatin A; PI, propidium iodide.
TSA blocks the G2/M cell cycle phase of keloid fibroblasts
The results demonstrating the impact of TSA treatment on keloid fibroblast proliferation prompted the investigation of the role of cell cycle progression in this phenotype. Following a 24 h incubation period, the average percentage of cells in the G2/M phase increased from 23.0% in the control group to 26.8% in the 1,000 nM TSA group (P=0.5287; Fig. 3). Notably, G2/M arrest was observed following longer incubation periods. After a 48 h incubation period, the average percentage of cells in the G2/M phase increased from 13.2% in the control group to 20.8% in the 1,000 nM TSA group (P=0.0064), while 72 h of TSA incubation was associated with an average increase in the percentage of cells in the G2/M phase from 16.2% in the control group to 33.0% in the 1,000 nM TSA group (P=0.0381; Fig. 3B). These results suggested that TSA may influence the cell cycle of keloid fibroblasts.
Figure 3.

TSA blocks the G2/M cell cycle phase of keloid fibroblasts. (A) PI staining was performed to analyze the cell cycle phase in response to 1,000 nM TSA treatment at 24, 48 or 72 h in culture. (B) Data are displayed as bar graphs. Results are presented as the mean ± standard deviation of three independent experiments (n=6). A two-tailed Student's t-test was used to compare the groups. *P<0.05 and **P<0.01 vs. respective control. TSA, trichostatin A; PI, propidium iodide.
TSA inhibits the expression of collagen and BCL2 in keloid fibroblasts
The ability of TSA to modulate the expression of collagen type Iα 1 chain (COL1A1) in keloid fibroblasts was investigated by RT-qPCR (Fig. 4A). COL1A1 expression, as detected using western blotting, exhibited an apparent decrease (Fig. 4B). In addition, the protein expression levels of COL1A1 and BCL2 in TSA-treated keloid fibroblasts were seemingly reduced compared with the controls.
Figure 4.

TSA decreases both the expression of collagen I and BCL2 in keloid fibroblasts. (A) COL1A1 mRNA expression was determined using reverse transcription-quantitative polymerase chain reaction following treatment with 1,000 nM TSA for 72 h in culture. (B) Western blotting was performed following treatment with 1,000 nM TSA for 72 h in culture. Results represent one of three independent experiments and were normalized to the β-actin control. n=4 for control (DMSO)- and TSA-treated groups.*P<0.05 vs. Control. TSA, trichostatin A; BCL2, B-cell lymphoma 2; DMSO, dimethyl sulfoxide.
TSA interferes with the miRNA expression profile in keloid fibroblasts
It has been previously reported that the expression of specific miRNAs is significantly altered following TSA treatment (17). The altered expression of specific miRNAs has been associated with ECM synthesis, fibroblast differentiation, epithelial-mesenchymal transition (EMT), in addition to cancer initiation, invasion and metastasis (17,18). Therefore, miRNA microarray analysis of keloid fibroblasts from three biological samples treated with 1,000 nM TSA for 72 h was performed in the present study. As illustrated in the heatmap presented in Fig. 5, a number of alterations in miRNA expression were observed. Specifically, of the miRNAs significantly altered by TSA treatment, 22 were upregulated (Table II) and 10 were downregulated (Table III). The predicted (TargetScan Human 7.1 and miRanda) or confirmed gene targets of these miRNAs are provided. Of particular relevance to the present study, the miR-125a and miR-17 sequences, which have been associated with fibroblast proliferation, were observed to be downregulated, while miR-30a-5p and miR-146a, which have been associated with transforming growth factor (TGF)-β signaling, were upregulated. Considering previous studies, a number of miRNAs were selected and alterations in their expression between treatment and control were evaluated by RT-qPCR. The miRNA microarray and RT-qPCR results verified the aforementioned observations, demonstrating that that miR-30a-5p exhibited a marked alteration during TSA-mediated inhibition of keloid fibroblast cell growth (Figs. 6 and 7). It has been reported that miR-30a-5p targets BCL2 mRNA and induces apoptosis in non-small cell lung cancer cells (19,20). Although the change observed was not the most evident, it was found that potential targets of this miR-30a-5p were more pertinent to the aims of the present study. Therefore, the role of miR-30a-5p in keloid fibroblast regulation was further investigated.
