

Abstract
Background
Hyperammonemia can result in hepatic encephalopathy, which in severe cases eventually can lead to coma and death. In dogs, congenital portosystemic shunts (CPSS) are the most common cause for hyperammonemia. Conservative treatment consists of a protein modified diet, nonabsorbable disaccharides, antibiotics, or some combinations of these. Sodium benzoate (SB) and sodium phenylbutyrate (SPB) both are used in the acute and long‐term treatment of humans with hyperammonemia caused by urea cycle enzyme deficiencies. Both treatments are believed to lower blood ammonia concentrations by promoting excretion of excess nitrogen via alternative pathways.
Objectives
To evaluate the efficacy and safety of PO treatment with SB and SPB on hyperammonemia and clinical signs in CPSS dogs.
Methods
Randomized, double‐blind, placebo‐controlled crossover trial. Concentrations of blood ammonia and bile acids were measured in CPSS dogs before and after a 5‐day treatment with SB, SPB, and placebo. A wash‐out period of 3 days was used between treatments. A standard questionnaire was developed and distributed to owners to evaluate clinical signs before and after each treatment.
Results
Blood ammonia concentrations were not influenced by any of the treatments and were comparable to those observed during placebo treatment. In addition, SB and SPB treatment did not result in improvement of clinical signs. Adverse effects during treatment included anorexia, vomiting, and lethargy.
Conclusions and Clinical Importance
Based on our results, we conclude that SB or SPB are not useful in the conservative treatment of hyperammonemia in dogs with CPSS.
Keywords: canine, hepatic encephalopathy, hyperammonemia
Abbreviations
- ANOVA
analysis of variance
- CPSS
congenital portosystemic shunts
- HA
hippuric acid
- HE
hepatic encephalopathy
- PAG
phenylacetylglycine
- SB
sodium benzoate
- SPA
sodium phenylacetate
- SPB
sodium phenylbutyrate
- UCED
urea cycle enzyme deficiency
1. INTRODUCTION
Ammonia is a key factor in the pathogenesis of hepatic encephalopathy (HE),1, 2, 3, 4 a syndrome that comprises a broad spectrum of neurodegenerative complications and in severe cases may even lead to coma and death.5 Ammonia detoxification is thus essential for maintaining homeostasis and occurs mainly by conversion of ammonia into urea by the urea cycle in the liver and by glutamine synthesis.6
Hyperammonemia in dogs is mostly secondary to congenital portosystemic shunts (CPSS).7, 8, 9 These CPSS are vascular anomalies that connect the portal vein to the systemic circulation, thereby bypassing the hepatic sinusoids.10 As a result, blood flow from the portal vein through the liver parenchyma can be decreased by >95%,11 carrying intestinal absorption products (eg, NH3) directly into the systemic circulation. This results in a decrease in ureagenesis and causes hyperammonemia.11 Decreased expression of the urea cycle enzymes, which has been shown to occur in CPSS dogs,12 also may play a role in the pathogenesis of hyperammonemia in affected dogs.
Surgical closure of the shunt is considered the ideal treatment of CPSS and was shown to result in a significantly longer survival time compared to conservatively treated dogs with CPSS.13 Conservative management is necessary when complete shunt closure is not achieved after surgical intervention, when surgery is medically contraindicated and in the event of financial constraints. Recommended conservative treatment is aimed at decreasing ammonia production and absorption from the gastrointestinal tract. Suggested treatments consist of a vegetable protein or protein‐restricted diet in combination with medical interventions such as administration of nonabsorbable disaccharides (eg, lactulose), antibiotics (eg, neomycin and metronidazole), or both.14
For many years, IV sodium benzoate (SB) and sodium phenylacetate (SPA) have been recommended for acute treatment of hyperammonemia and HE in human patients with urea cycle enzyme deficiencies (UCED). Both SB and SPA were reported to lower blood ammonia concentrations by providing alternative pathways for nitrogen excretion15, 16, 17 (Figure 1). In dogs, benzoate and phenylacetate form conjugates with glycine to form hippuric acid (HA) and phenylacetylglycine (PAG), respectively.18, 19, 20 Both HA and PAG are excreted by the kidneys, thereby each removing 1 mole of nitrogen for every mole of SB and SPA administered.21, 22
Figure 1.
