

SUMMARY
Chronic insomnia is highly prevalent and associated with significant morbidity (i.e., confers risk for multiple psychiatric and medical disorders, such as depression and hypertension). Therefore, it is essential to identify factors that perpetuate this disorder. One candidate factor in the neurobiology of chronic insomnia is hypothalamic-pituitary-adrenal-axis dysregulation, and in particular, alterations in circadian cortisol rhythmicity. Cortisol secretory patterns, however, fluctuate with both a circadian and an ultradian rhythm (i.e., pulses every 60e120 min). Ultradian cortisol pulses are thought to be involved in the maintenance of wakefulness during the day and their relative absence at night may allow for the consolidation of sleep and/or shorter nocturnal awakenings. It is possible that the wakefulness that occurs in chronic insomnia may be associated with the aberrant occurrence of cortisol pulses at night. While cortisol pulses naturally occur with transient awakenings, it may also be the case that cortisol pulsatility becomes a conditioned phenomenon that predisposes one to awaken and/or experience prolonged nocturnal awakenings. The current review summarizes the literature on cortisol rhythmicity in subjects with chronic insomnia, and proffers the suggestion that it may be abnormalities in the ultradian rather than circadian cortisol that is associated with the pathophysiology of insomnia.
Keywords: Insomnia, Cortisol, HPA-axis, Circadian rhythm, Ultradian rhythm
Introduction
Insomnia is an independent risk factor for psychiatric and medical disorders, including depression [1–5], hypertension [6–8], diabetes [9,10], and cardiovascular disease [11]. Insomnia-related morbidity, accidents and injuries, and absenteeism and presenteeism are estimated to cost approximately $5000 per affected individual per year [12]. These health risks and economic consequences are important given that approximately 32 million people in the United States have chronic insomnia (CI) [13]. Understanding the factors that perpetuate the disorder and subsequently increase risk for secondary health outcomes is therefore a major public health concern. While an extensive amount of research has been conducted on the behavioral factors that contribute to the development and maintenance of CI [14], little is known about the neurobiologic concomitants of insomnia. The present review summarizes the literature on the association between hypothalamic-pituitary-adrenal (HPA) axis functioning and CI, with a focus on the possibility that abnormalities in ultradian cortisol patterning may contribute to the incidence, severity, and/or chronicity of sleep continuity disturbance in the context of CI. Cortisol pulsatility has rarely been studied in this context and may reveal important information that is lost when examining only average nocturnal secretory levels and/or circadian patterns.
To date, the etiology of insomnia has been largely defined in behavioral rather than neurobiological terms. Acute insomnia (AI) is thought to become chronic in association with behavioral changes that result in sleep extension, sleep homeostasis dysregulation, and stimulus dyscontrol [14]. Sleep extension refers to the tendency to increase time in bed in an effort to recover lost sleep, which ultimately results in the mismatch between sleep opportunity and sleep ability. Sleep homeostasis dysregulation, in this context, refers to the tendency to reduce the amount of time spent awake during the day (i.e., resulting in reduced input to the “sleep homeostat”) but could also entail abnormal functioning of, or abnormal output from, the sleep homeostasis system [15]. Stimulus dyscontrol refers to the tendency to engage in behaviors other than sleep in the bedroom thereby weakening the association between the sleep environment (stimulus) and the physiologic state of sleep (response). These three factors are not only of theoretical relevance for the etiology of insomnia, they serve as the primary therapeutic targets of Cognitive Behavioral Therapy for Insomnia (CBT-I) [16], the indicated treatment for CI [17]. There may be, however, neurobiological (e.g., neuroendocrine) substrates or correlates of these phenomena, or completely independent neurobiological factors, that also play a role in the transition from acute to chronic insomnia. If so, these might provide additional, novel targets for intervention. Mainstream biological therapeutics have primarily focused on the treatment of insomnia with sedatives, which primarily exert therapeutic effects through the modulation of γ-aminobutyric acid (GABA) [18,19]. Despite widespread use of GABAergic medications [20], it was only recently that investigators posited that CI is related to GABA insufficiency [21,22]. The role of GABA insufficiency in the development and maintenance of CI, however, has a limited and mixed empirical basis [23]. In contrast, the alternative possibility is that CI occurs in association with excessive central nervous system (CNS) activation [24–28] and physiological hyperarousal [29,30]. Interestingly, the identified substrates for excessive CNS activation have not been theorized or experimentally shown to be related to glutamate (i.e., the neuro-transmitter that is the precursor for, and functionally the reciprocal of, GABA) but instead have been related to catecholamine functioning and more commonly HPA-axis-related hormones, in particular cortisol (i.e., insomnia occurs as a result of hypercortisolemia) [31,32].
The HPA-axis regulates a series of neuroendocrine responses to acute stressors that facilitate the mobilization of key adaptive physiological processes such as down-regulating digestive activity and maximizing glucose utilization [33,34]. Specifically, during stress, corticotropin-releasing hormone (CRH) is released from the hypothalamus, which subsequently triggers the release of adrenocorticotropic hormone (ACTH) from the anterior pituitary. ACTH stimulates the adrenal cortex, which produces and secretes glucocorticoids (i.e., cortisol in humans), the end-product of HPA-axis activation. Cortisol initially binds to high-affinity mineralocorticoid receptors (MRs). Once MRs have been sufficiently saturated, cortisol binds to glucocorticoid receptors (GRs), which then initiate a self-regulatory feedback system to shutdown the HPA-axis [35,36]. This process, from up-regulation to down-regulation, occurs in approximately 40‒60 min (at least as can be gleaned from the stress reactivity literature) [37,38]. Under non-stressful conditions, the HPA-axis follows a circadian rhythm. Specifically, glucocorticoids increase rapidly during the second part of the night, peak shortly after awakening (i.e., cortisol awakening response), and decline progressively throughout the day to reach their lowest levels (nadir) during the first half of the night, typically 1‒2 h after sleep onset (see Fig. 1A) [39,40]. In addition to the circadian rhythm of cortisol [41,42], cortisol release also follows an ultradian rhythm [43–48]. Specifically, cortisol is released in a pulsatile manner, with 15‒22 pulses per 24-h period (approximately one pulse every 60‒120 min), where the majority of the pulses occur during the diurnal phase [44,48–53]. It has been recently hypothesized that there may be a peripheral controller in the anterior pituitary that leads to the pulsatile release of cortisol [47,48,54]. That is, these ultradian oscillations in glucocorticoids may be related to the dynamic between CRH-induced ACTH (and subsequently glucocorticoid) secretion and glucocorticoid inhibition via the negative feedback loop. The pulse controller is believed to be located in the pituitary given that CRH pulse frequency is much greater (relative to ACTH and cortisol) [55], and because the timing and intensity of a glucocorticoid stress response is influenced by the timing of the stressor (i.e., whether the stressor occurs during the upward or downward phase of an ultradian pulse) [56], whereas CRH response does not necessarily vary by pulse phase [57]. Walker and colleagues demonstrated, however, that varying levels of CRH infusion [0.5‒2.5 μg/h] in adult male rats can modulate the ultradian oscillations in ACTH and glucocorticoids [54]. In this study, ultradian oscillations (during the circadian nadir) were observed at small doses of CRH infusion (0.5 mg/h), but not higher doses (e.g., 2.5 μg/ h). The oscillations observed following a low dose of CRH mimicked the endogenous ultradian pulses observed during the circadian peak in these hormones.
Fig. 1.
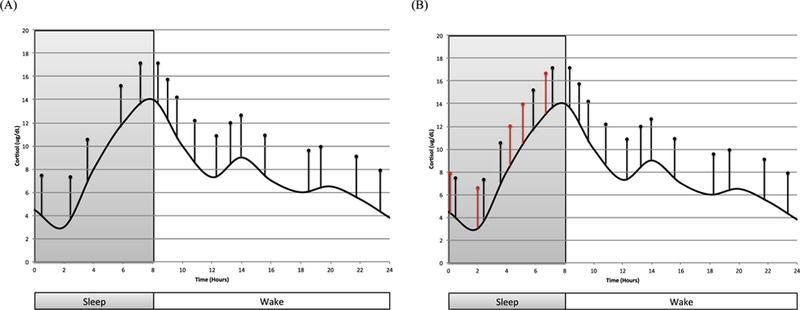
A‒B. Schematic representation of the circadian (base wave; black curve) and ultradian (pulses; black vertical lines) cortisol secretory patterns (Fig. 1A). This representation suggests that the “primary defect” in insomnia is more frequent pulses (additional red vertical lines in right panel; Fig. 1B). It is also possible that the pulse amplitudes will vary by group and time of day. Amplitude and phase shift (i.e., circadian) changes were not incorporated in this graphic, in order to highlight differences in the frequency of nocturnal cortisol pulses. (For interpretation of the references to color/colour in this figure legend, the reader is referred to the Web version of this article.)