Figure 5.

Hierarchically clustered (average linkage) heatmap of TSA induction of modification in miRNA expression in keloid fibroblasts. A total of 10 miRNAs were downregulated and 22 miRNAs upregulated as demonstrated by the heatmap. Red, significantly increased expression; green, significantly decreased expression. U6 small nuclear RNA was selected as a control. n=3 for C and T groups. miRNA/miR, microRNA; TSA, trichostatin A; C, control; T, TSA-treated.
Table II.
Upregulated microRNAs in keloid fibroblasts treated with Trichostatin A.
| miRs | Mean fold change ± SD | Notable targets | Key function |
|---|---|---|---|
| miR-1233 | 2.89±1.975 | HOXB3 | Invasion |
| miR-515-3p | 2.02±0.4761 | ||
| miR-132 | 2.05±0.4389 | RASA1 | Migration |
| miR-146a-5p | 4.89±3.199 | SMAD4, IRAK-1 | TGF-β signaling |
| miR-30d | 2.12±0.7582 | CDH1 | EMT |
| miR-1243 | 2.27±0.9789 | ||
| miR-1271 | 1.95±1.263 | PDK1, CDK1, FN1 | Proliferation, apoptosis |
| miR-1260 | 3.64±0.8275 | ||
| miR-30e-3p | 2.79±1.92 | SNAIL1 | Invasion, migration |
| miR-1274A | 3.84±2.318 | BMPR1B, FOXO4 | Proliferation, migration |
| miR-424 | 2.85±2.822 | MEK1, CCNE1 | Proliferation |
| miR-1274B | 3.83±2.457 | ||
| miR-766 | 5.56±3.752 | MDM4, SOX6, | p53 signaling |
| miR-378 | 3.60±3.019 | FOXG1, GLI3 | MAPK/TGF-β signaling |
| miR-30a-3p | 3.77±3.53 | IGF-1, PTEN, BAFF | Invasion, apoptosis |
| miR-31 | 3.83±3.626 | STK40, FIH-1 | Proliferation, differentiation |
| miR-664 | 3.93±3.863 | IRS1, SOX7, FOXO4 | Proliferation |
| miR-1290 | 5.86±4.125 | IRF2, LHX6, BCL2 | EMT |
| miR-720 | 7.48±4.239 | CDH1 | EMT |
| miR-30a-5p | 5.18±5.965 | BCL2, NEUROD1, Akt | Apoptosis |
| miR-24-2 | 7.44±6.375 | PKC-alpha, BCL2 | Apoptosis |
| miR-136 | 6.11±7.416 | E2F1, MIEN1, PMEL | Proliferation |
miR, microRNA; TGF, transforming growth factor; EMT, epithelial-mesenchymal transition.
Table III.
Downregulated microRNAs in keloid fibroblasts treated with Trichostatin A.
| miRs | Mean fold change ± SD | Notable targets | Key function |
|---|---|---|---|
| miR-10b | 0.22±0.1125 | CADM2, PIK3CA | Proliferation, migration |
| miR-339-3p | 0.42±0.1862 | PRL-1 | Proliferation |
| miR-125a-5p | 0.42±0.1962 | BAX, BMF | Apoptosis |
| miR-155 | 0.43±0.1884 | SKI, TGFβ2R | TGF-β signaling |
| miR-196b | 0.51±0.1001 | COL1A1, COL3A1 | ECM synthesis |
| miR-345 | 0.51±0.0151 | FOXQ1, YAP1, IRF1 | Metastasis, EMT |
| miR-15b | 0.44±0.1977 | RECK, ERK1 | Proliferation |
| miR-16 | 0.65±0.1977 | HGF, SESN1 | P53 signaling |
| miR-140-3p | 0.64±0.1564 | ATP6AP2, ATP8A1 | Proliferation, invasion |
| miR-339-5p | 0.66±0.166 | NACC1, MDM2 | P53 signaling |
miR, microRNA; TGF, transforming growth factor; ECM, extracellular matrix; EMT, epithelial-mesenchymal transition.
