

Abstract
A large library of fibrate-based N-acylsulphonamides was designed, synthesised, and fully characterised in order to propose them as zinc binders for the inhibition of human carbonic anhydrase (hCA) enzymatic activity. Synthesised compounds were tested against four hCAs (I, II, IX, and XII) revealing a promising submicromolar inhibitory activity characterised by an isozyme selectivity pattern. Structural modifications explored within this scaffold are: presence of an aryl ring on the sulphonamide, p-substitution of this aryl ring, benzothiazole or benzophenone as core nuclei, and an n-propyl chain or a geminal dimethyl at Cα carbon. Biological results fitted well with molecular modelling analyses, revealing a putative direct interaction with the zinc ion in the active site of hCA I, II and IX. These findings supported the exploration of less investigated secondary sulphonamides as potential hCA inhibitors.
Keywords: N-acylsulphonamides, carbonic anhydrase, PPAR antagonist, fibrate derivatives, benzothiazole, benzophenone
1. Introduction
Sulphonamides and their N-acyl derivatives represent common functional groups occurring in natural and synthetic drugs. The different biological activities played by these compounds attracted the attention of medicinal chemists, and a great number of lead compounds of pharmaceutical interest were discovered to date1–3. Indeed, N-acylsulphonamides have been widely used in medicinal chemistry as bioisosteres of carboxylic acids, given the similarity in terms of H-bond properties and acidity4. The bioisosteric replacement of carboxylic group with N-acylsulphonamide produced novel compounds showing pharmacological activity, sometimes improved with respect to the parent compounds. Paritaprevir (NS3 protease inhibitor)5, Venetoclax (Bcl-2 inhibitor)6, and Selexipag (prostacyclin receptor agonist)7 are N-acylsulphonamide drugs recently approved by FDA for the treatment of HCV infection, tumours, and pulmonary arterial hypertension, respectively. A number of N-acylsulphonamide derivatives, targeting different enzymes or receptors, have been submitted to clinical or preclinical evaluation: among them, inhibitors of bacterial enzymes8,9, asparagine synthetase inhibitors10, agonists and antagonists of nuclear receptor peroxisome proliferator-activated receptors (PPARs)11,12.
Historically, secondary sulphonamides represent an important class of drugs targeting carbonic anhydrases (CAs)13–16. These metalloenzymes, catalysing the CO2 hydration reaction, are involved in several physiologic functions, also depending on their distribution in different tissues. For these reasons, CA inhibitors present a wide biomedical application, interfering with acid/base regulation, respiration, gluconeogenesis, bone resorption, and tumorigenesis17. CA inhibitors, containing the sulphonamide moiety, are therapeutically used as diuretic, antiglaucoma, anticancer, and antiobesity drugs. In recent works, N-substituted sulphonamides, among other secondary sulphonamides, have been identified as strong human CA I and II inhibitors, showing nanomolar inhibition constants18,19.
They were characterised by an ionisable moiety as zinc binder directly interacting with the zinc ion in the active site, as demonstrated by X-ray crystallographic studies of human carbonic anhydrase (hCA) II-inhibitor adducts20. In the attempt to identify novel CA inhibitors, in this work, we describe the screening of N-acylsulphonamide derivatives previously synthesised and identified as PPAR antagonists21,22. These compounds belong to a library of benzothiazole derivatives of fibrates, known PPAR agonists, obtained by an agonist-antagonist switching programme. The inhibitory properties of these derivatives were evaluated on four selected human CA isozymes (I, II, IX, and XII). These isoforms were selected according to their pharmacological relevance for the discovery of innovative therapeutic approaches against hypoxic cancers (hCA IX and XII) and for their wide distribution in the human body (hCA I and II as off-targets). Starting from previously obtained results, a novel series of benzenesulphonamide derivatives were designed, synthesised, and submitted to CA inhibition assays. Furthermore, molecular modelling studies were carried out to explain the putative interactions between this library of heterocyclic compounds and the active sites of the four isozymes in order to rationalise future synthetic approaches within this scaffold and to further understand the structural requirements to obtain isoform selectivity among the tested CAs.
2. Experimental protocols
2.1. Chemistry
Melting points were determined with a Buchi Melting Point B‐450 and were uncorrected. NMR spectra were recorded on a Varian Mercury spectrometer at 300 MHz or on a Bruker spectrometer at 400 MHz. Proton chemical shifts were referenced to the TMS internal standard. Chemical shifts are reported in parts per million (ppm, δ units). Coupling constants are reported in units of Hertz (Hz). Splitting patterns are designed as s, singlet; d, doublet; t, triplet; q, quartet; dd, double doublet; m, multiplet; and b, broad. All commercial chemicals and solvents were reagent grade and were purchased from Sigma-Aldrich (St. Louis, MO); they are used without further purification, unless otherwise specified. Reactions were monitored by thin layer chromatography on silica gel plates (60 F‐254, Sigma-Aldrich) and the analysis of the plates was carried out using a UV lamp 254/365 nm. Flash chromatography was performed on silica gel 60 (Merck, Kenilworth, NJ). Elemental analyses for C, H, and N were recorded on a Perkin-Elmer 240 B microanalyzer obtaining analytical results within ±0.4% of the theoretical values for all compounds. The following solvents have been abbreviated: chloroform (CHCl3), dichloromethane (DCM), diethyl ether (Et2O), dimethyl sulphoxide (DMSO), ethanol (EtOH), methanol (MeOH), and tetrahydrofuran (THF).
2.1.1. General procedure for the synthesis of benzenesulphonamides (19–27)
To a stirred solution of fenofibric acid (1.0 mmol) in dry DCM (10 mL) at 0 °C, 1‐ethyl‐3‐[3‐dimethylaminopropyl]carbodiimide hydrochloride (EDC, 1.0 mmol), and 4‐dimethylaminopyridine (DMAP, 1.0 mmol) were added. After 15 min, the selected benzenesulphonamide (1.1 mmol) was added to the reaction mixture and the cold water/ice bath was removed. The reaction mixture was allowed to stir 24 h, diluted with DCM (10 mL), washed with 2N HCl (3 × 20 ml), dried on Na2SO4, filtered, and evaporated under reduced pressure. Crude products were purified by column chromatography.