It is hypothesized that a minimum level of cortisol in the blood stream is necessary for optimal functioning (i.e., a cortisol demand). From a signal processing/optimization point of view this is a time-varying demand that our controller needs to satisfy so that cortisol levels are always higher than the minimum necessary for optimal neuroendocrine function [47,83]. Inability to produce enough cortisol in the body can lead to a pathological state, such as adrenal insufficiency (Addison’s disease) [84]. Alternatively, excessive cortisol secretion can cause brain cell malfunction and even cell death [85,86]. This supports that cortisol in the blood stream also has an upper bound (i.e., maximum or holding cost). From a signal processing/optimization point of view this likely also has a time-varying holding cost that the ultradian controller needs to satisfy so that cortisol levels do not exceed this upper bound [47,83]. When cortisol secretion exceeds the holding cost, this too can create a pathological state (e.g., Cushing’s syndrome) [87–89]. Taken together, cortisol secretion may be regulated by a pulsatile controller, which assumes that the body needs to satisfy a cortisol demand (lower bound) and has a holding cost (upper bound). If the body has a circadian cortisol demand, and if the clearance rate by the liver does not change but the cortisol demand increases while the holding cost is the same, there will be more pulses but the amplitude of pulses won’t necessarily increase [47]. This scenario may be physiologically plausible for insomnia, in which the circadian changes in cortisol demand leads to the increased frequency of the ultradian rhythms. This is one of several possible pathways by which ultradian cortisol pulses become altered in CI. Other possible pathways include, but are not limited to, a reduced threshold (i.e., increased sensitivity to wakefulness), disinhibition (i.e., impairment of the negative feedback loop), and/or conditioning (i.e., strengthening the association between transient levels of wakefulness and cortisol pulses). These alternatives are discussed in detail later. Nevertheless, this increase in pulsatility during the nocturnal period may, in turn, represent the neuroendocrine substrate for the excessive CNS activation during the sleep period that is thought to account for incidence, severity, and chronicity of insomnia.
The focus on cortisol as a neuroendocrine substrate for CI derives from the association between the alerting and activating properties of this stress hormone (likely via the reciprocal connections between CRH and norepinephrine [58]) and the occurrence of sleeplessness in response to stress (perceived or real threat). For example, in mice, blue light exposure prior to sleep was associated with increases in glucocorticoids, and subsequently a delayed sleep onset [70]. The empirical evidence, particularly in humans, is mixed [59–69]. Some studies suggest that CI is associated with elevated circadian cortisol tone, particularly during the evening and early part of the night [61,64], yet others have failed to replicate these results [60,63,65]. Reasons for conflicting results are unknown, but methodological issues, ranging from types and subtypes of insomnia studied to the cortisol sampling rates utilized by each study, may play a role. In the case of the former, type and subtype of insomnia may be of relevance in a way that is not taken into account in most studies. For example, it may be that: 1) AI (variably defined as < 2 wk, < 4 wk, < 3 mo, and <6 mo time in past research) is associated with hypercortisolemia while CI is not; 2) patients with psychophysiologic but not paradoxical insomnia (i.e., sleep-state misperception) exhibit hypercortisolemia; and/or 3) patients that experience insomnia only during specific times of night (e.g., initial insomnia) exhibit elevated cortisol during those circumscribed periods of time (e.g., sleep onset). In the case of the latter, the sampling rate adopted for modeling patterns of cortisol secretion may have limited the generalizability of findings to the extent that different periods of the circadian phase were assessed and/or too few samples were acquired to obtain veridical averages or patterns.
In contrast to circadian cortisol secretion, there has been relatively little research into the association between insomnia and the ultradian rhythm of cortisol. While the underlying biological function of ultradian cortisol pulses is unknown, recent research suggests these pulses facilitate the regulation of glutaminergic transmission and hippocampal long-term potentiation [71]. These endogenous cortisol pulses (e.g., higher pulse frequency and higher pulse amplitude during the day) may also be responsible for increasing diurnal arousal and the maintenance of wakefulness during the day [54,72,73]. Given this perspective, the relative absence of cortisol pulses at night may allow for the consolidation of sleep and/or shorter nocturnal awakenings. Prior literature in healthy good sleeper subjects has indicated that it is the ultradian cortisol pulses, and not necessarily overall cortisol tone (i.e., the base component of the 24-hr oscillation), that are associated with sleep/wake processes [72,74,75]. Acute cortisol increases at night have been linked to less slow wave activity (decreased cortical synchronization) and more light sleep (greater amounts of Stage one and two sleep) and wakefulness at night [76–79]. More frequent cortisol pulses have also been observed following acute sleep deprivation, which supports that ultradian cortisol pulses may be involved in the promotion of wakefulness and/or the suppression of sleep [80–82]. This said, it follows that the ultradian cortisol rhythm may be overactive in CI (vs. more traditional amplitude changes or phase shifts in the 24-h secretory patterns). Please note, to our knowledge, no study has specifically demonstrated that increased ultradian pulses and maintenance of wakefulness during the day are causally related. This research, for example, would likely require an experimental study to show that the blockade of ultradian cortisol pulses induces increased sleepiness/drowsiness and/or polyphasic sleep cycles, and/or that the elicitation of cortisol pulses (perhaps via low-dose CRH infusion during the circadian nadir) increases alertness [54]. Rather, maintenance of wakefulness is a proposed function of ultradian cortisol pulses [72,73,80–82].
Summary of prior findings and limitations of previous studies
To date, fifteen studies have investigated diurnal and/or nocturnal cortisol patterning in subjects with insomnia-related sleep disturbance (see Table 1). Of these, only seven studies have found that cortisol patterning was significantly different in subjects with persistent insomnia relative to good sleeper controls. Two relatively consistent findings emerged from these studies. That is, subjects with persistent insomnia (relative to good sleeper controls) have 1) greater cortisol secretion in the evening and during the early part of the night [61,64], and 2) greater cortisol secretion in the morning [66–69]. Of the six studies that did not provide supportive data, two studies assessed plasma cortisol every 15‒30 min during the nocturnal period, and thus the failure to replicate is not likely to be attributable to a sampling confound [60,63]. No differences in diurnal or nocturnal salivary cortisol were observed when subjects were kept in controlled conditions either (i.e., constant routine protocol) [65]. Zhang and colleagues (2014) also showed no differences in evening salivary cortisol among a large sample of adults and adolescents with and without persistent insomnia [68]. One study even found that salivary cortisol at the time of awakening was significantly lower in subjects with insomnia compared to controls. This finding is not necessarily inconsistent with the studies cited above, given that the “morning” samples in this study were collected 30 min to several hours post-awakening and not immediately upon awakening. In fact, lower cortisol at awakening has been previously linked to greater post-awakening cortisol increases (i.e., a greater cortisol awakening response; CAR) [90]. The lower awakening cortisol among subjects with insomnia may be related to the timing of the awakening and/ or whether subjects woke up prior to anticipated, both of which are common in insomnia [91]. These lower levels at awakening, as well as the cortisol elevations in the evening and/or the early part of the night, may also be related to individual differences in circadian rhythms, particularly among subjects with initial insomnia. It is possible that the association between cortisol secretion and insomnia could be in part or in whole related to circadian misalignment and/or the interactive effect of circadian rhythms and sleep on 24-hr cortisol secretion. Future studies should therefore account for potential differences in the timing of awakening(s), the subject’s knowledge of when they are supposed to wake up (i.e., anticipation of awakening), and circadian preference (i.e., chronotype).