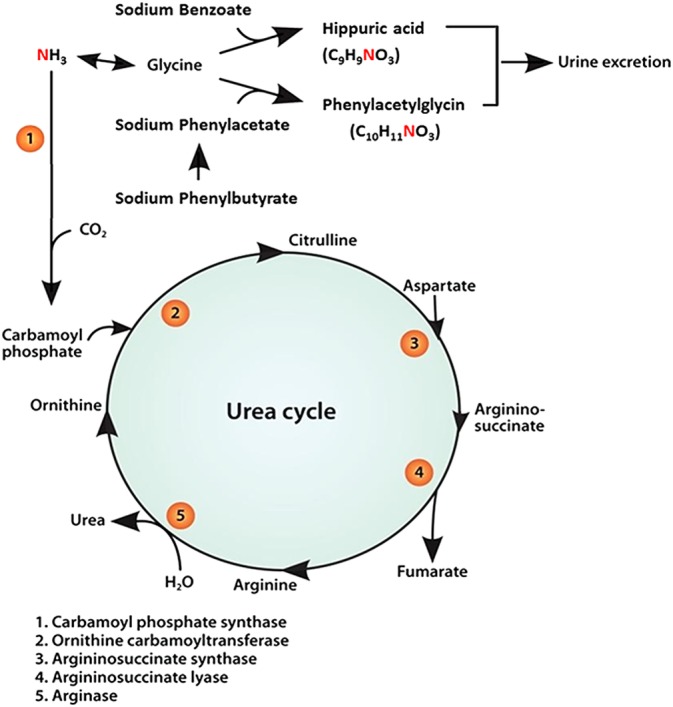
Schematic overview of SB and SPB metabolism. Sodium phenylbutyrate is oxidized into phenylacetate. Phenylacetate conjugates with glycine to form phenylacetylglycine (PAG) and SB conjugates with glycin to form hippuric acid (HA). Hippuric acid and PAG are excreted in the urine, each removing one nitrogen molecule. SB, sodium benzoate, SPB, sodium phenylbutyrate Modified from Misel et al23
In a recent study,18 the efficacy and safety of IV SB and SPA were evaluated in healthy and CPSS dogs. Elimination half‐lives of SB and SPA were 2.7 and 2.4 hours, respectively.18 These values are similar to those reported in humans. Both treatments were shown to have only mild adverse effects. However, treatments were only as effective as placebo treatment (ie, 0.9% NaCl) in decreasing ammonium concentrations when used as an IV bolus in treatment of hyperammonemia.
It is possible that prolonged treatment may be necessary to show efficacy in lowering ammonia concentrations. For PO treatment (ie, treatment for chronic hyperammonemia) the pro‐drug sodium phenylbutyrate (SPB) has been used.23 Sodium phenylbutyrate is rapidly oxidized to phenylacetate (Figure 1) and has the advantage of lacking the unpleasant odor of SPA, while maintaining its nitrogen removal efficacy.24 Our aim was to evaluate the efficacy and safety of monotherapy with SB and SPB in management of hyperammonemia and clinical signs in CPSS dogs.
2. ANIMALS AND METHODS
2.1. Animals
The study population consisted of dogs referred to the Department of Clinical Sciences of Companion Animals of the Utrecht University in which a definitive diagnosis of CPSS was made. Inclusion criteria were a confirmed diagnosis of CPSS based on increased concentrations of blood ammonia (reference interval, 15‐45 μmol/L) and bile acid concentrations (reference interval, 0‐10 μmol/L) and visualization of the shunt by ultrasonography, contrast computed tomography, or both. Dogs were excluded from the study when no definitive diagnosis of CPSS could be made, when blood ammonia concentration was <45 μmol/L at the start of the study or when dogs had received antibiotics, lactulose, or both within 2 weeks before the study. Written consent was obtained from each patient owner. All dogs were fed the same (commercial) hepatic diet.