Figure 6.

Confirmation of TSA-induced upregulation of miR-30a-5p, miR-146a, miR-766 and miR-769 in keloid fibroblasts. Results are presented the mean ± standard deviation of three independent experiments with triplicate wells, and were normalized to the control gene U6 small nuclear RNA (n=6–8). A two-tailed Student's t-test was used to compare the groups. *P<0.05 and **P<0.01 vs. respective control. miRNA/miR, microRNA; TSA, trichostatin A.
Figure 7.

Confirmation of TSA-induced downregulation of the miR-17-92 and miR-155 miRNA clusters in keloid fibroblasts. Results are presented the mean ± standard deviation of three independent experiments with triplicate wells, and were normalized to the control gene U6 small nuclear RNA. Analysis of relative gene expression data was performed using reverse transcription-quantitative polymerase chain reaction and the 2−ΔΔCq method (n=6–8). A two-tailed Student's t-test was used to compare the groups. *P<0.05, **P<0.01 and *** P<0.001 vs. respective control. miRNA/miR, microRNA; TSA, trichostatin A.
miR-30a-5p expression is downregulated in keloid tissues
Based on the observation that TSA altered the expression of miR-30a-5p in keloid fibroblasts, the expression of miR-30a-5p in keloid tissues was investigated. To achieve this, RT-qPCR was used to examine the expression levels of miR-30a-5p in five healthy skin and seven keloid tissue samples. As exhibited in Fig. 8, the mean expression levels of miR-30a-5p in keloid tissue samples were significantly decreased when compared with the healthy skin samples (P=0.0480). These results suggested that miR-30a-5p may serve an important role in inhibiting the pathological process of keloid formation.
Figure 8.
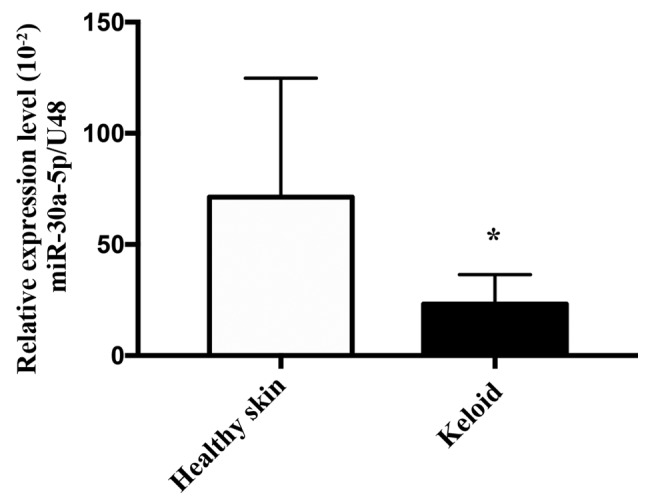
Expression of miR-30a-5p is downregulated in keloid tissue. Expression of miR-30a-5p in healthy skin (n=5) and keloid tissue (n=7) as determined by use of reverse transcription-quantitative PCR. miR-30a-5p PCR data were compared with small nucleolar RNA, C/D box 48, analysis of relative gene expression data was performed using the -ΔCT method. Results are presented the mean ± standard deviation, and a Mann-Whitney U-test was used to compare the groups. *P<0.05 vs. healthy control. miR, microRNA; PCR, polymerase chain reaction.