Table 1.
Existing literature on chronic insomnia and alterations in cortisol functioning.
| Citation | N | Sample Type | Insomnia Definition | PSG Assessed Sleep? | HPA-axis Outcome | Summary of Findings |
|---|---|---|---|---|---|---|
| Johns et al. (1971) [102] | 14 | Self-reported good and poor sleeper from a sample of 4th year, male medical students | Sleep questionnaire assessing TST, SL, and WASO. | No | 72-hr UFC | Poor sleepers excreted more UFC throughout the three phases of the day (i.e., morning, afternoon/evening, and overnight). Mean 24-hr UFC excretion was significantly higher among poor sleeper (527.9 ± 118.6 µg) compared to good sleepers (328.3 ± 57.1 µg). |
| Adam et al. (1986) [103] | 36 | Middle-age adults self-identified as poor sleepers compared to good sleepers | Single-item, self-rated poor sleep quality. | Yes | 48-hr UFC and five overnight samples. | 24-hr UFC and mean overnight UFC was greater among poor sleepers relative to good sleepers, but differences were n.s. Poor sleepers secreted approximately 10% more cortisol than good sleepers. |
| Vgontzas et al. (1998) [62] | 15 | Young adults with CI | Clinical interview; SL≥ 45 min and/or TST ≤ 6.5 hr for past 6 mo. | Yes | 24-hr UFC; two plasma cortisol samples (morning and night) | Mean 24-hr UFC was significantly associated with mean PSG-determined total wake time (r = 0.53); Plasma cortisol concentrations not significantly correlated with sleep variables. |
| Vgontzas et al. (2001) [61] | 24 | Young adults with CI compared to matched controls | Clinical interview; Sl ≥ 45 min and/or TST ≤ 6.5 hr for past 6 mo. SE < 80% during screening PSG. | Yes | Serial 24-hr plasma ACTH and cortisol (q30 min) | 24-h ACTH and cortisol secretion were significantly greater in patients with CI compared with controls, in particular during the evening and early part of the night and in those with short sleep duration i.e., <6 h; Patients with CI also had a greater number of cortisol peaks during the 24 recording period. |
| Rodenbeck et al. (2002) [64] | 14 | Seven males with CI and 7 age-and gender-matched controls | Clinical interview, using DSM-IV criteria for PI. | Yes | Serial 12-hr plasma cortisol (q60 min). Samples were taken beginning 4-hr before lights off until 1-hr after awakening | Evening (PI = 24.32 ± 10.33 ng/mL; C = 15.49 ± 4.49 ng/mL) and nocturnal AUCs (PI = 79.94 ± 15.88 ng/mL; C = 49.61 ± 6.67 ng/mL) were significantly increased in subjects with PI compared to controls. Among PI, evening cortisol was significantly correlated with SL (r = 0.82) and NWAK (r = 0.89), and nocturnal cortisol was correlated with SE (r = −0.82), WASO (r = 0.82) and %Stage2 Sleep (r = −0.83). |
| Riemann et al. (2002) [63] | 20 | Ten subjects with CI and 10 age-and gender-matched controls | Clinical interview, using DSM-IV criteria for PI. | Yes | Serial 12-hr plasma cortisol (q30 min) from 19:00 to 9:00. | There was no difference in plasma cortisol between subjects with PI (mean AUC ± SD = 214.4 ± 62.5 µg/ dL) and controls (mean AUC ± SD = 253.7 ± 75.9 µg/ dL). Statistically significant differences did not emerge when evaluating the data in 2-hr segments. |
| Shaver et al. (2002) [67] | 53 | Middle-aged women with Psychophysiological-Type Insomnia (n = 21; PP-type), Subjective Only-Type Insomnia (n = 18; SO-Type), and without Insomnia (n = 14; Control) | 2-week sleep diaries; had to report at least one of the following, 3 times/week: a) SL > 20 min, b) NWAK > 2, or c) EMA at least 2 times/week. PSG-based sleep quality. | Yes | Urinary cortisol assessed twice over three days, once 3-hr before bed and once immediately after awakening | Both of the insomnia groups had a greater AM-to-PM difference for cortisol than the control group (PP-type = 32.3 ± 28.45; SO-Type = 26.9 ± 16.3; Controls = 10.6 ± 13.4; units = µg/dL; p < .005). Women in the PP-type insomnia group had the highest levels of urinary cortisol in an early morning urine sample. |
| Rodenbeck et al. (2003) [104] | 10 | Seven male and three females with PI; Single infusions of placebo and 25 mg doxepin were applied following a double-blind, randomized cross-over design. Afterward, all patients received 25 mg doxepin for 3 wks. | Clinical interview, using DSM-IV criteria for PI. | Yes | Serial 12-hr plasma cortisol (q60 min) from 17:00 to 8:00. | Both doxepin application forms reduced mean cortisol levels from 9.0 ± 1.7 µg/l (single placebo i.v.) to 7.5 ± 1.6 µg/l (single doxepin i.v.) or 7.6 ± 2.0 µg/l (subchronic doxepin p.o.). Single doxepin i.v. significantly delayed both the time of cortisol nadir and the offset time of the quiescent period, while the subchronic doxepin p.o. significantly advanced the onset of the quiescent period. |
| Backhaus et al. (2004) [59] | 29 | Fourteen subjects with PI and 15 controls between 32 and 62 years of age | Clinical interview, using DSM-IV criteria for PI. | Yes, but only to screen out other sleep disorders. | Salivary cortisol on seven consecutive days (3 per day). Saliva samples were collected immediately after awakening (T1), 15 min later (T2), and just before bed (T3). | Cortisol immediately following awakening (T1) was significantly lower in subjects with PI compared to controls (p < .001). The change in salivary cortisol from awakening (T1) to evening (T3) was significantly lower for subjects with PI than for controls (PI: 6:4 ± 3:5; Controls: 10:8 ± 3:4). |
| Varkevisser et al. (2005) [65] | 24 | Eleven subjects with clinically diagnosed CI were compared with 13 matched controls. 24-hr constant-routine protocol was used. | Insomnia symptoms: (a) duration > 6 mo, (b) for ≥ 3 nights/week, and (c) be confirmed by PSG (SE < 85%; SL > 30 min or WASO > 45 min | Yes, but only to screen out other sleep disorders. | Serial 26-hr salivary cortisol (q180 min) from 11:00 to 13:00 (next day). | Non-statistically significant differences were found in the absolute level or the circadian parameters (amplitude, phase) of salivary cortisol between subjects with CI and controls (overall, PI = 4.78 ± 0.69 ng/mL, controls = 3.97 ± 0.64 ng/mL). |
| Backhaus et al. (2006) [60] | 29 | Sixteen subjects with PI and 13 age-matched controls. | Clinical interview, using DSM-IV criteria for PI. | Yes | Serial 8-hr plasma cortisol (q15 min) beginning at 22:00. | Plasma cortisol concentrations did not differ between groups for either the first or second half of the night. |
| Zhang et al. (2011) [105] | 96 | Healthy middle-aged adults from a community sample | Poor (SE < 85%) and good sleepers (SE ≥ 85%) assessed via actigraphy | No | Three days of salivary cortisol taken immediately after awakening | No difference was found in cortisol between poor and good sleepers. No correlation was found between actigraphy-measured sleep and salivary cortisol. |
| Xia et al. (2013) [66] | 90 | Thirty subjects with PI, 30 subjects with depression-comorbid insomnia (DCI), and 30 healthy controls | Clinical interview, using DSM-IV criteria for PI. PI must be present for >6 mo. | No | Single blood sample taken in the morning (between 07:30–08:00) to assess plasma CRH, ACTH, and cortisol | CRH and cortisol were significantly higher in both insomnia groups compared to the control group. ACTH was only higher in the DCI group (compared to controls). |
| Zhang et al. (2014) [68] | 449 | 244 adults (69 with PI) | Clinical interview, using DSM-IV criteria for PI. | No | Seven salivary cortisol samples were taken immediately upon awakening (T1), 30 (T2), 60 (T3), and 90 min (T4) after awakening, and at noon (T5), 16:00 h (T6), and 22:00 h (T7) | Adults with PI had larger CARi after awakening when compared with those without PI (PI = −2.88 nmol/dL; Controls = −6.30 nmol/dL, p = .009). There were no differences in other diurnal salivary cortisol variables between those with and without PI. |
| Floam et al. (2015) [69] | 48 | 29 subjects with PI and 19 healthy controls | Clinical Interview and PSQI, using DSM-5 criteria for insomnia Disorder. | No | Single blood sample taken at 11:30. | Compared with controls, individuals with Insomnia Disorder had a higher plasma cortisol (Insomnia = 12.20 ± 1.13 mg/dL, Controls = 9.19 ± 0.72 mg/dL, p < .05) |
ACTH = adrenocorticotropic hormone, AUC = area under the curve, CAR = cortisol awakening response, CI = chronic insomnia, CRH = corticotropin-releasing hormone, EMA = early morning awakenings, NWAK = nocturnal awakenings, PI = primary insomnia, PSG = polysomnography, PSQI = Pittsburgh Sleep Quality Index, SE = sleep efficiency, SL = sleep latency, TST = total sleep time, UFC = urinary free cortisol, WASO = wake after sleep onset.