2.2. Study design
The study was designed as a prospective, randomized, double‐blinded, placebo‐controlled 3‐arm crossover study. During the clinical trial, dogs were randomly assigned to receive 1 of the following treatment sequences: ABC, ACB, BCA, BAC, CBA, or CAB, in which A was placebo treatment, B was SPB treatment, and C was SB treatment. The dogs received a dosage of 250 mg/kg/day of SB and SPB, respectively, or placebo, all administered for a period of 5 days q8h. Between treatment periods, washout periods of 3 days were included in the protocol to avoid carry‐over effects. The short elimination half‐lives of SB and SPA result in attaining steady‐state concentrations within 2 doses and the wash‐out period represents >24 times the half‐lives, thus ensuring complete clearance of the drugs from the body. Immediate release capsules result in rapid absorption of SB and SPB with a Tmax of approximately 1‐1.5 hours. The Tmax of the active metabolite SPA is attained at approximately 3.5 hours in humans.25 All these pharmacokinetic parameters support the short washout period. Because the efficacy of parenteral treatment in patients responding to treatment was shown by mean ammonia concentrations decreasing significantly within 4 hours of initiation of treatment (SPC AMMONUL), treatment duration of 5 days for each drug was deemed sufficient to evaluate treatment outcomes. All study personnel as well as the owners of the dogs were blinded to treatment group assignment. The doses and treatment protocol were equivalent to protocols used in humans23 and a study evaluating the efficacy and safety of IV SB and SPA in dogs.18
2.3. Drug preparation
Placebo and SB 500 mg were manufactured by the pharmacy of the Faculty of Veterinary Medicine, Utrecht University, The Netherlands in no. 0 capsules, containing only microcrystalline cellulose (Bufa, Haarlem, The Netherlands) as placebo or containing SB (Bufa) and microcrystalline cellulose. The SPB 500 mg capsules (GMP pharmacy Mierlo‐Hout, Helmond, The Netherlands) were of equal size but white, also containing tricalcium phosphate, magnesium stearate, and siliciumdioxide as excipients. Although these capsules had a slightly different appearance, the owners and investigators did not know which substance the capsules contained. All treatments were dispensed weekly as the appropriate drug in the individual dose by the pharmacy, labeled with trial medication and dosing instructions, patient name, and subject number. Dispensing of the drugs was verified in the pharmacy, and returned bottles were collected by the researchers and checked for unused capsules.
2.4. Blood sampling and analysis
Venous blood samples for the determination of ammonia and bile acid concentrations were collected before treatment initiation (ie, pretreatment concentrations) and at the end of the 5‐day treatment period for each administered substance (ie, posttreatment concentrations). Blood was drawn from the jugular vein 2 and 4 hours after feeding. For ammonia measurements, blood was immediately placed in ice‐chilled sodium ethylenediaminetetraacetic acid (NaEDTA)‐coated tubes. Measurements of ammonia were performed within 10 minutes after blood sampling by a microdiffusion method using PocketChem (Menarini Benelux BV, Valkenswaard, The Netherlands). Bile acid concentrations were determined to evaluate liver function during treatment and to exclude that potential decreases in blood ammonia concentrations were a consequence of improved liver function. For bile acid measurements, blood was placed in 1.3‐mL heparin‐coated tubes. Bile acids were determined using a UniCel DxC 600 assay (Beckman Coulter Nederland BV, Woerden, The Netherlands).
2.5. Scoring system
The efficacy of the treatments also was evaluated by improvement in clinical signs. To do so, a standard questionnaire was developed and distributed to the owners to evaluate clinical signs before and after each treatment. Questions were intended to evaluate neurological signs (eg, decreased level of consciousness, weakness, decreased endurance, decreased playing behavior, decreased interaction with other dogs, convulsions, ataxia, disorientation, head pressing, circling, apparent blindness, aggression), gastrointestinal signs (eg, anorexia, vomiting, diarrhea), and urological signs (eg, polyuria, polydipsia, dysuria, hematuria). A scale from 0 to 4 was used for scoring of each clinical sign. A score 0 meant a sign was not seen at all during that period. A score of 1 was given when a sign was seen once, a score of 2 when a sign was seen >2 times but not every day, a score 3 when a sign was seen every day, and a score of 4 when a sign was present >1 time daily. Polyuria and polydipsia were scored 0, 2, or 4, meaning normal, slightly more than normal, and much more than normal, respectively. Endurance and willingness to play and interact with other dogs were scored 0, 2, or 4, with 0 meaning normal, 2 meaning less, and 4 meaning almost absent.