Predicted and confirmed mRNA targets
The results presented thus far suggested that miR-30a-5p may be associated with fibroblast proliferation and ECM deposition. The 3′-UTR of BCL2 was predicted to contain a miR-30a-5p binding site (Fig. 9A). To experimentally validate this miRNA-mRNA interaction, a luciferase assay was performed. As presented in Fig. 9B, miR-30a-5p significantly decreased BCL2 3′-UTR reporter activity when compared with the BCL2 3′-UTR negative control group. The inhibitory effect of miR-30a-5p was eliminated when the predicted miR-30a-5p binding site was mutated. TRAF6-3′ UTR was used as a positive control; the expression of luciferase in the group was significantly decreased (P<0.05), indicating that there was no problem in the transfection detection system.
Figure 9.

BCL2 is targeted by miR-30a-5p in keloid fibroblasts. (A) Nucleotide sequence of the predicted miR-30a-5p binding site in 3′-UTR of BCL2 mRNA. (B) 293T cells were transfected with luciferase reporter plasmids containing BCL2 3′-UTR and mutant BCL2 3′-UTR or BCL2 3′UTR NC. Luciferase activity was measured 48 h later. Results are presented the mean ± standard deviation of three independent experiments. One-way analysis of variance with Tukey's post-hoc test was used to compare the groups. ***P<0.001. BCL2, B-cell lymphoma 2; miR, microRNA; UTR, untranslated region; NC, negative control; TRAF6, TNF receptor associated factor 6.
Overexpression of miR-30a-5p inhibits apoptosis and the proliferation of keloid fibroblasts in vitro
To further investigate the role of miR-30a-5p in keloid fibroblasts treated with TSA, the transfection efficiency of miR-30a-5p mimics within keloid fibroblasts cultured for 72 h was determined. The effect of miR-30a-5p mimics on cell viability was monitored using MTT proliferation assays. The results demonstrated that the proliferation of cultured keloid fibroblasts was reduced by 8% when compared with the controls (P=0.0187; Fig. 10A). To study the inhibitory effect of miR-30a-5p on apoptosis, keloid fibroblast cultures were incubated with 60 nM miR-30a-5p for 72 h, and the level of apoptosis was ascertained using Annexin V-APC/PI staining and flow cytometry analysis. The results demonstrated that keloid fibroblast apoptosis was increased by 5.9% relative to the control (P=0.0031; Fig. 10B and C). However, no effects on cell cycle progression were observed (P=0.6303; Fig. 10D and E).
Figure 10.

Overexpression of miR-30a-5p promotes apoptosis. (A) The MTT assay indicated that an overexpression of miR-30a-5p inhibited keloid fibroblast cell growth compared with the negative control. (B) Flow cytometry results using APC staining performed to assess (C) cell apoptosis ratios at 72 h in culture following transfection of the miR-30a-5p mimic. (D) Flow cytometry results using PI staining to analyze the effect of miR-30a-5p on keloid fibroblast cell cycle. (E) The ratios demonstrated that miR-30a-5p did not affect the cell cycle progression of keloid fibroblasts. Results are presented the mean ± standard deviation of three independent experiments (n=3-4). A two-tailed Student's t-test was used to compare the groups. *P<0.05 and **P<0.01 vs. respective NC. miR, microRNA; PI, propidium iodide; APC, allophycocyanin; NC, negative control; ns, not significant.
Overexpression of miR-30a-5p inhibits the mRNA and protein expression levels of BCL2 and COL1A1 in keloid fibroblasts in vitro
To investigate potential interactions between the predicted target gene, BCL2, and miR-30a-5p, total RNA and protein from keloid fibroblasts transfected with negative control or miR-30a-5p mimics for 72 h was determined by RT-qPCR. Furthermore, western blot analyses were performed to determine the mRNA and protein expression levels of BCL2 and COL1A1. The mRNA and protein levels of BCL2 (Fig. 11A-C) and COL1A1 (Fig. 11D-E) were significantly decreased in keloid fibroblasts. When compared with the negative control mimic-transfected cells, BCL2 mRNA levels were decreased by 0.73-fold (P=0.0116) and COL1A1 mRNA by 0.68-fold (P=0.0118), and statistically significant reductions in BCL2 (P=0.0361) and COL1A1 (P=0.0012) protein expression levels were also observed.
Figure 11.