The failure to replicate across the six studies cited above may be related to methodological issues. While the definition and assessment of insomnia was relatively consistent across studies (most used DSM-IV criteria for Primary Insomnia). It should be noted that this definition is still generic with respect to type and subtype of insomnia. For example, it has been suggested that hypercortisolemia is stronger in CI with objective short sleep duration [92]. As (or more) important, the assessment of cortisol varied widely, that is, from plasma samples taken every 30 min for 24-h [61] to a single morning blood draw [66,69]. These differences in methodology may account for some of the variability in past findings but more importantly, they limit the ability to distinguish potential mechanistic pathways, especially given that most studies have assessed cortisol using narrow timeframes (e.g., morning cortisol only) and/or low sampling rates (e.g., single saliva sample). Finally, with one exception [61], no study has accounted for the relative influence ultradian cortisol pulses have on overall diurnal and nocturnal cortisol rhythms. Below, we propose an alternative model for understanding how cortisol may be related to the observed hyperarousal and conditioned wakefulness that occurs with chronic insomnia, and provide important considerations for how to evaluate this association in future research.
An alternative perspective on the association between chronic insomnia and hypercortisolemia
Theoretical considerations
While ultradian cortisol pulsatility is not a novel concept (research on this neuroendocrine phenomenon dates back as early as the 1980’s [48,50,77]), its role in the development and maintenance of sleep continuity disturbance has yet to be formally proposed or evaluated. That is, research on ultradian cortisol pulses has primarily been in relation to “normal” sleep and/or good sleepers and/or other species [48,74–76,78,79]. Whether alterations in the ultradian cortisol rhythm (e.g., more frequent cortisol pulses at night) confer risk for sleep disturbance, in particular insomnia, is unknown. There is theoretical basis, however, for why increased cortisol pulses at night would be implicated in chronic insomnia, given that one of the proposed functions of these ultradian pulses (every 60‒120 min) is to maintain wakefulness during the day and are a response to wakefulness during the night [72,73]. During acute insomnia there is a relative increase, compared to good sleep, in the number of awakenings or the amount of time spent awake during the night [93,94]. These AI-related awakenings are likely associated with increased nocturnal cortisol pulsatility (more awakenings, more cortisol pulses) [77,78,95]. With time, as the insomnia transitions from acute to chronic, the increased incidence of ultradian pulses at night may occur as a result of a reduced threshold, disinhibition, and/or conditioning. In the case of a reduced threshold, the amount or type of wakefulness required to trigger a cortisol pulse may decrease or change with time (as the association between wakefulness and cortisol pulsatility is conditioned) or with the transitions in brain states (i.e., hybrid state ‒ local wakefulness during otherwise more global sleep) during sleep [96]. In the case of disinhibition, processes that normally inhibit these cortisol pulses at night may be functioning abnormally. For example, it may be the case that the negative feedback system that shuts down the HPA axis, and subsequently the production of glucocorticoids, becomes dis-inhibited in CI. Rodenbeck and colleagues (2001) previously stated that this may be one mechanism by which elevated evening cortisol contributes to the transition from acute to chronic insomnia [32]. In the case of conditioning, it is possible that the cortisol pulses become conditioned to naturally occurring upward sleep stage shifts or to specific times of night [76,77]. Taken together, among patients with CI, the ultradian cortisol pulsatility rate during the nocturnal phase of the day may more closely resemble the pulsatility rate that occurs during the diurnal phase of the day (Fig. 1B). While these explanations seem plausible, the first step in this line of research is to determine whether increased cortisol pulsatility may be observed in CI. To date, only one study has assessed differences in cortisol pulsatility among patients with CI, and found a greater number of pulses during a 24-hr period among patients with CI, relative to good sleepers [61]. However, it is unclear, based on the data reported, whether these differences in the ultradian cortisol rhythm occurred during the day, at night, or both.
Methodological considerations
Past research has been mostly unable to evaluate ultradian cortisol pulses, given that a more frequent sampling rate is needed to maximize the likelihood that a plasma sample will capture one of the pulses. In fact, most studies investigating the association between cortisol and CI have used single-point assessments (e.g., one sample following awakening) [66] or random-interval sampling (e.g., during the sleep period) [59,60,63,64], and those that have used fixed-interval sampling procedures have used sampling rates that only allow for the assessment of the base circadian cortisol curve (e.g., q30 and q120 min intervals) [61,65]. Only Vgontzas and colleagues (2001) have reported data on cortisol pulsatility among patients with CI, and showed that the number of pulses during the 24-h recording period was greater among patients compared to good sleeper controls. This study, however, 1) used a sampling rate (q30 min) that underestimated the number of pulses relative to what has been observed in non-clinical human research using a q10 min sampling rate (i.e., 6 vs. 18 pulses/24 h) [61,75,76,78,79] and 2) did not take into account time of day effects (e.g., day vs. night pulse rates). Taken together, future studies will benefit from using more dense sampling rates (q10 min) in order to better characterize circadian and ultradian cortisol rhythms in patients with CI. Even at a sampling rate of every 10 min, it is still possible that this will lead to a systematic underestimation of pulse frequency, given the random and sparse nature of hormone pulsatility. Since there are practical limitations to collecting blood continuously, more advanced methodological (such as an automated blood sampling system [97,98]) and/or analytical approaches are needed to maximize the ability to detect and quantify the timing and amplitude of these pulses. For example, Faghih and colleagues (2014) have recently developed a technique using compressed sensing that allows for the estimation of pulses from data that is collected serially (e.g., every 10 min for 24 h), but not continuously [52,99]. Notably, while the data was collected every 10 min, using this compressed sensing deconvolution algorithm, the pulses were recovered with a one-minute resolution. The key here is the sparse nature of the underlying secretory events and that the time resolution of the recovered secretory events is further improved. These more advanced analytical approaches may also minimize the burden to patients, if less frequent samples are needed to model the cortisol time series data and reliably (and accurately) detect the timing and amplitude of pulses.
In addition, future research in this area should be conducted in combination with the careful assessment of nocturnal sleep/wake patterns (via polysomnography; PSG). The inclusion of PSG is critical both as a screening tool (i.e., studies of insomnia must rule out the presence of occult sleep disorders), but also to systematically evaluate the association between ultradian cortisol pulses and metrics related to spontaneous awakenings from sleep (e.g., frequency, timing, duration, and electroencephalography (EEG) spectral profile of the awakenings). Furthermore, including PSG would allow for the quantification of group differences in 1) the frequency of pre-vs. post-awakening cortisol pulses and 2) duration and EEG spectral profile for awakenings with and without pre-occurring cortisol pulses. That is, patients with CI may exhibit more frequent pre-awakening cortisol pulses, and/or longer duration awakenings and spectral profiles with greater amounts of high-frequency EEG activity, and that this effect may be more pronounced in awakenings preceded by pulses.