2.6. Statistical analysis
After testing the blood chemistry data for normality using the Kolmogorov‐Smirnov Z‐test, parametric tests were used to evaluate for differences between groups. A Kruskal‐Wallis test was used to evaluate differences between treatment groups at the outset. A 1‐way analysis of variance (ANOVA) for repeated measures was used to test for the presence of carryover effects and for differences between treatments. Post hoc, a pairwise comparison of the difference was performed using Tukey's test. The Wilcoxon signed‐rank test for repeated measurements was used to test for differences of the scoring systems. P‐values <.05 were considered significant. Results are expressed as a mean ± SD or as median and range when they were not normally distributed.
3. RESULTS
Ten dogs (5 males and 5 females) were enrolled in the study, ranging in age from 3 months to 10 years and weighing 1.5‐36 kg. The 10 dogs included 1 Nova Scotia Duck Tolling Retriever, 1 Doberman, 2 Chihuahuas, 1 Cairn Terrier, 1 Miniature Schnauzer, 1 Weimaraner, 1 Bernese Mountain dog, 1 Yorkshire Terrier, and 1 medium sized crossbreed dog. Nine dogs completed all treatments. One dog developed paraparesis because of discospondylitis and did not participate in the SB arm. Adverse effects that were noticed were anorexia (4 dogs on placebo and 2 dogs on SB treatment), vomiting (2 dogs on placebo, 3 dogs on SPB, and 2 dogs on SB treatment), and lethargy (3 dogs on placebo, 1 on SPB, and 3 on SB treatment). Adverse effects were noticed during both placebo and SB and SPB treatments. All adverse effects were temporary and resolved without treatment.
With 1‐way ANOVA for repeated measurements, it was first determined if there was a carryover effect. The test determined that the sequence of the treatments had no effect on the outcome of the variables recorded. No significant differences were found between plasma bile acid and blood ammonia concentrations at 2 and 4 hours after feeding. Therefore, the average of both time points (2 and 4 hours after feeding) was used to evaluate treatment effect on plasma ammonia and bile acid concentrations (Supporting Information Table 1 and Figure 1). Blood ammonia concentrations ranged between 60 and 286 μmol/L. No significant differences in blood ammonia concentrations were found between treatment groups at the start of the treatment. No significant differences in blood ammonia concentrations were found before and after treatment with placebo, SPB, or SB treatment. Ammonia concentrations before and after placebo treatment were 164 ± 86 μmol/L and 149 ± 78 μmol/L, respectively (P = .10). Pre‐ and post‐treatment ammonia concentrations with SB treatment were 172 ± 58 μmol/L and 187 ± 66 μmol/L (P = .21) and with SPB treatment were 154 ± 74.0 μmol/L and 155 ± 90 μmol/L (P = .64), respectively.
Plasma bile acid concentrations ranged between 39 and 630 μmol/L. No significant differences in plasma bile acid concentrations were found between treatment groups at the start of the treatment. No significant differences in plasma bile acid concentrations were found before or after any of the treatments. Pre‐ and post‐treatment bile acid concentrations with placebo were 303 ± 152 μmol/L and 264 ± 118 μmol/L, respectively (P = .08). Pre‐ and post‐treatment bile acid concentrations with SB were 344 ± 99 μmol/L and 241 ± 85 μmol/L (P = .37) and with SPB treatment were 293 ± 106 μmol/L and 229 ± 86 μmol/L (P = .18), respectively. Clinical signs present in dogs before treatment were polyuria, polydipsia, or both (n = 5), intermittent abnormal behavior (n = 5), lethargy (n = 6), decreased endurance (n = 8), anorexia (n = 6), vomiting (n = 6), and ataxia (n = 3). No significant differences in clinical sign scores were found before and after SB, SPB, and placebo treatment.