Overexpression of miR-30a-5p decreases the mRNA and protein levels of BCL2 and COL1A1 in keloid fibroblasts. (A) COL1A1 and BCL2 mRNA expression were determined using reverse transcription-quantitative polymerase chain reaction following transfection of keloid fibroblasts with miR-30a-5p mimic. (B) COL1A1 and BCL2 protein levels were determined with use of western blotting following transfection of keloid fibroblasts with miR-30a-5p mimic. (C) Grayscale value analysis of protein was performed by Image-Pro Plus 6.0 software. Results are presented as the mean ± standard deviation of three independent experiments (n=3-4). A two-tailed Student's t-test was used to compare the groups. *P<0.05 and **P<0.01 vs. respective NC. miR, microRNA; BCL2, B-cell lymphoma 2; COL1A1, collagen type I α 1.
Discussion
In the current study, an important inhibitory role of TSA and miR-30a-5p in the process of fibrosis in keloid fibroblasts was demonstrated. TSA is a broad-spectrum HDAC inhibitor that serves a key role in the epigenetic regulation of multiple genes involved in fibrosis and adverse remodeling (14,18–21). TSA effectively inhibits the EMT of hepatic stellate cells (22) and abrogates TGF-β1-induced, fibrosis-associated gene expression in skin fibroblasts (23). To date, a considerable body of evidence has provided an insight into the crosstalk among different signaling pathways in fibrosis, and has elucidated the molecular actions of TSA in attenuating fibrogenesis (22–25). Of particular note, one study demonstrated that TSA induces apoptosis in keloid fibroblasts (26). The results of the current study demonstrated that 1,000 nM TSA produced a time-dependent inhibition of keloid fibroblast proliferation and inhibited cell growth in the G2/M cell cycle phase. Treatment with 1,000 nM TSA also altered the miRNA expression profile of genes involved in cell proliferation, apoptosis and migration. Previous research has suggested that miRNAs serve important roles in fibrosis, and may be useful targets for the treatment of this disease (27). The present study tested and verified the differential expression of miRNAs, identified from an array, through RT-qPCR analysis, and the results were consistent with the TaqMan low-density miRNA array, RT-qPCR and miRNA analyses. Notably, the miR-17-92 cluster was downregulated, suggesting that it may contribute to cell proliferation and fibrosis (28,29). By contrast, specific miRNAs, reportedly associated with fibroblast differentiation, such as miR-146a, were observed to be upregulated (30).
miR-30a-5p is a multifunctional miRNA that has been implicated in numerous cell processes, including cell growth, proliferation and migration (20,31,32). To investigate the significance of miR-30a-5p in human keloid fibroblasts in the current study, the expression of miR-30a-5p in human keloid tissues and normal healthy tissue samples was first compared. The results demonstrated that keloid tissues exhibited decreased expression of miR-30a-5p when compared with healthy skin tissues. miR-30a-5p overexpression was also observed to induce apoptosis in keloid fibroblasts in vitro. These results provide evidence of a functional, mechanistic and clinically relevant role for this molecule. However, further research is required to elucidate the targets of miR-30a-5p, in addition to the molecular signaling pathways mediating these different biological effects in human keloid fibroblasts.
A previous report demonstrated that miR-30a-5p sensitizes non-small cell lung cancer cells to paclitaxel by inducing apoptosis via BCL2 inhibition (20). BCL2 regulates cell death by inhibiting apoptosis (20). BCL2 also serves as a phosphatase enzyme that is essential for the specific and effective termination of fibrosis (33). With the use of bioinformatics analysis software in the present study, BCL2 was identified as a predicted target of miR-30a-5p, a result that was verified using a dual-luciferase reporter assay. In addition, BCL2 was observed to be downregulated in keloid fibroblasts transfected with a miR-30a-5p mimics. As the miR-3a-5p mimic induced apoptosis and decreased the expression of COL1A1, it is likely that miR-30a-5p may induce apoptosis and inhibit the proliferation of keloid fibroblasts by inhibiting BCL2.