Potential benefits of the proposed research
The potential benefits of exploring ultradian cortisol pulsatility in the context of CI include: 1) the provision of an alternative perspective regarding how hypercortisolemia may be related to chronic sleep continuity disturbance. That is, a first demonstration that insomnia-related sleeplessness is not only related to amplitude and/or phase shifts in the circadian patterning of cortisol secretion but also related to the emergence and/or the intensification of nocturnal cortisol pulsatility (and related wake-promoting effects), the present perspective allows for future exploration of whether ultradian changes in cortisol tone are related to the transition from acute to chronic insomnia, and 3) the present perspective may suggest an alternative therapeutic approach to the medical management of CI. Presently, the approach is about de-potentiating cortical activation where the alternative perspective may be to de-potentiate factors associated with the elicitation/consolidation of wakefulness. For example, if cortisol pulsatility (as it does during wakefulness) is responsible for wake promotion during the sleep period, the inhibition of neuroendocrine substrates and/or glucocorticoid receptors associated with wake consolidation may prove to be as, or more, effective a treatment approach than substances that promote GABA-related sedation. The HPA-axis, however, is a complex system and efforts to modify it pharmacologically have not proven useful, translationally. It is possible, however, that delving deeper into the psychoneuroendocrinology, going beyond measuring “levels” and looking at details like ultradian rhythms, may prove to be more informative. In those details, we may find links that can lead to more specific ways to manipulate this system, by targeting specific aspects of it without altering its overall functioning that is so important to health.
Practice points.
Alterations in circadian cortisol secretion (both in amplitude and phase) is a leading candidate for the neuroendocrine substrate responsible for conditioned wakefulness and physiological hyperarousal. Yet, past research in this area is equivocal, mostly due to variability in methodological approach.
Ultradian cortisol pulses are an additional but neglected aspect of HPA-axis functioning, and examinations of how it relates to the pathophysiology of insomnia may prove to be important.
Transient nocturnal cortisol pulsatility may be the result of central nervous system activation associated with insomnia-related awakenings, however in the long term, cortisol pulses may become conditioned phenomena that precipitate and/or prolong a nocturnal awakening. That is, cortisol pulses may be the consequence of nocturnal awakenings in acute insomnia, and a precipitant of nocturnal awakenings in chronic insomnia.
The present perspective also has implications for the pharmacological treatment of insomnia. Such that, if in chronic insomnia, cortisol pulses are wake-promoting phenomena, the inhibition of these pulses may be an alternative treatment approach to traditional hypnotics.
Research agenda.
There are four clear research questions that extend from the present review:
Are alterations in the ultradian rhythm of other HPA-axis-related hormones (i.e., CRH and ACTH) also associated with chronic insomnia [100]? And are these alterations specific to the night or across the 24-h day? Cortisol represents the end-stage of the HPA-axis, yet CRH and ACTH play an equal role in determining the frequency, intensity, and duration of an HPA-axis response [40]. Despite this, these three hormones are rarely assessed concurrently, and therefore, future studies should aim to look at circadian and ultradian secretory patterns of all three substrates among patients with chronic insomnia, or at least cortisol and ACTH, which both can be reliably assessed in the periphery.
Is variability in circadian and/or ultradian cortisol rhythms a predisposing, perpetuating, or Pavlovian factor with respect to the etiology and/or pathophysiology of insomnia [101]? Subsequent research will need to use longitudinal designs to investigate whether greater cortisol pulsatility precedes chronic insomnia (and pre-disposes individuals to insomnia) and/or develops as a function of the disorder (and then perpetuates it).
Would a positive association between greater ultradian cortisol pulses (either in frequency, timing, or intensity) and chronic insomnia suggest an alternative therapeutic approach to the medical management of the disorder? Specifically, as compared to approaches that focus on GABA-related sedation, an alternative therapeutic target may be the inhibition of neuroendocrine hormones (e.g., cortisol) and/or the promotion of glucocorticoid receptor activation, which may, in turn, function as an antagonist to wake promotion.
What kind of wakefulness is associated with cortisol pulses? More specifically, do these pulses occur only with relatively prolonged awakenings or with all awakenings, regardless of duration? For example, is it possible that cortisol pulses occur with the micro-awakenings that are typical of sleep fragmentation? If cortisol pulses do occur with sleep fragmentation, this may have clinical implications for sleep disorders other than chronic insomnia, such as obstructive sleep apnea and periodic limb movement disorder.
Acknowledgement
The authors would like to thank Dr. Allan Pack and Dr. Elizabeth Klerman for providing useful feedback on earlier version of the manuscript.
Abbreviations
- ACTH
adrenocorticotropic hormone
- AI
acute insomnia
- CAR
cortisol awakening response
- CBT-I
cognitive behavioral therapy for insomnia
- CI
chronic insomnia
- CNS
central nervous system
- CRH
corticotropin-releasing hormone
- EEG
electroencephalography
- GABA
γ-aminobutyric acid
- GR
glucocorticoid receptors
- HPA
hypothalamic pituitary adrenal
- MR
mineralocorticoid receptors
- PSG
polysomnography
Footnotes
Conflicts of interest
The authors report no potential conflicts of interest or financial relationships with commercial interests that are relevant to this work.
References
- [1].Benca RM, Peterson MJ. Insomnia and depression. Sleep Med 2008;9:3–9. 10.1016/S1389-9457(08)70010-8. [DOI] [PubMed] [Google Scholar]
- [2].Neckelmann D, Mykletun A, Dahl A. Chronic insomnia as a risk factor for developing anxiety and depression. Sleep 2007;30:873–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Riemann D, Voderholzer U. Primary insomnia: a risk factor to develop depression? J Affect Disord 2003;76:255–9. https://doi.org/10.1016/S0165-0327(02)00072-1. [DOI] [PubMed] [Google Scholar]
- [4].Perlis ML, Smith LJ, Lyness JM, Matteson SR, Pigeon WR, Jungquist CR. Insomnia as a risk factor for onset of depression in the elderly insomnia. Behav Sleep Med 2010;4:85–103. 10.1207/s15402010bsm0402. [DOI] [PubMed] [Google Scholar]
- [5].Roane BM, Taylor DJ. Adolescent insomnia as a risk factor for early adult depression and substance abuse. Sleep 2008;31:1351–6. [PMC free article] [PubMed] [Google Scholar]
- [6].Vgontzas A, Liao D, Bixler E, Chrousos G. Insomnia with objective short sleep duration is associated with a high risk for hypertension. Sleep 2009. 10.2337/dc09-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Suka M, Yoshida K, Sugimori H. Persistent insomnia is a predictor of hypertension in Japanese male workers. J Occup Health 2003;45:344–50. 10.1539/joh.45.344. [DOI] [PubMed] [Google Scholar]