2.1.1.1. 2-[4-(4-Chlorobenzoyl)phenoxy]-2-methyl-N-[(4-methylphenyl)sulfonyl]propanamide (19)
White solid, 51% yield (silica gel, CHCl3/MeOH 98:2); m.p. 185–186 °C; 1H NMR (DMSO-d6) δ1.44 (s, 6H, C(CH3)2), 2.29 (s, 3H, CH3Ph), 6.66 (d, 2H, J 8.7 Hz, CHAr), 7.35 (d, 2H, J 8.1 Hz, CHAr), 7.51 (d, 2H, J 8.7 Hz, CHAr), 7.60–7.71 (m, 6H, CHAr), 12.40 (bs, 1H, NH, D2O exchange);13C NMR (DMSO-d6) δ 21.3, 24.3, 80.9, 117.9, 128.1, 129.1, 129.9, 130.2, 131.6, 132.1, 136.2, 136.5, 137.5, 144.8, 159.0, 172.9, 193.7. Anal. Calcd. for C24H22ClNO5S: C, 61.08; H, 4.70; N, 2.97. Found: C, 60.93; H, 4.69; N, 2.97.
2.1.1.2. 2-[4-(4-Chlorobenzoyl)phenoxy]-N-[(4-methoxyphenyl)sulfonyl]-2-methylpropanamide (20)
White solid, 54% yield (silica gel, DCM/MeOH 98:2); m.p. 188–189 °C; 1H NMR (CDCl3) δ1.51 (s, 6H, C(CH3)2), 3.87 (s, 3H, OCH3), 6.73 (d, 2H, J 8.7 Hz, CHAr), 6.99 (d, 2H, J 8.7 Hz, CHAr), 7.46 (d, 2H, J 8.7 Hz, CHAr), 7.62 (d, 2H, J 9.0 Hz, CHAr), 7.70 (d, 2H, J 9.0 Hz, CHAr), 7.93 (d, 2H, J 8.7 Hz, CHAr), 8.90 (bs, 1H, NH, D2O exchange); 13C NMR (CDCl3) δ 24.3, 55.7, 81.5, 114.1, 119.0, 128.6, 129.0, 130.9, 131.2, 131.9, 132.1, 135.8, 138.8, 157.4, 164.1, 171.9. Anal. Calcd. for C24H22ClNO6S: C, 59.07; H, 4.54; N, 2.87. Found: C, 59.19; H, 4.53; N, 2.88.
2.1.1.3. 2-[4–(4-Chlorobenzoyl)phenoxy]-N-[(4-chlorophenyl)sulfonyl]-2-methylpropanamide (21)
White solid, 69% yield (silica gel, DCM/MeOH 98:2); m.p. 204–205 °C (dec); 1H NMR (DMSO-d6) δ1.44 (s, 6H, C(CH3)2), 6.73 (d, 2H, J 9.0 Hz, CHAr), 7.46–7.69 (m, 9H, CHAr, and NH, D2O exchange), 7.78 (d, 2H, J 8.7 Hz, CHAr); 13 C NMR (DMSO-d6) δ 24.8, 81.3, 117.8, 128.0, 129.0, 129.3, 129.5, 129.8, 129.9, 131.5, 132.0, 136.7, 137.4, 159.5, 174.0, 193.6. Anal. Calcd. for C23H19Cl2NO5S: C, 56.11; H, 3.89; N, 2.84. Found: C, 56.24; H, 3.88; N, 2.84.
2.1.1.4. 2-[4–(4-Chlorobenzoyl)phenoxy]-2-methyl-N-[(4-nitrophenyl)sulfonyl]propanamide (22)
White solid, 56% yield (silica gel, DCM/MeOH 98:2); m.p. 196–198 °C; 1H NMR (DMSO-d6) δ1.45 (s, 6H, C(CH3)2), 6.71 (d, 2H, J 9.0 Hz, CHAr), 7.54 (d, 2H, J 9.0 Hz, CHAr), 7.61 (d, 2H, J 8.7 Hz, CHAr), 7.68 (d, 2H, J 8.7 Hz, CHAr), 8.03 (d, 2H, J 8.7 Hz, CHAr), 8.34 (d, 2H, J 8.7 Hz, CHAr); 13C NMR (DMSO-d6) δ24.8, 81.3, 117.8, 124.5, 129.0, 129.4, 130.0, 131.5, 132.0, 136.6, 137.5, 150.1, 159.4, 174.3, 193.6. Anal. Calcd. for C23H19ClN2O7S: C, 54.93; H, 3.81; N, 5.57. Found: C, 55.07; H, 3.79; N, 5.56.
2.1.1.5. N-{[4-(acetylamino)phenyl]sulfonyl}-2-[4–(4-chlorobenzoyl)phenoxy]-2-methylpropanamide (23)
White solid, 53% yield (silica gel, DCM/MeOH 98:2); m.p. 226–228 °C; 1H NMR (DMSO-d6) δ1.44 (s, 6H, C(CH3)2), 2.03 (s, 3H, CH3), 6.69 (d, 2H, J 8.7 Hz, CHAr), 7.52–7.78 (m, 10H, CHAr), 10.37 (bs, 1H, NHSO2, D2O exchange), 12.38 (bs, 1H, NHPh, D2O exchange); 13C NMR (DMSO-d6) δ24.3, 24.5, 80.9, 118.1, 118.6, 129.0, 129.4, 130.4, 131.6, 132.0, 136.5, 137.5, 144.3, 159.0, 169.4, 172.7, 173.4, 193.5. Anal. Calcd. for C25H23ClN2O6S: C, 58.31; H, 4.50; N, 5.44. Found: C, 58.44; H, 4.50; N, 5.43.