Similar to the effects observed in fibroblasts treated with 1,000 nM TSA, overexpression of miR-30a-5p resulted in the inhibition of keloid fibroblast cell proliferation in vitro, as well as apoptosis induction. However, the miR-30a-5p mimic exerted no observable effect on the cell cycle progression of active keloid fibroblasts. Thus, TSA may induce G2/M cycle arrest in keloid fibroblast through other gene networks that require further research. Moreover, miR-30a-5p mimics induced apoptosis and inhibited proliferation, which was analogous but not identical to the effects of TSA treatment in keloid fibroblasts. TSA may have downregulated the mRNA and protein expression levels of BCL2 and COL1A1 by upregulating the expression of miR-30a-5p.
In conclusion, possibly due to its capacity to upregulate the expression of miR-30a-5p, TSA was observed to induce apoptosis and inhibit the proliferation of keloid fibroblasts. Using this process, TSA may inhibit the synthesis of ECM in keloid fibroblasts. The results of the present study provide novel and noteworthy information regarding the mechanisms underlying the capacity for TSA to induce miR-30a-5p and thus regulate apoptosis in keloid fibroblasts in vitro. The results present a foundation for the potential use of TSA and miR-30a-5p in the diagnosis and treatment of patients with keloids. However, additional in vivo experiments are necessary to confirm these results.
Acknowledgements
The authors would like to acknowledge the Key Laboratory of Immunodermatology, Ministry of Health (China Medical University, Shenyang, China) for providing the space and equipment for conducting the experiments.
Funding
This study was supported by: The Science & Technology Fund of Liaoning Province (grant no. 201501013); the National Natural Science Fund of China (grant no. 81602741); the Distinguished Professor Foundation of Liaoning Province [grant no. Liao (2012) 145]; and the Major Science and Technology Platform of Liaoning Province [grant no. Liao (2010) 191].
Availability of data and materials
The data used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
CH and XJ conceived and designed the experiments; XJ performed the experiments; XJ, LQ, QZo, QZh, SC and YW analyzed the data; QZo, QZh, SC, YW, XG and HC contributed reagents/materials/analysis tools; XJ wrote the manuscript. HC and XG assessed the data and revised the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Informed consent was obtained from all individual participants included in the present study. The present study was approved by the Ethics Committee of the No.1 Hospital of China Medical University [approval no. (2016)71; Shenyang, China].
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Li Z, Jin Z. Comparative effect and safety of verapamil in keloid and hypertrophic scar treatment: A meta-analysis. Ther Clin Risk Manag. 2016;12:1634–1641. doi: 10.2147/TCRM.S118748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xue M, Jackson CJ. Extracellular matrix reorganization during wound healing and its impact on abnormal scarring. Adv Wound Care (New Rochelle) 2015;4:119–136. doi: 10.1089/wound.2013.0485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilgus TA, Wulff BC. The importance of mast cells in dermal scarring. Adv Wound Care (New Rochelle) 2014;3:356–365. doi: 10.1089/wound.2013.0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Younai S, Nichter LS, Wellisz T, Reinisch J, Nimni ME, Tuan TL. Modulation of collagen synthesis by transforming growth factor-beta in keloid and hypertrophic scar fibroblasts. Ann Plast Surg. 1994;33:148–151. doi: 10.1097/00000637-199408000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Suarez E, Syed F, Rasgado TA, Walmsley A, Mandal P, Bayat A. Skin equivalent tensional force alters keloid fibroblast behavior and phenotype. Wound Repair Regen. 2014;22:557–68. doi: 10.1111/wrr.12215. [DOI] [PubMed] [Google Scholar]