4. DISCUSSION
We evaluated both the efficacy and the safety of the PO treatment of chronic hyperammonemia using SB and SPB in dogs with CPSS. Orally administered SB and SPB did not result in significant decreases in blood ammonia concentrations or improvement in clinical signs related to CPSS. Although SB and SPA were reported to decrease ammonia concentrations in human patients, high‐level evidence (ie, systematic reviews of randomized controlled trials, findings of randomized, placebo‐controlled clinical trials) is lacking to prove the actual efficacy of these treatments. To our knowledge, ours is the first placebo‐controlled clinical trial assessing the efficacy of PO treatment with SB and SPB. The lack of efficacy of SB and SPB treatment found in our study is in accordance with results of previous studies reporting SB and SPA to be lacking in efficacy treatments as treatments for hyperammonemia. No decrease in blood ammonia concentration, despite significant excretion of HA in the urine, also was seen in a study in which rats with portocaval shunts were treated PO with SB.26 Furthermore, in other studies performed in UCED patients,27, 28, 29 treatment with SB and SPB did not result in a decrease of blood ammonia concentration in all patients, and in some patients, ammonia concentrations even increased after treatment. An increase in ammonia concentrations also was observed in some of the dogs in our study.
In a placebo‐controlled, randomized crossover study evaluating the efficacy of IV SB for the treatment of acute hyperammonemia in CPSS dogs,18 blood ammonia concentrations were significantly decreased (approximately 60%) after SB treatment. However, the same effect was achieved after treating the dogs with the same volume of IV 0.9% NaCl (ie, placebo treatment), and no significant differences in ammonia concentrations were found between SB and NaCl treatments. In the same study, administering NaCl to healthy dogs resulted in significantly lower ammonia concentrations compared to SB and SPA treatments. Volume expansion, rather than the excretion of nitrogen via alternative pathways, was suggested as a possible mechanism for the decrease in ammonia concentrations after SB and SPA bolus treatments. The fact that ammonia concentrations were not affected after PO treatment with SB, SPB, and placebo in our study strengthens this assumption because volume expansion does not occur with PO treatment.
Both SB and SPB treatments were shown to be safe and well‐tolerated in all dogs. The adverse effects (ie, anorexia, vomiting, and lethargy) observed were mild, of short duration, and comparable to those reported in previous studies in dogs18 and humans23, 30, 31 and with placebo treatment. However, vomiting, anorexia, and lethargy also occurred before treatment as a result of the CPSS and may be regarded as a consequence of unsuccessful treatment of CPSS.
In conclusion, SB and SPB did not alter ammonia concentrations in this cohort of dogs with CPSS or ameliorate clinical signs. Therefore, we do not recommend use of these substances, either as monotherapy or as part of combination therapy, to treat hyperammonemia in dogs with CPSS.
CONFLICT OF INTEREST DECLARATION
Authors declare no conflict of interest.
OFF‐LABEL ANTIMICROBIAL DECLARATION
Authors declare no off‐label use of antimicrobials.
INSTITUTIONAL ANIMAL CARE AND USE COMMITTEE (IACUC) OR OTHER APPROVAL DECLARATION
A written consent was obtained from each animal owner.
HUMAN ETHICS APPROVAL DECLARATION
Authors declare human ethics approval was not needed for this study.
Supporting information
Supplemental Figure 1 Whisker plot of ammonia concentrations before and after treatments in 10 CPSS dogs. Plac; Placebo, SB; sodium benzoate, SPB; sodium phenylbutyrate, BT; before treatment, AT; after treatment. Treatments were each of five‐day duration with wash‐out periods of three days. Each box and whisker plot illustrates the median (line in the middle), mean (+), 25th and 75th percentiles (top and bottom of each box); whiskers extend from the 5th and 95th percentile values.