- [8].Bathgate CJ, Edinger JD, Wyatt JK, Krystal AD. Objective but not subjective short sleep duration associated with increased risk for hypertension in individuals with insomnia. Sleep 2016;39:1037–45. 10.5665/sleep.5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mallon L, Broman JE, Hetta J. High incidence of diabetes in men with sleep complaints or short sleep duration: a 12-year follow-up study of a middle-aged population. 43 Diabetes Care 2005;28:2762–7. 10.2337/diacare.28.11.2762. [DOI] [PubMed] [Google Scholar]
- [10].Vgontzas AN, Liao D, Pejovic S, Calhoun S, Karataraki M, Bixler EO. Insomnia with objective short sleep duration is associated with type 2 diabetes: a population-based study. Diabetes Care 2009;32:1980–5. 10.2337/dc09-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sofi F, Cesari F, Casini A, Macchi C, Abbate R, Gensini GF. Insomnia and risk of cardiovascular disease: a meta-analysis. Eur J Prev Cardiol 2014;21: 57–64. 10.1177/2047487312460020. [DOI] [PubMed] [Google Scholar]
- [12].Daley M, Morin CM, LeBlanc M, Gregoire J-P, Savard J. The economic burden of insomnia: direct and indirect costs for individuals with insomnia syndrome, insomnia symptoms, and good sleepers. Sleep 2009;32:55–64. [PMC free article] [PubMed] [Google Scholar]
- [13].NIH. NIH state-of-the-science conference statement on manifestations and management of chronic insomnia in adults. NIH Consens State Sci Statements 2005. June 13‒15;22(2):1–30. [PubMed] [Google Scholar]
- [14].Perlis ML, Shaw PJ, Cano G, Espie CA. Models of insomnia. Princ. Pract. Sleep med 5th ed. 2011. p. 850–65. [Google Scholar]
- [15].Pigeon WR, Perlis ML. Sleep homeostasis in primary insomnia. Sleep Med Rev 2006;10:247–54. doi:S1087–0792(05)00097–3 [pii]\r 10.1016/j.smrv.2005.09.002. [DOI] [PubMed] [Google Scholar]
- [16].Morin CM. Cognitive-behavioral therapy of insomnia. Sleep Med Clin 2006;1:375–86. 10.1016/j.jsmc.2006.06.008. [DOI] [Google Scholar]
- [17].Qaseem A, Kansagara D, Forciea MA, Cooke M, Denberg TD, Barry MJ, et al. Management of chronic insomnia disorder in adults: a clinical practice guideline from the American college of physicians. Ann Intern Med 2016;165:125–33. 10.7326/M15-2175. [DOI] [PubMed] [Google Scholar]
- [18].Doble A New insights into the mechanism of action of hypnotics. J Psychopharmacol 1999;13:S11–20. [DOI] [PubMed] [Google Scholar]
- [19].Crestani F, Martin JR, Möhler H, Rudolph U. Mechanism of action of the hypnotic zolpidem in vivo. Br J Pharmacol 2000;131:1251–4. 10.1038/sj.bjp.0703717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Walsh JK, Schweitzer PK. Ten-year trends in the pharmacological treatment of insomnia. Sleep 1999;22:371–5. [PubMed] [Google Scholar]
- [21].Winkelman JW, Buxton OM, Jensen JE, Benson KL, O’Connor SP, Wang W, et al. Reduced brain GABA in primary insomnia: preliminary data from 4T proton magnetic resonance spectroscopy (1H-MRS). Sleep 2008;31: 1499–506. 10.1093/sleep/31.11.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Plante DT, Jensen JE, Schoerning L, Winkelman JW. Reduced g-aminobutyric acid in occipital and anterior cingulate cortices in primary insomnia: a link to major depressive disorder? Neuropsychopharmacology 2012;37: 1548–57. 10.1038/npp.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Spiegelhalder K, Regen W, Nissen C, Feige B, Baglioni C, Riemann D, et al. Magnetic resonance spectroscopy in patients with insomnia: a repeated measurement study. PLoS One 2016;11, e0156771 https://doi.org/10.1371/journal.pone.0156771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Nofzinger EA, Buysse DJ, Germain A, Price J, Miewald J, Kupfer DJ. Insomnia: functional neuroimaging evidence for hyperarousal. Sleep 2004;27:272 10.1176/appi.ajp.161.11.2126. [DOI] [PubMed] [Google Scholar]
- [25].Merica H, Blois R, Gaillard JM. Spectral characteristics of sleep EEG in chronic insomnia. Eur J Neurosci 1998;10:1826–34. 10.1046/j.1460-9568.1998.00189.x. [DOI] [PubMed] [Google Scholar]
- [26].Merica H, Gaillard JM. The EEG of the sleep onset period in insomnia: a discriminant analysis. Physiol Behav 1992;52:199–204. 10.1016/0031-9384(92)90258-4. [DOI] [PubMed] [Google Scholar]
- [27].Lamarche CH, Ogilvie RD. Electrophysiological changes during the sleep onset period of psychophysiological insomniacs, psychiatric insomniacs, and normal sleepers. Sleep 1997;20:724–33. [PubMed] [Google Scholar]
- [28].Perlis ML, Smith MT, Andrews PJ, Orff HJ, Giles DE. Beta/gamma EEG activity in patients with primary and and secondary insomnia and good sleeper controls. Sleep 2001;24:110–7. [DOI] [PubMed] [Google Scholar]
- [29].Bonnet MH, Arand DL. Hyperarousal and insomnia: state of the science. Sleep Med Rev 2010;14:9–15. 10.1016/j.smrv.2009.05.002. [DOI] [PubMed] [Google Scholar]
- [30].Basta M, Chrousos GP, Vela-Bueno A, Vgontzas AN. Chronic insomnia and stress system. Sleep Med Clin 2007;2:279–91. https://doi.org/10.1016/j.jsmc.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Perlis M, Gehrman P, Pigeon WR, Findley J, Drummond S. Neurobiologic mechanisms in chronic insomnia. Sleep Med Clin 2009;4:549–58. 10.1016/j.jsmc.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].*Rodenbeck A, Hajak G. Neuroendocrine dysregulation in primary insomnia. Rev Neurol (Paris) 2001;157:S57–61. [PubMed] [Google Scholar]
- [33].de Kloet ER. Brain corticosteroid receptor balance and homeostatic control. Front Neuroendocrinol 1991;12:95–164. 10.1080/09614520701469617. [DOI] [PubMed] [Google Scholar]
- [34].Johnson E, Kamilaris T, Chrousos G, Gold P. Mechanisms of stress: a dynamic overview of hormonal and behavioral homeostasis. Neurosci Bio-behav Rev 1992;16:115–30. [DOI] [PubMed] [Google Scholar]
- [35].Tsigos C, Chrousos GP. Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J Psychosom Res 2002;53:865–71. [DOI] [PubMed] [Google Scholar]
- [36].Gunnar M, Quevedo K. The neurobiology ofstress and development. Annu Rev Psychol 2007;58:145–73. https://doi.org/10.1146/annurev.psych.58.110405.085605. [DOI] [PubMed] [Google Scholar]
- [37].Lopez-Duran NL, Mayer SE, Abelson JL. Modeling neuroendocrine stress reactivity in salivary cortisol: adjusting for peak latency variability. Stress 2014. 10.3109/10253890.2014.915517. [DOI] [PubMed] [Google Scholar]
- [38].Kudielka BM, Buske-Kirschbaum A, Hellhammer DH, Kirschbaum C. HPA axis responses to laboratory psychosocial stress in healthy elderly adults, younger adults, and children: impact of age and gender. Psychoneur-oendocrinology 2004;29:83–98. [DOI] [PubMed] [Google Scholar]
- [39].Gunnar MR, Vazquez DM. Low cortisol and a flattening of expected day-time rhythm : potential indices of risk in human development. Dev Psychopathol 2001;13:515–38. [DOI] [PubMed] [Google Scholar]
- [40].Buckley TM, Schatzberg AF. On the interactions of the hypothalamic-pituitary-adrenal (HPA) axis and sleep: normal HPA axis activity and circadian rhythm, exemplary sleep disorders. J Clin Endocrinol Metab 2005;90:3106–14. 10.1210/jc.2004-1056. [DOI] [PubMed] [Google Scholar]
- [41].Van Cauter E, Leproult R, Kupfer DJ. Effects of gender and age on the levels and circadian rhythmicity of plasma cortisol. J Clin Endocrinol Metab 1996;81:2468–73. [DOI] [PubMed] [Google Scholar]
- [42].Dickmeis T. Glucocorticoids and the circadian clock. J Endocrinol 2009;200:3–22. 10.1677/JOE-08-0415. [DOI] [PubMed] [Google Scholar]