- 6.Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 7.Sonkoly E, Stahle M, Pivarcsi A. MicroRNAs: Novel regulators in skin inflammation. Clin Exp Dermatol. 2008;33:312–315. doi: 10.1111/j.1365-2230.2008.02804.x. [DOI] [PubMed] [Google Scholar]
- 8.Igoucheva O, Alexeev V. MicroRNA-dependent regulation of cKit in cutaneous melanoma. Biochem Biophys Res Commun. 2009;379:790–794. doi: 10.1016/j.bbrc.2008.12.152. [DOI] [PubMed] [Google Scholar]
- 9.Molnar V, Tamasi V, Bakos B, Wiener Z, Falus A. Changes in miRNA expression in solid tumors: An miRNA profiling in melanomas. Semin Cancer Biol. 2008;18:111–122. doi: 10.1016/j.semcancer.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. 2015;20:3898–3941. doi: 10.3390/molecules20033898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schneider MR. MicroRNAs as novel players in skin development, homeostasis and disease. Br J Dermatol. 2012;166:22–28. doi: 10.1111/j.1365-2133.2011.10568.x. [DOI] [PubMed] [Google Scholar]
- 12.Zhao LM, Zhang JH. Histone deacetylase inhibitors in tumor immunotherapy. Curr Med Chem. 2017 doi: 10.2174/0929867324666170801102124. (Epub ahead of prin) [DOI] [PubMed] [Google Scholar]
- 13.Singh A, Patel P, Jageshwar, Patel VK, Jain DK, Kamal M, Rajak H. The safety, efficacy and therapeutic potential of histone deacetylase inhibitors with special reference to panobinostat in gastrointestinal tumors: A Review of Preclinical and Clinical Studies. Curr Cancer Drug Targets. 2018;18:720–736. doi: 10.2174/1568009617666170630124643. [DOI] [PubMed] [Google Scholar]
- 14.Yang L, Qu M, Wang Y, Duan H, Chen P, Wang Y, Shi W, Danielson P, Zhou Q. Trichostatin a inhibits transforming growth Factor-β induced reactive oxygen species accumulation and myofibroblast differentiation via enhanced NF-E2-related factor 2-antioxidant response element signaling. Mol Pharmacol. 2013;83:671–680. doi: 10.1124/mol.112.081059. [DOI] [PubMed] [Google Scholar]
- 15.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 16.Rao X, Huang X, Zhou Z, Lin X. An improvement of the 2ˆ(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat Bioinforma Biomath. 2013;3:71–85. [PMC free article] [PubMed] [Google Scholar]
- 17.Rhodes LV, Nitschke AM, Segar HC, Martin EC, Driver JL, Elliott S, Nam SY, Li M, Nephew KP, Burow ME, Collins-Burow BM. The histone deacetylase inhibitor trichostatin A alters microRNA expression profiles in apoptosis-resistant breast cancer cells. Oncol Rep. 2012;27:10–16. doi: 10.3892/or.2011.1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams SM, Golden-Mason L, Ferguson BS, Schuetze KB, Cavasin MA, Demos-Davies K, Yeager ME, Stenmark KR, McKinsey TA. Class I HDACs regulate angiotensin II-dependent cardiac fibrosis via fibroblasts and circulating fibrocytes. J Mol Cell Cardiol. 2014;67:112–125. doi: 10.1016/j.yjmcc.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bodas M, Mazur S, Min T, Vij N. Inhibition of histone-deacetylase activity rescues inflammatory cystic fibrosis lung disease by modulating innate and adaptive immune responses. Respir Res. 2018;19:2. doi: 10.1186/s12931-017-0705-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu X, Jin S, Ma Y, Fan Z, Yan Z, Li W, Song Q, You W, Lyu Z, Song Y. miR-30a-5p enhances paclitaxel sensitivity in non-small cell lung cancer through targeting BCL-2 expression. J Mol Med (Berl) 2017;95:861–871. doi: 10.1007/s00109-017-1539-z. [DOI] [PubMed] [Google Scholar]