Supplemental Table 1 AMmonia and bile acid concentrations before and after treatments in 10 CPSS dogs.
van Straten G, van Dalen D, Mesu SJ, et al. Efficacy of orally administered sodium benzoate and sodium phenylbutyrate in dogs with congenital portosystemic shunts. J Vet Intern Med. 2019;33:1331–1335. 10.1111/jvim.15477
REFERENCES
- 1. Perazzo JC, Tallis S, Delfante A, et al. Hepatic encephalopathy: an approach to its multiple pathophysiological features. World J Hepatol. 2012;4:50‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Butterworth RF. Pathophysiology of hepatic encephalopathy: a new look at ammonia. Metab Brain Dis. 2002;17:221‐227. [DOI] [PubMed] [Google Scholar]
- 3. Butterworth RF, Giguere JF, Michaud J, Lavoie J, Layrargues GP. Ammonia: key factor in the pathogenesis of hepatic encephalopathy. Neurochem Pathol. 1987;6:1‐12. [DOI] [PubMed] [Google Scholar]
- 4. Butterworth RF. Effects of hyperammonaemia on brain function. J Inherit Metab Dis. 1998;21(Suppl 1):6‐20. [DOI] [PubMed] [Google Scholar]
- 5. American Association for the Study of Liver Diseases, European Association for the Study of the Liver . Hepatic encephalopathy in chronic liver disease: 2014 practice guideline by the European Association for the Study of the liver and the American Association for the Study of Liver Diseases. J Hepatol. 2014;61:642‐659. [DOI] [PubMed] [Google Scholar]
- 6. Meijer AJ, Lamers WH, Chamuleau RAFM. Nitrogen metabolism and ornithine cycle function. Physiol Rev. 1990;70:701‐748. [DOI] [PubMed] [Google Scholar]
- 7. van Straten G, Spee B, Rothuizen J, van Straten M, Favier RP. Diagnostic value of the rectal ammonia tolerance test, fasting plasma ammonia and fasting plasma bile acids for canine portosystemic shunting. Vet J. 2015;204:282‐286. [DOI] [PubMed] [Google Scholar]
- 8. Ruland K, Fischer A, Hartmann K. Sensitivity and specificity of fasting ammonia and serum bile acids in the diagnosis of Portosystemic shunts in dogs and cats. Vet Clin Pathol. 2010;39:57‐64. [DOI] [PubMed] [Google Scholar]
- 9. Gerritzen‐Bruning MJ, Van Den Ingh TSGAM, Rothuizen J. Diagnostic value of fasting plasma ammonia and bile acid concentrations in the identification of portosystemic shunting in dogs. J Vet Intern Med. 2006;20:13‐19. [DOI] [PubMed] [Google Scholar]
- 10. Van den Ingh TS, Rothuizen J, Meyer HP. Portal hypertension associated with primary hypoplasia of the hepatic portal vein in dogs. Vet Rec. 1995;137:424‐427. [DOI] [PubMed] [Google Scholar]
- 11. Meyer HP, Rothuizen J, Van Den Brom WE, et al. Quantitation of portosystemic shunting in dogs by ultrasound‐guided injection of 99MTc‐macroaggregates into a splenic vein. Res Vet Sci. 1994;57:58‐62. [DOI] [PubMed] [Google Scholar]
- 12. van Straten G, van Steenbeek FG, Grinwis GC, et al. Aberrant expression and distribution of enzymes of the urea cycle and other ammonia metabolizing pathways in dogs with congenital portosystemic shunts. PLoS One. 2014;9:e100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Greenhalgh SN, Reeve JA, Johnstone T, et al. Long‐term survival and quality of life in dogs with clinical signs associated with a congenital portosystemic shunt after surgical or medical treatment. J Am Vet Med Assoc. 2014;245:527‐533. [DOI] [PubMed] [Google Scholar]
- 14. Ettinger SJ. Textbook of Veterinary Internal Medicine: Diseases of the Dog and Cat. Philadelphia: Saunders; 1975. [Google Scholar]
- 15. Batshaw ML, Brusilow SW. Treatment of hyperammonemic coma caused by inborn errors of urea synthesis. J Pediatr. 1980;97:893‐900. [DOI] [PubMed] [Google Scholar]