- [43].*Van Cauter E, Spiegel K. Hormones and metabolism during sleep. In: Schwartz W, editor. Sleep sci. Integr. Basic res. Clin. Pract, basel: karger; 1997. p. 144–74. [Google Scholar]
- [44].*Young EA, Abelson JL, Lightman SL. Cortisol pulsatility and its role in stress regulation and health. Front Neuroendocrinol 2004;25:69–76. 10.1016/j.yfrne.2004.07.001. [DOI] [PubMed] [Google Scholar]
- [45].Waite EJ, Mckenna M, Kershaw Y, Walker JJ, Cho K, Piggins HD, et al. Ultradian corticosterone secretion is maintained in the absence of circadian cues. Eur J Neurosci 2012;36:3142–50. 10.1111/j.1460-9568.2012.08213.x. [DOI] [PubMed] [Google Scholar]
- [46].McMaster A, Jangani M, Sommer P, Han N, Brass A, Beesley S, et al. Ultradian cortisol pulsatility encodes a distinct, biologically important signal. PLoS One 2011;6 10.1371/journal.pone.0015766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].*Faghih RT, Dahleh MA, Brown EN. An optimization formulation for characterization of pulsatile cortisol secretion. Front Neurosci 2015;9:1–10. 10.3389/fnins.2015.00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].*Lightman SL, Wiles CC, Atkinson HC, Henley DE, Russell GM, Leendertz JA, et al. The significance of glucocorticoid pulsatility. Eur J Pharmacol 2008;583:255–62. 10.1016/j.ejphar.2007.11.073. [DOI] [PubMed] [Google Scholar]
- [49].Lavie P, Kripke DF. Ultradian circa 1½ hour rhythms: a multioscillatory system. Life Sci 1981;29:2445–50. [DOI] [PubMed] [Google Scholar]
- [50].Follenius M, Simon C, Brandenberger G, Lenzi P. Ultradian plasma corticotropin and cortisol rhythms: time-series analyses. J Endocrinol Invest 1987;10:261–6. 10.1007/BF03348128. [DOI] [PubMed] [Google Scholar]
- [51].Brown EN, Meehan PM, Dempster AP, Emery N, Meehan PM, Arthur P. A stochastic differential equation model of diurnal cortisol patterns. Am J Physiol Endocrinol Metab 2001;2114:450–61. [DOI] [PubMed] [Google Scholar]
- [52].Faghih RT, Dahleh MA, Adler GK, Klerman EB, Brown EN. Deconvolution of serum cortisol levels by using compressed sensing. PLoS One 2014;9: 1–12. 10.1371/journal.pone.0085204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Veldhuis JD, Iranmanesh A, Lizarralde G, Johnson ML. Amplitude modulation of a burstlike mode of cortisol secretion subserves the circadian glucocorticoid rhythm. Am J Physiol 1989;257:E6–14. [DOI] [PubMed] [Google Scholar]
- [54].*Walker JJ, Spiga F, Waite E, Zhao Z, Kershaw Y, Terry JR, et al. The origin of glucocorticoid hormone oscillations. PLoS Biol 2012;10 10.1371/journal.pbio.1001341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ixart G, Barbanel G, Nouguier-Soulé J, Assenmacher I. A quantitative study of the pulsatile parameters of CRH-41 secretion in unanesthetized free-moving rats. Exp Brain Res 1991;87:153–8. 10.1007/BF00228516. [DOI] [PubMed] [Google Scholar]
- [56].Rankin J, Walker JJ, Windle R, Lightman SL, Terry JR. Characterizing dynamic interactions between ultradian glucocorticoid rhythmicity and acute stress using the phase response curve. PLoS One 2012;7 10.1371/journal.pone.0030978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Windle RJ, Wood SA, Lightman SL, Ingram CD. The pulsatile characteristics of hypothalamo-pituitary-adrenal activity in female Lewis and Fischer 344 rats and its relationship to differential stress responses. Endocrinology 1998;139:4044–52. 10.1210/endo.139.10.6238. [DOI] [PubMed] [Google Scholar]
- [58].Tsigos C, Chrousos GP. Physiology of the hypothalamic-pituitary-adrenal axis in health and dysregulation in psychiatric and autoimmune disorders. Endocrinol Metab Clin N Am 1994;23:451–66. [PubMed] [Google Scholar]
- [59].Backhaus J, Junghanns K, Hohagen F. Sleep disturbances are correlated with decreased morning awakening salivary cortisol. Psychoneur-oendocrinology 2004;29:1184–91. 10.1016/j.psyneuen.2004.01.010. [DOI] [PubMed] [Google Scholar]
- [60].Backhaus J, Junghanns K, Born J, Hohaus K, Faasch F, Hohagen F. Impaired declarative memory consolidation during sleep in patients with primary insomnia: influence of sleep architecture and nocturnal cortisol release. Biol Psychiatr 2006;60:1324–30. 10.1016/j.biopsych.2006.03.051. [DOI] [PubMed] [Google Scholar]
- [61].*Vgontzas AN, Bixler EO, Lin HM, Prolo P, Mastorakos G, Vela-Bueno A, et al. Chronic insomnia is associated with nyctohemeral activation of the hypothalamic-pituitary-adrenal Axis: clinical implications. J Clin Endo-crinol Metab 2001;86:3787–94. [DOI] [PubMed] [Google Scholar]
- [62].Vgontzas AN, Tsigos C, Bixler EO, Stratakis CA, Zachman K, Kales A, et al. Chronic insomnia and activity of the stress system: a preliminary study. J Psychosom Res 1998;45:21–31. 10.1016/S0022-3999(97)00302-4. [DOI] [PubMed] [Google Scholar]
- [63].Riemann D, Klein T, Rodenbeck A, Feige B, Horny A, Hummel R, et al. Nocturnal cortisol and melatonin secretion in primary insomnia. Psychiatr Res 2002;113:17–27. 10.1016/S0165-1781(02)00249-4. [DOI] [PubMed] [Google Scholar]
- [64].Rodenbeck A, Huether G, Rüther E, Hajak G. Interactions between evening and nocturnal cortisol secretion and sleep parameters in patients with severe chronic primary insomnia. Neurosci Lett 2002;324:159–63. 10.1016/S0304-3940(02)00192-1. [DOI] [PubMed] [Google Scholar]
- [65].Varkevisser M, Van Dongen HP a, Kerkhof G a. Physiologic indexes in chronic insomnia during a constant routine: evidence for general hyper-arousal? Sleep 2005;28:1588–96. [PubMed] [Google Scholar]
- [66].Xia L, Chen GH, Li ZH, Jiang S, Shen J. Alterations in hypothalamus-pituitary-adrenal/thyroid axes and gonadotropin-releasing hormone in the patients with primary insomnia: a clinical research. PLoS One 2013;8: 1–6. 10.1371/journal.pone.0071065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Shaver JL, Johnston SK, Lentz MJ, Landis CA. Stress exposure, psychological distress, and physiological stress activation in midlife women with insomnia. Psychosom Med 2002;64:793–802. 10.1097/01.PSY.0000024235.11538.9A. [DOI] [PubMed] [Google Scholar]
- [68].Zhang J, Lam S-PP, Li SX, Ma RCW, Kong APS, Chan MHM, et al. A community-based study on the association between insomnia and hypothalamic-pituitary-adrenal axis: sex and pubertal influences. J Clin Endocrinol Metab 2014;99:2277–87. 10.1210/jc.2013-3728. [DOI] [PubMed] [Google Scholar]
- [69].Floam S, Simpson N, Nemeth E, Scott-Sutherland J, Gautam S, Haack M. Sleep characteristics as predictor variables of stress systems markers in insomnia disorder. J Sleep Res 2015;24:296–304. https://doi.org/10.1111/jsr.12259. [DOI] [PubMed] [Google Scholar]
- [70].Pilorz V, Tam SKE, Hughes S, Pothecary CA, Jagannath A, Hankins MW, et al. Melanopsin regulates both sleep-promoting and arousal-promoting responses to light. PLoS Biol 2016;14:1–25. https://doi.org/10.1371/journal.pbio.1002482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sarabdjitsingh RA, Jezequel J, Pasricha N, Mikasova L, Kerkhofs A, Karst H, et al. Ultradian corticosterone pulses balance glutamatergic transmission and synaptic plasticity. Proc Natl Acad Sci 2014;111:14265–70. 10.1073/pnas.1411216111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].*Gronfier C, Brandenberger G. Ultradian rhythms in pituitary and adrenal hormones: their relations to sleep. Sleep Med Rev 1998;2:17–29. 10.1016/S1087-0792(98)90051-X. [DOI] [PubMed] [Google Scholar]