2.1.1.6. N-{4-[({2-[4–(4-chlorobenzoyl)phenoxy]-2-methylpropanoyl}amino)sulfonyl]phenyl} benzamide (24)
White solid, 51% yield (silica gel, CHCl3/MeOH 98:2); m.p. 232–234 °C; 1H NMR (DMSO-d6) δ1.46 (s, 6H, C(CH3)2), 6.71 (d, 2H, J 8.7 Hz, CHAr), 7.51–7.68 (m, 9H, CHAr), 7.82 (d, 2H, J 8.7 Hz, CHAr), 7.84–7.98 (m, 4H, CHAr), 10.64 (bs, 1H, NHSO2, D2O exchange), 12.45 (bs, 1H, NHPh, D2O exchange); 13C NMR (DMSO-d6) δ24.4, 81.0, 118.2, 120.1, 126.9, 128.3, 128.9, 129.3, 130.5, 131.6, 132.4, 134.7, 136.5, 137.6, 139.2, 142.6, 144.4, 159.0, 166.5, 172.8, 193.6. Anal. Calcd. for C30H25ClN2O6S: C, 62.44; H, 4.37; N, 4.85. Found: C, 62.35; H, 4.36; N, 4.86.
2.1.1.7. 2-[4-(4-Chlorobenzoyl)phenoxy]-2-methyl-N-({4-[(phenylacetyl)amino]phenyl}sulphonyl) propanamide (25)
White solid, 47% yield (silica gel, DCM/MeOH 98:2); m.p. 170–172 °C; 1H NMR (DMSO-d6) δ1.43 (s, 6H, C(CH3)2), 3.64 (s, 2H, CH2), 6.72 (d, 2H, J 8.1 Hz, CHAr), 7.19–7.30 (m, 5H, CHAr), 7.55–7.76 (m, 10H, CHAr), 10.63 (bs, 1H, NHSO2, D2O exchange); 13C NMR (DMSO-d6) δ24.4, 43.7, 81.0, 118.3, 118.9, 127.1, 128.8, 129.1, 129.5, 129.6, 131.6, 132.0, 135.8, 136.5, 144.2, 159.0, 170.3, 172.8, 193.6. Anal. Calcd. for C31H27ClN2O6S: C, 62.99; H, 4.60; N, 4.74. Found: C, 62.89; H, 4.61; N, 4.75.
2.1.1.8. 2-(4-Benzoylphenoxy)-N-(phenylsulphonyl)acetamide (26)
White solid, 59% yield (silica gel, DCM); m.p. 61–62 °C; 1H NMR (CDCl3) δ4.55 (s, 2H, CH2), 6.95 (d, 2H, J 8.7 Hz, CHAr), 7.45–7.83 (m, 5H, CHAr), 7.66–7.83 (m, 5H, CHAr), 8.10 (d, 2H, J 7.5 Hz, CHAr), 9.06 (bs, 1H, NH, D2O exchange); 13C NMR (CDCl3) δ77.2, 114.4, 126.4, 128.3, 129.0, 129.1, 129.8, 132.3, 132.6, 132.8, 137.6, 142.0, 159.7, 195.2. Anal. Calcd. for C21H17NO5S: C, 63.79; H, 4.33; N, 3.54. Found: C, 64.02; H, 4.32; N, 3.53.
2.1.1.9. 2-(4-Benzoylphenoxy)-N-(phenylsulphonyl)pentanamide (27)
White solid, 49% yield (silica gel, DCM); m.p. 140–142 °C; 1H NMR (CDCl3) δ0.87 (t, 3H, J 6.9 Hz, CH3), 1.36–1.43 (m, 2H, CH2), 1.82–1.91 (m, 2H, CH2), 4.58 (t, 1H, J 6.0 Hz, CH), 6.83 (d, 2H, J 9.0 Hz, CHAr), 7.46–7.65 (m, 6H, CHAr), 7.71–7.76 (m, 4H, CHAr), 7.95 (d, 2H, J 7.2 Hz, CHAr), 8.81 (bs, 1H, NH, D2O exchange); 13 C NMR (CDCl3) δ13.5, 17.8, 34.1, 78.6, 114.7, 126.4, 128.3, 129.0, 129.8, 132.0, 132.3, 132.6, 134.2, 137.6, 137.9, 159.6, 169.3, 195.2. Anal. Calcd. for C24H23NO5S: C, 65.89; H, 5.30; N, 3.20. Found: C, 65.91; H, 5.29; N, 3.21.
2.1.2. General procedure for the synthesis of esters (29 and 31)
A solution of sodium (2.5 mmol) in absolute EtOH (10 ml) was added to 4-hydroxybenzophenone (2.5 mmol), dissolved in absolute EtOH (10 ml), at room temperature, and under nitrogen atmosphere. Ethyl 2-bromoacetate (or ethyl 2-bromovalerate) (2.5 mmol) in absolute EtOH (5 ml), was added to the mixture, and the resulting solution was refluxed for 20 h. After evaporation of solvent under reduced pressure, the residue was poured into water (20 ml) and extracted with Et2O (3 × 20 mL). The organic layer was dried over Na2SO4 and concentrated under reduced pressure.
2.1.2.1. Ethyl (4-benzoylphenoxy)acetate (29)
White solid, 63% yield. Characterisation data were in agreement with those reported in the literature23.
2.1.2.2. Ethyl 2–(4-benzoylphenoxy)pentanoate (31)
Pale yellow oil, 69% yield (silica gel, DCM); 1H NMR (CDCl3) δ0.98 (t, 3H, J 6.9 Hz, CH3), 1.25 (t, 3H, J 6.9 Hz, CH3), 1.49–1.63 (m, 2H, CH2), 1.83–2.08 (m, 2H, CH2), 4.23 (q, 2H, J 7.2 Hz, OCH2), 4.70 (dd, 1H, J 7.8 Hz, J 7.5 Hz, CH), 6.92 (d, 2H, J 8.7 Hz, CHAr), 7.43–7.48 (m, 2H, CHAr), 7.52–7.59 (m, 1H, CHAr), 7.75 (d, 2H, J 7.2 Hz, CHAr), 7.79 (d, 2H, J 7.2 Hz, CHAr); 13C NMR (CDCl3) δ 13.6, 14.1, 18.5, 34.6, 61.4, 76.2, 114.4, 128.2, 129.7, 130.7, 131.9, 132.5, 138.0, 161.4, 171.2, 195.5. Anal. Calcd. for C20H22O4: C, 73.60; H, 6.79; Found: C, 73.52; H, 6.81.