- 16. Batshaw ML, Brusilow S, Waber L, et al. Treatment of inborn errors of urea synthesis: activation of alternative pathways of waste nitrogen synthesis and excretion. N Engl J Med. 1982;306:1387‐1392. [DOI] [PubMed] [Google Scholar]
- 17. Brusilow S, Tinker J, Batshaw ML. Amino acid acylation: a mechanism of nitrogen excretion in inborn errors of urea synthesis. Science. 1980;207:659‐661. [DOI] [PubMed] [Google Scholar]
- 18. van Straten G, de Sain‐van der Velden MGM, van Geijlswijk IM, et al. Saline is as effective as nitrogen scavengers for treatment of Hyperammonemia. Sci Rep. 2017;7:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moldave K, Meister A. Synthesis of phenylacetylglutamine by human tissue. J Biol Chem. 1957;229:463‐476. [PubMed] [Google Scholar]
- 20. James MO, Smith RL, Williams RT, Reidenberg M. The conjugation of phenylacetic acid in man, sub‐human primates and some non‐primate species. Proc R Soc Lond B Biol Sci. 1972;182:25‐35. [DOI] [PubMed] [Google Scholar]
- 21. Tremblay GC, Qureshi IA. The biochemistry and toxicology of benzoic acid metabolism and its relationship to the elimination of waste nitrogen. Pharmacol Ther. 1993;60:63‐90. [DOI] [PubMed] [Google Scholar]
- 22. Iannitti T, Palmieri B. Clinical and experimental applications of sodium phenylbutyrate. Drugs R D. 2011;11:227‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Haberle J, Boddaert N, Burlina A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:1‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brusilow SW. Phenylacetylglutamine may replace urea as a vehicle for waste nitrogen excretion. Pediatr Res. 1991;29:147‐150. [DOI] [PubMed] [Google Scholar]
- 25. Phuphanich S, Baker SD, Grossman SA, et al. Oral sodium phenylbutyrate in patients with recurrent malignant Gliomas: a dose escalation and pharmacologic study. Neuro Oncol. 2005;7:177‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Steele RD. Hyperammonemia and orotic aciduria in portacaval‐shunted rats. J Nutr. 1984;114:210‐216. [DOI] [PubMed] [Google Scholar]
- 27. Brusilow SW, Danney M, Waber LJ, et al. Treatment of episodic hyperammonemia in children with inborn errors of urea synthesis. N Engl J Med. 1984;310:1630‐1634. [DOI] [PubMed] [Google Scholar]
- 28. Green TP, Marchessault RP, Freese DK. Disposition of sodium benzoate in newborn infants with Hyperammonemia. J Pediatr. 1983;102:785‐790. [DOI] [PubMed] [Google Scholar]
- 29. Praphanphoj V, Boyadjiev SA, Waber LJ, Brusilow SW, Geraghty MT. Three cases of intravenous sodium benzoate and sodium phenylacetate toxicity occurring in the treatment of acute hyperammonaemia. J Inherit Metab Dis. 2000;23:129‐136. [DOI] [PubMed] [Google Scholar]
- 30. Misel ML, Gish RG, Patton H, Mendler M. Sodium benzoate for treatment of hepatic encephalopathy. Gastroenterol Hepatol (N Y). 2013;9:219‐227. [PMC free article] [PubMed] [Google Scholar]
- 31. Sushma S, Dasarathy S, Tandon RK, Jain S, Gupta S, Bhist MS. Sodium benzoate in the treatment of acute hepatic encephalopathy: a double‐blind randomized trial. Hepatology. 1992;16:138‐144. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1 Whisker plot of ammonia concentrations before and after treatments in 10 CPSS dogs. Plac; Placebo, SB; sodium benzoate, SPB; sodium phenylbutyrate, BT; before treatment, AT; after treatment. Treatments were each of five‐day duration with wash‐out periods of three days. Each box and whisker plot illustrates the median (line in the middle), mean (+), 25th and 75th percentiles (top and bottom of each box); whiskers extend from the 5th and 95th percentile values.
Supplemental Table 1 AMmonia and bile acid concentrations before and after treatments in 10 CPSS dogs.