- 21.Tung CW, Hsu YC, Cai CJ, Shih YH, Wang CJ, Chang PJ, Lin CL. Trichostatin A ameliorates renal tubulointerstitial fibrosis through modulation of the JNK-dependent Notch-2 signaling pathway. Sci Rep. 2017;7:14495. doi: 10.1038/s41598-017-15162-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaimori A, Potter JJ, Choti M, Ding Z, Mezey E, Koteish AA. Histone deacetylase inhibition suppresses the transforming growth factor beta1-induced epithelial-to-mesenchymal transition in hepatocytes. Hepatology. 2010;52:1033–45. doi: 10.1002/hep.23765. [DOI] [PubMed] [Google Scholar]
- 23.Rombouts K, Niki T, Greenwel P, Vandermonde A, Wielant A, Hellemans K, De Bleser P, Yoshida M, Schuppan D, Rojkind M, Geerts A. Trichostatin A, a histone deacetylase inhibitor, suppresses collagen synthesis and prevents TGF-beta(1)-induced fibrogenesis in skin fibroblasts. Exp Cell Res. 2002;278:184–197. doi: 10.1006/excr.2002.5577. [DOI] [PubMed] [Google Scholar]
- 24.Sun J, Wang Y, Cui W, Lou Y, Sun G, Zhang D, Miao L. Role of epigenetic histone modifications in diabetic kidney disease involving renal fibrosis. J Diabetes Res. 2017;7:242–384. doi: 10.1155/2017/7242384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghosh AK, Mori Y, Dowling E, Varga J. Trichostatin A blocks TGF-beta-induced collagen gene expression in skin fibroblasts: involvement of Sp1. Biochem Biophys Res Commun. 2007;354:420–426. doi: 10.1016/j.bbrc.2006.12.204. [DOI] [PubMed] [Google Scholar]
- 26.Diao JS, Xia WS, Yi CG, Wang YM, Li B, Xia W, Liu B, Guo SZ, Sun XD. Trichostatin A inhibits collagen synthesis and induces apoptosis in keloid fibroblasts. Arch Dermatol Res. 2011;303:573–580. doi: 10.1007/s00403-011-1140-1. [DOI] [PubMed] [Google Scholar]
- 27.Babalola O, Mamalis A, Lev-Tov H, Jagdeo J. The role of microRNAs in skin fibrosis. Arch Dermatol Res. 2013;305:763–76. doi: 10.1007/s00403-013-1410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu H, Han C, Wu T. MiR-17-92 cluster promotes hepatocarcinogenesis. Carcinogenesis. 2015;36:1213–22. doi: 10.1093/carcin/bgv112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dakhlallah D, Batte K, Wang Y, Cantemir-Stone CZ, Yan P, Nuovo G, Mikhail A, Hitchcock CL, Wright VP, Piper MG, Marsh CB. Epigenetic regulation of miR-17~92 contributes to the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2013;187:397–405. doi: 10.1164/rccm.201205-0888OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li YL, Wang J, Zhang CY, Shen YQ, Wang HM, Ding L, Gu YC, Lou JT, Zhao XT, Ma ZL, Jin YX. MiR-146a-5p inhibits cell proliferation and cell cycle progression in NSCLC cell lines by targeting CCND1 and CCND2. Eur Respir J. 2016;7:59287–59298. doi: 10.18632/oncotarget.11040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He R, Yang L, Lin X, Chen X, Lin X, Wei F, Liang X, Luo Y, Wu Y, Gan T, et al. MiR-30a-5p suppresses cell growth and enhances apoptosis of hepatocellular carcinoma cells via targeting AEG-1. Int J Clin Exp Pathol. 2015;8:15632–15641. [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Z, Dai X, Chen Y, Sun C, Zhu Q, Zhao H, Liu G, Huang Q, Lan Q. MiR-30a-5p is induced by Wnt/β-catenin pathway and promotes glioma cell invasion by repressing NCAM. Biochem Biophys Res Commun. 2015;465:374–380. doi: 10.1016/j.bbrc.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 33.Safaeian L, Abed A, Vaseghi G. The role of Bcl-2 family proteins in pulmonary fibrosis. Eur J Pharmacol. 2014;741:281–9. doi: 10.1016/j.ejphar.2014.07.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used and/or analyzed during the current study are available from the corresponding author on reasonable request.