2.1.3. General procedure for the synthesis of acids (30 and 32)
2N NaOH (12.5 mmol) was added to esters 29 and 31 (1.5 mmol) in THF (10 ml) and the mixture was stirred at r.t. overnight. THF was removed under reduced pressure; the solution was acidified by 4N HCl, obtaining a precipitate that was collected by filtration under vacuum.
2.1.3.1. (4-Benzoylphenoxy)acetic acid (30)
White solid, 72% yield. Characterisation data were in agreement with those reported in the literature23.
2.1.3.2. 2-(4-Benzoylphenoxy)pentanoic acid (32)
White solid, 99% yield; m.p. 75–77 °C; 1H NMR (CDCl3) δ0.98 (t, 3H, J 7.2 Hz, CH3), 1.50–1.63 (m, 2H, CH2), 1.96–2.05 (m, 2H, CH2), 4.75 (dd, 1H, J 7.8 Hz, J 7.8 Hz, CH), 6.93 (d, 2H, J 8.7 Hz, CHAr), 7.43–7.48 (m, 2H, CHAr), 7.53–7.59 (m, 1H, CHAr), 7.74 (d, 2H, J 7.2 Hz, CHAr), 7.81 (d, 2H, J 7.2 Hz, CHAr); 13C NMR (CDCl3) δ 13.6, 18.5, 34.5, 75.6, 114.4, 128.2, 129.8, 130.9, 132.1, 132.6, 137.9, 161.2, 175.9, 195.7. Anal. Calcd. for C18H18O4: C, 72.47; H, 6.08; Found: C, 72.51; H, 6.07.
2.2. CA inhibition assays
An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activity. Phenol red (0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5, for α-CAs) as buffer, and 20 mM NaClO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalysed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. In particular, CO2 was bubbled in distilled deionised water for 30 min till saturation. A CO2 kit (Sigma, Milan, Italy) was used to measure the concentration in serially diluted solutions from the saturated one at the same temperature. For each inhibitor at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalysed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (1 µM) were prepared in distilled–deionised water and dilutions up to 0.1 nM were done thereafter with the assay buffer. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E–I complex or for the eventual active site mediated hydrolysis of the inhibitor. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3 and the Cheng–Prusoff equation and represent the average from at least three different determinations. All recombinant CA isoforms were obtained in-house as previously reported24,25.
2.3. Molecular modelling studies
Crystal structures of hCA I (pdb: 3lxe, 1.9 Å, in complex with topiramate), hCA II (pdb: 4e3d, 1.6 Å, in complex with 2,5-dihydroxybenzoic acid), hCA IX (pdb: 3iai, 2.2 Å, in complex with acetazolamide [AAZ]) and XII (pdb: 1jd0, 1.5 Å, in complex with AAZ) were obtained from the Brookhaven Protein Data Bank. All ligands (AAZ, topiramate and 2,5-dihydroxybenzoic acid) and the zinc-bound water molecule of the hCA II structure were retained and all other non-protein atoms were deleted. Chain A was retained if more than one protein structure was present in the crystal structure. Hydrogen atoms were added with the “protonate 3 D” tool and subsequently a steepest-descent energy minimisation was performed using the AMBER14:EHT force field (MOE software package, version 2018.0101, chemical computing group, Inc., Montreal, Canada)26. The four protein structures were superposed on the backbone atoms of hCA I (Cα atoms, RMSD: 1.395 Å, for 236 residues). The coordinates of the hCA II zinc-bound water molecule were copied into the other hCA structures.
The molecular structures of the ligands were prepared with the MOE software package. All stereoisomers were generated. Subsequently, the ligand structures were energy minimised (MMFF94x force field) and the ligands were saved as multi-mol2 files.
Docking calculations were performed with the GOLD software package version 5.6.1 (CCDC, Cambridge, UK) using the ChemScore scoring function (25 genetic algorithm runs per ligand) and default settings. The binding pocket was defined as within 14 Å around a centroid (x: −18.899; Y: 36.167; z: 45.640; corresponds to the AAZ O3 atom of the hCA IX structure after superposition upon the hCA I structure). Dockings into the active site were performed either with or without a zinc-bound water molecule (obtained from hCA II structure 4e3d)27.
3. Results and discussion
3.1. Synthetic approach and in vitro CA inhibition studies
A wide group of benzothiazole compounds, bearing an N-acylsulphonamide portion, have been firstly synthesised by our research group and tested as PPAR antagonists. Some of them were also studied for assessing antitumor effects, mainly in cancer models overexpressing PPARα28,29. We decided to evaluate their inhibitory effect on four selected human CA isoenzymes (I, II, IX, and XII) and AAZ was used as reference compound. A preliminary screening of compounds 1–7, methane and benzenesulphonamide derivatives, was carried out, and the results are reported in Table 1. The greater affinities showed by benzenesulphonamide derivatives prompted us to further explore benzenesulphonamide derivatives such as p-substitutions on the aromatic ring (8–14). In addition, we also tested compounds 15–18, previously synthesised starting from the parent compounds clofibrate, gemfibrozil, bezafibrate, and fenofibrate30.
Table 1.
Inhibitory activity of derivatives 1–18 and reference compound acetazolamide (AAZ) against four selected hCA isoforms (I, II, IX, and XII) by stopped-flow CO2 hydrase assay31–35.
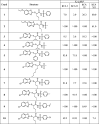 
|
The three methanesulphonamide derivatives (compounds 2, 4, 6) showed no activity against CA except for compound 2 which had a slight activity against hCA XII (Ki = 61.0 μM), whereas corresponding benzenesulphonamides (1, 3, 5) moderately inhibited CA I, II, and IX, with a slight preference for CA II. Compounds 1–4 and 6–7, bearing an alkylic chain on Cα (adjacent to carbonyl group), produced an improved CA inhibition compared to phenyl derivative 5.
The series of compounds 8–14 includes p-substituted benzenesulphonamides, obtained starting from compound 7. The best inhibition profiles against CA were found for p-methoxy derivative 9 and p-chloro derivative 10. They produced a selective inhibition with submicromolar Ki values against hCA IX and hCA II respectively. The insertion of methyl (8), nitro (11), acetylamino (12), benzoylamino (13) and phenylacetylamino (14) groups did not improve their inhibition properties.