- [73].*Balbo M, Leproult R, Van Cauter E. Impact of sleep and its disturbances on hypothalamo-pituitary-adrenal axis activity. Internet J Endocrinol 2010;1: 1–16. 10.1155/2010/759234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Gronfier C, Simon C, Piquard F, Ehrhart J, Brenberger G. Neuroendocrine processes underlying ultradian sleep regulation in man. J Clin Endocrinol Metab 1999;84:2686–90. 10.1210/jc.84.8.2686. [DOI] [PubMed] [Google Scholar]
- [75].Gronfier C, Chapotot F, Weibel L, Jouny C, Brandenberger G. Pulsatile cortisol secretion and EEG delta waves are controlled by two independent but synchronized generators 2009. p. 94–100. [DOI] [PubMed] [Google Scholar]
- [76].Gronfier C, Luthringer R, Follenius M, Schaltenbrand N, Macher JP, Muzet A, et al. Temporal relationships between pulsatile cortisol secretion and electroencephalographic activity during sleep in man. Electro-encephalogr Clin Neurophysiol 1997;103:405–8. 10.1016/S0013-4694(97)00013-1. [DOI] [PubMed] [Google Scholar]
- [77].Born J, Kern W, Bieber K, Fehm-Wolfsdorf G, Schiebe M, Fehm H. Night-time plasma cortisol secretion is associated with specific sleep stages. Biol Psychiatr 1986;21:1415–24. [DOI] [PubMed] [Google Scholar]
- [78].Follenius M, Brandenberger G, Bandesapt JJ, Libert JP, Ehrhart J. Nocturnal cortisol release in relation to sleep structure. Sleep 1992;15:21–7. [DOI] [PubMed] [Google Scholar]
- [79].Weibel L, Follenius M, Spiegel K, Ehrhart J, Brandenberger G. Comparative effect of night and daytime sleep on the 24-hour cortisol secretory profile. Sleep 1995;18:549–56. [PubMed] [Google Scholar]
- [80].Chapotot F, Buguet A, Gronfier C, Brandenberger G. Hypothalamo-pituitary-adrenal axis activity is related to the level of central arousal: effect of sleep deprivation on the association of high-frequency waking electroencephalogram with cortisol release. Neuroendocrinology 2001;73:312–21. 10.1159/000054648. [DOI] [PubMed] [Google Scholar]
- [81].Leproult R, Copinschi G, Buxton O, Van Cauter E. Sleep loss results in an elevation of cortisol levels the next evening. Sleep 1997;20:865–70. [PubMed] [Google Scholar]
- [82].Von Treuer K, Norman TR, Armstrong SM. Overnight human plasma melatonin, cortisol, prolactin, TSH, under conditions of normal sleep, sleep deprivation, and sleep recovery. J Pineal Res 2007;20:7–14. [DOI] [PubMed] [Google Scholar]
- [83].Faghih RT. From physiological signals to pulsatile dynamics: a sparse system identification approach. Dyn. Neurosci., Cham Springer International Publishing; 2018. p. 239–65. 10.1007/978-3-319-71976-4_10. [DOI] [Google Scholar]
- [84].Lennernäs H, Skrtic S, Johannsson G. Replacement therapy of oral hydrocortisone in adrenal insufficiency: the influence of gastrointestinal factors. Expet Opin Drug Metabol Toxicol 2008;4:749–58. 10.1517/17425255.4.6.749. [DOI] [PubMed] [Google Scholar]
- [85].Sapolsky RM. Stress, the aging brain, and the mechanisms of neuron death Cambridge, MA: The MIT Press; 1992. [Google Scholar]
- [86].De Kloet ER, Joëls M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci 2005;6:463–75. 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- [87].Yanovski JA, Cutler GB. Glucocorticoid action and the clinical features of Cushing’s syndrome. Endocrinol Metab Clin N Am 1994;23:487–509. [PubMed] [Google Scholar]
- [88].Raff H, Raff JL, Findling JW. Late-night salivary cortisol as a screening test for cushing ‘s syndrome. J Clin Endocrinol Metab 1998;83:2681–6. 10.1210/jcem.83.8.4936. [DOI] [PubMed] [Google Scholar]
- [89].Starkman MN, Giordani B, Gebarski SS, Berent S, Schork MA, Schteingart DE. Decrease in cortisol reverses human hippocampal atrophy following treatment of Cushing’s disease. Biol Psychiatr 1999;46: 1595–602. [DOI] [PubMed] [Google Scholar]
- [90].Vargas I, Lopez-Duran NL. Dissecting the impact of sleep and stress on the cortisol awakening response among healthy young adults. Psychoneur-oendocrinology 2014;40:10–6. [DOI] [PubMed] [Google Scholar]
- [91].Born J, Hansen K, Marshall L, Mölle M, Fehm HL. Timing the end of nocturnal sleep. Nature 1999;397:29–30. 10.1038/16166. [DOI] [PubMed] [Google Scholar]
- [92].Vgontzas AN, Fernandez-Mendoza J, Liao D, Bixler EO. Insomnia with objective short sleep duration: the most biologically severe phenotype of the disorder. Sleep Med Rev 2013;17:241–54. https://doi.org/10.1016/j.smrv.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Ellis JG, Gehrman P, Espie CA, Riemann D, Perlis ML. Acute insomnia: current conceptualizations and future directions. Sleep Med Rev 2012;16: 5–14. 10.1016/j.smrv.2011.02.002. [DOI] [PubMed] [Google Scholar]
- [94].Ellis JG, Perlis ML, Neale LF, Espie CA, Bastien CH. The natural history of insomnia: focus on prevalence and incidence of acute insomnia. J Psychiatr Res 2012;46:1278–85. 10.1016/j.jpsychires.2012.07.001. [DOI] [PubMed] [Google Scholar]
- [95].Spath-Schwalbe E, Gofferje M, Kern W, Born J, Fehm HL. Sleep disruption alters nocturnal ACTH and cortisol secretory patterns. Biol Psychiatr 1991;29:575–84. 10.1016/0006-3223(91)90093-2. [DOI] [PubMed] [Google Scholar]
- [96].Buysse DJD, Germain A, Hall M, Monk T. A neurobiological model of insomnia. Drug Discov Today 2012:1–15. 10.1016/j.ddmod.2011.07.002.A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Bhake RC, Leendertz J a, Linthorst a CE, Lightman SL. Automated 24-hours sampling of subcutaneous tissue free cortisol in humans. J Med Eng Technol 2013;37:180–4. 10.3109/03091902.2013.773096. [DOI] [PubMed] [Google Scholar]
- [98].Henley DE, Leendertz JA, Russell GM, Wood SA, Taheri S, Woltersdorf WW, et al. Development of an automated blood sampling system for use in humans. J Med Eng Technol 2009;333:309–1902. 10.1080/03091900802185970. [DOI] [PubMed] [Google Scholar]
- [99].Faghih RT, Dahleh MA, Adler GK, Klerman EB, Brown EN. Quantifying pituitary-adrenal dynamics and deconvolution of concurrent cortisol and adrenocorticotropic hormone data by compressed sensing. IEEE Trans Biomed Eng 2015;62:2379–88. https://doi.org/10.1109/TBME.2015.2427745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Richardson G Human physiological models of insomnia. Sleep Med 2007;8:9–14. 10.1016/j.sleep.2007.04.011. [DOI] [PubMed] [Google Scholar]
- [101].Perlis ML, Smith MT, Pigeon WR. Etiology and pathophysiology of insomnia. Princ Pract Sleep Med 2005:714–25. 10.1016/B0-72-160797-7/50067-7. [DOI] [Google Scholar]
- [102].Johns MW, Gay TJ, Masterton JP, Bruce DW. Relationship between sleep habits, adrenocortical activity and personality. Psychosom Med 1971;33: 499–508. [DOI] [PubMed] [Google Scholar]
- [103].Adam K, Tomeny M, Oswald I. Physiological and psychological differences between good and poor sleepers. J Psychiatr Res 1986;20:301–16. 10.1016/0022-3956(86)90033-6. [DOI] [PubMed] [Google Scholar]
- [104].Rodenbeck A, Cohrs S, Jordan W, Huether G, Rüther E, Hajak G. The sleep-improving effects of doxepin are paralleled by a normalized plasma cortisol secretion in primary insomnia: a placebo-controlled, double-blind, randomized, cross-over study followed by an open treatment over 3 weeks. Psychopharmacology (Berl) 2003;170:423–8. 10.1007/s00213-003-1565-0. [DOI] [PubMed] [Google Scholar]
- [105].Zhang J, Ma R, Kong A, So W, Li A, Lam S, et al. Relationship of sleep quantity and quality with 24-hour urinary catecholamines and salivary awakening cortisol in healthy middle-aged adults. Sleep 2011;34:225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]