Benzenesulphonamides 15–18, obtained by their carboxylic precursors, were all inactive against CA XII. Compounds 15 and 16 (clofibrate and gemfibrozil derivatives) were completely inactive against all tested isoenzymes, whereas 17 and 18 showed a moderate inhibition against CA I, II and IX. The best inhibition values were found for 18 (fenofibrate derivative), that showed submicromolar Ki inhibition against I and IX isoenzymes.
According to the above reported results, we selected fenofibrate derivative 18 as a novel lead compound to develop novel chemical derivatives, by adding substituents in para position to the benzenesulphonamide moiety (Figure 1) such as methyl, methoxy, chlorine, nitro, acetylamino, benzoylamino, and phenylacetylamino groups. Compounds 19–25 were easily synthesised by reacting fenofibric acid, obtained from hydrolysis of fenofibrate, with p-substituted benzenesulphonamides, in the presence of 1-ethyl-3–(3-dimethylaminopropyl)carbodiimide (EDC) and 4-dimethylaminopyridine (DMAP), in dry dichloromethane and under nitrogen atmosphere (Scheme 1).
Figure 1.

From lead compound 18 to novel benzenesulphonimides 19–27.
Scheme 1.

Reagents and conditions: (a) p-substituted benzenesulphonamide, EDC, DMAP, dry dichloromethane, 0 °C-r.t., 24 h.
With the aim to evaluate the effect of steric hindrance on Cα, we planned the synthesis of 26 and 27 (Figure 1), where the Cα was unsubstituted or substituted with an n-propylic chain; both derivatives are lacking of chlorine substitution on benzophenone ring.
Finally, products 26 and 27 were synthesised as depicted in Schemes 2–3. Esters 29 and 31 were obtained by nucleophilic displacement of proper ethyl 2-bromoalkanoates by 4-hydroxybenzophenone; hydrolysis by 2N NaOH afforded acids 30 and 32, that were coupled with benzenesulphonamide as previously described.
Scheme 2.

Reagents and conditions: (a) ethyl 2-bromoacetate, sodium, absolute ethanol, reflux, 20 h; (b) 2N NaOH, THF, r.t., 24 h; (c) benzenesulphonamide, EDC, DMAP, dry dichloromethane, 0 °C-r.t., 24 h.
Scheme 3.

Reagents and conditions: (a) ethyl 2-bromovalerate, sodium, absolute ethanol, reflux, 20 h; (b) 2N NaOH, THF, r.t., 24 h; (c) benzenesulphonamide, EDC, DMAP, dry dichloromethane, 0 °C-r.t., 24 h.
Compounds 19–27 were also screened against CA I, II, IX, and XII to derive robust SAR within this scaffold: results are shown in Table 2.
Table 2.
Inhibitory activity of derivatives 19–27 and reference compound acetazolamide (AAZ) against four selected hCA isoforms (hCA I, II, IX, and XII) by stopped-flow CO2 hydrase assay31,32.
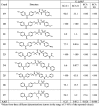 |
Overall, the p-substitution of aromatic ring with respect to parent derivative 18 did not improve hCA inhibition, except for compounds 22 and 24, showing both a marked inhibition of hCA II (Ki =16 and 77 nM, respectively), and a good selectivity versus this isoform. The presence of an acetylamino group was detrimental for hCA inhibition (23), whereas the introduction of an additional aromatic ring (24) produced a marked effect on both hCA I and hCA II. The further elongation of the substituent (25) decreased the inhibitory effect. Compounds 26 and 27, lacking of the chlorine atom on benzophenone structure and showing a different steric hindrance on Cα, did not induce a sensible inhibition of tested isoenzymes.
3.2. Molecular modelling studies
3.2.1. Docking studies into the active site of hCA I
The ligands were docked into the active site of hCA I with and without a zinc-bound water molecule. This water molecule was obtained from the hCA II structure (pdb: 4e3d) as described in the Materials and Methods section. Compounds 3, 18, 21, and 24 show lower Ki values than 10 μM (Tables 1 and 2). Two different poses have been identified in which the ligands can directly interact with the active site zinc ion. In the first pose, the carbonyl group located between the two phenyl moieties forms an interaction with the active site zinc ion (Figure 2(A)). The two phenyl groups form hydrophobic interactions with His64, Ala121, Val143, Leu198, His200, and Val207. A second hydrogen bond is formed between the other carbonyl atom of the ligand and the side chain of His67. The terminal unsubstituted phenyl group forms a hydrophobic interaction with the side chain of Val62. In addition, hydrogen atoms of His64 and Leu198 point towards the centroids of the phenyl rings resulting in aromatic-H bonds. The sulphonamide group is solvent exposed. This pose is observed for many compounds, including compounds 18 and 24.
Figure 2.

(A) The docked pose of compound 18 (turquoise), (B) compound 21 (purple) and (C) compound 1 (R-isomer; green) in the active site of hCA I (pdb: 3lxe). Hydrogen bonds and interactions to the active site zinc ion are indicated in red dashed lines. Aromatic system – H bonds are indicated in yellow dashed lines.
In the second docked pose, the sulphonamide moiety is located close the active site zinc ion (Figure 2(B)). One of the sulphonamide oxygen atoms forms an interaction with the zinc ion, while the other oxygen atom forms a hydrogen bond with the backbone of Thr199. The chlorine-substituted phenyl group forms hydrophobic interactions with Ala121, Leu141, Val143, and Leu198. One of the ligand’s hydrogen atoms points towards the centre of His94 to form aromatic-H bonds. No additional hydrogen bonds are observed between the ligand and the enzyme, however, the amide bond and the carbonyl group of the ligand are solvent accessible and may form interaction with water. This pose is observed for many compounds, including compounds 3, 18, and 21.
Docked poses have been identified in which the ligands directly interact with the zinc-bound water molecule. For example, the R isomer of compound 1 forms a hydrogen bond with the water molecule through its sulphonamide oxygen atom (Figure 2(C)). The carbonyl group of the ligand forms a hydrogen bond with the side chain of Gln92. A third hydrogen bond is formed between the ligand and the side chain of Trp5. The propyl group of the ligand is located in a hydrophobic pocket formed by Val62, His64, His67, and His200. The phenyl group of the ligand forms hydrophobic interactions with Phe91, Leu131, Leu141, and Leu198. Interestingly, similar docked poses have not been obtained for the S isomer of compound 1. Similar observations have been made for the other compounds in which some isoforms may bind to one stereoisomer but not to the other.
3.2.2. Docking studies into the active site of hCA II
The docked pose of compound 22 (Ki =16 nM) suggests that the nitro group could be located near the zinc ion (Figure 3(A)). The partially negatively charged oxygen atoms of the nitro group may form a hydrogen bond with the backbone of Thr199 and electrostatic interactions with the zinc ion. One of the sulphonamide oxygen atoms forms a hydrogen bond with the side chain of Gln92, while the other oxygen atom is water-accessible. The ligand amide group forms a hydrogen bond with the side chain of Asn67 and the chlorine substituent is close enough to the side chain of Lys170 for electrostatic interactions (distance = 4.5 Å). Hydrophobic interactions are formed mainly with Leu198. All hydrogen bond donors and acceptors of the ligand that do not interact with the active site are water-accessible.
Figure 3.

The docked pose of (A) compound 22 (turquoise) and (B) compound 18 (purple) in the active site of hCA II (pdb: 4e3d). Hydrogen bonds and interactions to the active site zinc ion are indicated in red dashed lines. Aromatic system – H bonds are indicated in yellow dashed lines.
The docked pose of compound 18 (Ki = 1.3 μM) indicates that the sulphonamide moiety of the ligand is located near the zinc ion (Figure 3(B)). One of the sulphonamide oxygen forms a direct interaction with the zinc ion, while the other oxygen atom may form hydrogen bonds with Leu198 (backbone) and/or Thr200 (backbone and side chain). One of the side chain hydrogen atoms of Asn67 points towards the centroid of the ligand phenyl group, which could lead to aromatic-H interactions. The chlorine substituent may form electrostatic interactions with Lys170 (distance =5.4 Å).
Many docked poses of the other ligands show a similar position for the sulphonamide group as obtained for compound 18.
3.2.3. Docking studies into the active site of hCA IX
The docked pose for compound 18 (Ki = 0.44 μM) indicates that the sulphonamide may be located close to the zinc ion, enabling one oxygen to form an interaction with the zinc ion (tetrahedral orientation around zinc) and the other oxygen atom to form hydrogen bonds with Thr200 (Figure 4(A)). The carbonyl group adjacent to the sulphonamide moiety is located close to the zinc ion and interaction may occur, resulting in a distorted trigonal bipyramidal orientation around zinc. The terminal phenyl moiety forms hydrophobic interactions with Val121, Val143, and Leu198. The central phenyl group forms hydrophobic interactions with Trp5, while the substituted phenyl group forms hydrophobic interactions with Pro202. A similar docked pose as described for compound 18 or other docked poses in which the ligand directly interacts with the zinc ion are not observed for most of the compounds.
Figure 4.

The docked pose of (A) compound 18 (purple) and (B) alternative docked pose of compound 18 (purple) in the active site of hCA IX (pdb: 3iai). Hydrogen bonds and interactions to the active site zinc ion are indicated in red dashed lines. Aromatic system – H bonds are indicated as yellow dashed lines.
An alternative docked pose has been obtained for compound 18 in which it interacts with the zinc-bound water molecule via one of its sulphonamide oxygen atoms (Figure 4(B)). The other oxygen atom forms a hydrogen bond to the side chain of Gln92. A second hydrogen bond is formed with the side chain of Trp5. The ligand phenyl group forms hydrophobic interactions with Pro202, while the two methyl groups are located in a hydrophobic cavity formed by Val131, Leu135, Leu141, and Leu198.
3.2.4. Docking studies into the active site of hCA XII
No docking poses have been obtained in which the ligands interact directly with the zinc ion or the zinc-bound water molecule.
4. Conclusions
This library of fibrate-based N-acylsulphonamides supported the exploration of other zinc binders for the inhibition of human CAs with respect to the widely studied primary sulphonamides. These secondary sulphonamides maintained the possibility to interact with the zinc ion through several binding modes. As also reported in previous crystallographic studies, they present a reduced submicromolar inhibitory activity, but they gained an interesting isoform selectivity against the four tested hCAs. With the support of molecular modelling studies, we assessed the structural requirements within this scaffold to further improve the biological activity such as the presence of an aryl ring on the sulphonamide, p-substitution of this aryl ring, benzothiazole or benzophenone as tailing moieties, and an n-propyl chain or a geminal dimethyl at Cα carbon.
Funding Statement
This work was supported by FAR funds (Italian Ministry for Instruction, University and Research) of Alessandra Ammazzalorso.
Disclosure statement
The authors state no conflict of interest and they have received no payment in preparation of this manuscript.
References
- 1.(a) Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–81. [DOI] [PubMed] [Google Scholar]; (b) Carta F, Supuran CT, Scozzafava A. Sulfonamides and their isosters as carbonic anhydrase inhibitors. Future Med Chem 2014;6:1149–65. [DOI] [PubMed] [Google Scholar]
- 2.Khan FA, Mushtaq S, Naz S, et al. Sulfonamides as potential bioactive scaffolds. Curr Org Chem 2018;22:818–30. [Google Scholar]
- 3.Ammazzalorso A, De Filippis B, Giampietro L, Amoroso R. N-acylsulfonamides: synthetic routes and biological potential in medicinal chemistry. Chem Biol Drug Des 2017;90:1094–105. [DOI] [PubMed] [Google Scholar]
- 4.Meanwell NA. Synopsis of some recent tactical application of bioisosteres in drug design. J Med Chem 2011;54:2529–91. [DOI] [PubMed] [Google Scholar]
- 5.Pilot-Matias T, Tripathi R, Cohen D, et al. In vitro and in vivo antiviral activity and resistance profile of the hepatitis C virus NS3/4A protease inhibitor ABT-450. Antimicrob Agents Chemother 2015;59:988–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Souers AJ, Leverson JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 2013;19:202–8. [DOI] [PubMed] [Google Scholar]
- 7.Asaki T, Kuwano K, Morrison K, et al. An oral and selective IP prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. J Med Chem 2015;58:7128–37. [DOI] [PubMed] [Google Scholar]
- 8.Winton VJ, Aldrich C, Kiessling LL. Carboxylate surrogates enhance the antimycobacterial activity of UDP-galactopyranose mutase probes. ACS Infect Dis 2016;2:538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patil V, Kale M, Raichurkar A, et al. Design and synthesis of triazolopyrimidine acylsulfonamides as novel anti-mycobacterial leads acting through inhibition of acetohydroxyacid synthase. Bioorg Med Chem Lett 2014;24:2222–5. [DOI] [PubMed] [Google Scholar]
- 10.Koroniak L, Ciustea M, Gutierrez JA, Richards N. Synthesis and characterization of an N-acylsulfonamide inhibitor of human asparagine synthetase. Org Lett 2003;5:2033–6. [DOI] [PubMed] [Google Scholar]
- 11.Ohashi M, Oyama T, Miyachi H. Different structures of the two peroxisome proliferator-activated receptor gamma (PPARγ) ligand-binding domains in homodimeric complex with partial agonist, but not full agonist. Bioorg Med Chem Lett 2015;25:2639–44. [DOI] [PubMed] [Google Scholar]
- 12.Ammazzalorso A, De Lellis L, Florio R, et al. Cytotoxic effect of a family of peroxisome proliferator-activated receptor antagonists in colorectal and pancreatic cancer cell lines. Chem Biol Drug Des 2017;90:1029–35. [DOI] [PubMed] [Google Scholar]
- 13.(a) Sapegin A, Kalinin S, Angeli A, et al. Unprotected primary sulfonamide group facilitates ring-forming cascade en route to polycyclic [1,4]oxazepine-based carbonic anhydrase inhibitors. Bioorg Chem 2018;76:140–6. [DOI] [PubMed] [Google Scholar]; (b) Supuran CT. Structure-based drug discovery of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2012;27:759–72. [DOI] [PubMed] [Google Scholar]; (c) Melis C, Meleddu R, Angeli A, et al. Isatin: a privileged scaffold for the design of carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2017;32:68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Petrou A, Geronikaki A, Terzi E, et al. Inhibition of carbonic anhydrase isoforms I, II, IX and XII with secondary sulfonamides incorporating benzothiazole scaffolds. J Enzyme Inhib Med Chem 2016;31:1306–11. [DOI] [PubMed] [Google Scholar]; (b) Gul HI, Mete E, Taslimi P, et al. Synthesis, carbonic anhydrase I and II inhibition studies of the 1,3,5-trisubstituted-pyrazolines. J Enzyme Inhib Med Chem 2017;32:189–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) D’Ascenzio M, Guglielmi P, Carradori S, et al. Open saccharin-based secondary sulfonamides as potent and selective inhibitors of cancer-related carbonic anhydrase IX and XII isoforms. J Enzyme Inhib Med Chem 2017;32:51–9. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pustenko A, Stepanovs D, Žalubovskis R, et al. 3H-1,2-benzoxathiepine 2,2-dioxides: a new class of isoform-selective carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem 2017;32:767–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alp C, Maresca A, Alp NA, et al. Secondary/tertiary benzenesulfonamides with inhibitory action against the cytosolic human carbonic anhydrase isoforms I and II. J Enzyme Inhib Med Chem 2013;28:294–8. [DOI] [PubMed] [Google Scholar]
- 17.(a) Alterio V, Di Fiore A, D’Ambrosio K, et al. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms? Chem Rev 2012;112:4421–68. [DOI] [PubMed] [Google Scholar]; (b) Supuran CT. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin Ther Pat 2018;28:709–12. [DOI] [PubMed] [Google Scholar]; (c) Krasavin M, Korsakov M, Ronzhina O, et al. Primary mono- and bis-sulfonamides obtained via regiospecific sulfochlorination of N-arylpyrazoles: inhibition profile against a panel of human carbonic anhydrases. J Enzyme Inhib Med Chem 2017;32:920–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yıldırım A, Atmaca U, Keskin A, et al. N-Acylsulfonamides strongly inhibit human carbonic anhydrase isoenzymes I and II. Bioorg Med Chem 2015;23:2598–605. [DOI] [PubMed] [Google Scholar]
- 19.(a) Awadallah FM, Bua S, Mahmoud WR, et al. Inhibition studies on a panel of human carbonic anhydrases with N1-substituted secondary sulfonamides incorporating thiazolinone or imidazolone-indole tails. J Enzyme Inhib Med Chem 2018;33:629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zengin Kurt B, Sonmez F, Durdagi S, et al. Synthesis, biological activity and multiscale molecular modeling studies for coumaryl-carboxamide derivatives as selective carbonic anhydrase IX inhibitors. J Enzyme Inhib Med Chem 2017;32:1042–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Di Fiore A, Maresca A, Alterio V, et al. Carbonic anhydrase inhibitors: x-ray crystallographic studies for the binding of N-substituted benzenesulfonamides to human isoform II. Chem Commun (Camb) 2011;47:11636–8. [DOI] [PubMed] [Google Scholar]; (b) Briganti F, Pierattelli R, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Part 37. Novel classes of carbonic anhydrase inhibitors and their interaction with the native and cobalt-substituted enzyme: kinetic and spectroscopic investigations. Eur J Med Chem 1996;31:1001–10. [Google Scholar]
- 21.Ammazzalorso A, Giancristofaro A, D’Angelo A, et al. Benzothiazole-based N-(phenylsulfonyl)amides as a novel family of PPARα antagonists. Bioorg Med Chem Lett 2011;21:4869–72. [DOI] [PubMed] [Google Scholar]
- 22.Ammazzalorso A, D'Angelo A, Giancristofaro A, et al. Fibrate-derived N-(methylsulfonyl)amides with antagonistic properties on PPARα. Eur J Med Chem 2012;58:317–22. [DOI] [PubMed] [Google Scholar]
- 23.Giampietro L, D’Angelo A, Giancristofaro A, et al. Effect of stilbene and chalcone scaffolds incorporation in clofibric acid on PPARα agonistic activity. Med Chem 2014;10:59–65. [DOI] [PubMed] [Google Scholar]
- 24.D’Ascenzio M, Carradori S, Secci D, et al. Selective inhibition of human carbonic anhydrases by novel amide derivatives of probenecid: synthesis, biological evaluation and molecular modelling studies. Bioorg Med Chem 2014;22:3982–8. [DOI] [PubMed] [Google Scholar]
- 25.De Monte C, Carradori S, Secci D, et al. Cyclic tertiary sulfamates: selective inhibition of the tumor-associated carbonic anhydrases IX and XII by N- and O-substituted acesulfame derivatives. Eur J Med Chem 2014;84:240–6. [DOI] [PubMed] [Google Scholar]
- 26.Labute P. Protonate3D: assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins 2009;75:187–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) Akdemir A, De Monte C, Carradori S, Supuran CT. Computational investigation of the selectivity of salen and tetrahydrosalen compounds towards the tumor-associated hCA XII isozyme. J Enzyme Inhib Med Chem 2015;30:114–8. [DOI] [PubMed] [Google Scholar]; (b) Güzel-Akdemir Ö, Angeli A, Demir K, et al. Novel thiazolidinone-containing compounds, without the well-known sulphonamide zinc-binding group acting as human carbonic anhydrase IX inhibitors. J Enzyme Inhib Med Chem 2018;33:1299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ramya PVS, Angapelly S, Angeli A, et al. Discovery of curcumin inspired sulfonamide derivatives as a new class of carbonic anhydrase isoforms I, II, IX, and XII inhibitors. J Enzyme Inhib Med Chem 2017;32:1274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Florio R, De Lellis L, di Giacomo V, et al. Effects of PPARα inhibition in head and neck paraganglioma cells. PLoS One 2017;12:e0178995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benedetti E, d'Angelo M, Ammazzalorso A, et al. PPARα antagonist AA452 triggers metabolic reprogramming and increases sensitivity to radiation therapy in human glioblastoma primary cells. J Cell Physiol 2017;232:1458–66. [DOI] [PubMed] [Google Scholar]
- 30.Ammazzalorso A, Carrieri A, Verginelli F, et al. Synthesis, in vitro evaluation, and molecular modeling investigation of benzenesulfonimide Peroxisome Proliferator-Activated Receptors α antagonists. Eur J Med Chem 2016;114:191–200. [DOI] [PubMed] [Google Scholar]
- 31.Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–73. [PubMed] [Google Scholar]
- 32.Mocan A, Carradori S, Locatelli M, et al. Bioactive isoflavones from Pueraria lobata root and starch: Different extraction techniques and carbonic anhydrase inhibition. Food Chem Toxicol 2018;112:441–7. [DOI] [PubMed] [Google Scholar]
- 33.(a) Supuran CT.Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert Opin Drug Discov 2017;12:61–88. [DOI] [PubMed] [Google Scholar]; (b) Supuran CT.Structure and function of carbonic anhydrases. Biochem J 2016;473:2023–32. [DOI] [PubMed] [Google Scholar]; (c) Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–77. [DOI] [PubMed] [Google Scholar]; (d) Supuran CT, Vullo D, Manole G, et al. Designing of novel carbonic anhydrase inhibitors and activators. Curr Med Chem Cardiovasc Hematol Agents 2004;2:49–68. [PubMed] [Google Scholar]; (e) Akocak S, Lolak N, Bua S, Supuran CT. Discovery of novel 1,3-diaryltriazene sulfonamides as carbonic anhydrase I, II, VII, and IX inhibitors. J Enzyme Inhib Med Chem 2018;33:1575–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.(a) Supuran CT. How many carbonic anhydrase inhibition mechanisms exist? J Enzyme Inhib Med Chem 2016;31:345–60. [DOI] [PubMed] [Google Scholar]; (b) Rehman SU, Chohan ZH, Gulnaz F, Supuran CT. In-vitro antibacterial, antifungal and cytotoxic activities of some coumarins and their metal complexes. J Enzyme Inhib Med Chem 2005;20:333–40. [DOI] [PubMed] [Google Scholar]; (c) Clare BW, Supuran CT. Carbonic anhydrase activators. 3: structure‐activity correlations for a series of isozyme II activators. J Pharm Sci 1994;83:768–73. [DOI] [PubMed] [Google Scholar]; (d) Ozensoy Guler O, Capasso C, Supuran CT. A magnificent enzyme superfamily: carbonic anhydrases, their purification and characterization. J Enzyme Inhib Med Chem 2016;31:689–94. [DOI] [PubMed] [Google Scholar]; (e) De Simone G, Langella E, Esposito D, et al. Insights into the binding mode of sulphamates and sulphamides to hCA II: crystallographic studies and binding free energy calculations. J Enzyme Inhib Med Chem 2017;32:1002–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.(a) Supuran CT. Carbonic anhydrase inhibitors in the treatment and prophylaxis of obesity. Expert Opin Ther Pat 2003;13:1545–50. [Google Scholar]; (b) Winum JY, Temperini C, El Cheikh K, et al. Carbonic anhydrase inhibitors: clash with Ala65 as a means for designing inhibitors with low affinity for the ubiquitous isozyme II, exemplified by the crystal structure of the topiramate sulfamide analogue. J Med Chem 2006;49:7024–31. [DOI] [PubMed] [Google Scholar]; (c) Gul HI, Mete E, Eren SE, et al. Designing, synthesis and bioactivities of 4-[3-(4-hydroxyphenyl)-5-aryl-4,5-dihydro-pyrazol-1-yl]benzenesulfonamides. J Enzyme Inhib Med Chem 2017;32:169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Alper Türkoğlu E, Şentürk M, Supuran CT, Ekinci D. Carbonic anhydrase inhibitory properties of some uracil derivatives. J Enzyme Inhib Med Chem 2017;32:74–7. [DOI] [PMC free article] [PubMed] [Google Scholar]